
Abstract
Allotetraploid cotton species (Gossypium hirsutum and Gossypium barbadense) have long been cultivated worldwide for natural renewable textile fibers. The draft genome sequences of both species are available but they are highly fragmented and incomplete1,2,3,4. Here we report reference-grade genome assemblies and annotations for G. hirsutum accession Texas Marker-1 (TM-1) and G. barbadense accession 3–79 by integrating single-molecule real-time sequencing, BioNano optical mapping and high-throughput chromosome conformation capture techniques. Compared with previous assembled draft genomes1,3, these genome sequences show considerable improvements in contiguity and completeness for regions with high content of repeats such as centromeres. Comparative genomics analyses identify extensive structural variations that probably occurred after polyploidization, highlighted by large paracentric/pericentric inversions in 14 chromosomes. We constructed an introgression line population to introduce favorable chromosome segments from G. barbadense to G. hirsutum, allowing us to identify 13 quantitative trait loci associated with superior fiber quality. These resources will accelerate evolutionary and functional genomic studies in cotton and inform future breeding programs for fiber improvement.
Similar content being viewed by others

Main
Cotton represents the largest source of natural textile fibers in the world. Over 90% of annual fiber production comes from allotetraploid cotton (G. hirsutum and G. barbadense), which originated from an allopolyplodization event approximately 1–2 million year ago, followed by millennia of asymmetric subgenome selection5,6. G. hirsutum is cultivated all over the world because of its high yield and G. barbadense is prized for its superior fiber quality. To cultivate G. hirsutum that produces longer, finer and stronger fibers, one approach is to introduce the superior fiber traits from G. barbadense into G. hirsutum. A genomics-enabled breeding strategy requires a detailed and robust understanding of genomic organization.
Here we applied single-molecule real-time sequencing technology (PacBio RSII) to assemble de novo the genome sequences of G. hirsutum accession TM-1 and G. barbadense accession 3–79. We generated 194.01 gigabases (Gb) of genome sequences for G. hirsutum and 210.98 Gb for G. barbadense, with an estimated depth of coverage of at least 80× for each genome (Supplementary Tables 1 and 2). This allowed us to assemble each genome into contigs, with contig L50 (the minimum length of sequences accounting for half of total assemblies) of 1.89 megabases (Mb) for G. hirsutum and 2.15 Mb for G. barbadense (Table 1). We used Illumina paired-end data to correct low-quality nucleotides and insertions/deletions (InDels) from the PacBio sequencing. These polished contigs were processed for a hybrid assembly by using high-resolution optical mapping (BioNano Genomics Irys) data from the same accessions (Supplementary Table 3). This allowed us to assemble 3,434 scaffolds for G. hirsutum and 3,919 for G. barbadense, with a scaffold L50 of 5.22 Mb and 6.89 Mb respectively (Supplementary Table 4). To construct chromosome-scale scaffolds, we used high-throughput chromosome conformation capture (Hi-C) data from each accession to categorize and order these assemblies obtained by optical mapping (Supplementary Fig. 1; Supplementary Tables 5 and 6)6,7. The final assemblies include 2,190 scaffolds for G. hirsutum and 3,032 for G. barbadense, of which the largest 26 super-scaffolds representing all chromosomes occupied 98.94% and 97.68% of all sequences respectively (Supplementary Figs. 2–5; Table 1).
To validate our assembly, both genomes were compared with the previously published genetic map8. Results indicate high consistency for each chromosome (Supplementary Figs. 6–9; Supplementary Table 7). The assembly accuracy and completeness were supported by perfect matches with 36 completely sequenced bacterial artificial chromosome sequences (Supplementary Table 8) and alignment of Illumina short-read data from mate-pair libraries for both accessions (Supplementary Table 9). The assembly completeness in genic regions is supported by the identification of 1,415 (98.2%) of those 1,440 highly conserved core proteins in the BUSCO dataset for G. hirsutum and 1,420 (98.6%) for G. barbadense (Supplementary Fig. 10)9. Compared with previously published draft genomes1,3, these sequences represent a significant improvement in contiguity (55-fold against G. hirsutum and 90-fold against G. barbadense) (Supplementary Table 10). Further improvement of these sequences might be achieved in genome gap-filling and accurate assembly of the highly complex regions; even so, these new genome sequences will serve as reference assemblies for tetraploid cotton.
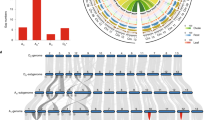
In the current assembly, we predict 70,199 genes in G. hirsutum and 71,297 genes in G. barbadense (Table 1; Supplementary Tables 11 and 12). We generated PacBio single-molecule long-read isoform sequencing data to annotate 115,835 transcription isoforms for G. hirsutum and used full-length transcripts to update 109,778 isoforms for G. barbadense10. The global landscape of genes on chromosomes was integrated with epigenetic modifications (Fig. 1). The global N6-methyldeoxyadenine (6mA) level is estimated to be 0.21% of all adenines for G. hirsutum and 0.22% for G. barbadense by using PacBio data. Interestingly, 6mA modification exhibits a nearly even distribution pattern along each chromosome, distinct from the relatively low levels of 5-methylcytosine (5mC) in chromosome arms (Fig. 1).

a, Gene density in each chromosome. b, Transposable element (TE) content in each chromosome. For a and b, green lines represent data in G. hirsutum and blue lines represent data in G. barbadense. c, SNP density between G. hirsutum and G. barbadense. d, InDel density between G. hirsutum and G. barbadense. e, 5mC DNA methylation levels. f, 6mA DNA methylation levels. g, Histone modification levels of H3K4me3 (yellow histogram) and H3K9me2 (purple histogram). For c–g, the outer tracks show data in G. hirsutum and inner tracks show data in G. barbadense. All these data are shown in 1 Mb windows sliding 200 kb. The inner lines show syntenic blocks in homoeologous chromosomes between the A and D subgenomes.
The high contiguity and completeness of the genome assemblies represent major improvements in regions with high contents of repeat sequences. We successfully assembled centromeric regions for each chromosome, identified by analysis of centromere-related long terminal repeat (LTR) retrotransposons (Supplementary Tables 13–16), most of which are missing in previous assemblies of G. hirsutum based on Illumina short-read sequencing (Supplementary Fig. 11)1. These regions have a markedly high content of LTR retrotransposons (Supplementary Fig. 12).
Sequence comparisons between the two cotton genomes were made to determine genomic divergence between the two representative accessions of G. hirsutum and G. barbadense. We identified a total of 12,816,698 single nucleotide polymorphisms (SNPs), averaging 5.89 per kilobase (kb; Supplementary Table 17). The SNP frequency of 8,131,276 (5.95 per kb) in the A subgenome (At) is slightly larger than that in the D subgenome (Dt), with 4,685,422 (5.81 per kb; two-sided Wilcoxon rank sum test, P < 2.2 × 10−16; Supplementary Fig. 13). The chromosomal distribution of SNPs is similar to the findings in a comparative population genome study11, including the notable decrease of genome variants in chromosome A01 (Fig. 1). We also identified 2,682,689 small InDels with an average of 1.2 per kb (Supplementary Table 18 and Supplementary Fig. 14). These SNPs and InDels are expected to have large functional effects on a total of 14,076 genes in G. hirsutum and 14,880 genes in G. barbadense (Supplementary Fig. 15). We also identified 4,039 genes with signals of positive selection (Ka/Ks >1) in the two genomes by using these variants (Supplementary Table 19). These genes are over-represented in several biological pathways including Ras/ARF protein signal transduction (Supplementary Fig. 16). Of note is the observation that 6.5% of SNPs and 7.2% of InDels were identified in the missing regions of the draft sequence of G. hirsutum1, representing previously undetected genetic variations for tetraploid cotton.
The high-quality reference genomes allowed us to identify large structural variants by a direct comparative genome analysis of the two accessions (Supplementary Figs. 17–19). We found 170.2 Mb of genome sequence identified as inversions between G. hirsutum and G. barbadense, including 120.4 Mb of At subgenome and 49.8 Mb of Dt (Supplementary Table 20). Interestingly, four chromosomes exhibit paracentric inversions and eleven chromosomes exhibit pericentric inversions in heterochromatin (Supplementary Table 21). We found that four large inversions, including three paracentric inversions (in1, in3 and in4) and one pericentric inversion (in2) in the A06 chromosome, are present with discrete chromatin interactions around breakpoints by inter-species mapping of Hi-C data (Fig. 2a), highlighting the power of the Hi-C technique in identifying large-scale chromosome rearrangements12,13. These data are also supported by BioNano optical maps covering inversion breakpoints (Supplementary Figs. 20–23). Another example shows one large pericentric inversion in chromosome D12 (Supplementary Figs. 24 and 25). This unexpected number of paracentric/pericentric inversions in cotton suggests a need for future exploration of their biological function, as described in Arabidopsis, wheat and humans14,15,16. We also detected 3,820 translocations (1,074 intra-chromosome translocations occupying 3.8 Mb and 2,746 inter-chromosome translocations occupying 6.8 Mb) (Supplementary Table 22).

a, Identification of large inversions in chromosome A06. The upper three heatmaps show a chromatin interaction matrix, including mapping Hi-C data in G. barbadense against the G. barbadense genome (Gb_map_Gb), mapping Hi-C data in G. hirsutum against the G. barbadense genome (Gh_map_Gb) and mapping Hi-C data in G. barbadense against reordered genome sequences of G. barbadense based on these inversions (Gb_map_Gb_reorder). The middle panel shows genome alignment between G. hirsutum and G. barbadense. The four regions exhibiting large inversions are shown by arrows. The lower three heatmaps show a chromatin interaction matrix with mapping Hi-C data against the G. hirsutum genome. The data mapping strategy is similar to those in the upper track. The Hi-C heatmaps are shown at 100 kb resolution. The triangle on each chromosome shows the centromeric region. BioNano contigs which supported the PacBio assembly around these inversion breakpoints are shown in Supplementary Figs. 20–23. b, The PAVs in G. hirsutum and G. barbadense genomes. For each chromosome, the left heatmap shows ratios of presence sequences in G. hirsutum. The right heatmap shows ratios of presence sequences in G. barbadense. Each heatmap is represented in 1 Mb windows sliding 200 kb. c, Genome alignment of the D subgenomes in G. hirsutum (Gh_Dt) and G. barbadense (Gb_Dt) with G. raimondii (Gr_D5). Lines between chromosomes (Chr) show syntenic regions.
The direct genome comparison analysis allowed a systematic characterization of presence/absence variations (PAVs) between the two accessions. We identified 9,135 segments in G. hirsutum with a total length of 179.9 Mb that are absent in G. barbadense and 7,710 segments in G. barbadense with a total length of 139.8 Mb that are absent in G. hirsutum (Fig. 2b; Supplementary Tables 23 and 24). We found that 1,844 genes in G. hirsutum and 1,614 genes in G. barbadense are in these PAV regions (Supplementary Table 25). Of those genes, 220 unique to G. barbadense are highly expressed during fiber development (Supplementary Fig. 26). We also found a 450 bp segment absent in the third exon of an EXPANSIN gene in G. barbadense, which gives rise to the loss of a polysaccharide-binding domain (Supplementary Figs. 27 and 28). Interestingly, the truncated expansin protein is known to be functional in the formation of superior fiber quality in G. barbadense17.
The reference-grade genome assemblies for both tetraploids enabled us to explore differences in genome organization between subgenomes and their diploid ancestors. We started this analysis by re-sequencing 13 diploid species with D genomes (Supplementary Fig. 29 and Supplementary Table 26). We show that both D subgenomes in the tetraploids are probable to have the same diploid ancestor species G. raimondii (D5 genome; Supplementary Figs. 30 and 31). This allows us to carry out a direct genome comparison with G. raimondii18. We found that each tetraploid has some unique structural variants when compared with G. raimondii, such as the large pericentric inversions in chromosome D05 for G. barbadense and D12 for G. hirsutum (Fig. 2c), suggesting that these variants have arisen after polyploidization. We also observed that the two tetraploids have shared some structural variants relative to G. raimondii, such as the large inversion in the D09 chromosome for both tetraploids (Fig. 2c). We then aligned the Hi-C data in G. arboreum (A2 genome), a proposed diploid ancestor species for tetraploid cotton, against both A subgenomes of G. hirsutum and G. barbadense to investigate genome variants between diploid and tetraploid cotton. These Hi-C matrix data indicate that chromosome rearrangements occur in all 13 chromosomes (Supplementary Fig. 32) and the majority of those variants are shared by both A subgenomes in the two tetraploids. We found that the largest pericentric inversion (in2) in chromosome A06 is unique in G. hirsutum (Fig. 2a; Supplementary Fig. 33), suggesting that it probably occurred after polyploidization. We conclude that the A genome in diploid cotton was reorganized following allopolyploidization, leading to large chromosome inversions in different tetraploids.
On the basis of this observation that there is wide genetic variation between G. hirsutum accession TM-1 and G. barbadense accession 3–79, it is anticipated that some of these variations could be responsible for phenotypic differences that include fiber traits. To manipulate these variations for directional breeding, we constructed an introgression line population aimed at introducing favorable variants that control the formation of important agronomic traits such as fiber quality from G. barbadense to G. hirsutum (Supplementary Fig. 34). We sequenced 168 introgression lines determined by molecular markers and identified 466 introgression segments that covered all 26 chromosomes (Fig. 3a; Supplementary Tables 27 and 28). We found that an introgression line containing an introgression segment in chromosome D12 had limited fuzz fibers, similar to its donor parent G. barbadense accession 3–79 (Fig. 3a,b). This location of introgression segment is the same as that suggested from mapping-by-sequencing of the fuzz-less natural mutant G. hirsutum Xuzhou142fl, for which the genetic basis was previously not well understood (Fig. 3c). These results indicate that the genetic variant underlying the fuzz-less mutant in G. hirsutum is co-localized with the quantitative trait locus (QTL) in G. barbadense. Characterization of this introgression segment, together with natural fiber mutants, will facilitate a comparative analysis of the mechanism of fuzz fiber initiation between G. barbadense and G. hirsutum.

a, Construction of an introgression line population by using G. hirsutum Emain22 (as a recurrent parent) and G. barbadense 3–79 (as a donor parent). The upper track shows the fiber characteristics of both cotton accessions. The lower track shows the distribution of introgression segments identified from the 168 introgression lines along the 26 chromosomes in G. hirsutum. The x-axis shows the 26 chromosomes and the y-axis shows the 168 introgression lines. b, Fiber characteristics for introgression line N29. The right circle shows the introgression segment in the chromosome D12 (from 47.4 Mb to 54.6 Mb) which is indicated by the red box. c, Mapping-by-sequencing of Xuzhou142fl. Ratio of SNPs in chromosomes A12 and D12 between two different pools from the F2 population (one pool consisting of cotton plants with the same fiber characteristics of Xuzhou142 and one pool consisting of cotton plants exhibiting the same phenotype with Xuzhou142fl). d, Distribution of fiber-quality related quantitative trait loci (QTLs) in chromosomes of G. hirsutum. Each QTL is indicated by a red box. The specific location of each QTL is shown in Supplementary Table 30. e, QTL mapping for the trait of fiber length (mm). f, QTL mapping for the trait of fiber elongation rate (%). For e and f, the x-axes show all the introgression segments and the y-axes show the logarithm of odds (LOD) score. The physical locations of QTLs are shown with arrows.
To identify beneficial alleles for superior fiber quality in G. barbadense, we carried out a QTL analysis for traits related to fiber quality in this introgression population (Supplementary Table 29). In total, 13 QTLs were identified for five traits, including 2 QTLs for fiber length, 4 for fiber strength, 2 for micronaire value, 2 for fiber elongation rate and 3 for fiber uniformity (Fig. 3d–f; Supplementary Table 30). Of these QTLs, 9 are previously uncharacterized. By examining the expression levels of genes located in the 13 QTLs, we detected 235 genes that are highly expressed during fiber development (Supplementary Table 31). We also integrated genome variant data to predict candidate genes that deserve further efforts in fine-mapping to confirm genes with major effects on these traits (Supplementary Table 32). We found that one QTL at chromosome A02 is associated with fiber length. In this QTL, one uncharacterized gene (Ghir_A02G003440), encoding a predicted glycosylphosphatidylinositol anchored lipid transfer protein, exhibits a negatively correlative relationship between gene expression level at the elongation stage and fiber length in the introgression line population; it is potentially associated with the development of long fiber in G. barbadense (Supplementary Fig. 35). These QTL data provide a framework for detailed functional analysis of genomic segments in G. barbadense and should be further exploited for cultivating cotton with superior fiber by introgression breeding in the future.
To further investigate the possible mechanism of transcriptional regulation for these genes, we sequenced the transcriptomes of fibers at 10 days post anthesis (DPA) for these 168 introgression lines (Supplementary Table 33). We then identified expression QTLs (eQTLs) for 125 of those 235 genes (Supplementary Table 34). We found that the QTL at the chromosome A02 described above was associated with the expression of two genes (Ghir_D09G014120 and Ghir_D09G014460) in chromosome D09, encoding an ubiquitin extension protein and a microtubule-associated protein respectively; these two genes are possible candidate genes for fiber strength (Supplementary Tables 30 and 32). These eQTLs suggest that expression of these genes may be associated with certain genomic loci over a long distance or in an inter-chromosome manner.
By assembly of reference genomes for two cultivated cotton accessions, we have been able to identify extensive variations. These variations should be integrated with those from genome analyses of other accessions to fully exploit genome divergence between the two species in the future. We explored beneficial genome sequences underlying superior fiber quality between two representative accessions of species by constructing introgression lines that can be used for the cultivation of desirable traits in intensive cotton breeding. These resources will be of great importance to the community for facilitating functional and evolutionary genomics studies and will inform genome-enabled cotton fiber improvement.
URLs
Genome assemblies and annotation, http://cotton.hzau.edu.cn/EN/download.php; CottonGen database, https://www.cottongen.org/; PacBio SMRT-Analysis, https://www.pacb.com/products-and-services/analytical-software/smrt-analysis/; IrysSolve pipeline, https://bionanogenomics.com/support-page/bionano-solve/; LACHESIS software, http://shendurelab.github.io/LACHESIS/; MUMmer software, http://mummer.sourceforge.net/; BUSCO embryophyta_odb9 dataset, https://busco.ezlab.org/frame_wget.html.
Methods
Materials
The highly homozygous Upland cotton (Gossypium hirsutum) accession Texas Marker-1 (TM-1) and Sea-Island cotton (Gossypium barbadense) accession 3–79 were cultivated in the field in Wuhan, China in 2016. Both accessions were sequenced to assemble the draft genomes using Illumina short-read technology previously1,2,3. To improve genome assembly quality using single-molecule real-time (SMRT) sequencing technology (Pacific Biosciences), fresh young leaves were collected from one single plant for each species and immediately frozen in liquid nitrogen.
Library construction and PacBio sequencing
Genomic DNA for PacBio Sequencing was extracted using the CTAB method. To construct sequencing libraries, genomic DNA was fragmented by g-TUBE centrifuged at 2,000 r.p.m. for 2 min, treated with end-repair, adapter ligation and exonuclease digestion as recommended by Pacific Biosciences. DNA fragments at about 10–50 kb were selected by BluePippin electrophoresis (Sage Sciences). DNA libraries were sequenced on the PacBio RS II platform (Pacific Biosciences) with the P6–C4 chemistry. A total of 189 SMRT cells were sequenced for G. hirsutum producing 204 Gb raw data and 200 cells for G. barbadense producing 219 Gb raw data (Supplementary Table 1).
Genome assembly
The PacBio SMRT-Analysis package (see URLs) was used for processing raw polymerase reads (parameters: readScore, 0.75; minSubReadLength, 500), including removing sequencing adapters and filtering reads with low quality and short length. Because of the high error rate of PacBio reads, we first corrected those by using an error correction module embedded in Canu (version 1.3) with parameter correctedErrorRate 0.045 (ref. 19). The high-quality PacBio sub-reads were then used for genome assembly by using Canu19. The assembled contigs were supported by mapping 96.5% of clean sub-reads (with sequence length >10 kb) for G. hirsutum and 98.4% for G. barbadense using BLASR (version 1.3.1)20. Then, consensus sequences of assemblies were subject to mapping of around 50× Illumina pair-end reads for the same cotton species using BWA (version 0.7.10-r789)21 and were polished using Pilon (version 1.22) software (parameters: --mindepth 10 --changes --fix bases)22. This allowed the correction of 884,720 SNPs and 3,428,790 InDels for G. hirsutum and 832,474 SNPs and 3,193,534 InDels for G. barbadense. The genome assembly process was conducted on a Linux cluster (CentOS release 6.5) in parallel (100 qsub jobs). Each job requires 8 CPU and 50 GB peak memory (RAM) usage. These assemblies show no evidence of contamination by alignment with the UniVec and RefSeq microbial genome databases in the National Center for Biotechnology Information (NCBI).
Scaffolding by using optical maps of the BioNano system
Cotton plants TM-1 and 3–79 were cultivated in a growth chamber at Brigham Young University. Young leaves were collected after two days of dark treatment. High-molecular-weight DNA was isolated and labeled according to standard BioNano protocols with the single-stranded nicking endonuclease Nt. BssSI (ref. 23). The labeled DNA sample was loaded onto the IrysChip nanochannel array. The stretched DNA molecules were imaged with the BioNano Irys system. Raw image data were converted into bnx files; from these, the AutoDetect software generated basic labeling and DNA length information.
After filtering by molecule length and label density, a total of 213.2 Gb single-molecule data for G. hirsutum and 373.7 Gb for G. barbadense were produced. High-quality labeled molecules were pairwise aligned, clustered and assembled into contigs with the BioNano Genomics assembly pipeline, IrysSolve23,24. A physical map was assembled for each genome (a total length of 2,185.8 Mb for G. hirsutum and a total length of 2,119.6 Mb for G. barbadense; Supplementary Table 3).
To create hybrid scaffolds, optical maps were aligned to PacBio assembled contigs and scaffolded with BioNano’s hybrid-scaffold tool25. The process includes comparing BioNano genome nick-based maps to in silico nick maps of the genome sequence to find their best matches and potential reciprocal scaffolding of each dataset. If there were conflict sites between the sequence and physical maps, both of them were cut at the conflict sites and assembled again (the software Hybrid-scaffold parameter of ‘-B 2 -N 2’).
Chromosome assembly using Hi-C
In previous studies6,7, we have generated high-quality Hi-C data (based on HindIII) for G. hirsutum accession TM-1 and G. barbadense accession 3–79. These clean data were used to assist in constructing chromosome-level assemblies. This assembly approach is shown in Supplementary Fig. 1. In this study, we first performed a pre-assembly for error correction of scaffolds which required the splitting of scaffolds into segments of 50 kb on average. The Hi-C data were mapped to these segments using BWA (version 0.7.10-r789) software21. The uniquely mapped data were retained to perform assembly by using LACHESIS software26. Detailed information for Hi-C read mapping is shown in Supplementary Table 5. Any two segments which showed inconsistent connection with information from the raw scaffold were checked manually. These corrected scaffolds were then assembled with LACHESIS. Parameters for running LACHESIS included: CLUSTER_MIN_RE_SITES, 225; CLUSTER_MAX_LINK_DENSITY, 2; ORDER_MIN_N_RES_IN_TRUN, 105; ORDER_MIN_N_RES_IN_SHREDS, 105. In this step, 2,168 scaffolds (representing 98.94% total length) were anchored to chromosomes in G. hirsutum, and 1,966 scaffolds (representing 97.68% total length) were anchored to chromosomes in G. barbadense (Supplementary Table 6). To assess the quality of assembly, Hi-C data were mapped to chromosomes using HiC-Pro software (version 2.7.1)27 and placement and orientation errors exhibiting obvious discrete chromatin interaction patterns were manually adjusted. The interaction matrix of each chromosome was visualized with heatmaps at the 100 kb resolution. In addition, a published genetic map with 4,049 bins between G. hirsutum and G. barbadense was aligned to both genomes after Hi-C directed assembly (Supplementary Table 7)8. To evaluate the completeness of genome assemblies, the 1,440 conserved protein models in the BUSCO embryophyta_odb9 dataset (see URLs) were searched against both genomes by using the BUSCO (version 2) program with default settings9. This gave 1,378 (95.7%) and 1,385 (96.2%) complete BUSCO hits for G. hirsutum and G. barbadense, respectively (Supplementary Fig. 10).
Identification of centromeric regions
The relatively conserved and centromere-related 5′ LTR sequences in cotton have been identified previously8,28 and named as GhCR1-5′LTR, GhCR2-5′LTR, GhCR3-5’LTR and GhCR4-5′LTR. Here, to identify the centromeric regions, these LTR sequences were aligned with the TM-1 and 3–79 genomes by blastn, with sequence similarity ≥80% and e-value ≤1e–20. After filtering alignments, we used the SPSS software (version 17.0) to calculate the 95% confidence interval for the median representing the centromeric region for each chromosome.
Genome alignment and gene synteny analysis
Genome alignment between G. hirsutum and G. barbadense was performed using the MUMmer (version 3.23)29 program with parameters settings –maxmatch –c 90 –l 40. The alignments were filtered by running delta-filter with parameter −1. SNPs and InDels in the two accessions were extracted by running show-snp in the one-to-one alignment blocks. We also mapped DNA sequencing data (>20×) from the Illumina HiSeq platform of each accession against the other genome using BWA (version 0.7.10-r789) software. The unique mapping data were used for identifying SNPs and InDels using the Genome Analysis Toolkit (GATK, version 3.1.1) and Samtools (version 0.1.19) programs as described previously6,30,31. Those variants which were supported by at least two software tools from MUMmer, Samtools and GATK were used for further analysis. All these variants were annotated using the ANNOVAR program32.
To identify syntenic gene blocks between tetraploid subgenomes and diploids, we conducted an All-vs-All blastp (e-value <1e-10, -v 5, -b 5) for each genome pair. The homologous genes were analyzed by the MCScanX package33 with default settings except for gap_penalty -3. Syntenic blocks were defined as those with at least five syntenic genes.
Identification of inversions and translocations
To identify inversions and translocations, the genome of G. hirsutum was aligned with G. barbadense using MUMmer3 (version 3.23). The alignment blocks exhibiting inversions were extracted for manual checking. Alignment blocks identified in different positions were extracted to check their flanking blocks. If alignment blocks had non-colinear flanking sequences, those were retained as putative translocations. The translocations were further divided into inter-chromosomal translocations and intra-chromosomal translocations. Both inversions and translocations were identified with a length of >100 bp and identity of >90%.
An analysis of genome regions showed that 8,528 (97.4%) candidate inversions (8,753 candidates in total) were identified within intact PacBio contigs in both genomes. Another analysis of BioNano optical maps showed that 8,513 (97.2%) of those candidates were supported at the breakpoints of inversions for at least one genome (88.2% for G. hirsutum and 87.6% for G. barbadense). Additional mapping of clean PacBio sub-reads with a length >10 kb (with a genome coverage of 25×) showed that 8,409 (96.0%) of candidates had evidence from spanning sub-reads at inversion breakpoints. To obtain highly confident inversions, 480 sequences which were identified around assembly gaps and had no evidence from BioNano data or PacBio reads were excluded from further analysis. The same method was applied to filter low-quality translocations using PacBio reads and BioNano data. Besides, both 20 kb flanking regions around breakpoints of each putative translocation between the two genomes were aligned to filter out the false positives which might be originated from paralogous sequences (coverage >50%).
Identification of PAVs
The putative PAVs were identified by extracting unaligned regions between G. hirsutum and G. barbadense from the ‘show-diff’ command in MUMmer3 (version 3.23). This gave sequences of 400 Mb for G. hirsutum and 301 Mb for G. barbadense. These sequences were then filtered by discarding those overlapping with gap regions in the respective genome. To identify putatively unique presence regions, the remaining sequences above (391 Mb for G. hirsutum and 271 for G. barbadense) were then filtered by alignment with the other genome using blastn (1e-5). The candidate PAV regions were retained by filtering those with coverage >50% and identity >90% in each genome (238 Mb for G. hirsutum and 167 Mb for G. barbadense). Additionally, the sequencing reads were aligned to the other species using blastn with coverage >30% and identity >80%, to further exclude those segments that were probably from un-assembled reads. Finally, we obtained unique sequences totalling 179.9 Mb in G. hirsutum and 139.8 Mb in G. barbadense.
Integration of re-sequencing data for diploid cotton and identification of SNPs
Cotton leaves for 13 diploid species with D genomes were collected from the National Wild Cotton Nursery, Sanya, China. For each species, DNA was extracted from leaves using the CTAB method. For each species, at least 5 µg DNA was used for library construction using the Illumina TruSeq DNA Sample Prep Kit with insertion size of about 350 bp. All these libraries were sequenced on an Illumina HiSeq 2000 platform with genome coverage of at least 15× (pair-end 100 bp; Supplementary Table 26). After trimming of low-quality bases using Trimmomatic (version 0.32), the clean data were mapped to the D subgenomes of both tetraploids and D5 genome of G. raimondii using BWA software18. The re-sequencing data for G. herbaceum (A1 genome) and G. arboreum (A2 genome) were downloaded from the NCBI Sequence Read Archive (SRA) database (PRJNA349094)34. The clean data were mapped to the A subgenomes of both tetraploids using BWA software. All the unique mapping data were extracted to identify SNPs using GATK (version 3.1.1) and Samtools (version 0.1.19) programs30,31. SNPs supported by both programs with a sequencing coverage of at least 8 were retained for further analysis.
Bulked-segregant analysis
Cotton accession Xuzhou 142 and its natural mutant Xuzhou 142fl were hybridized to construct the F2 population with self-crossing seeds from 30 F1 plants. In the F2 generation, DNA samples from 30 plants exhibiting the same phenotype as the wild parent were mixed with equal amounts from each and used as the wild pool. The mutant DNA pool was constructed by using DNA samples from 30 plants exhibiting the same phenotype as the mutant parent. For each plant, DNA extraction was performed using the Plant Genome Extraction Kit (TIANGEN Biotech). DNA libraries were constructed using an Illumina TruSeq DNA Sample Prep Kit with insertion size of about 350 bp. Both DNA pools were sequenced by an Illumina platform (HiSeq 4000) with data of at least 75 Gb (pair-end 150 bp). These DNA sequencing data were mapped to the genome of G. hirsutum using BWA software (version 0.7.10-r789) and the unique mapping data were used to identify SNPs with Samtools and GATK programs as described above. The method for bulked-segregant analysis was the same as that described previously35. SNP ratios were calculated in 1 Mb windows sliding 100 kb.
Construction of chromosome segment substitution lines (CSSLs)
Emian22, as the recipient parent, is an elite Upland cotton cultivar with moderate fiber quality and no resistance to Verticillium wilt. The donor parent 3–79 is a genetic and cytogenetic standard line for G. barbadense with super fiber quality and high resistance to Verticillium wilt. The BC1 backcross population, (Emian22 × 3–79) × Emian22, was constructed in 2005 in Wuhan; an inter-specific genetic linkage map based on it was constructed previously36. Over subsequent years, BC1 underwent systematic backcrosses using the recurrent parent Emian22. In 2007, the BC4 generation was obtained and subjected to genotyping analysis using 254 markers across the 26 chromosomes36. In 2009, the high-density genetic linkage map was updated in our laboratory and was released in 2011 (ref. 37). Based on the updated genetic map, a total of 515 SSR markers with an average interval of 10 cM between adjacent markers were selected for this study. During the period of molecular marker-assisted selection, only the plants that harbored target segments (without non-target segments) were retained; donor segments covered the whole cotton genome as far as possible. When one plant contained less than three donor segments from Sea-Island cotton 3–79, it was self-pollinated to produce a homozygous line. Finally, a total of 325 CSSLs were obtained to represent the GhILs-Gb population; among them, 177 lines harbored only one donor segment (Supplementary Fig. 34).
Identification of SNPs and construction of a bin map
The CSSL population was cultivated in the field in Wuhan, China. Leaf samples were collected for DNA extraction with the Plant Genome Extraction Kit (TIANGEN Biotech). The sequencing library of each line was constructed using the Illumina TruSeq DNA Sample Prep Kit with insertion size of about 350 bp. A total of 168 CSSLs and their parents were sequenced on an Illumina HiSeq platform with at least 6× coverage in this study (pair-end 150 bp; Supplementary Table 27). All clean reads were mapped to the G. hirsutum genome using BWA software (version 0.7.10-r789) and the unique mapping data were retained for further analysis. To identify SNPs, the GATK and Samtools software programs were applied as described above. Only those SNPs that were supported by both programs were retained. These putative SNPs were further filtered based on the following criteria: (1) the quality of SNPs should be over 100; (2) each SNP was supported by at least five reads; and (3) the adjacent SNPs should have a distance of at least 10 bp.
To identify introgression segments from G. barbadense to G. hirsutum, a sliding-window approach was applied38. Firstly, the G. hirsutum genome was divided into 422,164 bins consisting of consecutive 30 SNPs between two parents (with a total of 12,657,873 SNPs and an average of 5.6 per kb). Then, all the alleles represented by SNPs in each CSSL were filtered using SNPs from both parents. Only those that had the same allele as one of the parents were retained. Based on the allele ratio (G. barbadense/G. hirsutum), each bin was then defined as having a homozygous G. barbadense 3–79 genotype (larger than 25:5) or having a homozygous G. hirsutum Emian22 genotype (smaller than 5:25). Bins with allele ratios between these cutoffs were defined as having a heterozygous genotype. In this analysis, bins that had larger numbers (more than 20) of missing SNP genotype were discarded. Consecutive bins with the same genotype were combined into segments. The recombination breakpoints were assumed when two segments had different genotypes.
Identification of quantitative trait loci (QTLs) and expression QTLs (eQTLs) using CSSLs
The introgression population was planted in the field of Shihezi, China in 2015−2017, Huanggang, China in 2015 and Jingzhou, China in 2017 for collection of traits related to fiber quality, including fiber length, fiber strength, micronaire value, fiber elongation rate, fiber uniformity and short fiber rate. These mature fiber traits for each CSSL were measured using a high-volume instrument (HFT9000; Premier). To identify QTLs, the introgression segments were divided into non-overlapping blocks in all introgression lines, which gave rise to a total of 535 blocks. The identification of QTLs for each trait related to fiber quality was performed with QTL IciMapping (version 4.0)39. In this analysis, the RSETP-LRT-ADD mapping method was applied with a logarithm of odds (LOD) threshold of 2.5.
Fiber samples at 10 days post anthesis (DPA) for the 168 introgression lines and parents were collected from the field in Wuhan, China. For each sample, fibers of at least 20 cotton bolls from different plants were mixed together. Total RNA for each line was extracted by using a guanidine thiocyanate method. The Illumina TruSeq Stranded RNA Library Preparation Kit was used to construct libraries which were then sequenced on an Illumina HiSeq platform (pair-end 150 bp; Supplementary Table 33). High-quality clean data were mapped to the G. hirsutum genome by TopHat2 (version 2.0.13) and the expression levels (FPKM) were calculated using Cufflinks (version 2.2.1)40,41. To identify eQTLs, the expression levels of those genes (FPKM >1) were normalized using a normal quantile transformation. The method for eQTL identification was similar to a previous study42. Briefly, the STRUCTURE (version 2.3) software was run to estimate the population structure for the introgression lines43. Multiple linear regression embedded in the TASSEL software (version 5.0) was used to identify eQTLs for genes in chromosome segments associated with traits related to fiber quality44.
Statistical analysis
The comparison of SNP density between the At and Dt subgenomes was carried out using a two-sided Wilcoxon rank sum test. Gene Ontology (GO) enrichment analysis was carried out using Blast2GO software with a two-sided Fisher’s exact test; only GO terms with a false discovery rate of less than 0.05 (FDR <0.05) were retained.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All the raw sequencing data generated during the current study are available in the BioProject database under accession number PRJNA433615. The genome assemblies and annotation files are available at the website http://cotton.hzau.edu.cn/EN/download.php and the CottonGen database (https://www.cottongen.org/). All the materials in this study, including introgression lines, are available upon reasonable request.
References
Zhang, T. et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537 (2015).
Li, F. et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 33, 524–530 (2015).
Yuan, D. et al. The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 5, 17662 (2015).
Liu, X. et al. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci. Rep. 5, 14139 (2015).
Senchina, D. S. et al. Rate variation among nuclear genes and the age of polyploidy in. Gossypium. Mol. Biol. Evol. 20, 633–643 (2003).
Wang, M. et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 49, 579–587 (2017).
Wang, M. et al. Evolutionary dynamics of 3D genome architecture following polyploidization in cotton. Nat. Plants 4, 90–97 (2018).
Wang, S. et al. Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol. 16, 108 (2015).
Simao, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Wang, M. et al. A global survey of alternative splicing in allopolyploid cotton: landscape, complexity and regulation. New Phytol. 217, 163–178 (2018).
Fang, L. et al. Genomic insights into divergence and dual domestication of cultivated allotetraploid cottons. Genome Biol. 18, 33 (2017).
Harewood, L. et al. Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. Genome Biol. 18, 125 (2017).
Dixon, J. R. et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 50, 1388–1398 (2018).
Fransz, P. et al. Molecular, genetic and evolutionary analysis of a paracentric inversion in Arabidopsis thaliana. Plant J. 88, 159–178 (2016).
Ma, J. et al. Identification of genes bordering breakpoints of the pericentric inversions on 2B, 4B, and 5A in bread wheat (Triticum aestivum L.). Genome 58, 385–390 (2015).
Ciuladaite, Z., Preiksaitiene, E., Utkus, A. & Kučinskas, V. Relatives with opposite chromosome constitutions, rec(10)dup(10p)inv(10)(p15.1q26.12) and rec(10)dup(10q)inv(10)(p15.1q26.12), due to a familial pericentric inversion. Cytogenet. Genome Res. 144, 109–113 (2014).
Li, Y. et al. GbEXPATR, a species-specific expansin, enhances cotton fibre elongation through cell wall restructuring. Plant Biotechnol. J. 14, 951–963 (2015).
Paterson, A. H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427 (2012).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Chaisson, M. J. & Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13, 238 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Lam, E. T. et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 30, 771–776 (2012).
Cao, H. et al. Rapid detection of structural variation in a human genome using nanochannel-based genome mapping technology. Gigascience 3, 34 (2014).
Valouev, A., Schwartz, D. C., Zhou, S. & Waterman, M. S. An algorithm for assembly of ordered restriction maps from single DNA molecules. Proc. Natl Acad. Sci. USA 103, 15770–15775 (2006).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Zhang, W. et al. Identification of centromeric regions on the linkage map of cotton using centromere-related repeats. Genomics 104, 587–593 (2014).
Delcher, A. L., Phillippy, A., Carlton, J. & Salzberg, S. L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 30, 2478–2483 (2002).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Du, X. et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 50, 796–802 (2018).
Soyk, S. et al. Bypassing negative epistasis on yield in tomato imposed by a domestication gene. Cell 169, 1142–1155 (2017).
Zhang, Y., Lin, Z., Xia, Q., Zhang, M. & Zhang, X. Characteristics and analysis of simple sequence repeats in the cotton genome based on a linkage map constructed from a BC1 population between Gossypium hirsutum and G. barbadense. Genome 51, 534–546 (2008).
Yu, Y. et al. Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypium hirsutum and G. barbadense. BMC Genomics 12, 15 (2011).
Huang, X. et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 19, 1068–1076 (2009).
Li, H., Ye, G. & Wang, J. A modified algorithm for the improvement of composite interval mapping. Genetics 175, 361–374 (2007).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Fu, J. J. et al. RNA sequencing reveals the complex regulatory network in the maize kernel. Nat. Commun. 4, 2832 (2013).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587 (2003).
Bradbury, P. J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Acknowledgements
We thank K. Wang (Institute of Cotton Research of the Chinese Academy of Agricultural Sciences, Anyang, China) for providing leaf samples of wild cotton species. This project was financially supported by the National Natural Science Foundation of China (31830062) and National Transgenic Plant Research of China (2016ZX08005-001) to X.Z. This project was also supported by National Postdoctoral Program for Innovative Talents (BX201700094) and the Young Elite Scientists Sponsorship Program by China Association for Science and Technology to M.W. Funding was also provided by the National Science Foundation Plant Genome Research Program (1339412) to J.U. Additional support was provided from Cotton Incorporated. We thank Rise Services for office accommodations in Orem, UT, USA.
Author information
Authors and Affiliations
Contributions
X.Z., J.U., Z. Lin and K.L. conceived and designed the project. Z. Lin and L.T. constructed the introgression lines and collected materials. J.U. and D.Y. generated and analyzed the BioNano data. D.Z., L.P., P.W., G.Z., Z.Y., H.H., F.Y., J.Y., Y.Y., Z. Liu and F.H. performed experiments. M.W., L.T., D.Y., M.L., Q.Z. and H.Z. performed PacBio and Illumina sequencing. M.W., D.Y., J.L., C.S., L.P., F.L., Y.M., L. Zhang, B.L. and P.Q. analyzed the data. K.L., L. Zhu, S.J., X.Y., L.M., G.L. and L.C. contributed to project discussion. M.W. wrote the manuscript draft and X.Z., K.L. and J.U. revised it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–35, Supplementary Tables 1–18, 21, 26 and 30, and Supplementary Note
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, M., Tu, L., Yuan, D. et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat Genet 51, 224–229 (2019). https://doi.org/10.1038/s41588-018-0282-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-018-0282-x
This article is cited by
-
Precise fine-turning of GhTFL1 by base editing tools defines ideal cotton plant architecture
Genome Biology (2024)
-
Pangenome analysis reveals transposon-driven genome evolution in cotton
BMC Biology (2024)
-
Rapidly mining candidate cotton drought resistance genes based on key indicators of drought resistance
BMC Plant Biology (2024)
-
Drought response revealed by chromatin organization variation and transcriptional regulation in cotton
BMC Biology (2024)
-
Genome-wide analyses of member identification, expression pattern, and protein–protein interaction of EPF/EPFL gene family in Gossypium
BMC Plant Biology (2024)
