
Abstract
COVID-19 presents with a wide range of severity, from asymptomatic in some individuals to fatal in others. Based on a study of 1,051,032 23andMe research participants, we report genetic and nongenetic associations with testing positive for SARS-CoV-2, respiratory symptoms and hospitalization. Using trans-ancestry genome-wide association studies, we identified a strong association between blood type and COVID-19 diagnosis, as well as a gene-rich locus on chromosome 3p21.31 that is more strongly associated with outcome severity. Hospitalization risk factors include advancing age, male sex, obesity, lower socioeconomic status, non-European ancestry and preexisting cardiometabolic conditions. While non-European ancestry was a significant risk factor for hospitalization after adjusting for sociodemographics and preexisting health conditions, we did not find evidence that these two primary genetic associations explain risk differences between populations for severe COVID-19 outcomes.
Similar content being viewed by others


Main
The COVID-19 pandemic has caused unprecedented disruption to modern societies throughout the world. Since the emergence of the disease, it has become clear that the course of the disease can vary considerably between individuals1, with some experiencing mild or nonexistent symptoms and others experiencing severe outcomes, including hospitalization or even death. It has been well documented that several host factors are correlated with disease progression, with primary risk factors including sex, age, ancestry and the presence of underlying medical conditions2.
Much less is known about the genetic basis of COVID-19 risk, both in terms of susceptibility to infection and severity of outcomes after infection. Genetics plays a role in host susceptibility to infection and disease pathogenesis in humans3. Notable examples include the protective effects of the CCR5Δ32 mutation on infection with the human immunodeficiency virus type 1 (ref. 4) and the sickle-cell-causing mutation in the HBB gene offering protection against the malaria-causing Plasmodium falciparum5. Over the past decade, genome-wide association studies (GWAS) have proved to be a useful tool for uncovering new infectious disease susceptibility loci, identifying loci associated with pathogen clearance or persistence and providing supporting evidence for the role of certain host factors implicated in disease progression and severity6,7.
Given the rapid emergence of COVID-19, preexisting genetic cohorts offer a path to rapid data collection that can address questions surrounding the relationship between host genetics and COVID-19 in a timely fashion. Among the largest preexisting genetic cohorts are those that have been developed via direct-to-consumer genetic testing. 23andMe is a direct-to-consumer genetic testing company with over 10 million genotyped customers. As part of the 23andMe service, customers are genotyped on SNP microarrays and offered the opportunity to participate in scientific research; approximately 80% of customers consent to do so. In general, research participation is conducted via online surveys, which research participants can complete at any time. Research participants are recontactable and can be invited to participate in new surveys that are developed over time.
In this article, we describe the engagement of the 23andMe research cohort to address questions surrounding COVID-19 risk factors and host genetics. Having collected data from over 1 million research participants, we identified 15,434 individuals who reported a positive COVID-19 test, of whom 1,131 reported hospitalization with COVID-19 symptoms. We first investigated nongenetic risk factors associated with COVID-19 severity and found that lower socioeconomic status, African American ancestry, obesity and preexisting conditions were associated with a higher risk of hospitalization. We subsequently conducted GWAS of phenotypes related to both COVID-19 diagnosis and severity. We performed GWAS separately in samples of European, Latino and African American ancestries and used the resulting data to perform a trans-ancestry meta-analysis. We identified a strong association with the ABO gene, which appears to be connected with testing positive for SARS-CoV-2, and another strong association within a gene-rich locus at chromosome 3p21.31, which appears to be connected with COVID-19 severity.
Results
Respondent characteristics
Due to the geographically localized nature of the COVID-19 outbreak during the study period, we geo-targeted the email recruitment campaign to follow the outbreak as it moved through the United States (Fig. 1). As of 25 July 2020, 1,051,032 research participants had taken the COVID-19 baseline survey. Respondents were included in this analysis if they had consented to research and had a non-missing response to the question, ‘Have you been tested for COVID-19?’. Of those, 15,434 self-reported a positive SARS-CoV-2 test result. Of that group, 1,131 reported hospitalization (Table 1). Most respondents were currently based in the United States (93.2%), followed by the United Kingdom (2.4%), with the remainder responding from other countries around the world (4.4%). Most respondents were of European ancestry (80.3%), although the study also included substantial representation from Latino (11.3%, n = 118,787) and African American or Black (2.7%, n = 28,592; hereafter referred to as African American) ancestries. Study participants were 63% female with a median age of 51 years.

Email recruitment was conducted at the indicated time points for the states highlighted in red.
Those reporting a positive SARS-CoV-2 test were more likely to be male (odds ratio (OR) = 1.22, 95% confidence interval (CI) 1.18–1.26, P < 2.2 × 10−16), younger on average (43.0 years versus 51.0 years, P < 0.001) and less likely to be of European ancestry (70.3% compared to 80.3% of all study participants, P < 0.001; Table 1). Living in an urban environment was associated with a higher likelihood of reporting a positive test (within the test positive population, 95% were urban residents versus 90.5% of all respondents; P < 0.001) as was employment as a healthcare professional (within the test positive population, 21.7% were healthcare professionals versus 9.2% of all respondents, P < 0.001).
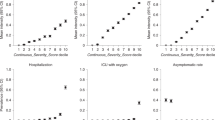
In addition to being more likely to report a positive SARS-CoV-2 test (1.7% versus 1.4%, P < 2.2 × 10−16; chi-squared test), male respondents were more likely to report hospitalization (10.1% of males versus 7.4% of females with a positive test, P = 4.3 × 10−8; chi-squared test). While the proportion of individuals reporting a positive SARS-CoV-2 test declined as a function of age, hospitalization rates increased dramatically with age (Fig. 2a). Generally, non-European ancestry was associated with higher rates of self-reported SARS-CoV-2 infection and higher proportions of hospitalization. For Latinos, the higher proportion of hospitalization was consistent with a higher proportion of individuals reporting a positive SARS-CoV-2 test compared to other groups (observed to expected ratio (O/E) = 0.93, P = 0.27; chi-squared test). However, for African Americans, the proportion reporting hospitalization was almost twice as high as expected from the proportion reporting a positive SARS-CoV-2 test (O/E = 1.96, P = 3 × 10−11; chi-squared test), implying either more severe outcomes for those who became infected or an underreporting of positive test status (Fig. 2b).

a, Age distribution of those reporting a positive SARS-CoV-2 test result (light gray) or hospitalization with COVID-19 (dark gray). b, Age-standardized percentage of COVID-19 cases reporting hospitalization by ancestry group.
Combining common risk factors into a single multivariable logistic regression model (obesity, type 2 diabetes, fatty liver disease and high blood pressure), the most significant risk factor for hospitalization within the population reporting a positive test for COVID-19 was obesity (defined as BMI > 30), which accounted for a doubling in the risk of hospitalization (adjusted OR = 2.07, 95% CI 1.67–2.57) after adjusting for age, sex, ancestry, education, household income and other cardiometabolic preexisting conditions (Table 2). In this model, African Americans were 82% more likely to be hospitalized for COVID-19 (adjusted OR = 1.82, 95% CI 1.33–2.50). Socioeconomic status was inversely associated with hospitalization risk, with a 4% decrease in hospitalization per US$10,000 increase in median income in the zip code of residence. High school or lower education conferred a 38% increased risk in hospitalization (adjusted OR = 1.38, 95% CI 1.10–1.74). Results were robust to stratification on the basis of data collection date range as well as stratification on the basis of geographical region (Supplementary Table 1).
The prevalence of infection, estimated as the number of reported positive tests in a state relative to the number of study participants in the state, varied across geographical regions. The highest proportions of positive SARS-CoV-2 tests were reported in New York (4.4%) and New Jersey (3.3%) and the lowest proportions were reported in Maine (0.4%) and West Virginia (0.4%). Since most of the case data were collected between late April and early June 2020, hotspots that developed earlier in the pandemic are better represented compared to those that arose later in the course of the pandemic (Extended Data Fig. 1a,b). Nonetheless, the self-reported prevalence of positive SARS-CoV-2 tests at the US state level was reasonably well correlated with the number of positive SARS-CoV-2 tests reported per capita8 as of July 2020 (Extended Data Fig. 1c; Pearson r = 0.85). However, the prevalence of self-reported positive SARS-CoV-2 test status was higher in the 23andMe database than the per-capita estimates, likely reflecting differences in the composition of the 23andMe database and the general population and potential selection bias arising from individuals with a positive test potentially being more likely to choose to participate in the study.
GWAS
Our GWAS analyses included one phenotype aimed at understanding SARS-CoV-2 infection and four phenotypes aimed at understanding aspects of COVID-19 severity. For the ‘diagnosis’ phenotype contrasting test-positive and test-negative individuals, our trans-ancestry meta-analysis included 12,972 cases and 101,268 controls. This analysis identified two loci with robust associations (Fig. 3a and Supplementary Table 2). The first locus is located in proximity to the ABO gene and the second association is within a region of chromosome 3p21.31. Conversely, the 4 ‘severity’ phenotypes included between 636 and 1,447 cases and between 796,151 and 797,180 controls, depending on the exact phenotype used (Supplementary Table 3). The chromosome 3p21.31 locus appeared to be robustly associated at genome-wide significance across all phenotypes, whereas the ABO locus only achieved genome-wide significance in the test-positive versus test-negative phenotype (Fig. 3b, Extended Data Fig. 2 and Supplementary Table 2).

a, The ‘test-positive versus test-negative’ phenotype. b, The ‘severe respiratory symptoms’ phenotype. SNPs achieving genome-wide significance are highlighted in red. The nearest gene to each index SNP is indicated above the relevant association peaks. While the nearest gene to the index SNP at the chromosome 3 locus differs between the two panels, this likely represents statistical fluctuations between SNPs in tight LD rather than differences in association signal (Fig. 6).
ABO blood group
In our phenotype contrasting SARS-CoV-2 test-positive and test-negative individuals, we identified an association at chromosome 9q34.2, with the index SNP rs9411378 (P = 5.3 × 10−20, C allele OR = 0.857; Fig. 4a and Extended Data Fig. 3a). While the precision of effect size estimates varied between populations, we observed no evidence of effect size heterogeneity between populations (Fig. 4b and Supplementary Table 2). This index SNP is in linkage disequilibrium (LD) with a functional variant in the ABO gene, specifically rs8176719 (r2 = 0.57, 0.57 and 0.40 in the European, Latino and African American populations, respectively), which is a well-known single-nucleotide deletion that usually confers a type O blood group when present in the homozygous form. While multiple rare variants elsewhere within the ABO gene can contribute to blood group determination, individuals heterozygous for the deletion are most likely to have blood groups A or B, whereas individuals without any copies of the deletion are most likely to have blood groups A, B or AB.

a, Regional plot around the ABO locus. The colors indicate the strength of LD relative to the index SNP (rs9411378). Imputed variants are indicated with ‘+’ symbols or ‘×’ symbols for coding variants. Where imputed variants were not available, directly genotyped variants are indicated by ‘o’ symbols or diamond symbols for coding variants. b, ORs and allele frequencies for the rs9411378 index SNP compared across populations. The error bars indicate the 95% CIs.
To further understand the relationship between positive SARS-CoV-2 test status and ABO blood group, we used genetically determined blood group assignments (Methods) and estimated the contribution to risk by comparing each blood group against each of the others. For all COVID-19 phenotypes, we found that the O blood group was protective compared to the other blood groups, whereas blood groups A, B and AB did not differ from each other (Fig. 5, Extended Data Fig. 4 and Supplementary Table 4). Direction of effect was broadly consistent across populations (Extended Data Fig. 5). We observed a similar effect size in both diagnosis and severity phenotypes, albeit with severity phenotypes not achieving statistical significance. However, the effect size estimates at the ABO locus with the severity phenotypes appeared somewhat sensitive to choice of control population and were attenuated when contrasting individuals with severe outcomes to test-positive individuals without severe outcomes (Supplementary Note and Extended Data Fig. 6), which is consistent with a role in susceptibility rather than severity. We also noted that the association with SARS-CoV-2 test status was in contrast to the ABO results obtained when considering individuals that reported influenza symptoms in the years before the COVID-19 pandemic, where the O blood group appeared to be a risk factor.

The maximum likelihood point estimates are indicated by square points, with statistically significant associations (P < 0.001; likelihood ratio test) highlighted in red. The error bars indicate the 95% CIs. The AB blood group did not show differentiation from groups A or B (Extended Data Fig. 4).

a, Regional plot around the chromosome 3 locus. The colors indicate the strength of LD relative to the index SNP (rs13078854). The imputed variants are indicated with ‘+’ symbols or ‘×’ symbols for coding variants. Where imputed variants were not available, directly genotyped variants are indicated by ‘o’ symbols or diamond symbols for coding variants. b, ORs and allele frequencies for the rs13078854 index SNP compared across populations. The error bars indicate the 95% CIs.
To understand how the association differentiated by test-positive or test-negative status, we tested rs9411378 for association contrasting test-positive individuals to individuals who did not report having taken a SARS-CoV-2 test and repeated this analysis for test-negative individuals. We found that association remained strong for the test-positive contrast (P = 1.91 × 10−13, OR = 0.878) but was much reduced in the test-negative contrast (P = 0.002, OR = 1.093). This suggests that the association at ABO is likely driven by differentiation between the test-positive population and the general population, rather than the test-negative population.
Given preliminary reports that the ABO locus has not been observed as being associated in other studies of COVID-19 host genetics, we investigated the robustness of our association to population stratification (Supplementary Note). We did not observe evidence for heterogeneity of effect across geographical regions or ancestral backgrounds (Extended Data Fig. 7).
Preliminary reports suggested that the rhesus factor can also contribute to differences in susceptibility and severity9. We did not detect a genetic association at the RHD locus, which suggests that rhesus factor is not a major risk factor by itself independent of ABO blood group. To investigate further, we also compared positive and negative forms of each ABO blood group and detected no significant difference in any comparison (Supplementary Table 5).
Chromosome 3p21.31
We identified an association at chromosome 3p21.31, which was shared across all phenotypes (Fig. 6a and Extended Data Figs. 3b and 8). The association was strongest in our phenotypes related to respiratory symptoms, with the lowest P value observed in the severe respiratory symptoms phenotype (index SNP rs13078854, alleles A/G, P = 1.6 × 10−18) and with a relatively large estimated effect size (G allele OR = 0.592, 95% CI 0.527–0.665). Most of the support for this association comes from the European population (Fig. 6b), likely reflecting the larger sample size for this cohort, but we identified no evidence of effect size heterogeneity between populations (Supplementary Table 2). However, the risk allele is also more common in the European population, with the rs13078854 A allele having a 7.8%, 5.8% and 2.7% frequency in the European, Latino and African American populations, respectively. As for the ABO locus, we did not observe evidence for heterogeneity of effect across geographical regions or ancestral backgrounds (Extended Data Fig. 9).
The credible set for this locus overlaps the LZTFL1 gene, although none of the variants in the credible set alter the resulting protein. The locus also contains other nearby genes that could plausibly be driving the association, including SLC6A20, CCR9, FYCO1, CXCR6 and XCR1.
Given the reported differences in outcome severity between males and females, we tested for a difference in effect at the chromosome 3p21.31 locus. Testing rs13078854 separately in males and females for the severe respiratory symptoms phenotype gave an OR = 0.49 (95% CI 0.41–0.59) in males and OR = 0.69 (95% CI 0.58–0.82) in females, with the difference being moderately significant (P = 0.003; z-test). On the basis of the association between ABO and SARS-CoV-2 test status, we further hypothesized that the chromosome 3p21.31 locus may show a difference in effect size depending on ABO blood type. Conditioning on blood type O, we estimated the OR of rs13078854 to be 0.63 (95% CI 0.52–0.77), whereas conditioning on any other blood type gave an OR of 0.57 (95% CI 0.49–0.66). Therefore, we conclude that ABO blood type does not modulate the effect at the chromosome 3p21.31 locus (P = 0.80; z-test).
Other associations
In addition to the two main associations, we observed five weaker associations that, while achieving genome-wide significance, typically only included a small number of low-frequency variants within the association peak and may represent false positive associations that should likely only be considered further on independent validation (Supplementary Table 2).
Discussion
The COVID-19 pandemic represents a unique emergency in recent human history and has dramatically accelerated the pace of scientific investigation into the effects of the virus on human health. In this article, we utilized a direct-to-consumer research platform to collect data regarding experiences of COVID-19 on a large scale and over a compressed timeline.
In this study, both Latino and African American groups reported a higher rate of SARS-CoV-2 infection and, as described elsewhere2,10,11, were more likely to report hospitalization relative to Europeans. For Latino respondents, the higher rate of hospitalization in our data was broadly consistent with a higher rate of infection. However, for African Americans, the risk of hospitalization was higher than expected relative to the infection rate and remained so after adjusting for socioeconomic characteristics (income and education), age, sex and other comorbidities. These results highlight the central importance of systemic social factors in altering the risk of severe outcomes within minority communities.
Our study adds to the growing body of literature around the role of host genetics in COVID-19 and the data from this study provide validation for a number of associations that have been reported elsewhere (Supplementary Tables 6 and 7). In particular, our data strengthen the evidence for a role for ABO in COVID-19 host genetics. The ABO blood group has been reported as a risk factor for both COVID-19 susceptibility12 and severity13 and is notable given the reported links between COVID-19 and blood clotting complications14,15. Our data support a role for ABO in susceptibility to infection, suggesting that blood type O is protective unlike non-O blood types. Whereas previous reports suggested protection was limited to the rhesus-positive group9, our data do not support that conclusion.
The mechanism by which ABO is associated with COVID-19 is unclear but ABO blood groups can play a direct role in pathogen infection by serving as receptors and/or coreceptors16. SARS-CoV-2 is an enveloped virus that carries ABO antigens on the viral spike glycoprotein and host envelope glycolipids. Recent work has shown the SARS-CoV-2 spike protein interacts with multiple host C-type lectin receptors in a glycosylation-dependent manner17,18, which is similar to previous work on the SARS-CoV virus from the earlier severe acute respiratory syndrome outbreak19,20. Differential glycosylation of the spike protein or envelope glycolipids from expression of different ABO glycosyltransferases may then impact the binding and propagation of SARS-CoV-2 viral particles in the host. Others have speculated that the lower susceptibility of blood type O could be linked to anti-A blood antibodies inhibiting the adhesion of coronavirus to angiotensin converting enzyme 2 (ACE2)-expressing cells, thereby providing protection21.
The ABO locus is also highly pleiotropic22 and exhibits complex population structure23. Interestingly, while older literature regarding the association between ABO and influenza is inconsistent24, our own data suggest that blood type O is actually a risk factor for seasonal flu. This is notable because SARS-CoV-2 testing in the United States was largely restricted to individuals with flu-like symptoms at the time we were collecting most of our data. As such, it is possible that the population of individuals receiving SARS-CoV-2 tests was enriched for influenza cases and the apparent protective nature of ABO for COVID-19 could arise from a subtle form of collider bias.
Likewise, our data strengthen the evidence of association at the chromosome 3p21.31 gene cluster, first identified by Ellinghaus et al.13. The locus contains multiple genes (SLC6A20, LZFTL1, CCR9, CXCR6, XCR1, FYCO1) that could be functionally implicated in COVID-19 pathology. In particular, SLC6A20 has been noted13 as potentially forming a complex with ACE2, the cell surface receptor for SARS-CoV-2 viral entry25,26. It is possible that increased SLC6A20 expression leads to increased ACE2 protein levels and greater viral uptake. LZFTL1 has been implicated in ciliogenesis and intracellular trafficking of ciliary proteins, which may impact airway epithelial cell function. As noted elsewhere13, CXCR6 promotes natural killer T cell and tissue-resident memory CD8+ T cells residence in the lung27 and plays a role in the trafficking of T lymphocytes to the bronchial epithelia during respiratory infection and inflammatory lung disease. CCR9 predominantly regulates T cell homing to the gut, which may indirectly impact the response in the lung; however, it has also been shown to regulate eosinophil recruitment to the lung28. Recent studies have identified elevated chemokines and eosinophilia as hallmarks of severe disease29,30,31 but additional work is required to define any functional contribution of these genes to the genetic association with COVID-19.
In phenotypes contrasting individuals with severe COVID-19 symptoms to controls without a COVID-19 diagnosis, the risk variants at the chromosome 3p21.31 locus achieved ORs of approximately 2.0 in our data, which is relatively large in the context of GWAS studies. Given that risk alleles are also relatively common (approximately 3–8% frequency, depending on the population), it is likely that this locus makes a meaningful contribution to determining why some individuals experience severe COVID-19 outcomes. However, while the population sample sizes in our study differ considerably, we found little evidence to suggest that allele frequency differences at this locus could account for the higher rate of severe outcomes from COVID-19 for non-European ancestry groups. In fact, the primary risk allele at the chromosome 3p21.31 locus is most common in European populations and less common in Latino and African American populations.
In under four months, over one million research participants contributed to this study of a new disease via online surveys. However, while this represents a large sample, there are notable caveats to relying on self-reported data for a disease with lethal outcomes. First, cases identified in this study were healthy enough to respond to the survey and therefore likely biased toward a healthier case population than otherwise exists. Likewise, survey data cannot easily ascertain information regarding exposure, which limits our ability to analyze true virus susceptibility. In addition, 23andMe research participants are a self-selected group and may not reflect the general population. Furthermore, the scarcity of testing in the United States during our data collection period likely further obscured the true picture of SARS-CoV-2 infections, potentially resulting in misestimation of risk factors compared to a sample randomly drawn from the broader population with perfect case and control classification.
Methods
Overview of study recruitment and data collection
Participants in this study were recruited from the customer base of 23andMe, a personal genetics company. All individuals included in the analyses provided informed consent and answered surveys online according to our human participants research protocol, which was reviewed and approved by Ethical and Independent Review Services, a private institutional review board (http://www.eandireview.com).
Primary recruitment was carried out by email to approximately 6.7 million 23andMe research participants over 18 years of age and living in the United States or United Kingdom. Additionally, preexisting customers were invited to participate in the study through promotional materials on the 23andMe website, the 23andMe mobile application and via social media. Study participation consisted solely of web-based surveys, including an initial baseline survey and three follow-up surveys fielded one month after completion of the baseline survey. Because enrollment is ongoing, not all participants would have received or completed all of the follow-up surveys. All surveys included questions about symptoms of cold or flu-like illnesses from February 2020 onward, COVID-19 diagnosis and testing, hospitalization, severity of illness, COVID-19 diagnosis of first- and second-degree family members and potential sources of exposure to COVID-19. Other respondent characteristics, such as age, sex, preexisting conditions, educational attainment, zip code and smoking status had been collected via previously deployed surveys for most participants but were also queried in the COVID-19 baseline survey if the data were missing. All questions asked within the surveys are reported in the Supplementary Note.
We geo-targeted the email recruitment campaign to follow the outbreak as it moved through the United States. Emails to each state/country were batched into tranches on the basis of the anticipated timing of the hospitalization demand peak within each region as assessed from the Institute for Health Metrics and Evaluation (IHME) prediction model32. Each tranche was recruited via email a minimum of two weeks after the estimated peak hospitalization demand, as determined by the IHME predictive models. IHME predictions varied over the course of the study; the order in which regions were targeted was adjusted accordingly. The email send dates are detailed in Supplementary Table 8.
Social and demographic risk factors for COVID-19 hospitalization
To explore nongenetic factors associated with hospitalization for COVID-19, we explored age, sex, ancestry, median household income of the residential zip code, educational attainment, body mass index (BMI) and preexisting conditions as risk factors within US respondents using bivariate and multivariate logistic regression models. Ancestry was inferred via a previously described genetic ancestry classification algorithm33,34. Since social and demographic factors are strongly associated with COVID-19 hospitalization and many of the preexisting conditions, final models estimating the relationship between the preexisting condition and COVID-19 hospitalization were adjusted for age (10-year increments), sex, ancestry, high school or lower education and median household income of the residential zip code (in US$10,000 increments). Covariates explored but not found to be significant in the final model included alcohol intake, urban/rural residence, red meat intake and current smoking.
A final multivariate logistic regression model was specified to quantify the risk of hospitalization for COVID-19 within the SARS-CoV-2 test-positive population. Individuals with missing data were excluded. To ensure that the associations observed in the model were robust to variation arising from regional differences, we also fitted the hospitalization model having stratified the data into that collected during the first and second phases of the study period (April and May versus June and July) and separately by stratifying on the basis of US region (south, west, north central, and northeast).
The relationship between age and COVID-19 and hospitalization was determined by categorizing cases by 10-year age increments between 30 and 80 and then calculating the percentage of cases in each age group and the percentage of cases that reported hospitalization. To describe differences in hospitalization by ancestry, age-standardized estimates were calculated by applying the percentage of cases hospitalized within the age strata for European, African American and Latino respondents and applying that percentage to the age structure of all cases in the study population. This resulted in an age-adjusted estimate of COVID-19-related hospitalization reflective of this study population, rather than the broader US population. While our data also included respondents of other ancestries, such as East and South Asian, the sample sizes for these populations were too small for robust inferences to be made. All statistical analyses and mapping were conducted in R v.3.3.
Phenotype definitions for GWAS
Using the information derived from the surveys, we defined a set of phenotypes that aimed to capture aspects of COVID-19 diagnosis and severity. After preliminary analyses, we defined one ‘diagnosis’ phenotype that contrasted positive and negative outcomes from a SARS-CoV-2 test and four phenotypes that captured aspects of COVID-19 ‘severity’, and compared individuals experiencing pneumonia, hospitalization or the need for respiratory support in the form of supplementary oxygen or ventilation to individuals without a COVID-19 diagnosis (Supplementary Table 3). Our phenotype definitions were motivated from data collected in the early weeks of the study, when we performed preliminary analysis to define phenotypes that appeared best powered within a GWAS context. From these preliminary analyses, we concluded that cases were best defined by requiring a positive SARS-CoV-2 test, which reduced the probability of false positive cases (for example, from influenza) arising from symptom-based diagnoses alone. Likewise, we determined that large-scale population-based controls (who had neither been diagnosed with COVID-19 nor tested positive for SARS-CoV-2) performed well within the GWAS context for severity phenotypes, which we believe is justified because most individuals within the population are not expected to have severe outcomes and more sophisticated control definitions relying on exposure information were unlikely to be accurately captured from self-reported survey information. Given that data collection occurred early in the pandemic, it may be assumed that the population controls represent a largely unexposed population. Full derivations of phenotypes from the original survey questions are given in the Supplementary Note.
Genotyping and SNP imputation
DNA extraction and genotyping were performed on saliva samples by Clinical Laboratory Improvement Amendments-certified and College of American Pathologists-accredited clinical laboratories of Laboratory Corporation of America. Samples were genotyped on one of five genotyping platforms. The V1 and V2 platforms were variants of the Illumina HumanHap550 BeadChip and contained a total of about 560,000 SNPs, including about 25,000 custom SNPs selected by 23andMe. The V3 platform was based on the Illumina OmniExpress BeadChip and contained a total of about 950,000 SNPs and custom content to improve the overlap with our V2 array. The V4 platform was a fully custom array of about 950,000 SNPs and included a lower redundancy subset of V2 and V3 SNPs with additional coverage of lower-frequency coding variation. The V5 platform was based on the Illumina Global Screening Array, consisting of approximately 654,000 preselected SNPs and approximately 50,000 custom content variants. Samples that failed to reach a 98.5% call rate were reanalyzed. Individuals whose analyses failed repeatedly were recontacted by the 23andMe customer service to provide additional samples, as is done for all 23andMe customers.
Participant genotype data were imputed using the Haplotype Reference Consortium panel35, augmented by the Phase 3 1000 Genomes Project panel36 for variants not present in the Haplotype Reference Consortium. We phased and imputed data for each genotyping platform separately. For the non-pseudoautosomal region of the X chromosome, males and females were phased together in segments, treating males as already phased; pseudoautosomal regions were phased separately. We then imputed males and females together, treating males as homozygous pseudodiploids for the non-pseudoautosomal region.
GWAS
Genotyped participants were included in GWAS analyses on the basis of ancestry as determined by a genetic ancestry classification algorithm34. For each phenotype, we selected a set of unrelated individuals so that no two individuals shared more than 700 cM of DNA identical by descent. For case-control phenotypes, if a case and a control were identified as having at least 700 cM of DNA identical by descent, we preferentially discarded the control from the sample.
For case-control comparisons, we tested for association using logistic regression, assuming additive allelic effects. For tests using imputed data, we used the imputed dosages rather than best-guess genotypes. We included covariates for age, age squared, sex and sex:age interaction, the top ten principal components to account for residual population structure and dummy variables to account for genotyping platform. The association test P value was computed using a likelihood ratio test, which in our experience is better behaved than a Wald test on the regression coefficient. Results for the X chromosome were computed similarly, with males coded as if they were homozygous diploid for the observed allele. Association testing for the Y chromosome was considered separately (Supplementary Note).
We ran the GWAS for each phenotype separately and combined both genotyped and imputed data. When choosing between imputed and genotyped GWAS results, we favored the imputed result unless the imputed variant was unavailable or failed quality control. For imputed variants, we removed variants with low imputation quality (r2 < 0.5 averaged across batches or a minimum r2 < 0.3) or with evidence of batch effects (analysis of variance (ANOVA) F-test across batches, P < 10−50). For genotyped variants, we removed variants only present on our V1 or V2 arrays (due to small sample size) that failed a Mendelian transmission test in trios (P < 10−20), failed a Hardy–Weinberg test in Europeans (P < 10−20), failed a batch effect test (ANOVA P < 10−50) or had a call rate < 90%.
We repeated the GWAS analysis separately in each population cohort for which we had sufficient data (European, Latino, African American; Extended Data Fig. 10) and the resulting summary statistics were corrected for inflation using genomic control when the inflation factor was estimated to be greater than 1 (Supplementary Table 9). We then performed a trans-ancestry meta-analysis using a fixed effects model (inverse variance method37), restricting to variants of at least 1% minor allele frequency in the pooled sample and again corrected for inflation using genomic control where necessary. We tested associated loci for evidence of heterogeneity between populations using Cochran’s Q test.
Within each GWAS, we identified regions with genome-wide significant associations. We defined the region boundaries by identifying all SNPs with P < 10−5 within the vicinity of a genome-wide significant association and then grouping these regions into intervals so that no two regions were separated by <250 kilobases (kb). We considered the SNP with the smallest P value within each interval to be the index SNP. Within each region, we calculated a credible set using the method by Maller et al.38.
We also annotated our findings based on LD with results from published GWAS, coding variation and expression quantitative trait loci specifically by finding annotations with r2 > 0.5 and within 500 kb of the index SNP.
Blood group analyses
We classified haplotypes into blood groups on the basis of genotypes at three SNPs (rs8176747, rs41302905 and rs8176719)13,39. A deletion at rs8176719 confers a type O haplotype, as does a T allele at rs41302905. If neither rs8176719 nor rs41302905 confers type O, then rs8176747 distinguishes between types A and B. This assignment paradigm is described in Supplementary Table 10.
Given the haplotype assignments, individuals were assigned a blood type on the basis of their diploid combination of haplotypes, with type O being recessive, so that individuals with two O haplotypes were assigned type O, individuals with one O and one A haplotype were assigned type A, and so on.
We note that the blood group assignment methodology described above is incomplete and there are other rare variants that can influence blood group39. To understand the accuracy of the genetic blood group assignments, we compared them to self-reported blood groups from over 1.47 million research participants. We found that the genetic assignments achieved 86–94% precision and 63–95% recall compared to the self-reported data, depending on population and blood group (Supplementary Table 11).
We tested for the association between ABO blood group and COVID-19 phenotypes in each population using logistic regression, testing blood group pairs separately (for example, individuals with blood group O versus individuals with blood group A) and only testing unrelated individuals. We included covariates for age, age squared, sex, sex:age interaction and the top ten principal components. We meta-analyzed across populations using a fixed effects model. We repeated these analyses using self-reported blood group assignments in place of genetically determined assignments and found the results to be qualitatively similar.
We performed a similar analysis between ABO blood groups and experience of influenza by considering research participants who answered the question: Have you had influenza (flu) in the past 12 months? Common symptoms of flu are fever over 100° F (38° C), muscle aches, chills and sweats, headache, dry cough, fatigue, nasal congestion and sore throat. Compared to the common cold, symptom onset for influenza is faster, more severe and can last 1–2 weeks. To avoid overlap with individuals reporting experiences with COVID-19, we tested for association between influenza and the ABO blood groups using a sample of individuals who answered the question during either the 2017–2018 flu season, defined as starting in October 2017 and ending in September 2018, or the 2018–2019 flu season, defined as starting in October 2018 and ending in September 2019.
To test for differences between rhesus-positive and rhesus-negative blood groups, we used the structural variant esv3585521 to obtain rhesus type. This variant, located within the RHD gene, has a 39.4% frequency in European populations and associates very strongly with self-reported rhesus type in 23andMe data (OR = 22.1, P = 1.8 × 10−298). We took individuals imputed as homozygous for the deletion as being rhesus-negative. Within each blood group, we tested for an association between rhesus type and COVID-19 phenotypes in the European ancestry population using logistic regression. We included covariates for age, age squared, sex, sex:age interaction and the top ten principal components.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The full set of de-identified summary statistics can be made available to qualified investigators who enter into an agreement with 23andMe that protects participant confidentiality. Interested investigators should visit the COVID-19 Dataset Access Program at https://research.23andme.com/covid19-dataset-access/. Summary statistics for the SNPs with P < 1 × 10−4 within the trans-ancestry meta-analysis are provided as a Supplementary Data file.
References
Guan, W.-J. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720 (2020).
Williamson, E. J. et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 584, 430–436 (2020).
Chapman, S. J. & Hill, A. V. S. Human genetic susceptibility to infectious disease. Nat. Rev. Genet. 13, 175–188 (2012).
Dean, M. et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 273, 1856–1862 (1996).
Allison, A. C. Protection afforded by sickle-cell trait against subtertian malarial infection. Br. Med. J. 1, 290–294 (1954).
Tian, C. et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat. Commun. 8, 599 (2017).
Band, G. et al. Insights into malaria susceptibility using genome-wide data on 17,000 individuals from Africa, Asia and Oceania. Nat. Commun. 10, 5732 (2019).
Dong, E., Du, H. & Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 20, 533–534 (2020).
Zietz, M., Zucker, J. & Tatonetti, N. P. Associations between blood type and COVID-19 infection, intubation, and death. Nat. Commun. 11, 5761 (2020).
Rodriguez-Diaz, C. E. et al. Risk for COVID-19 infection and death among Latinos in the United States: examining heterogeneity in transmission dynamics. Ann. Epidemiol. 52, 46–53.e2 (2020).
Millett, G. A. et al. Assessing differential impacts of COVID-19 on black communities. Ann. Epidemiol. 47, 37–44 (2020).
Zhao, J. Relationship between the ABO blood group and the COVID-19 susceptibility. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa1150 (2020).
Ellinghaus, D. et al. Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med. 383, 1522–1534 (2020).
Levi, M., Thachil, J., Iba, T. & Levy, J. H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 7, e438–e440 (2020).
Rapkiewicz, A. V. et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine 24, 100434 (2020).
Cooling, L. Blood groups in infection and host susceptibility. Clin. Microbiol. Rev. 28, 801–870 (2015).
Amraie, R. et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2 and are differentially expressed in lung and kidney epithelial and endothelial cells. Preprint at bioRxiv https://doi.org/10.1101/2020.06.22.165803 (2020).
Gao, C. et al. SARS-CoV-2 spike protein interacts with multiple innate immune receptors. Preprint at bioRxiv https://doi.org/10.1101/2020.07.29.227462 (2020).
Valverde, P. et al. Molecular insights into DC-SIGN binding to self-antigens: the interaction with the blood group A/B antigens. ACS Chem. Biol. 14, 1660–1671 (2019).
Jeffers, S. A. et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl Acad. Sci. USA 101, 15748–15753 (2004).
Guillon, P. et al. Inhibition of the interaction between the SARS-CoV spike protein and its cellular receptor by anti-histo-blood group antibodies. Glycobiology 18, 1085–1093 (2008).
Chesmore, K., Bartlett, J. & Williams, S. M. The ubiquity of pleiotropy in human disease. Hum. Genet. 137, 39–44 (2018).
Mourant, A. E., Kopec, A. C. & Domaniewska-Sobczak, K. in The Distribution of the Human Blood Groups, and Other Polymorphisms (Oxford Univ. Press, 1976).
Horby, P., Nguyen, N. Y., Dunstan, S. J. & Baillie, J. K. The role of host genetics in susceptibility to influenza: a systematic review. PLoS ONE 7, e33180 (2012).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020).
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Wein, A. N. et al. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med. 216, 2748–2762 (2019).
López-Pacheco, C., Soldevila, G., Du Pont, G., Hernández-Pando, R. & García-Zepeda, E. A. CCR9 is a key regulator of early phases of allergic airway inflammation. Mediators Inflamm. 2016, 3635809 (2016).
Lucas, C. et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469 (2020).
Mathew, D. et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 369, eabc8511 (2020).
Zhang, X. et al. Viral and host factors related to the clinical outcome of COVID-19. Nature 583, 437–440 (2020).
COVID-19 Projections (IHME, 2020); https://covid19.healthdata.org/projections
Bryc, K., Durand, E. Y., Macpherson, J. M., Reich, D. & Mountain, J. L. The genetic ancestry of African Americans, Latinos, and European Americans across the United States. Am. J. Hum. Genet. 96, 37–53 (2015).
Durand, E. Y., Do, C. B., Mountain, J. L. & Macpherson, J. M. Ancestry composition: a novel, efficient pipeline for ancestry deconvolution. Preprint at bioRxiv https://doi.org/10.1101/010512 (2014).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Maller, J. B. et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat. Genet. 44, 1294–1301 (2012).
Bugert, P., Rink, G., Kemp, K. & Klüter, H. Blood group ABO genotyping in paternity testing. Transfus. Med. Hemother. 39, 182–186 (2012).
Acknowledgements
We thank the 23andMe research participants who made this study possible. We thank A. Ewing, A. Petrakovitz, A. Park, A. Silk, A. Collins, B. Macintosh, C. Kao, C. Ball, C. Pai, D. Hinds, D. Parry, E. Ratcliff, E. Bullis, E. Hall, F. Alam, J. Haggarty, J. Christenson, J. Lawrence, J. Chau, J. Shaw, J. Cackler, K. Heilbron, K. Kukar, K. Watson, M. Frendo, O. Valenti, R. Workman, R. Lopatin, R. Bell, R. Eckert, S. Rodgers, S. Rys, S. Averbeck, S. Fuller, V. Lane and Y. Jiang for contributions and insights. We also thank the 23andMe research team: B. Hicks, C. Tian, D. Dhamija, E. Babalola, E. S. Noblin, E. M. Jewett, G. D. Poznik, G. Cuellar Partida, J. O’Connell, J. Shi, J. L. Mountain, J. Y. Tung, K. Bryc, K. E. Huber, K.-H. Lin, K. F. McManus, K. Fletez-Brant, M. K. Luff, M. H. McIntyre, M. Lowe, M. E. Moreno, P. Wilton, P. Fontanillas, P. Nandakumar, S. V. Mozaffari, S. L. Elson, S. Das, S. J. Micheletti, S. Shringarpure, V. Tran, W. Wang, W. Freyman and X. Wang.
Author information
Authors and Affiliations
Consortia
Contributions
J.F.S., A.J.S., C.H.W., C.Y., S.A. and A.A. designed the study. J.F.S. developed the survey. J.F.S., A.J.S., C.H.W., C.Y., S.A. and A.A. developed the recruitment and participant engagement strategy. The 23andMe COVID-19 team acquired and processed the data. J.F.S., C.H.W., D.C., T.F.-S. and A.A. analyzed the data. J.F.S., A.J.S., C.H.W., S.A., A.S., J.E.-G. and A.A. interpreted the data. J.F.S., A.J.S., A.S. and A.A. wrote the first draft of the manuscript. All authors participated in preparing the manuscript by reading and commenting on drafts before submission.
Corresponding author
Ethics declarations
Competing interests
J.F.S., A.J.S., C.Y., C.H.W., T.F.-S., D.C., A.S., S.A. and A.A. are current or former employees of 23andMe and hold stock or stock options in 23andMe. J.E.-G. is an employee of GlaxoSmithKline and owns company stock.
Additional information
Peer review information Nature Genetics thanks Jeffrey Barrett and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 SARS-CoV-2 prevalence in the 23andMe study and comparison to nationally reported data.
a, Proportion of SARS-CoV-2 test positive study respondents in the continental United States combining all reported cases from February-July 2020. The map has been smoothed using a weighted local sum for each county combining data from 15 neighboring counties and weighting by sample size. b, Unsmoothed estimated prevalence of COVID-19 test positive individuals on a per-county basis within the continental United States. c, Scatter plot of state-level COVID-19 test positive prevalence as assessed in the 23andMe database compared to that obtained from national statistics. The blue line denotes a line of best fit, with the greyed area indicating the 95% confidence band.
Extended Data Fig. 2 Manhattan and QQ plots from the trans-ethnic meta-analysis for three phenotypes.
a, The ‘hospitalized’ phenotype. b, The ‘respiratory support’ phenotype. c, The ‘pneumonia’ phenotype. The nearest gene to each index SNP is indicated above each association peak. SNPs achieving genome-wide significance are highlighted in red.
Extended Data Fig. 3 Regional plots of association at the ABO and chr3 loci for each population.
a,b, Regional plots within each population around the ABO locus within the ‘test positive vs. negative’ phenotype (a) and the chr3 locus within the ‘severe respiratory symptoms’ phenotype (b). Colors indicate strength of linkage disequilibrium relative to the index SNP from the trans-ethnic meta-analysis. Imputed variants are indicated with ‘+’ symbols or ‘x’ symbols for coding variants. Where imputed variants were not available, directly genotyped variants are indicated by ‘o’ symbols or diamond symbols for coding variants.
Extended Data Fig. 4 Comparison of AB blood group to other ABO blood groups in the trans-ethnic meta-analysis.
Maximum likelihood point estimates are indicated by square points, with statistically significant associations (P < 0.001; likelihood ratio test) highlighted in red.
Extended Data Fig. 5 Comparison of blood groups across phenotypes and across populations (European, African American, and Latino).
Maximum likelihood point estimates are indicated by square points, with statistically significant associations (P < 0.001; likelihood ratio test) highlighted in red. Note that the estimate for the O vs. A comparison within the ‘respiratory support’ phenotype and African American population did not converge and was therefore excluded.
Extended Data Fig. 6 Effect size estimates at the chr3 and ABO loci within the severity phenotypes with different control definitions.
For each phenotype, we estimated the effect size using both the original control definition (phenotypes with the _dx_negative_controls suffix), and again using controls that report a SARS-CoV-2 positive test but not a severe outcome (phenotypes with the _test_positive_controls suffix). Maximum likelihood point estimates are indicated by square points, with error bars indicating 95% confidence intervals.
Extended Data Fig. 7 Comparison of ABO index SNP effect sizes within different strata.
a-c, The index SNP (rs9411378) effect sizes in the ‘test positive vs. negative’ phenotype as estimated within European ancestry groups (a), US states (b), and US regions (c). For all plots, points indicate the maximum likelihood estimate, error bars indicate 95% confidence intervals, and numbers within brackets indicate the numbers of cases and controls within the given group.
Extended Data Fig. 8 Forest plot of chr3p21.31 index SNP (rs13078854) in the trans-ethnic analysis and for each population (European, Latino, and African American).
Square points indicate the fixed effect estimate from the meta-analysis, with error bars indicating 95% confidence intervals, and genome-wide significant associations highlighted in red.
Extended Data Fig. 9 Comparison of chr3p21.31 index SNP effect sizes within different strata.
a,b, The index SNP (rs13078854) effect sizes in the ‘severe respiratory symptoms’ phenotype as estimated within US states (a) and US regions (b). For all plots, points indicate the maximum likelihood estimate, error bars indicate 95% confidence intervals, and numbers within brackets indicate the numbers of cases and controls within the given group.
Extended Data Fig. 10 Manhattan plots for all phenotypes and each population prior to trans-ethnic meta-analysis.
The nearest gene to each index SNP is indicated above each association peak. SNPs achieving genome-wide significance are highlighted in red.
Supplementary information
Supplementary Information
Supplementary Note and Tables 1–11
Supplementary Data 1
Summary statistics for SNPs with P < 1 × 10−4 within the trans-ethnic meta-analysis.
Rights and permissions
About this article

Cite this article
Shelton, J.F., Shastri, A.J., Ye, C. et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat Genet 53, 801–808 (2021). https://doi.org/10.1038/s41588-021-00854-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-021-00854-7
This article is cited by
-
Priority index for critical Covid-19 identifies clinically actionable targets and drugs
Communications Biology (2024)
-
Role of the Neanderthal Genome in Genetic Susceptibility to COVID-19: 3p21.31 Locus in the Spotlight
Biochemical Genetics (2024)
-
Association between SUMF1 polymorphisms and COVID-19 severity
BMC Genomic Data (2023)
-
Associations between polygenic risk score and covid-19 susceptibility and severity across ethnic groups: UK Biobank analysis
BMC Medical Genomics (2023)
-
Genome-wide cross-trait analysis and Mendelian randomization reveal a shared genetic etiology and causality between COVID-19 and venous thromboembolism
Communications Biology (2023)
