
Abstract
The koala, the only extant species of the marsupial family Phascolarctidae, is classified as ‘vulnerable’ due to habitat loss and widespread disease. We sequenced the koala genome, producing a complete and contiguous marsupial reference genome, including centromeres. We reveal that the koala’s ability to detoxify eucalypt foliage may be due to expansions within a cytochrome P450 gene family, and its ability to smell, taste and moderate ingestion of plant secondary metabolites may be due to expansions in the vomeronasal and taste receptors. We characterized novel lactation proteins that protect young in the pouch and annotated immune genes important for response to chlamydial disease. Historical demography showed a substantial population crash coincident with the decline of Australian megafauna, while contemporary populations had biogeographic boundaries and increased inbreeding in populations affected by historic translocations. We identified genetically diverse populations that require habitat corridors and instituting of translocation programs to aid the koala’s survival in the wild.
Similar content being viewed by others


Main
The koala is an iconic Australian marsupial, instantly recognizable by its round, humanoid face and distinctive body shape. Fossil evidence identifies as many as 15–20 species, following the divergence of koalas (Phascolarctidae) from terrestrial wombats (Vombatidae) 30–40 million years ago1,2 (Supplementary Fig. 1). The modern koala, Phascolarctos cinereus, which first appeared in the fossil record ~350,000 years ago, is the only extant species of the Phascolarctidae. Like other marsupials, koalas give birth to underdeveloped young. Birth occurs after just 35 d of gestation, with young lacking immune tissues or organs. Their immune system develops while they are in the pouch, meaning survival during early life depends on immunological protection provided by mothers’ milk.
A specialist arboreal folivore feeding almost exclusively from Eucalyptus spp., the koala has a diet that would be toxic or fatal to most other mammals3. Due to the low caloric content of this diet, the koala rests and sleeps up to 22 h a day4. A detailed understanding of the mechanisms by which koalas detoxify eucalyptus and protect their young in the pouch has been elusive, as there are no koala research colonies and access to milk and tissue samples is opportunistic. The genome enables unprecedented insights into the unique biology of the koala, without having to harm or disturb an animal of conservation concern.
The genome also enables a holistic, scientifically grounded approach to koala conservation. Australia has the highest mammal extinction record of any country during the Anthropocene5, and koala numbers have plummeted in northern parts of its range since European settlement of the continent6, but increased in southern sections of the range, notably in parts of Victoria and South Australia. The uneven response of koala populations throughout its range is one of the most difficult issues in its management7. The species was heavily exploited by a pelt trade (1870s to late 1920s), which harvested millions of animals6,8,9. Today, the threats are primarily due to loss and fragmentation of habitat, urbanization, climate change and disease. Current estimates put the number of koalas in Australia at only 329,000 (range 144,000–605,000), and a continuing decline is predicted6. Koalas present a complex conservation conundrum: in the north, causes of decline include ongoing habitat fragmentation, urbanization and disease. However, decline in the south has followed a different path10, with widespread, often sequential, translocations (1920–1990s) from a limited founder population, which has resulted in genetically bottlenecked populations that are overabundant to the point of starvation in some areas11. There are marked differences in the degree to which threats affect each population, thereby cautioning against one prescription for population recovery.
Adding to the complexity of koala conservation is the impact of disease, specifically koala retrovirus (KoRV) and Chlamydia. KoRV is thought to have arrived in Australia via a putative murine vector before cross-species transmission12,13. It is now prevalent in northern koalas and appears to be spreading to southern populations14. Some strains appear to be more virulent than others and are putatively associated with an increase in neoplastic disease15. Similarly, Chlamydia, which in some individuals causes severe symptoms yet in others remains asymptomatic, may have crossed the species barrier from introduced hosts such as domestic sheep and cattle following European settlement16. A complete koala genome offers insights into the species’ genetic susceptibility to these diseases, provides the genomic basis for innovative vaccines, and can underpin new conservation management solutions that incorporate the species’ population and genetic structure, such as facilitating gene flow via habitat connectivity or translocations.
Results
Genome landscape
Koalas have 16 chromosomes, differing from the ancestral marsupial 2n = 14 karyotype by a simple fission of ancestral chromosome 2 giving rise to koala chromosomes 4 and 717. We sequenced the complete genome using 57.3-fold PacBio long-read coverage, generating a 3.42 Gb reference assembly. The primary contigs from the FALCON assembly (representing homozygous regions of the genome) yielded genome version phaCin_unsw_v4.1. This comprised 3.19 Gb, including 1,906 contigs with an N50 of 11.6 Mb and the longest at 40.6 Mb. The heterozygous regions of the genome (representing the alternate contigs from the assembly) totaled 230 Mb, with an N50 of 48.8 kb (Table 1, Supplementary Tables 1–3 and Methods). Approximately 30-fold coverage of Illumina short reads was used to polish the assembly. BioNano optical maps plus additional conserved synteny information for marsupials were used for scaffolding18 to assemble long-read contigs into ‘virtual’ chromosome scaffolds (‘super-contigs’) (Supplementary Tables 4 and 5 and Supplementary Note). The largest super-contig spanned approximately half of koala chromosome 7 (Supplementary Fig. 2).
Our long-read-based sequence presented the opportunity to identify and study centromeres, which are multi-megabase regions that are challenging to construct in eutherian (for example, human and mouse)19 genome assemblies due to intractable higher order arrays of satellites. Centromeres are smaller in marsupials than in eutherians, and as such are more amenable to analysis20. Chromatin immunoprecipitation and sequencing using antibodies to centromeric proteins (CENP-A and CREST)21 enabled the identification of scaffolds containing putative centromeric regions (Supplementary Fig. 3) and the characterization of known and new repeats, including composite elements within koala centromeric domains (Supplementary Table 6–10) that lack the previously annotated retroelement, kangaroo endogenous retrovirus (KERV), found in some tammar wallaby centromeres22. Koala centromeres span a total of 2.6 Mb of the koala haploid genome, equivalent to an average of 300 kb of centromeric material per chromosome. Like those of other species with small centromeres19,20,23,24, koala centromeres lack higher order satellite arrays (Supplementary Tables 7–10). Among the newly identified repeats, some are similar to composite elements recently described in gibbon centromeres25, where absence of higher order satellite arrays accompanied the evolution of new composite elements with putative centromere function. The composition of the koala centromere therefore supports mounting evidence that transposable elements represent a major, functional component of small centromeres when higher order satellite arrays are absent20,24,25.
Interspersed repeats account for approximately 47.5% of the koala genome; 44% of these are transposable elements (Supplementary Table 11). As in other mammalian genomes, short interspersed nuclear elements (SINEs) and long interspersed nuclear elements (LINEs) are the most numerous elements (35.2% and 28.9% of total number of elements, respectively), with LINEs making up 32.1% of the koala genome. The long-read sequence assembly also enabled full characterization and annotation of repeat-rich long noncoding RNAs, including RSX, which mediates X chromosome inactivation in female marsupials26. Koala RSX represents the first marsupial RSX to be fully annotated and to have its structure predicted (Supplementary Fig. 4 and Supplementary Note). As expected, it was expressed in all female tissues, but in no male tissues27.
The assembled koala genome has very high coverage of coding regions: we recovered 95.1% of 4,104 mammalian benchmarking universal single-copy orthologs (BUSCOs)28, the highest value for any published marsupial genome (Supplementary Table 5) and comparable with that of the human assembly (GRCh38, which scores 94.1% of orthologs). Analysis of gene family evolution using a maximum-likelihood framework identified 6,124 protein-coding genes in 2,118 gene families with at least two members in koala. Among these, 1,089 have more gene members in koala than in any of the other species (human, mouse, dog, tammar wallaby, Tasmanian devil, gray short-tailed opossum, platypus, chicken; Supplementary Fig. 5).
Having characterized the genome, we undertook detailed analyses of key genes and gene families to gain insights into the genomic basis of the koala’s highly specialized biology. Gene families of particular interest were those that encode proteins involved in induced ovulation, those proteins involved in the complex lactation process, those proteins responsible for immunity, and those enzymes that enable the koala to subsist on a toxic diet.
Ability to tolerate a highly toxic diet
The koala’s diet of eucalyptus leaves contains high levels of plant secondary metabolites29, phenolic compounds30 and terpenes (for example, ref. 31) that would be lethal to most other mammals32. Koalas thus experience little competition for food resources. Eucalyptus grandis shows substantial expansion in terpene synthase genes relative to other plant genomes33. Eucalypt toxicity is therefore likely to have exerted selection pressure on the koala’s ability to metabolize such xenobiotics, so we searched for genes encoding enzymes with a detoxification function and investigated sequence evolution at these loci.
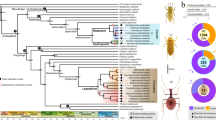
Cytochrome P450 monooxygenase (CYP) genes represent a multi-gene superfamily of heme-thiolate enzymes that play a role in detoxification through phase 1 oxidative metabolism of a range of compounds including xenobiotics34. These genes have been identified throughout the tree of life, including in plants, animals, fungi, bacteria and viruses35. In the koala genome we found two lineage-specific monophyletic expansions of the cytochrome P450 family 2 subfamily C (CYP2Cs, 31 members in koala) (Fig. 1a). The functional importance of these CYP2C genes was further demonstrated through analysis of expression in 15 koala transcriptomes from two koalas, showing particularly high expression in the liver, consistent with a role in detoxification (Supplementary Fig. 6).

a, Phylogenetic tree of CYP2 gene family in koala (Pcin; 31 CYP2 members), compared with marsupials: tammar wallaby (Meug), Tasmanian devil (Shar), gray short-tailed opossum (Mdom); eutherian mammals: human (Hsap), rat (Rnor), mouse (Mmus), dog (Cfam); monotreme: platypus (Oana); and outgroup chicken (Ggal). Two independent monophyletic expansions are seen in koala in the CYP2C subfamily (highlighted by red arcs). b, CYP synteny map showing expansion of CYP2C genes in koala as compared to eutherians (human, dog, rat, mouse) and another marsupial (opossum). c–h, Selection analysis of CYP gene expansion. c, Normalized dN-dS (SLAC (single-likelihood ancestor counting) method) across the alignment of 152 CYP sequences (only sites with data in koala and at least one other species; red bars show sequence depth). Points indicate statistically significant (threshold α = 0.1) evidence for codons under selection. Four sites show positive selection across entire tree (SLAC; green points); 70 sites show episodic selection (MEME (mixed effects model of evolution); blue diamonds). d, Comparison of episodic selection on particular codons across koala CYP genes (n = 31); x axis shows codons with evidence of statistically significant selection anywhere on the tree (identified in c). e, Comparison of mean episodic selection among koala CYP genes (n = 70). Points indicate mean empirical Bayes factor (EBF) for sites under selection for each sequence; error bars, 95% confidence interval. f–h, Mean EBF (natural log transformed, EBF values of 0 excluded) for koala tree tips (n = 31; red) relative to all others (n = 121 in 9 species (see Methods), blue). Points show mean, error bars ± 95% confidence interval, evaluated as 1.96 × s.e.m. (using sequence depth as sample size; red bars in c). Codon positions on x axis refer to multispecies alignment from c. Symbols above each point indicate that the mean value for koala site falls outside the 95% confidence interval for all other species (above, “ + ”; below, “–”; two-tailed test at α = 0.05). Raw statistics shown (unadjusted for multiple comparisons).
Comparing CYP2C gene context in mouse versus koala identified conserved flanking markers strongly suggestive of tandem duplication (Fig. 1b). Further sequence-level analysis of the CYP expansions indicated that most conserved regions are under strong purifying selection (Fig. 1c). However, there is evidence that individual CYP codons have experienced episodic diversifying selection while purifying selection shapes the rest of the gene (Fig. 1c–h, Supplementary Note and Supplementary Tables 12 and 13). Adaptive expansion of CYP2C and maintenance of duplicates appear to have worked in concert, resulting in higher enzyme levels for detoxification while the interplay between purifying and diversifying selection resulted in neofunctionalization within the CYPs. Such adaptations enable koalas to detoxify their highly specialized diet rich in plant secondary metabolites.
The characterization of koala CYP2Cs has significant therapeutic potential. The high expression levels of CYP2C genes in the liver helps to explain why meloxicam, a nonsteroidal anti-inflammatory drug (NSAID) known to be metabolized by the protein product of CYP2C in humans36,37 and frequently used for pain relief in veterinary care, is so rapidly metabolized in the koala and a handful of other eucalypt-eating marsupials (common brushtail possum and eastern ringtail possum) compared with eutherian species37,38. It is expected that other NSAIDs are also rapidly metabolized in koalas and have little efficacy at suggested doses39. Anti-chlamydia antibiotics such as chloramphenicol are degraded rapidly by koalas; treatment with a single dose applicable to humans is insufficient in koalas, which require a daily dose for up to 30 to 45 d. This discovery of CYP2C gene expression levels will inform new research into the pharmacokinetics of medicines in koalas.
Taste, smell and food choice
Like many specialist folivores, koalas are notoriously selective feeders, making food choices both to target nutrients and to avoid plant secondary metabolites40. Koalas have been observed to sniff leaves before tasting them41, and their acute discrimination has been correlated with the complexity and concentration of plant secondary metabolites42. This suggests an important role for olfaction and vomerolfaction, as well as taste. While most herbivores circumvent plant chemical defenses by detoxifying one or a few compounds43, the complexity of eucalyptus plant secondary metabolites, in combination with the terpene expansion in eucalypts, led us to hypothesize that the koala requires enhanced capabilities both in specialist detection and in plant secondary metabolite detoxification. We therefore investigated the genomic basis of the koala’s taste and smell senses, finding multiple gene family expansions that could enhance its ability to make food choices.
We report an expansion of one lineage of vomeronasal receptor type 1 (V1R) genes associated with the detection of nonvolatile odorants (Supplementary Note). There are six such genes in koala, compared with only one in the Tasmanian devil and gray short-tailed opossum, and none found in tammar wallaby, human, mouse, dog, platypus or chicken. The expansion of one lineage of V1R genes is consistent with the koala’s ability to discriminate among diverse plant secondary metabolites.
Surprisingly, given the degree of its dietary specialization, the olfactory receptor genes (n = 1,169) characterized in koala had a gene repertoire that was slightly smaller than that of gray short-tailed opossum (1,431 genes), tammar wallaby (1,660 genes) and Tasmanian devil (1,279 genes) (Supplementary Note). This may be understood in the context of relaxed selection on olfactory receptors among dietary specialists44.
We also report genomic evidence of expansions within the taste receptor families that would enable the koala to optimize ingestion of leaves with a higher moisture and nutrient content in concert with the concentration of toxic plant secondary metabolites in their food plants. The koala’s ability to ‘taste water’ is potentially enhanced by an apparent functional duplication of the aquaporin 5 gene45,46,47 (Supplementary Table 14 and Supplementary Note).
The TAS2R family has a role in ‘bitter’ taste, enabling recognition of structural toxins such as terpenes, phenols and glycosides. These are found in various levels in eucalypts as plant secondary metabolites3,30,31,48. In marsupials, the TAS2R family includes the orthologous repertoires from eutherians, as well as three specific expansions in the last common ancestor shared by all marsupials49,50 (Fig. 2). Large koala-specific duplications in four marsupial orthologous groups have produced a large koala TAS2R repertoire of 24 genes (Fig. 2). The koala has more TAS2Rs than any other Australian marsupial, and among the most of all mammal species49,50, including paralogs of human and mouse receptors whose agonists are toxic glycosides (Supplementary Table 15 and Supplementary Note). The TAS1R gene families, responsible for sweet taste and umami amino acid perception, have previously been reported as pseudogenized in eutherians with highly specialized diets, such as the giant panda51. In the koala, however, we found that all TAS1R genes are putatively functional (Supplementary Fig. 7).

TAS2R genes are responsible for bitter taste perception. a, Maximum-likelihood tree of TAS2Rs (including pseudogenes) in the four marsupials, where the sequences contained 250 amino acids. 28 representative TAS2Rs of orthologous gene groups (OGGs) in eutherians (red circles) and 7 platypus TAS2Rs (gray circles) were also used. There were 27 distinct marsupial OGGs (supported by ≥99% bootstrap values), where the nodes of OGG clades are indicated by white open circles. Bootstrap values of ≥70% in the nodes connecting OGG clades are indicated by asterisks. There are three marsupial-specific clusters (I, II and III) where massive expansion events occurred in the common ancestor of marsupials after their split from eutherian ancestors. b–e, Reconstructed maximum-likelihood trees of TAS2R orthologs in which there are more than two duplicates of koala TAS2Rs: b, TAS2R41; c, TAS2R705; d, TAS2R710; and e, TAS2R720. Genomic structures of the umami and sweet taste receptor TAS1Rs were also analyzed and found to be functional in koala (see Supplementary Note).
Genomics of an induced ovulator
Koala reproduction is of particular interest because the koala is an induced ovulator52, with key genes controlling female ovulation (LHB, FSHB, ERR1, ERR2), as well as prostaglandin synthesis genes important in parturition and ejaculation (PTGS1, PTGS2, PTGS3) (Supplementary Note). We identified genes putatively involved in the induction of ovulation in the female by male seminal plasma (NGF), and in coagulation of seminal fluid (ODC1, SAT1, SAT2, SMOX, SRM, SMS) (Supplementary Note), which may function to prevent sperm leakage from the female reproductive tract in this arboreal species.
Genomic characterization of koala milk
A koala young is about the size of a kidney bean and weighs < 0.5 g. It crawls into the mother’s posteriorly opening pouch and attaches to a teat, where it remains for 6–7 months. It continues to suck after it has left the pouch until about a year old.
Analysis of the genome, in conjunction with a mammary transcriptome and a milk proteome, enabled us to characterize the main components of koala milk (Supplementary Fig. 8, Supplementary Table 16, Supplementary Note and ref. 53). The high-quality assembly of the genome allowed both the identification of marsupial-specific genes and determination of their evolutionary origins based on their genomic locations. For instance, we found that there are four Late Lactation Protein (LLP) genes tightly linked to both trichosurin and β-lactoglobulin (Supplementary Fig. 8), potentially allowing marsupials to fine-tune milk protein composition across the stages of lactation to meet the changing needs of their young. Additionally, the koala marsupial milk 1 (MM1) gene, a novel marsupial gene, is located close to the gene encoding very early lactation protein (VELP), an ortholog of Glycam1 (or PP3) that encodes a eutherian antimicrobial protein53 (Supplementary Fig. 8). In eutherians, this region contains an array of short glycoproteins that have antimicrobial properties and are found in secretions such as milk, tears and sweat. We propose that MM1 has an antimicrobial role in marsupial milk, along with three other short novel genes located in the same region. We also detected expansions in another antimicrobial gene family, the cathelicidins.
Koala immunome and disease
At the time of European settlement, koalas were widespread in eastern mainland Australia, from north Queensland to the southeastern corner of South Australia. Today they are mainly confined to the east coast and are listed as ‘vulnerable’ under Australia’s Environment Protection and Biodiversity Conservation Act 199954. There is strong evidence to suggest that some fragmented populations of koalas are already facing extinction, particularly in formerly densely populated koala territories in southeast Queensland and northern New South Wales. A major challenge for the conservation of these declining koala populations is the high prevalence of disease, especially that caused by the obligate intracellular bacterial pathogen Chlamydia pecorum, which is found across the geographic range, with the exception of some offshore islands55. A main challenge for managing these populations has been the lack of knowledge about the koala immune response to disease. Recent modeling suggests the best way to stabilize heavily affected koala populations is to target disease56.
The long-read-based genome enabled the de novo assembly of complex, highly duplicated immune gene families and comprehensive annotation of immune gene clusters53,57,58. These include the major histocompatibility complex (MHC)59, as well as T cell receptors (TCR), immunoglobulin (IG) (Supplementary Fig. 9, Supplementary Tables 17 and 18, and Supplementary Note), natural killer cell (NK) receptor58 and defensin60 gene clusters. Together these findings provide a starting point for new disease research and allow us to interrogate the immune response to the most significant pathogen of the koala, C. pecorum.
Of the more than 1,000 koalas arriving annually at wildlife hospitals in Queensland and New South Wales, 40% have late-stage chlamydial disease and cannot be rehabilitated. Annotation of koala immune genes enabled us to study variation within candidate genes known to play a role in resistance and susceptibility to chlamydia infection in other species (Supplementary Tables 18–20). Preliminary case/control association tests for five koalas involved in a chlamydia vaccination trial showed that the MHCII DMA and DMB genes, as well as the CD8-a gene, may be involved in differential immune responses to chlamydia vaccine (Supplementary Table 21 and Supplementary Note). We also conducted differential expression analysis of RNA sequencing (RNA-seq) data from conjunctival tissue collected from koalas at necropsy, both with and without signs of ocular chlamydiosis, showing that in diseased animals, 1,508 of the 26,558 annotated genes (5.7%) were twofold upregulated, while 685 (2.6%) were downregulated by greater than twofold when compared with healthy animals (Supplementary Fig. 9 and Supplementary Note). In diseased animals, upregulated genes were associated with Gene Ontology (GO) terms for a range of immunological processes, including signatures of leukocyte infiltration (Supplementary Fig. 9). Immune responses in the affected conjunctivas were directed at TH1 rather than TH2 responses. Proinflammatory mediators such as CCL20, IL1α, IL1β, IL6 and SSA1 were also upregulated. As in human trachoma, this cascade of proinflammatory products may help to clear the infection but may also lead to tissue damage in the host61. Furthermore, resolution of human trachoma infection is thought to require a IFN-γ driven TH1 response62, and in diseased koalas we found that IFN-γ was upregulated 4.7-fold in the conjunctival tissue. These annotated koala immune genes will now help us to define features of protective versus pathogenic immunological responses to the disease and may be invaluable for effective vaccine design.
Koala genomes are undergoing genomic invasion by koala retrovirus (KoRV)63, which is spreading from the north of the country to the south. Both endogenous (germline transmission) and exogenous (infectious ‘horizontal’ transmission) forms are extant64. Our results provide a comprehensive view of KoRV insertions in the koala genome. We found a total of 73 insertions in the phaCin_unsw_4.1 assembly (Supplementary Table 22). It is likely that most of these 73 loci are endogenous, consistent with our observation of integration breakpoint sequences that are shared with one or both of the other koala genomes reported (Supplementary Tables 23 and 24).
We investigated the sites of KoRV insertion to define their proximity to protein-coding genes and explore possible disruptions. This analysis identified insertions into 24 protein-coding genes (Supplementary Table 25). However, none is likely to disrupt protein-coding capacity, since 22 insertions are in introns and the other two are in 3′ untranslated regions. Transcription proceeding from the proviral long terminal repeat (LTR) could possibly affect the transcription of the host genes.
Understanding the genetics of host resistance to chlamydia and the etiology of the retrovirus will help inform the development of vaccines against both diseases, as well as translocation strategies.
Genome-informed conservation
Broad-scale population management of koalas is critical to conservation efforts. This is challenging because distribution models are not easily generalized across bioregions, and further complicated by the unique regional conservation issues described above. Since it is not possible to generalize management, it is imperative that decisions are informed by empirical data relevant to each bioregion.
Analysis of the koala genome provided the unique opportunity to combine historical evolutionary data with high-resolution contemporary population genomic markers to address these management challenges. To infer the ancient demographic history of the species, we analyzed the long-read reference genome and short-read data from two other koalas, using the pairwise sequentially Markovian coalescent (PSMC) method65 (Fig. 3a, Supplementary Fig. 10 and Methods). The data show that the modern koala, which appeared in the fossil record 350,000 years ago2, underwent an initial increase in population, followed by a rapid and widespread decrease in population size ~30,000–40,000 years ago. This is consistent with fossil evidence of rapid declines in multiple Australian species, including the extinct megafauna, 40,000–50,000 years ago66 and 30,000–40,000 years ago67. The koala was thus one of a number of species affected by decline during this time that did not ultimately become extinct67.

a, Top: plot of surface temperature (temp.) over past 3 million years based on a five-point running mean of δ18O data76. Bottom: population demographic history inferred from diploid sequences of three koalas (females ‘Pacific Chocolate’ and ‘Bilbo’, male ‘Birke’) using the pairwise sequential Markovian coalescent (PSMC) method. Koala silhouette indicates earliest fossil record of modern koala2. Gray shading, human arrival in Australia77 (see ref. 78); red circles, estimated extinction times of 16 megafaunal genera in mainland Australia79; aqua area, last glacial period; vertical dashed green line, last glacial maximum; vertical solid black line, first koala population declines 40,000 years ago. Dark colored lines are estimated from genome data; lighter lines, plots inferred from 100 bootstrap replicates. A mutation rate of 1.45 × 10−8 mutations per site per generation and 7-year generation time were assumed. b, Right, principal component (PC) analysis (including 95% inertia ellipses) of 1,200 SNPs in 49 wild koalas from throughout Australia. Left, geographic clustering of wild koalas in eastern Australia in relation to proposed biogeographic barriers68,72, highlighting known historic barriers to gene flow, the Brisbane and Clarence River Valleys, but also suggesting a role for the Hunter Valley. The cluster of genetically similar southern koalas reflects a recent history of widespread translocation8. c, Average inbreeding coefficient (F) (calculated by TrioML80,81) of 49 wild koalas. Qld, Queensland; SE, southeast; NSW, New South Wales. P values arising from linear modeling represent significant differences in mean F between regions (***P < 0.001; **P < 0.01). There is a high correlation between geographic distance and genetic distance (Mantel test: r2 = 0.4898), indicating that genetic rescue between populations is feasible. Center lines, median; box limits, upper and lower quartiles. Upper whisker = min(max(x), Q_3 + 1.5 × IQR), lower whisker = max(min(x), Q_1 – 1.5 × IQR); i.e., upper whisker = upper quartile + 1.5 × box length, lower whisker = lower quartile – 1.5 × box length; circles, outliers. Linear modeling indicated that mean F differed significantly between several regions (Mid-coast NSW–Southern Australia, P = 0.000524; Qld–Southern NSW, P = 0.00237; Qld–Southern Australia, P = 0.00000107; SE Qld–Southern Australia, P = 0.006596).
Distinct PSMC profiles of the koalas from two geographic areas and their failure to coalesce suggests some regional differences in koala populations, including impediments to gene flow (Fig. 3a). Regional differentiation was also detected in analyses of mtDNA68,69, although over a shorter time scale.
We analyzed populations of recent koala samples using 1,200 SNPs derived from targeted capture libraries mapped to the koala genome (Supplementary Note). We found notable levels of genetic diversity with limited fine-scale differentiation consistent with long-term connectivity across regions. We found evidence of low genetic diversity in southern koalas, consistent with a recent history of sequential translocations8,68,70,71 (Fig. 3b,c). At a continental scale, we show biogeographic barriers to gene flow associated with the Brisbane Valley and Clarence River, as identified by mtDNA studies68,72, and find a barrier associated with the Hunter Valley, which was not previously known in koalas (Fig. 3b). Levels of inbreeding varied across regions (Fig. 3c), but the northern populations most under threat in New South Wales and Queensland show high levels of genetic diversity.
The information generated here provides a foundation for a conservation management strategy to maintain gene flow regionally while incorporating the genetic legacy of biogeographic barriers. Furthermore, the contrast in genome-wide levels of diversity between southern and northern populations highlights the detrimental consequences of the unmonitored use of small isolated populations as founders for reestablishing and/or rescuing of populations on genome-wide levels of genetic diversity. Low levels of genetic diversity in southern koalas have been associated with genetic abnormalities consistent with inbreeding depression, including testicular abnormalities73.
Now that we understand the consequences of past translocations, and the existing genetic structure, it is clear that maintaining and facilitating gene flow via habitat connectivity will be the most effective means of ensuring genetically healthy koala populations over the long term. However, where more intensive measures such as translocation are required to rescue genetically depauperate southern populations, these tools and data provide the basis for decisions that maximize benefits while minimizing risks74,75. Future utility of these SNPs will also include tracking of individual pedigrees in captive koala populations and in those wild populations being intensively monitored.
The koala genome offers insights into historic and contemporary population dynamics, providing evolutionary and genetic context for a species that is the focus of considerable management actions and resources. By providing a deeper understanding of disease dynamics and population genetic processes, including the maintenance and monitoring of gene flow, this genomic information will enable the development of strategies necessary to preserve the species, from the preservation of habitat corridors through to the genetic rescue of isolated populations. As members of government advisory committees, some of the authors have initiated inclusion of genomic information into the New South Wales Koala Strategy. This will be used to inform koala management in the state with the goal of securing koalas in the wild for the future.
Discussion
The koala genome provides the highest quality marsupial genome to date. This assembly has enabled insights into the colonization of the koala genome by an exogenous retrovirus and revealed the architecture of the immune system, necessary to study and treat emerging diseases that threaten koala populations. A greater understanding of genetic diversity across the species will guide the selection of individuals from genetically healthy northern populations to augment genetically restricted populations in the south, bearing in mind that chlamydia has not been detected on some offshore islands, so risk assessment should be carried out before embarking on translocations. Sequencing the genome has advanced our understanding of the unique biology of the koala, including detoxification pathways and innovations in taste and smell to enable food choices in an obligate folivore. Long-term survival of the species depends on understanding the impacts of disease and management of genetic diversity, as well as the koala’s ability to source moisture and select suitable foraging trees. This is particularly important given the koala’s narrow food range, which makes it especially vulnerable to a changing climate. The genome provides a springboard for conservation of this biologically unique and iconic Australian species.
URLs
FALCON assembly algorithm, https://github.com/PacificBiosciences/FALCON-integrate/; FALCON (v 0.3.0), http://falconframework.org/; RepeatMasker (v 4.0.3), http://www.repeatmasker.org/; RepeatModeler, http://www.repeatmasker.org/RepeatModeler/; RepBase (v 2015-08-07), http://www.girinst.org/repbase/; MAKER, http://www.yandell-lab.org/software/maker.html; Trinity (v 2.3.2), https://github.com/trinityrnaseq/trinityrnaseq/; SNAP, http://archive.broadinstitute.org/mpg/snap/; GeneMark, http://opal.biology.gatech.edu/GeneMark/; Augustus, http://bioinf.uni-greifswald.de/augustus/; NCBI Blast (v 2.3.0), https://blast.ncbi.nlm.nih.gov/Blast.cgi; OrthoMCL (v 2.0.9), http://orthomcl.org/orthomcl/; MAFFT (v 7.2.71), https://mafft.cbrc.jp/alignment/software/; TreeBeST (v 1.9.2), http://treesoft.sourceforge.net/treebest.shtml; HyPhy, https://veg.github.io/hyphy-site/; Datamonkey, http://www.datamonkey.org/; STAR, http://star.mit.edu/genetics/; featureCounts, http://bioinf.wehi.edu.au/featureCounts/; DESeq2, https://bioconductor.org/packages/release/bioc/html/DESeq2.html; SARTools, https://github.com/PF2-pasteur-fr/SARTools/; Dotter, https://sonnhammer.sbc.su.se/Dotter.html; GATK (v 3.3-0-g37228af), https://software.broadinstitute.org/gatk/; KAT comp, https://github.com/TGAC/KAT/; BUSCO (v 2), http://busco.ezlab.org/; Trimmomatic (v 0.36 PE), http://www.usadellab.org/cms/?page = trimmomatic; Bowtie2 (v 2.2.4), http://bowtie-bio.sourceforge.net/bowtie2/index.shtml; MACS2 (v 2.0.10.20131216), https://github.com/taoliu/MACS/; R (v 3.2.5), https://www.r-project.org/; gplots (v 3.0.1), https://cran.r-project.org/web/packages/gplots/index.html; bedtools (v 2.25.0), http://bedtools.readthedocs.io/en/latest/; kSamples (v 1.2-4), https://cran.r-project.org/web/packages/kSamples/index.html; ggbiplot (v 0.55), https://github.com/vqv/ggbiplot/; Tandem Repeats Finder, https://tandem.bu.edu/trf/trf.html; seqLogo, https://bioconductor.org/packages/release/bioc/html/seqLogo.html; RNAfold, http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi; UniProt/Swiss-Prot, http://www.uniprot.org/; dammit!, https://dammit.readthedocs.io/en/refactor-1.0/; Transfuse, https://github.com/cboursnell/transfuse/; GMAP, http://research-pub.gene.com/gmap/; Trim Galore!, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/; Kallisto, https://pachterlab.github.io/kallisto/; Sleuth, https://pachterlab.github.io/sleuth_walkthroughs/trapnell/analysis.html; All-vsl-all BLASTP (version 2.2.30+ ), https://blast.ncbi.nlm.nih.gov/Blast.cgi; MUSCLE (v 3.8.31), https://www.drive5.com/muscle/; HMMER suit (v 3.1b1 May 2013), http://hmmer.org/; FASTASEARCH (v 36.8.8), https://www.ebi.ac.uk/Tools/sss/fasta/; Integrative Genomics Viewer (IGV) (v 2.3.97), https://github.com/ssadedin/IGV-CRAM/; MEGA (v 7.0.18), https://www.megasoftware.net/; RAxML (v 8.2.11), https://sco.h-its.org/exelixis/web/software/raxml/index.html; Burrows-Wheeler aligner (v 0.7.15), http://bio-bwa.sourceforge.net/; Samtools (v 1.3), http://www.htslib.org/; Geneious (v 10.2.3), https://www.geneious.com/; Coancestry, https://www.zsl.org/science/software/coancestry/; PLINK (v 1.07), http://zzz.bwh.harvard.edu/plink/.
Methods
General methods
A full description of the Methods can be found in the Supplementary Note. No statistical methods were used to predetermine sample size.
Genome sequencing and assembly of the koala reference genome
Sequencing
Samples were obtained as part of veterinary care at the Port Macquarie Koala Hospital and Australia Zoo Wildlife Hospital, and from the Australian Museum Tissue Collection. Sample collection was performed in accordance with methods approved by the Australian Museum Animal Ethics Committee (permit numbers 11–03 and 15–05). “Pacific Chocolate” (Australian Museum registration M.45022), a female from Port Macquarie in northeast New South Wales, was sampled immediately after euthanasia by veterinary staff at the Port Macquarie Koala Hospital (27 June 2012), following unsuccessful treatment of severe chlamydiosis. Two koalas from southeast Queensland—a female, “Bilbo” (Australian Museum registration M.47724), from Upper Brookfield, and a male, “Birke”, from Birkdale—were sampled following euthanasia due to severe chlamydiosis (20 August 2015) and severe injuries (26 August 2012), respectively. High molecular weight (HMW) DNA was extracted from heart tissue for Pacific Chocolate and kidney tissue for Birke using the DNeasy Blood and Tissue kit (Qiagen), with RNaseA (Qiagen) treatment. HMW DNA from Bilbo was extracted for PacBio sequencing from spleen tissue using Genomic-Tip 100/G columns (Qiagen), DNA Buffer set (Qiagen) and RNaseA (Qiagen) treatment. Fifteen SMRTbell libraries were prepared (RCG) as per the PacBio 20-kb template preparation protocol, with an additional damage repair step performed after size selection. A minimum size cutoff of 15 or 20 kb was used in the size selection stage using the Sage Science BluePippin system. The libraries were sequenced on the Pacific Biosciences RS II platform (Pacific Biosciences) employing P6 C4 chemistry with either 240 min or 360 min movie lengths. A total of 272 SMRT Cells were sequenced to give an estimated overall coverage of 57.3 × based on a genome size of 3.5 Gbp. A TruSeq DNA PCR free library was constructed with a mean library insert size of 450 bp. 400,473,997 paired-end reads were generated yielding a minimum coverage of 34 × . HMW gDNA was sequenced on an Illumina 150bpPE HiSeq X Ten sequencing run (Illumina)
Assembly
An overlapping layout consensus assembly algorithm, FALCON (v 0.3.0) (see URLs), was used to generate the draft genome using PacBio reads. Total genome coverage before assembly was estimated by total bases from reads divided by 3.5 Gbp genome size. The estimated total coverage is 57.3 × . FALCON leverages error-corrected long seed reads to generate an overlapping layout consensus representation of the genome. Approximately 23 × of long reads are required by FALCON as seed reads, and the rest are used for error correction. The seed read length of the reads at the 60% percentile was calculated as 10,889 bp. The FALCON assembly was run on Amazon Web Service Tokyo region using r3.8xlarge spot instances as compute node, with the number of instances varying from 12 to 20 depending on availability.
After filtering low-quality and duplicate reads, approximately 57.3-fold long-read coverage was used for assembly. The primary contigs from the FALCON v 0.3.0 assembly (representing homozygous regions of the genome) yielded genome version phaCin_unsw_v4.1. This comprised 3.19 Gb, including 1,906 contigs with an N50 of 11.6 Mb and sizes ranging up to 40.6 Mb. The heterozygous regions of the genome (representing the alternative contigs from the assembly) were a total of 230 Mb, with an N50 of 48.8 Kb (Supplementary Table 2). Approximately 30-fold coverage of Illumina short reads was used to polish the assembly with Pilon86.
BUSCO analysis on the draft assembly was run against the mammalian ortholog database with the –long parameter on all genomes under comparison. This initial analysis showed the assembly only reached about 60% of genome completeness, suggesting a high number of indels in the draft genome. The genome polishing tool Pilon86 was employed to improve draft assembly from FALCON. About 30 × of 150 bp paired-end Illumina X Ten short reads from Bilbo was used as an input for this polishing process, which was run on a compute cluster provided by Intersect Australia Limited.
We implemented the method of Deakin et al.18 for super-scaffolding. Briefly, tables of homologous genes were generated using the physical order of genes on the chromosomes of gray short-tailed opossum and tammar wallaby as references and koala phaCin_unsw_v4.1 (Bilbo) as target (Supplementary Table 4).
Analysis of centromeric regions and repeat structure
Repeat content was called using RepeatMasker with combined RepBase libraries (v 2015-08-07) and RepeatModeller calls generated from the genome assemblies. The resulting calls were then filtered using custom Python scripts to remove short fragments (see “Code availability”) and combine tandem or overlapping repeat calls. To characterize the centromeric regions of the genome, chromatin immunoprecipitation (ChIP) was performed using the Invitrogen MAGnify Chromatin Immunoprecipitation System (Revision 6). Repeat content of the centromeric regions was determined using RepBase annotated marsupial repeats and output from RepeatModeller analysis of koala. RepeatMasker was used to locate repeats. Candidate centromeric segments were identified using two sliding window analyses, with window sizes of 200 kb and 20 kb and step sizes of 100 kb and 10 kb, respectively. Small tandem repeats were discovered in koala RSX sequence using the Tandem Repeat Finder program87, using +2, –3, and –7 as scores for match, mismatch and gap opening, respectively. Alignments of consensus repeat units with the RSX sequence were processed to obtain nucleotide frequency at each position.
Genome annotation and gene family analysis
Annotations were generated using the automated genome annotation pipeline MAKER88,89]. We masked repeats in the assembly by providing MAKER with a koala-specific repeat library generated with RepeatModeler90, against which RepeatMasker (v 4.0.3)91 queried genomic contigs. Gene annotations were made using a protein database combining the UniProt/Swiss-Prot92 protein database, all sequences for human (Homo sapiens), gray short-tailed opossum (Monodelphis domestica), Tasmanian devil (Sarcophilus harrisii) and tammar wallaby (Notamacropus eugenii) from the NCBI protein database93, and a curated set of marsupial and monotreme immune genes94. We downloaded all published koala mRNAseq reads from SRA (PRJNA230900, PRJNA327021) and reassembled de novo male, female and mammary transcriptomes using the default parameters of Trinity v 2.3.295. Each assembly was filtered such that contigs accounting for 90% of mapped reads were passed to MAKER as homologous transcript evidence. Ab initio gene predictions were made using the programs SNAP96, Genemark97 and Augustus98. Three iterative runs of MAKER were used to produce the final gene set.
Gene families were called using NCBI Blast (2.3.0) OrthoMCL (2.0.9)99. The protein sequences of genes belonging to orthogroups identified by OrthoMCL were aligned using MAFFT (7.2.71)100 and the gene tree was inferred using TreeBeST (1.9.2)101 providing a species tree to guide the phylogenetic reconstruction. Custom scripts (see “Code availability”) were applied to identify families with expansion within the koala, Diprotodontia, Australidelphia and marsupial lineages.
Sequence evolution
Sequence evolution on specific gene families was conducted on the cytochrome P450 (CYP), vomeronasal receptor (V1R), olfactory receptor (OR), aquaporin and taste receptor genes (Supplementary Note). Genes involved in koala development and reproduction and lactation were also characterized (Supplementary Note). Koala MHC, TCR and IGG genes were annotated and analyzed for expression between diseased and healthy animals (Supplementary Note). Evidence of selection across CYP and V1R genes was evaluated (Supplementary Note) using multispecies alignments (N = 152 and 8 sequences, respectively) in HyPhy102, hosted by the Datamonkey webserver103.
RNA-seq analysis of koala conjunctival tissue samples
Conjunctival tissue samples were collected from 26 koalas euthanized due to injury or disease by veterinarians at Australia Zoo Wildlife Hospital, Currumbin Wildlife Hospital and Moggill Koala Hospital. The collection protocol was approved by the University of the Sunshine Coast Animal Ethics Committee (AN/S/15/36). Health assessments of the eye were performed by an experienced veterinarian and classified as either ‘healthy’ (N = 13) or ‘diseased’ (N = 13) based on evidence of gross pathology consistent with ocular chlamydiosis55. Conjunctival tissue samples from each animal were placed directly in RNALater (Qiagen, Germany) buffer overnight at 4 °C before storing at –80 °C for later use. RNA was extracted using an RNeasy Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions, with an on-column DNase treatment to eliminate contaminating DNA from the sample. The concentration and quality of the isolated RNA was determined using a NanoDrop ND-1000 160 Spectrophotometer and Agilent BioAnalyzer (Agilent, USA). Library construction and sequencing were performed by the Ramaciotti Centre (UNSW, Kensington, NSW) with TruSeq stranded mRNA chemistry on a NextSeq500 (Illumina, USA). Reads were mapped to the phCin_unsw_v4.1 assembly using the default parameters of STAR104 and counts summed over features using featureCounts105. Differentially expressed genes were called using DESeq2106 as implemented in the SARTools package107.
Koala retrovirus (KoRV)
We searched for KoRV sequences within the scaffolds of the phaCin_unsw v4.1 assembly of the Bilbo genome sequence, and also within alternative contig sequences before their correction by Pilon (since we noticed that in a few cases KoRV sequences were removed in the course of the sequence polishing process). KoRV sequences were found by using the program blastn108 to search with KoRV genome reference sequences (GenBank AF151794 and AB721500). Search results were converted to BED format and the KoRV and recKoRV components of each read were merged with the program mergeBed. KoRV insertions within genes were identified using the program intersectBed109. Pre-integration allelic sequences were found by using blastn108 to search the phaCin_unsw v4.1 genome sequence assembly with sequences flanking KoRV/recKoRV integrations as queries. In two cases the expected allelic sequence was not present in the Bilbo genome, but was found by searching the genome of another koala (Pacific Chocolate). To check the expected relationship between pairs of allelic sequences, we inspected dot plot alignments of representative sequences (not shown) created with the program dotter110.
Koala population genomics: historical population size
Demographic history was inferred from the diploid sequence of each of the three koalas, using a pairwise sequential Markovian coalescent (PSMC) method65. We conducted a range of preliminary analyses and found that PSMC plots were not sensitive to the values chosen for the maximum number of iterations (N), the number of free atomic time intervals (p), the maximum time to the most recent common ancestor (t), and the initial value of ρ. Based on these investigations, our final PSMC analyses of the three genome sequences used values of N = 25, t = 5, ρ = 1 and p = 4 + 25 × 2 + 4 + 6. The number of atomic time intervals is similar to that recommended for analyses of modern human genomes65, which are similar in size to the koala genomes. We determined the variance in estimates of Ne using 100 bootstrap replicates. Replicate analyses in which we varied the values of p, t and ρ produced PSMC plots that were broadly similar to those using our chosen ‘optimal’ settings (Supplementary Fig. 10).
The plots of demographic history were scaled using a generation length of 7 years, corresponding to the midpoint of the range of 6 to 8 years estimated for the koala111 and the midpoints of the estimates of the human mutation rate (1.45 × 10−8 mutations per site per generation; summarized by ref. 112) and mouse mutation rate (5.4 × 10−9 mutations per site per generation113) were applied in the absence of a mutation rate estimate for koala (Supplementary Fig. 10). The koala mutation rate is likely to be closer to that of humans, based on greater similarity in genome size, life history, and effective population size, relative to mouse112.
Koala population genomics: contemporary population analysis
Forty-nine koalas were sampled throughout the distribution using a hierarchical approach to allow examination of genetic relationships at a range of scales, from familial to range-wide. All individuals were sequenced using a target capture approach described in ref. 114, with a kit targeting 2,167 marsupial exon sequences. Illumina sequence reads were quality-filtered and trimmed (see ref. 114 for details) and mapped to the koala genome (Bowtie2, v2.2.4115). A panel of 4,257 SNP sites was identified (using GATK version 3.3-0-g37228af116) that showed expected levels of relatedness and differentiation among the sampled individuals. A panel of 1,200 SNPs (obtained by mapping to targets, filtering, and selecting one SNP per target) showed fine-scale regional differentiation consistent with evolutionary history and recent population management (Fig. 3).
Statistics and reproducibility
In Fig. 1e, points shown indicate the mean empirical Bayes factor (EBF) for sites under selection; error bars, 95% confidence interval. In Fig. 1f–h, 95% confidence intervals are calculated as 1.96 × s.e.m. (sample size is sequence depth, as indicated by red bars in Fig. 1c).
In Fig. 3c, center lines indicate median and box limits indicate upper and lower quartiles. Upper whisker = min(max(x), Q_3 + 1.5 × IQR), lower whisker = max(min(x), Q_1 – 1.5 × IQR); i.e., upper whisker = upper quartile + 1.5 × box length, lower whisker = lower quartile – 1.5 × box length. Circles indicate outliers. Linear modeling indicated that mean F differed significantly between several regions (Midcoast New South Wales–Southern Australia, P = 0.000524; Queensland–Southern New South Wales, P = 0.00237; Queensland–Southern Australia, P = 0.00000107; Southeast Queensland–Southern Australia, P = 0.006596).
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Code availability
(1) Custom scripts to identify gene families with expansion within the koala, Diprotodontia, Australidelphia and marsupial lineages; (2) custom scripts to identify refined repeat calls; and (3) code used to generate SNP genotypes from exon capture data are available at https://github.com/DrRebeccaJ/KoalaGenome.
Data availability
The Phascolarctos cinereus BioSamples are as follows: Bilbo 61053, SAMN06198159; Pacific Chocolate, SAMEA91939168; Birke. SAMEA103910665. Koala Genome Consortium Projects for the Koala Whole Genome Shotgun project and genome assembly are registered under the umbrella BioProject PRJEB19389 (union of PRJEB5196 and PRJNA359763).
Transcriptome data are submitted under PRJNA230900 (adrenal, brain, heart, lung, kidney, uterus, liver and spleen) and PRJNA327021 (milk and mammary gland). Illumina short-read data for Birke is submitted under PRJEB19982.
The Bilbo 61053 assembly described in this paper is version MSTS01000000 and consists of sequences MSTS01000001–MSTS01001906. For the Bilbo assembly Illumina X Ten reads are submitted under PRJEB19457 and PacBio reads under PRJEB19889.
ChIP-seq data have been deposited under BioProject PRJNA415832 and GEO GSE111153.
References
Meredith, R. W., Krajewski, C., Westerman, M. & Springer, M. S. Relationships and divergence times among the orders and families of Marsupialia. Mus. North. Ariz. Bull. 65, 383–406 (2009).
Black, K. H., Price, G. J., Archer, M. & Hand, S. J. Bearing up well? Understanding the past, present and future of Australia’s koalas. Gondwana Res. 25, 1186–1201 (2014).
Gleadow, R. M., Haburjak, J., Dunn, J. E., Conn, M. E. & Conn, E. E. Frequency and distribution of cyanogenic glycosides in Eucalyptus L’Hérit. Phytochemistry 69, 1870–1874 (2008).
Nagy, K. & Martin, R. Field metabolic rate, water flux, food consumption and time budget of koalas, Phascolarctos cinereus (Marsupialia: Phascolarctidae) in Victoria. Aust. J. Zool. 33, 655–665 (1985).
Woinarski, J. C., Burbidge, A. A. & Harrison, P. L. Ongoing unraveling of a continental fauna: decline and extinction of Australian mammals since European settlement. Proc. Natl. Acad. Sci. USA 112, 4531–4540 (2015).
Adams-Hosking, C. et al. Use of expert knowledge to elicit population trends for the koala (Phascolarctos cinereus). Divers. Distrib. 22, 249–262 (2016).
McAlpine, C. et al. Conserving koalas: a review of the contrasting regional trends, outlooks and policy challenges. Biol. Conserv. 192, 226–236 (2015).
Martin, R. & Handasyde, K. A. The Koala: Natural History, Conservation and Management. (UNSW Press: Sydney, New South Wales, Australia (1999).
Hrdina, F. & Gordon, G. The koala and possum trade in Queensland, 1906–1936. Aust. Zool. 32, 543 (2004).
Menkhorst, P. Hunted, marooned, re-introduced, contracepted: a history of koala management in Victoria. in Too Close for Comfort: Contentious Issues in Human–Wildlife Encounters (eds. Lunney, D. et al.) 73–92 (Royal Zoological Society of NSW, Mosman, New South Wales, Australia, 2008).
Seymour, A. M. et al. High effective inbreeding coefficients correlate with morphological abnormalities in populations of South Australian koalas (Phascolarctos cinereus). Anim. Conserv. 4, 211–219 (2001).
Simmons, G., Clarke, D., McKee, J., Young, P. & Meers, J. Discovery of a novel retrovirus sequence in an Australian native rodent (Melomys burtoni): a putative link between gibbon ape leukemia virus and koala retrovirus. PLoS One 9, e106954 (2014).
Alfano, N. et al. Endogenous gibbon ape leukemia virus identified in a rodent (Melomys burtoni subsp.) from Wallacea (Indonesia). J. Virol. 90, 8169–8180 (2016).
Tarlinton, R. E., Meers, J. & Young, P. R. Retroviral invasion of the koala genome. Nature 442, 79–81 (2006).
Xu, W. et al. An exogenous retrovirus isolated from koalas with malignant neoplasias in a US zoo. Proc. Natl. Acad. Sci. USA 110, 11547–11552 (2013).
Taylor-Brown, A. & Polkinghorne, A. New and emerging chlamydial infections of creatures great and small. New Microbes New Infect. 18, 28–33 (2017).
Hayman, D. Marsupial cytogenetics. Aust. J. Zool. 37, 331–349 (1989).
Deakin, J. E. et al. Anchoring genome sequence to chromosomes of the central bearded dragon (Pogona vitticeps) enables reconstruction of ancestral squamate macrochromosomes and identifies sequence content of the Z chromosome. BMC Genomics 17, 447 (2016).
Brown, J.D. & O’Neill, R.J. The evolution of centromeric DNA sequences. Encyclopedia of Life Sciences https://doi.org/10.1002/9780470015902.a0020827.pub2 (Wiley, Hoboken, NJ, USA, 2014).
Carone, D. M. et al. A new class of retroviral and satellite encoded small RNAs emanates from mammalian centromeres. Chromosoma 118, 113–125 (2009).
Earnshaw, W. C. & Rothfield, N. Identification of a family of human centromere proteins using autoimmune sera from patients with scleroderma. Chromosoma 91, 313–321 (1985).
O’Neill, R. J. W., O’Neill, M. J. & Graves, J. A. M. Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393, 68–72 (1998).
Nagaki, K. et al. Sequencing of a rice centromere uncovers active genes. Nat. Genet. 36, 138–145 (2004).
Zhang, Y. et al. Structural features of the rice chromosome 4 centromere. Nucleic Acids Res. 32, 2023–2030 (2004).
Carbone, L. et al. Centromere remodeling in Hoolock leuconedys (Hylobatidae) by a new transposable element unique to the gibbons. Genome Biol. Evol. 4, 648–658 (2012).
Grant, J. et al. Rsx is a metatherian RNA with Xist-like properties in X-chromosome inactivation. Nature 487, 254–258 (2012).
Hobbs, M. et al. A transcriptome resource for the koala (Phascolarctos cinereus): insights into koala retrovirus transcription and sequence diversity. BMC Genomics 15, 786 (2014).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Foley, W. J. & Moore, B. D. Plant secondary metabolites and vertebrate herbivores–from physiological regulation to ecosystem function. Curr. Opin. Plant Biol. 8, 430–435 (2005).
Eschler, B. M., Pass, D. M., Willis, R. & Foley, W. J. Distribution of foliar formylated phloroglucinol derivatives amongst Eucalyptus species. Biochem. Syst. Ecol. 28, 813–824 (2000).
Pass, G. J., McLean, S., Stupans, I. & Davies, N. Microsomal metabolism of the terpene 1,8-cineole in the common brushtail possum (Trichosurus vulpecula), koala (Phascolarctos cinereus), rat and human. Xenobiotica 31, 205–221 (2001).
Ngo, S. N. T., McKinnon, R. A. & Stupans, I. Cloning and expression of koala (Phascolarctos cinereus) liver cytochrome P450 CYP4A15. Gene 376, 123–132 (2006).
Myburg, A. A. et al. The genome of Eucalyptus grandis. Nature 510, 356–362 (2014).
Kirischian, N., McArthur, A. G., Jesuthasan, C., Krattenmacher, B. & Wilson, J. Y. Phylogenetic and functional analysis of the vertebrate cytochrome P450 2 family. J. Mol. Evol. 72, 56–71 (2011).
Nelson, D. R. The cytochrome P450 homepage. Hum. Genomics 4, 59–65 (2009).
Miners, J. O. & Birkett, D. J. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br. J. Clin. Pharmacol. 45, 525–538 (1998).
Davies, N. M. & Skjodt, N. M. Clinical pharmacokinetics of meloxicam. A cyclo-oxygenase-2 preferential nonsteroidal anti-inflammatory drug. Clin. Pharmacokinet. 36, 115–126 (1999).
Kimble, B. et al. In vitro hepatic microsomal metabolism of meloxicam in koalas (Phascolarctos cinereus), brushtail possums (Trichosurus vulpecula), ringtail possums (Pseudocheirus peregrinus), rats (Rattus norvegicus) and dogs (Canis lupus familiaris). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 161, 7–14 (2014).
Blanshard, W. & Bodley, K. Koalas. in Medicine of Australian Mammals (eds. Vogelnest, L. & Woods, R.) 307–327 (Csiro Publishing, Melbourne, Victoria, Australia, 2008).
Villalba, J. J., Provenza, F. D. & Bryant, J. Consequences of the interaction between nutrients and plant secondary metabolites on herbivore selectivity: benefits or detriments for plants? Oikos 97, 282–292 (2002).
Kratzing, J. E. The anatomy and histology of the nasal cavity of the koala (Phascolarctos cinereus). J. Anat. 138, 55–65 (1984).
Moore, B. D., Foley, W. J., Wallis, I. R., Cowling, A. & Handasyde, K. A. Eucalyptus foliar chemistry explains selective feeding by koalas. Biol. Lett. 1, 64–67 (2005).
Freeland, W.J. & Janzen, D.H. Strategies in herbivory by mammals: the role of plant secondary compounds. Am. Nat. 108, 269–289 https://doi.org/10.1086/282907 (1974).
McBride, C. S. Rapid evolution of smell and taste receptor genes during host specialization in Drosophila sechellia. Proc. Natl. Acad. Sci. USA 104, 4996–5001 (2007).
Watson, K. J. et al. Expression of aquaporin water channels in rat taste buds. Chem. Senses 32, 411–421 (2007).
Rosen, A. M., Roussin, A. T. & Di Lorenzo, P. M. Water as an independent taste modality. Front. Neurosci. 4, 175 (2010).
Gilbertson, T. A., Baquero, A. F. & Spray-Watson, K. J. Water taste: the importance of osmotic sensing in the oral cavity. J. Water Health 4, 35–40 (2006).
Meyerhof, W. et al. The molecular receptive ranges of human TAS2R bitter taste receptors. Chem. Senses 35, 157–170 (2010).
Hayakawa, T., Suzuki-Hashido, N., Matsui, A. & Go, Y. Frequent expansions of the bitter taste receptor gene repertoire during evolution of mammals in the Euarchontoglires clade. Mol. Biol. Evol. 31, 2018–2031 (2014).
Li, D. & Zhang, J. Diet shapes the evolution of the vertebrate bitter taste receptor gene repertoire. Mol. Biol. Evol. 31, 303–309 (2014).
Li, R. et al. The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317 (2010).
Johnston, S. D., McGowan, M. R., O’Callaghan, P., Cox, R. & Nicolson, V. Studies of the oestrous cycle, oestrus and pregnancy in the koala (Phascolarctos cinereus). J. Reprod. Fertil. 120, 49–57 (2000).
Morris, K. M. et al. Characterisation of the immune compounds in koala milk using a combined transcriptomic and proteomic approach. Sci. Rep. 6, 35011 (2016).
Department of the Environment. Phascolarctos cinereus (combined populations of Queensland, New South Wales and the Australian Capital Territory) in Species Profile and Threats Database (Department of the Environment, Canberra, Australian Capital Territory, 2016).
Polkinghorne, A., Hanger, J. & Timms, P. Recent advances in understanding the biology, epidemiology and control of chlamydial infections in koalas. Vet. Microbiol. 165, 214–223 (2013).
Rhodes, J. R. et al. Using integrated population modelling to quantify the implications of multiple threatening processes for a rapidly declining population. Biol. Conserv. 144, 1081–1088 (2011).
Morris, K. et al. The koala immunological toolkit: sequence identification and comparison of key markers of the koala (Phascolarctos cinereus) immune response. Aust. J. Zool. 62, 195–199 (2014).
Morris, K. M. et al. Identification, characterisation and expression analysis of natural killer receptor genes in Chlamydia pecorum infected koalas (Phascolarctos cinereus). BMC Genomics 16, 796 (2015).
Cheng, Y. et al. Characterisation of MHC class I genes in the koala. Immunogenetics 70, 125–133 (2018).
Jones, E. A., Cheng, Y., O’Meally, D. & Belov, K. Characterization of the antimicrobial peptide family defensins in the Tasmanian devil (Sarcophilus harrisii), koala (Phascolarctos cinereus), and tammar wallaby (Macropus eugenii). Immunogenetics 69, 133–143 (2017).
Burton, M. J. et al. Pathogenesis of progressive scarring trachoma in Ethiopia and Tanzania and its implications for disease control: two cohort studies. PLoS Negl. Trop. Dis. 9, e0003763 (2015).
Derrick, T., Roberts, C., Last, A. R., Burr, S. E. & Holland, M. J. Trachoma and ocular chlamydial infection in the era of genomics. Mediators Inflamm. 2015, 791847 (2015).
Stoye, J. P. Koala retrovirus: a genome invasion in real time. Genome Biol. 7, 241 (2006).
Hobbs, M. et al. Long-read genome sequence assembly provides insight into ongoing retroviral invasion of the koala germline. Sci. Rep. 7, 15838 (2017).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Roberts, R. G. et al. New ages for the last Australian megafauna: continent-wide extinction about 46,000 years ago. Science 292, 1888–1892 (2001).
Field, J., Wroe, S., Trueman, C. N., Garvey, J. & Wyatt-Spratt, S. Looking for the archaeological signature in Australian megafaunal extinctions. Quat. Int. 285, 76–88 (2013).
Neaves, L. E. et al. Phylogeography of the koala, (Phascolarctos cinereus), and harmonising data to inform conservation. PLoS One 11, e0162207 (2016).
Tsangaras, K. et al. Historically low mitochondrial DNA diversity in koalas (Phascolarctos cinereus). BMC Genet. 13, 92 (2012).
Taylor, A. C., Graves, J. A., Murray, N. D. & Sherwin, W. B. Conservation genetics of the koala (Phascolarctos cinereus). II. Limited variability in minisatellite DNA sequences. Biochem. Genet. 29, 355–363 (1991).
Taylor, A. C. et al. Conservation genetics of the koala (Phascolarctos cinereus): low mitochondrial DNA variation amongst southern Australian populations. Genet. Res. 69, 25–33 (1997).
Dennison, S. et al. Population genetics of the koala (Phascolarctos cinereus) in north-eastern New South Wales and south-eastern Queensland. Aust. J. Zool. 64, 402–412 (2017).
Cristescu, R. et al. Inbreeding and testicular abnormalities in a bottlenecked population of koalas (Phascolarctos cinereus). Wildl. Res. 36, 299–308 (2009).
Frankham, R. et al. Predicting the probability of outbreeding depression. Conserv. Biol. 25, 465–475 (2011).
Frankham, R. et al. Genetic Management of Fragmented Animal and Plant Populations (Oxford University Press, Oxford, 2017).
Hansen, J., Sato, M., Russell, G. & Kharecha, P. Climate sensitivity, sea level and atmospheric carbon dioxide. Philos. Trans. A Math. Phys. Eng. Sci. 371, 20120294 (2013).
O’Connell, J. F. & Allen, J. The process, biotic impact, and global implications of the human colonization of Sahul about 47,000 years ago. J. Archaeol. Sci. 56, 73–84 (2015).
Clarkson, C. et al. Human occupation of northern Australia by 65,000 years ago. Nature 547, 306–310 (2017).
Saltré, F. et al. Climate change not to blame for late Quaternary megafauna extinctions in Australia. Nat. Commun. 7, 10511 (2016).
Wang, J. Triadic IBD coefficients and applications to estimating pairwise relatedness. Genet. Res. 89, 135–153 (2007).
Wang, J. COANCESTRY: a program for simulating, estimating and analysing relatedness and inbreeding coefficients. Mol. Ecol. Resour. 11, 141–145 (2011).
Warren, W. C. et al. Genome analysis of the platypus reveals unique signatures of evolution. Nature 453, 175–183 (2008).
Mikkelsen, T. S. et al. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 447, 167–177 (2007).
Renfree, M. B. et al. Genome sequence of an Australian kangaroo, Macropus eugenii, provides insight into the evolution of mammalian reproduction and development. Genome Biol. 12, R81 (2011).
Murchison, E. P. et al. Genome sequencing and analysis of the Tasmanian devil and its transmissible cancer. Cell 148, 780–791 (2012).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Yandell, M. & Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 13, 329–342 (2012).
Smit, A., Hubley, R. & Green, P. RepeatModeler Open-1.0. 2008–2015 (2014).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0. 2013–2015 (2015).
Boutet, E. et al. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: how to use the entry view. in Plant Bioinformatics: Methods and Protocols (ed. Edwards, D.) 23–54 (2016).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Wong, E. S., Papenfuss, A. T. & Belov, K. Immunome database for marsupials and monotremes. BMC Immunol. 12, 48 (2011).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Borodovsky, M. & Lomsadze, A. Gene identification in prokaryotic genomes, phages, metagenomes, and EST sequences with GeneMarkS suite. Curr. Protoc. Bioinformatics 4, 4.5.1–4.5.17 (2011).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Li, L., Stoeckert, C. J. Jr & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Vilella, A. J. et al. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19, 327–335 (2009).
Pond, S.L.K. & Muse, S.V. HyPhy: hypothesis testing using phylogenies. in Statistical Methods in Molecular Evolution 125–181 (Springer, New York, 2005).
Delport, W., Poon, A. F., Frost, S. D. & Kosakovsky Pond, S. L. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455–2457 (2010).
Dobin, A. & Gingeras, T. R. Mapping RNA‐seq reads with STAR. Curr. Protoc. Bioinformatics 11, 11.14.1–11.14.19 (2015).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Varet, H., Brillet-Guéguen, L., Coppée, J.-Y. & Dillies, M.-A. SARTools: a DESeq2- and edgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS One 11, e0157022 (2016).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Sonnhammer, E. L. & Durbin, R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167, GC1–GC10 (1995).
Phillips, S. S. Population trends and the koala conservation debate. Conserv. Biol. 14, 650–659 (2000).
Lynch, M. et al. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 17, 704–714 (2016).
Uchimura, A. et al. Germline mutation rates and the long-term phenotypic effects of mutation accumulation in wild-type laboratory mice and mutator mice. Genome Res. 25, 1125–1134 (2015).
Bragg, J. G., Potter, S., Bi, K. & Moritz, C. Exon capture phylogenomics: efficacy across scales of divergence. Mol. Ecol. Resour. 16, 1059–1068 (2016).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Acknowledgements
R.N.J. and the Australian Museum acknowledge the Australian Museum Foundation, Bioplatforms Australia, New South Wales Environmental Trust grant 2014/RD/0015, Australian Research Council LIEF Grant LE160100154, the University of Sydney HPC service and Amazon Web Services for support; and C. Staples from Featherdale Wildlife Park, C. Flanagan from Port Macquarie Koala Hospital, J. Hangar, E. Hynes, J. Reed, S. Ingleby, A. Divljan and S. Ginn for assistance with sample acquisition. K.B. acknowledges support from the Australian Research Council and Bioplatforms Australia. M.R.W. and the Ramaciotti Centre for Genomics acknowledge support from the Australian Research Council, from the Australian Government NCRIS scheme via Bioplatforms Australia, the New South Wales State Government RAAP scheme and the University of New South Wales. W.H. and W.J.N. were supported by strategic BBSRC funding (Institute Strategic Programme Grant BB/J004669/1) and by the NBI Computing Infrastructure for Science (CiS) group. A.D.G., K.M.H and K.T. were supported by grant R01GM092706 from the National Institute of General Medical Sciences (NIGMS) and A.D.G. had additional support from Morris Animal Foundation grant D14ZO-94. T.N.H., Z.D. and R.J.O. were supported by awards from the National Science Foundation 1613806 and the facilities within the Center for Genome Innovation at the University of Connecticut. C.E.H. thanks CSIRO National Research Collections Australia funding. K.B. and A.P. thank the veterinary staff at Australia Zoo Wildlife Hospital, Currumbin Wildlife Hospital and Moggill Koala Hospital for their assistance in the collection of samples for the koala conjunctival transcriptome study. T.H. acknowledges the Kyoto University Research Administration Office (KURA) for support and was financed by JSPS KAKENHI grant number 16K18630 and the Sasakawa Scientific Research Grant from the Japan Science Society. A.P. and P.T. acknowledge financial support from the Australian Research Council and A.G. financial support via Australian Research Council Discovery Grant DP110104377. C.M.W. is supported by a University of Sydney research fellowship from the estate of Mabs Melville. All authors thank Bioplatforms Australia and Pacific Biosciences. The authors thank T. Haydon for valuable editorial input; S. Potter for expert technical assistance; and R. Gleadow, C. Frere, D. Lunney and D. Alvarez-Ponce for valuable discussions on content.
Author information
Authors and Affiliations
Contributions
R.N.J., K.B., P.T. and M.R.W. designed the original concept and scientific objectives and oversaw the project and analyses. R.N.J., D.C., M.D.B.E., A.K.G., D.O., A.K. and P.T. acquired samples for sequencing. T.L.R., M.R.W., Z.C., D.O., G.J.E. and F.D.P. performed library preparation, genome sequencing, assembly and annotation. S.Y.W.H. performed PSMC analysis. A.Y.-Y.C. characterized repetitive sequences. R.J.O., T.N.H. and Z.D. characterized centromeric and telomeric regions. C.M.W. and M.B.R. annotated and analyzed reproductive and developmental genes. K.M.M. annotated and analyzed lactation genes. T.H. and D.C. annotated and analyzed TAS1R and TAS2R genes. H.R.P. annotated and analyzed OR genes. D.C. annotated and analyzed aquaporin genes. K.B., Y.C., P.A.B., E.A.J., D.O. and E.P. annotated and analyzed MHC, Ig, TCR, NK and antimicrobial genes. A.P., K.B., D.O. and D.M. analyzed the ocular RNA-seq data. P.A.B., B.W., C.E.G., P.T., K.B. and A.P investigated candidate genes for chlamydia vaccine response. P.T., M.R.W., R.S., M.H., A.K., A.D.G. and K.T. characterized retrovirus insertions and wrote the KoRV sections of the manuscript. J.G.B., S.D., M.D.B.E., G.J.F., L.E.N., R.N.J., B.W. and C.J.H. contributed to analyses and interpretation of exon capture sequence data. P.D.W., S.A.W. and H.R.P. annotated and analyzed RSX data. W.J.N., C.E.G., Y.C., W.H., F.D., M.G., K.M.E., B.W. and C.R. analyzed CYP genes. C.E.G. and C.M.W. analyzed V1R genes. J.E.D., A.G. and H.R.P. constructed super-scaffolds. J.A.M.G., V.A., F.D., C.J.H., K.M.H., A.P., B.W., D.C., M.H., D.E.A.-P., P.A.B., L.E.N., C.E.G., S.A.W. and C.E.H. provided constructive feedback on data analysis and interpretation. R.N.J., P.T., K.B., M.R.W., A.P., M.D.B.E. and G.J.F. obtained funding and other resources. R.N.J. and K.B. wrote the manuscript with input from all other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Gondwanan origin (Australia in yellow) and phylogenetic representation of the extant Marsupialia, depicting American orders (yellow branches), Australian orders (green branches) and their divergence in the Eocene.
Included in greater detail is the order Diprotodontia (shaded green) - represented as the three superorders Macropodiformes, Phalangeriformes and Vombatiformes, the latter including families Vombatidae (wombats) and Phascolarctidae (koalas). Included is diversity of extinct koala fossil locations and estimated extinction dates (depicted in maroon on the map). There is evidence for up to 16 species in the family Phascolarctidae, with ~3 species coexisting at any one time 1,2,*,†. The genus Phascolarctos first appeared during the Pliocene (4.5-2 mya) and the modern koala more recently with fossil evidence dating from 350 ka (depicted in on the map green with current range in purple). Graph depicts estimated population numbers of P. cinereus over last 100,000 years.
*Munemasa, M. et al. Phylogenetic analysis of diprotodontian marsupials based on complete mitochondrial genomes. Genes Genet. Syst. 81, 181–191 (2006).
†May-Collado, L.J., Kilpatrick, C.W. & Agnarsson, I. Mammals from ‘down under’: a multi-gene species-level phylogeny of marsupial mammals (Mammalia, Metatheria). PeerJ 3, e805 (2015).
Supplementary Figure 2 Predicted koala chromosome 7 using arrangement of koala supercontigs compared to gray short-tailed opossum and tammar wallaby.
Homology amongst marsupial chromosomes was determined previously from cross-species chromosome painting, which divided marsupial genomes into 19 conserved segments* and could be extrapolated to all previously G-banded marsupial karyotypes†. Koala chromosome 7 corresponds to conserved segments C7 and C8, which are located on the short arm of wallaby chromosome 7 and long arm of tammar wallaby chromosome 1 respectively, and on the short arm of opossum chromosome 1 (insert). The contigs (indicated by different colours) making up each koala supercontig and the size of the supercontigs are indicated. The tammar wallaby scaffold identifier numbers have also been provided. Gray short-tailed opossum chromosome 1 has been used as a reference as the sequence has been oriented on the gray short-tailed opossum chromosome 83.
*Rens, W. et al. Reversal and convergence in marsupial chromosome evolution. Cytogenet. Genome Res. 102, 282–290 (2003).
†Deakin, J.E., Graves, J.A. & Rens, W. The evolution of marsupial and monotreme chromosomes. Cytogenet. Genome Res. 137, 113–129 (2012).
Supplementary Figure 3 Summary of data used to infer koala centromeric regions.
a-b, Graphs of the percentage of marsupial annotated repeats between a, CENP-A-IP (purple) and Input (teal) and b, CREST-IP (purple) and Input (teal). Equal enrichment between IP and Input is represented by 50% distribution for each repeat and deviation from 50% indicates enrichment in either IP or Input. c, Heatmap of sequence divergence for the ChIP-Seq Peaks and whole genome. The color in the legend represents the percent divergence from the model sequence. Both rows and columns were clustered using Euclidean distance, with each row representing a different region of the genome and each column representing a particular repetitive sequence. The ChIP-Seq peaks are labeled by appending –Bed1 along the right of the heatmap and blue along the left, while the genomic scaffolds are labeled by appending –bed2 along the region labels on the right and with a red mark on the left of the heatmap. d-g, A principal component analysis was performed using the length of repeat normalised to the length of the region (d and f of each pair) and each region's average divergence from the model of each repeat in that region (e and g of each pair). For these analyses, the top 2,000 ChIP-seq peaks (for both the CENP-A and CREST ChIP experiments), containing 48,090 unique repeats, were compared to the genome assembly broken into 33,209 regions/windows (200kb in size), containing 7,038,290 unique repeats. Regions derived from the ChIP-Seq peaks are labelled with red/Bed1 while the whole genome scaffolds are labelled with blue/Bed2. The scatter of points from each set of regions is also shown with the ChIP-Seq regions being show with a light red circle, the regions from the whole genome shown with a blue circle and the total diversity of all of the regions shown in a dark red circle. d-e, marsupial repeat models; f-g, de novo repeat models.
Supplementary Figure 4 Assembly of koala RSX.
RSX mediates marsupial X-chromosome inactivation 26. It is functionally analogous to the eutherian specific Xist gene, but is independently evolved. a, Koala scaffolds aligned to the gray short-tailed opossum X. Red lines are reciprocal best hits of protein coding genes. RSX is located on a 5.9 Mbp scaffold (scaff00196), on which gene order is completely conserved with gray short-tailed opossum. Gene mapping and chromosome painting* has demonstrated that gene content of the X is conserved across all marsupials studied to date, but gene order is not. The koala X is no exception, being rearranged relative to the X of both wallaby and gray short-tailed opossum. b, Sequence similarity plot of koala RSX to itself. There were three repeat arrays detected (coloured arrows). Sequence logos show base frequency of the consensus 33 bp repeat in the 5’ 12kb array (red arrow), and a 31 bp repeat detected in the middle 5.2 kb array (blue arrow). The third 1.75 kb repeat array (green arrow) had a 152 bp repeat with 8 copies. These three repeat arrays are conserved in gray short-tailed opossum RSX (Supplementary information: Fig. 1). c, Alignment of the consensus koala 33 bp repeat from the 12kb array, with the consensus 34 bp repeat from opossum. Above the alignment is the minimum free energy predicted† stem-loop formed by the palindromes (shaded on the alignment) in koala, and below the alignment the stem-loop predicted in gray short-tailed opossum. Each base is coloured according to base-pairing probability.
*Deakin, J.E. et al. Reconstruction of the ancestral marsupial karyotype from comparative gene maps. BMC Evol. Biol. 13, 258 (2013). †Gruber, A.R., Lorenz, R., Bernhart, S.H., Neuböck, R. & Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 36, W70–W74 (2008).
Supplementary Figure 5 Gene family expansion in the annotated ‘Bilbo’ koala genome (phaCin_unsw_v4.1) and in Australian marsupials.
a Gene families that contained the largest number of genes in the koala. b Gene families that contained more genes in the koala than in any other member of the 9 species included in the OrthoMCL analysis (human, mouse, dog, tammar wallaby, Tasmanian devil, gray short-tailed opossum, platypus, chicken). c Gene families that contained more genes in the Australian marsupials included in the analysis (koala, tammar wallaby, Tasmanian devil, gray short-tailed opossum), than in the eutherian mammals (human, mouse, dog) or out-groups (platypus, chicken).
Supplementary Figure 6 Analysis of CYP2C expression in transcriptomes from two koalas.
Expression (TPM) across 15 transcriptomes from two koalas showing an overall higher expression of the CYP2 genes in the livers of the 2 koalas, with correlation between the two individuals (spearman's rank correlation coefficient = 0.928, P< 2.2e-16).
Supplementary Figure 7 Genomic structure of the umami and sweet taste receptor TAS1R genes in marsupial genome assemblies.
a Each box depicts an exon with black vertical bars indicating a sequencing error causing frameshift mutation. In the koala assembly, TAS1R1 and TAS1R2 have a frameshift mutation, respectively, but these are attributed to PacBio assembly error (see below). In the tammar wallaby assembly, TAS1R1 and TAS1R3 have a frameshift mutation, respectively, which is due to Sanger sequencing error (apparently miscalled in the trace data sequences of the corresponding sites (e.g., NCBI TI numbers: 1378971959 and 1718787462 for TAS1R1 and 1437875519 and 1634206001 for TAS1R3)). The 1st exon of TAS1R2 in the gray short-tailed opossum assembly and the 6th exon of TAS1R2 in the Tasmanian devil assembly are completely truncated (missing in the data). The inside of the 6th exon in the Tasmanian devil assembly is truncated but intact in the alternative Tasmanian devil assembly (GCA_000219685.1). Therefore, any TAS1Rs do not appear to be pseudogenized in these marsupials. b Mapping results of Illumina short reads from the koala against the PacBio assembly in the corresponding site of each frameshift mutation of TAS1R1 and TAS1R2, which would be due to PacBio assembly error because all Illumina short reads insert one base to restore the open reading frame. Noted that these inserts are allelic variations, which could cause such PacBio assembly error.
Supplementary Figure 8 Schematic maps of two lactation-expressed gene families and phylogeny of the complex family of late lactation proteins.
a Two highly expressed milk proteins with no known function or eutherian homologs, Very Early Lactation Protein (VELP) and Marsupial Milk 1 (MM1), were identified in the koala genome. We previously found that koala VELP shows homology to a eutherian antimicrobial protein, Glycam1 53. In the koala genome VELP is located in the region syntenic to Glycam1 confirming orthology. The MM1 gene is likely located close to VELP. In the syntenic region in the human genome is an array of short glycoproteins including lacritin, dermicidin, Glycam1 and mucin-like 1 – all antimicrobial proteins found in secretions such as milk, tears and sweat. In koala, three short novel genes were also seen in this region, including one that shows homology to lacritin and dermicidin. Although direct orthology to any of the human proteins was not seen, their location and similar length and structure indicate that MM1 and the additional novel genes may be related to this group of human antimicrobials, and may also have an antimicrobial role. b Koala milk-expressed lipocalins (Late Lactation Proteins (LLPs), B-lactoglobulin (LGB), Lipocalin-2 and Trichosurin (TRSN)) are clustered together on a scaffold together with a large number of related lipocalin genes. Several of these genes are highly expressed in koala milk and may serve nutritional roles. The group of lipocalin genes appears to have expanded through gene duplications within the marsupial lineage, particularly within the marsupial-specific LLP (4 genes) and Trichosurin-related (7 genes) groups. These duplications may allow marsupials to fine tune milk protein composition across the extended lactation. * denotes partial gene. c Phylogeny of Late Lactation Protein (LLP) sequences across marsupials. LLP genes have a complex evolutionary history with duplications and deletions. One to four LLP proteins have been identified across marsupial species. This tree shows that some LLP genes are conserved among Australian marsupials, while lineage specific duplications occur in several species, including koala. The phylogeny was constructed using the Maximum Likelihood method based on the JTT matrix-based model. Scale units are number of amino acid substitutions per site.
Supplementary Figure 9 Schematic maps of koala major histocompatibility complex (MHC) and T cell receptors (TCR) loci, and RNA-seq analysis of gene regulation in chlamydial ocular disease in the koala.
a The core MHC region on scaffold#255 contains 138 genes. Out of 23 annotated class I loci, eight are located in this region, including one classical and three nonclassical genes 59 and four pseudogenes. b The koala is the first Australian marsupial to have its TCR loci fully assembled and annotated. The koala TCR system contains the conventional α, β, δ, and γ chains, and an additional isotype class known as the TCR µ chain (encoded by TRM), which is unique to marsupials and monotremes and missing in eutherian mammals. The koala TRA/D, TRB, TRG, and TRM loci are each located on a single scaffold (scaffold1, 71, 153, and 59, respectively) in the PacBio assembly. The TRA/D locus contains 94 putatively functional gene segments, including 52 Vα, two Dδ, three Jδ, one Cδ, 35 Jα, and a Cα. The TRB locus contains 33 Vβ segments, and three sets of Dβ, Jβ, and Cβ segments arranged in tandem cassettes, each comprising one Dβ, two to four Jβ, and one Cβ. The TRG locus consists of four Vγ, four Jγ, and one Cγ. Four sets of TRM gene segment clusters, each consisting of one V, one or two D, one J, one Vj (joined VJ segment specific to TCR µ chain), and one C, are found in the koala; two sets are likely functional with all segments containing an intact open reading frame. c Koala with healthy left eye while right eye displays clinical signs of chronic active chlamydial keratoconjunctivitis. d Distribution of upregulated genes in diseased koalas mapping to ‘GO:0002376 Immune System Process’ annotated at the CateGOrizer.
Supplementary Figure 10 Summary of koala demographic history inferred using the pairwise sequential Markovian coalescent (PSMC) method.
a-b Depiction of koala population demographic history inferred using the PSMC method from the diploid sequences of three koalas (a generation time of 7 years was assumed). We applied the midpoint of the mutation rate estimate for either a, human (1.45×10-8 mutations per site per generation) or b, mouse (5.4×10-9 mutations per site per generation). c Depiction of koala population demographic history for each individual koala (Pacific Chocolate, Bilbo, and Birke). A generation time of 7 years and a mutation rate of 1.45×10-8 mutations per site per generation was assumed. PSMC analyses were performed using the 'optimal' settings described in Fig. 3 of the main text, as well as using alternative values for the number of free atomic time intervals (p=44, 54, or 74), initial value of rho (ρ=2, 5, or 10), and the maximum time to the most recent common ancestor (t=10 or 20).
Supplementary information
Supplementary Figures and Text
Supplementary Figures 1–10, Supplementary Note and Supplementary Tables 1–6, 11, 14, 17, 19–21, 23, 24
Supplementary Data 2
Gene Ortholog list. These groups include the 1:1 orthologs and also the genes families. For clarity, a pattern has been ascribed to each group (1: single gene, m: multiple species, 0: absent).
Supplementary Tables 7–10
De novo and Mars summary statistics for ChIP-seq. Tables showing the results of testing if the mean value for each repeat is significantly different between the regions defined by the ChIP-seq experiments and the whole genome. The table includes the number of observations for each repeat in each region and the unadjusted P-value for the Kolmogorov–Smirnov test and Anderson–Darling test are reported for when values are continuous (KS.boot.p.value / AD.Cont.p.value) and discontinuous (KS.p.value / AD.Disc.p.value). Yellow highlights are the repeats that demarcate CENP-A peaks specifically.
Supplementary Table 12
SLAC and MEME codon-based output. Excel spreadsheet containing full codon-based output for SLAC and MEME analysis of 152 cytochrome P450 sequences.
Supplementary Table 13
MEME output by codon and by branch. Excel spreadsheet containing full by-codon and by-branch output from MEME analysis of 152 cytochrome P450 sequences.
Supplementary Table 15
List of annotated taste receptor genes in koala and other marsupial assemblies.
Supplementary Table 16
List of annotated genes involved in koala lactation. Key milk proteins that form a major part of the protein content of milk across mammalian species have been identified in the koala genome, mammary transcriptome and milk proteomes. This includes proteins involved in nutrition and transport such as caseins, β-lactoglobulin, α-lactalbumin and lactotransferrin. While duplications are seen within the three casein families (α, κ and β) in the monotreme and eutherian lineage, in the koala genome, like other marsupials investigated, only single copies of each casein gene were identified. Milk-expressed lipocalins were clustered together and linked to a large number of other lipocalin genes. The genomic location of two marsupial-specific novel milk proteins (VELP and MM1) was also identified.
Supplementary Table 18
List of annotated koala immune genes.
Supplementary Table 22
Locations of KoRV loci in phaCin_unsw_v4.1 assembly. The data is in BED format (with 0-based numbering of the start field).
Supplementary Table 25
Genes with KoRV insertions.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Johnson, R.N., O’Meally, D., Chen, Z. et al. Adaptation and conservation insights from the koala genome. Nat Genet 50, 1102–1111 (2018). https://doi.org/10.1038/s41588-018-0153-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-018-0153-5
This article is cited by
-
Identification of the RSX interactome in a marsupial shows functional coherence with the Xist interactome during X inactivation
Genome Biology (2024)
-
Expanding the known distribution of phascolartid gammaherpesvirus 1 in koalas to populations across Queensland and New South Wales
Scientific Reports (2024)
-
Contrasting segregation patterns among endogenous retroviruses across the koala population
Communications Biology (2024)
-
Genome of the endangered eastern quoll (Dasyurus viverrinus) reveals signatures of historical decline and pelage color evolution
Communications Biology (2024)
-
Effects of 6-methoxybenzoxazolinone (6-MBOA) on animals: state of knowledge and open questions
The Science of Nature (2024)
