
Abstract
Decentralized manufacture of thermostable mRNA vaccines in a microneedle patch (MNP) format could enhance vaccine access in low-resource communities by eliminating the need for a cold chain and trained healthcare personnel. Here we describe an automated process for printing MNP Coronavirus Disease 2019 (COVID-19) mRNA vaccines in a standalone device. The vaccine ink is composed of lipid nanoparticles loaded with mRNA and a dissolvable polymer blend that was optimized for high bioactivity by screening formulations in vitro. We demonstrate that the resulting MNPs are shelf stable for at least 6 months at room temperature when assessed using a model mRNA construct. Vaccine loading efficiency and microneedle dissolution suggest that efficacious, microgram-scale doses of mRNA encapsulated in lipid nanoparticles could be delivered with a single patch. Immunizations in mice using manually produced MNPs with mRNA encoding severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein receptor-binding domain stimulate long-term immune responses similar to those of intramuscular administration.
Similar content being viewed by others

Main
Unvaccinated communities in low- and middle-income countries are at high risk for repeated outbreaks of Coronavirus Disease 2019 (COVID-19) and other infectious diseases1, which increase mortality, promote the emergence of more dangerous variants and negatively impact the economy2. Mass vaccination in these communities has been hampered by issues such as inadequate cold-chain-compatible storage and transport infrastructure and an insufficient number of healthcare personnel3,4. Distributed, local systems for manufacturing suitable vaccines offer a potential solution. A promising vaccine format in these regions is thermostable microneedle patches (MNPs)5,6,7,8. MNPs can be self-applied, are less painful than intramuscular (IM) injection, produce no sharps waste, can be formulated to remain shelf stable for months and have been used with several types of vaccines, including various nucleic acids9,10,11,12,13,14. In the context of COVID-19, lipid nanoparticle (LNP)-encapsulated mRNA vaccines, such as those produced by Moderna and Pfizer-BioNTech, have proven highly effective in preventing severe disease. To our knowledge, intradermal (ID) delivery of an mRNA vaccine in an LNP vehicle using an MNP with long-term thermostability has not been reported previously.
Manufacturing of MNPs introduces new challenges in fabrication, loading and scalability that have slowed their development, despite being ideal for deployment in low-resource areas15,16,17,18. For accurate dosing and adequate skin penetration, microneedles must be sharp and consistent in size from batch to batch19. MNPs are limited by the small volume available for vaccine loading, especially when excipients are required to stabilize labile antigens20. MNPs are typically handmade individually with labor-intensive, manual and imprecise steps, such as centrifugation, which makes consistent, automated manufacturing using these methods challenging21.
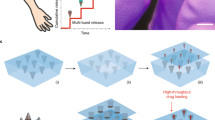
Here we describe a microneedle vaccine printer (MVP) to fabricate dissolvable MNPs loaded with LNP-encapsulated mRNA vaccines or other cargos (Fig. 1a–c). Integrating a microneedle fabrication process within a standalone, modular device presents unique challenges. Microneedle formation, which is typically achieved through molding22, droplet fabrication23, inkjet printing24,25 or 3D printing26,27,28, must produce microneedles with sharp, accurate and micron-scale features. Mold filling must be driven by a repeatable process that minimizes waste, reduces moving parts, requires no user interaction and integrates into an automatable machine-driven workflow. In each run, the MVP dispenses vaccine ink fills microneedle molds without disrupting the ink using vacuum to remove air through the mold, and accelerates drying using an automated workflow with minimal human intervention. The automated workflow integrates a high-precision robotic dispenser, programmable vacuum chamber and modular motion stages containing reusable microneedle molds. The process used in the device is based on vacuum application, compatible with a wide range of MNP designs, and optimized to minimize vaccine waste.

a, Modular inks containing mRNA, lipids and polymer can be customized for microneedle vaccine printing. b,c, The MVP (b) can be distributed to remote areas to provide local manufacturing capability (c) of thermostable MNPs. d, After automated dispensing, vacuum is applied through PDMS to load the polymer–vaccine solution into the microneedle mold. Molds are then transferred to a drying station for accelerated drying. e, The time needed to load the polymer–vaccine solution into the PDMS mold was measured for various design and process parameters. Unpaired, two-tailed t-tests were used for all comparisons, except for polymer type, where an ordinary one-way ANOVA was used (n = 3 independent samples). f, Drying rate for different drying strategies. ANCOVA was used to compare drying rate estimates, which were derived from a linear regression of drying rate data (n = 3 independent samples). g, Total drop area (n = 3 independent samples) and patch coverage (n = 100 independent samples) are a function of polymer solution used for dispensing. h, Imaging of MNPs fabricated with the device: MNP on acrylic solid backing (top left), SEM image of MNPs with conical (top right) and pyramid (bottom left) geometries and a single pyramid MNP (bottom right). i, Images of tip-loaded MNPs (outlined in white) co-dispensed with the device using red (top) or blue (bottom) dye as model cargo. j, MNP production throughput. k, MNP throughput as a function of drying time and printing tray length. Data represent the mean ± s.d (e,g) or mean ± 95% confidence interval (f). P values are represented by: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. NS, not significant.
Microneedle printing requires incorporation of a stabilizing dissolvable polymer into a consistently dispensable mRNA-LNP ink. After extensively screening inks in vitro, we determined that a combination of dissolvable polymers could successfully deliver active LNP-encapsulated mRNA and maintain stability for at least 6 months at room temperature. Using mRNA encoding the receptor-binding domain (RBD) of the SARS-CoV-2 spike (S) protein, we show that MNPs produced by the MVP have adequate mechanical properties and successfully penetrate porcine epidermis upon ex vivo application. In vivo testing of manually produced MNPs demonstrated an immune response similar to that of IM administration.
Results
Developing a microneedle vaccine printer
We developed a vacuum-based technique for driving viscous vaccine inks into molds. The process is based on air’s permeability and solubility in polydimethylsiloxane (PDMS)29. By applying vacuum either directly to the bottom of the PDMS mold during filling (Fig. 1d and Extended Data Fig. 1a) or by pre-degassing the PDMS mold immediately before filling (Extended Data Fig. 1b), viscous solutions of polymer and vaccine could be loaded into molds (Supplementary Videos 1 and 2).
The effect of various process and design parameters on loading time and MNP formation were studied for both approaches (Extended Data Fig. 1c–j). When applying vacuum directly to the mold, loading time depends on the thickness and composition of the PDMS sheet, the pattern used to apply vacuum below the PDMS mold and the pressure gradient applied (Fig. 1e). Loading of increasingly viscous inks (Extended Data Fig. 1k) required less than 20 min. Although vacuum application during microneedle fabrication is common, vacuum is typically applied to the atmosphere above the mold, resulting in the formation of bubbles in the vaccine and polymer solution30,31. Applying vacuum directly to the PDMS mold prevents bubble formation and eliminates the need for centrifugation, providing a repeatable process enabling automation, and it can be scaled up or down to meet any MNP size or quantity.
To reduce vaccine waste and drying time, we dispensed the minimum possible volume of vaccine ink needed to fill the microneedles. We tuned the polymer concentration to yield a thin, homogenous film of dried polymer with mechanical strength sufficient to survive demolding (Extended Data Fig. 2a). Dispensing was characterized by drop area of ink dispensed and the circularity of the backing formed after drying. By increasing viscosity to minimize the Marangoni effect—which drives polymer and vaccine to the edges of the drying droplet—we maximized circularity and mold coverage, measured in needles filled per mold (Extended Data Fig. 2b,c). Using thermogravimetric analysis to evaluate drying time, we determined that simultaneous application of vacuum below, and a desiccated vacuum atmosphere above, the MNP molds accelerated patch drying without any bubble formation (Fig. 1f and Supplementary Note 1).
To minimize user interaction and the need for on-site training, the above processes were automated using programmable components. A custom x–y translating stage with vacuum below (loading) and above (drying), accommodating up to 100 MNP molds at once, was manufactured (Supplementary Video 3 and Extended Data Fig. 3a–e). The vacuum loading and vacuum drying processes were combined with a robotic arm for repeatable and programmable dispensing with microliter-scale precision (Supplementary Videos 4 and 5 and Extended Data Fig. 3f). Parameters such as dispensing pattern, volume dispensed and dispensing height were optimized to minimize vaccine waste and yield MNPs with an ultrathin backing (Extended Data Fig. 3g–k). With one set of dispensing parameters, three different dissolvable polymer systems were dispensed with greater than 80% mold coverage (Fig. 1g). The MVP enabled the automated fabrication of various microneedle designs, including 10 × 10 arrays (Extended Data Fig. 3l) of pyramid and conical microneedles up to 1,500 µm tall made from a selection of dissolvable polymers common for vaccine delivery using MNPs (Fig. 1h).
Vaccine that dries in the backing of the MNP creates additional vaccine waste. To reduce waste, we implemented a two-step tip-loading process that has previously been used to concentrate vaccine in microneedle tips8,32. First, a vaccine ink containing the minimum amount of polymer necessary to stabilize the vaccine is loaded into the mold and dried. Then, a polymer-only ink is dispensed and dried to form the backing (Extended Data Fig. 4a,b). The MVP can also be used to tip load multiple cargos simultaneously as demonstrated by red and blue dye loading in different MNPs (Fig. 1i). Tip-loaded MNPs printed with the MVP have 100% correctly formed, sharp microneedles (Extended Data Fig. 4c).
A mathematical model was developed to quantify the number of patches fabricable per day as a function of different design parameters (Supplementary Notes 2 and 3 and Extended Data Fig. 4d–g). The throughput of the MVP is constrained by the slowest process step, which can be either the dispensing step or the drying step depending on the device size, demonstrating the importance of understanding both (Fig. 1j). Continuous processing is possible when the component processes are always running, and dispensing and drying throughputs are equal. Various design contours were plotted to capture throughput for any device considering parameters such as drying time and loading tray dimension (Fig. 1k) or patch size (Extended Data Fig. 4d).
Although a first-generation printer is capable of manufacturing 100 patches in 48 h, this can be increased by changing the size and complexity of the dispensing stage and drying area. The model can be an integrative tool for informing design considerations toward increasing throughput. This can be achieved efficiently by stacking modular microneedle tray molds vertically within the MVP (Extended Data Fig. 5a,b) or continuously fabricating MNPs using a pre-treatment stage to de-gas trays of microneedle molds before dispensing (Extended Data Fig. 5c). Patch removal and packaging could also potentially be automated in future designs using a robotic arm (Extended Data Fig. 5d–g).
Loading protein, DNA and mRNA vaccines in MNPs
The MVP was then used to load various biologics, specifically protein, DNA and mRNA-loaded LNPs. Two-step tip loading with BSA markedly increases loading efficiency—defined as the percent of cargo used for fabrication that is measured in the microneedle tips—compared to one-step MNP fabrication (Fig. 2a). The mass of the vaccine ink that is used during the first step of the two-step process was then optimized to yield high loading efficiency (Extended Data Fig. 6a,b). We observed that loading efficiency increases with decreasing formulation mass (Extended Data Fig. 6c). Using the best loading procedure, 32 µg of DNA and 90 µg of BSA were tip loaded and dispensed simultaneously using the MVP, with similar loading efficiency (Fig. 2b). The loading achieved with the MVP matched what is obtained when the MNPs are fabricated manually by standard laboratory techniques, further confirming the successful automation of the fabrication process for various antigens and vectors (Fig. 2b). Dispensing height was decreased to reduce loading variance (Extended Data Fig. 6d). Loading a DNA vaccine is consistent (s.d. = 1.6 µg) across the surface of a tray with 100 MNP molds with no detectable trends as a function of position (Fig. 2c and Extended Data Fig. 6e).

a, One-step and two-step microneedle loading efficiency (n = 6 independent samples). b, DNA and protein loading efficiency in MNPs made manually are similar to those made using the MVP (n = 4–9 independent samples). c, Heat map showing the consistent loading of DNA in MNPs across a 10 × 10 tray of MNP molds. Each square is an individual patch. Black arrows indicate the movement of the vaccine dispenser. d, MNPs consist of stabilizing polymer and LNPs, which have four lipid components. e,f, LNPs encapsulating mRNA that encodes for fLuc were mixed with various water-soluble polymers and dried. Protein expression in HeLa cells was measured after transfection with re-dissolved polymer to screen for a combination of polymers that produces protein expression similar to LNPs in suspension (n = 4 technical replicates). g, Microneedles dissolve and deliver mRNA-LNP vaccines to the intradermal space. h, Luminescence 6 h after footpad application of MNPs in mice for LNPs made with Lipid 5 or cKK-E12 (n = 5 biologically independent samples). i, Comparison of IM, manually fabricated MNP and printed MNP administration of Lipid 5 LNPs containing 1 µg of fLuc mRNA (n = 5 biologically independent samples). Data represent the mean ± s.d (a,b,e,f) or mean ± s.e.m. (h,i). Unpaired, two-tailed t-tests (a,b), ordinary two-way ANOVA (h) or ordinary one-way ANOVA (i) (Sidak’s multiple comparisons test) were used. P values are represented by: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. ave., average; dev., deviation; NS, not significant.
Next, we tested printing of MNPs that incorporate mRNA-LNPs stabilized in a dissolvable polymer matrix. LNPs are especially difficult to dry in a solid matrix because both chemical and colloidal stability must be preserved33. Additionally, the volume of polymer available in the microneedles is limited, making it challenging to prevent LNP aggregation. To investigate dissolvable polymers for stabilizing LNPs, we compared various biocompatible polymers that are commonly used to fabricate dissolvable microneedles based on their ability to maintain LNP stability at decreasing polymer-to-mRNA mass ratios. LNPs of approximately 147 nm diameter and −2.7 ± 0.6 mV surface potential encapsulating mRNA encoding firefly luciferase (fLuc) were fabricated using ionizable lipid, phospholipid, cholesterol and pegylated lipid (Fig. 2d and Extended Data Fig. 7a–c)34.
LNPs were then mixed with various soluble polymers and dried. After redissolution of the dry matrix, LNPs were used to transfect HeLa cells, and both cell viability and fLuc expression were measured relative to fresh, undried LNPs. None of the formulations tested impaired cell viability compared to LNPs alone (Extended Data Fig. 7d). Formulations containing more than 50% polyvinyl alcohol (PVA) of the total mass are best for stabilizing LNPs (Fig. 2e), whereas other common dissolvable MNP materials perform poorly (Fig. 2f). PVA’s slow drying rate (Extended Data Fig. 7e), hygroscopicity and viscosity (Extended Data Fig. 1k) can be overcome by blending it with polyvinylpyrrolidone (PVP—a faster-drying polymer with desirable mechanical properties (Extended Data Fig. 7f,g)—without sacrificing stability. LNPs are intact after dissolution of MNP polymer matrix—showing a moderate increase in diameter (Extended Data Fig. 7h), typical structure on transmission electron microscopy (TEM) (Extended Data Fig. 7i–k) and preserved mRNA size (Extended Data Fig. 7l).
We sought to select an LNP formulation suitable for ID administration via MNP (Fig. 2g) from two different leading ionizable lipids with very different structures and pKa: cKK-e12 and Lipid 5 (refs. 34,35,36). cKK-e12 was extensively reported efficient at delivering oligonucleotides via intravenous (IV) administration, whereas Lipid 5 was selected from a family of lipids studied for both IM and IV delivery. LNPs of each type encapsulating fLuc mRNA were characterized (Extended Data Fig. 7a–c) and loaded in the microneedles of MNPs using the best stabilizing dissolvable polymer formulation and most efficient loading method—that is, PVP:PVA 1:1 mass ratio and two-step fabrication. The encapsulated mRNA loading in each one of the 100 microneedles of an MNP was measured. We found that, although the mRNA distribution within the MNP is heterogenous, the batch-to-batch variation is low, with approximately 1.0 µg of mRNA tip loaded per MNP (Extended Data Fig. 7m–p). Protein expression after HeLa transfection with LNPs or LNPs redissolved from the microneedle patch showed that the MNP fabrication process does not affect LNP activity in vitro (Extended Data Fig. 7q). MNPs were applied to the footpad of mice, and fLuc expression was measured using luminescence37. LNPs with Lipid 5 have much greater protein expression in dose response than cKK-e12 (Fig. 2h), showing promise for further use in ID delivery.
To investigate ID administration of an LNP-based mRNA vaccine using MNPs, we again measured protein expression in vivo using fLuc mRNA encapsulated in LNPs, this time comparing ID footpad application of MNP with IM administration of an equivalent amount of fLuc mRNA in LNPs (Fig. 2i). Protein expression at 24 h is significantly higher when LNPs are delivered via MNPs rather than IM injection. We also compared the protein expression when MNPs were fabricated manually versus printed with the MVP. The automated printing process does not affect LNP activity in vivo (Fig. 2i).
MNP dissolution, immunogenicity and stability
We evaluated the mechanical and functional properties of MNPs produced with the combination of polymers best capable of stabilizing LNPs. Both conical and pyramid needles were considered as potentially viable geometries that offer varying deliverable volumes and tip angle (Extended Data Fig. 8a and Supplementary Table 1), which have been shown to influence skin penetration and dissolution38. In terms of both peak force (Fig. 3a) and stiffness (Fig. 3b), pyramid MNPs made of PVP:PVA outperform conical MNPs of the same composition, and all MNPs meet the minimum requirements for piercing skin39. For both geometries, ex vivo pig skin puncture (Fig. 3c) was evaluated by histology, and microneedle dissolution was evaluated by optical microscopy image analysis (Fig. 3d,e and Extended Data Fig. 8b). Although both geometries can access the dermis, 36% of the pyramid microneedle volume dissolved in 10 min compared to only 8% of the conical microneedle volume, allowing for greater vaccine delivery—potentially due to their smaller, sharper tip angle40. Pyramid MNPs were, therefore, used for all the following experiments. Increasing the fraction of PVP (Extended Data Fig. 8c–e) and LNPs (Extended Data Fig. 8f) in the blend can be used to increase the dissolved volume.

a,b, Pyramid MNPs made from PVP:PVA are stiffer (a) and stronger (b) than similarly sized conical MNPs (n = 10 independent samples). Unpaired two-tailed t-tests. c, Both conical and pyramid MNPs are capable of penetrating (black arrows) the epidermis (e) and accessing the dermis (d) for ID delivery. d,e, Pyramid MNPs dissolve significantly faster than conical MNPs using optical imaging e before and after application (n = 3 independent samples). Unpaired two-tailed t-tests. f, GMTs are similar between IM and MNP for SARS-CoV-2 RBD mRNA (n = 5 biologically independent samples). Ordinary two-way ANOVA (Sidak’s multiple comparisons test). g, Anti-RBD protein IgG titers through week 11, showing the different kinetics of humoral responses between MNP and IM administration and similar responses for SARS-CoV-2 RBD mRNA (n = 5 biologically independent samples). h, Electrochemiluminescence serum anti-RBD binding assay responses for SARS-CoV-2 variants showing similar responses for IM and MNP with SARS-CoV-2 RBD mRNA (n = 5 biologically independent samples). Ordinary two-way ANOVA (Sidak’s multiple comparisons test). i, PVP:PVA stabilizes mRNA-LNPs in MNPs for high protein expression after 6 months of storage at room temperature (n = 5 biologically independent samples). Each dataset is plotted with a linear fit, and the 95% confidence interval is shaded. j, Design relationship for meeting dosing requirements in humans for common mRNA vaccines, illustrating the relationship among single microneedle volume, microneedles per MNP and dose delivered in a single MNP. Black arrows indicate the size (in microneedles per MNP) where a human dose of mRNA is satisfied for the constant single microneedle volume used in these studies. Data represent mean ± s.d. (a,b,d) or mean ± s.e.m. (f–i). P values are represented by: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. MPa, megapascal; N, Newton; NS, not significant.
To develop a SARS-CoV-2 vaccine for administration by MNP, we used an mRNA encoding the RBD, modified with a T4 trimerization motif, similar to the construct in the BNT162b1 vaccine (Extended Data Fig. 9a)41. Before two-step loading, LNPs containing SARS-CoV-2 mRNA were dialyzed in water and concentrated using centrifugal filtration to increase MNP loading capacity (Extended Data Fig. 9b,c)42. For LNPs, loading efficiency was also increased by making smaller MNPs, which confines the Marangoni effect to the area covered by microneedles instead of the perimeter of the patch (Extended Data Fig. 9d,e). Otherwise, MNPs were fabricated according to the two-step loading described in Figs. 1 and 2, using PVP:PVA 1:1 at a 0.32 mass ratio (mg µg –1) to stabilize LNPs and ionizable lipid Lipid 5 to maximize intradermal protein expression. We used HEK cells to confirm expression of the SARS-CoV-2 mRNA-based vectors (Extended Data Fig. 9f).
Based on previous reports, we used a mouse model to assess immunogenicity of the SARS-CoV-2 vaccine43. Vaccines were administered at a dose of 6 μg encapsulated mRNA (Extended Data Fig. 9g–i) via MNP to the footpad of C57BL/6 mice, and an MNP boost was administered 28 days later (Extended Data Fig. 10a). As a control, a suspension of mRNA-LNPs at a 10-μg mRNA dose was administered via IM injection to the hind limb on the same schedule. Serum was collected every 2 weeks and analyzed for binding anti-RBD IgG titers. At 3 weeks after boost, we measured similar geometric mean titers (GMTs) between IM and MNP for the RBD mRNA vaccine (Fig. 3f). Post-prime responses are lower than those reported for licensed SARS-CoV-2 mRNA-LNP vaccines in a C57BL/6 model, which use different mRNA and lipids, but post-boost responses are similar43,44.
Although all mice receiving the mRNA-LNPs ultimately produce high GMTs, those receiving them IM respond within 1 week to the booster, whereas those receiving them via MNP respond within 3 weeks to the booster (RBD mRNA; Fig. 3g). We also observed that the kinetics of FLuc expression after MNP administration are slower than IM administration (Extended Data Fig. 10b). This is expected given that mRNA-LNPs are dissolving from a solid matrix into the skin via MNP, but a delay in protein expression may help explain the delay in immune response compared to dosing of liquid suspension. Given the slightly lower, yet similar, peak GMTs (Fig. 3f) and their correlates of protection with SARS-CoV-2 infection, RBD mRNA MNPs nonetheless yielded a robust immune response within a feasible amount of time after administration45. We used a multiplexed electrochemiluminescence assay to survey serum anti-RBD binding responses to SARS-CoV-2 S protein variants with different mutations of interest to demonstrate the breadth of protection offered by the MNP vaccine (Fig. 3h).
We compared the PVP:PVA blend that was developed for microneedle mRNA-LNP delivery to a conventional mRNA-LNP suspension administered IM at the same dose, using fLuc mRNA as a model for protein expression in mice. Both were stored at room temperature and at 4 °C and assessed 1 month, 3 months and 6 months later in vivo. Although the IM suspension’s potency decreases over 6 months, MNPs produce consistently high luminescence over the entire storage period (Fig. 3i), even when stored at room temperature. MNPs also maintain mRNA-LNPs stable when stored at 37 °C for 1 month, which matches the storage time between the prime and the boost (Extended Data Fig. 10c). In a similar study, RBD mRNA MNPs are immunogenic after at least 3 months of storage at room temperature and 4 °C (Extended Data Fig. 10d), when used as a booster for BALB/c mice primed with mRNA-LNP suspension.
Delivering a sufficient dose of LNP vaccine with MNPs is challenging due to the polymer mass needed to stabilize the LNPs. Using the needle volume and MNP loading efficiency from the anti-RBD titers peak study, we designed a model (Fig. 3j and Supplementary Note 4) that predicts the combination of volume and number of microneedles necessary to deliver the full Moderna (mRNA-1273) or Pfizer-BioNTech (BNT162b1) COVID-19 vaccine doses. The model predicts that 360 and 108 of the studied pyramid microneedles and formulation would deliver the full dose of the Moderna and Pfizer-BioNTech vaccines, respectively. MNPs containing a sufficient dose are less than 2 cm across.
Discussion
Although the magnitudes of humoral responses are similar, we observed interesting differences between IM administration of mRNA-LNP suspension and ID delivery of mRNA-LNPs using MNPs. mRNA-LNPs incorporating Lipid 5 outperformed those with cKK-E12 in terms of protein expression (Fig. 2h), suggesting that MNPs may be sensitive to ionizable lipid composition. Ionizable lipids govern protein expression and targeting and have been optimized for various organs and administration routes34,35,46,47. Humoral responses also develop faster IM (Fig. 3g). Previous research on microneedle-based vaccines suggest a connection between administration method and kinetics of the humoral response48,49. This may reflect the slower dissolution of MNP matrix (Extended Data Fig. 10b) or fundamental differences between ID and IM administration. These differences highlight opportunities to further investigate and engineer the potency of mRNA-LNPs delivered by MNPs. Additionally, RBD mRNA constructs may warrant further study as a target for vaccines with increased stability50.
MNPs containing mRNA-LNPs offer new opportunities to streamline vaccine administration and improve vaccine efficacy. ID administration of attenuated virus, virus-like particle and other vaccines may allow dose sparing compared to the IM or subdermal routes51,52. ID delivery of vaccines was recommended at a meeting of the World Health Organization Strategic Advisory Group of Experts as a means to reduce cost of the polio vaccine, and a DNA vaccine delivered ID via jet injector was authorized for emergency use in India53,54. Room-temperature-stable vaccines would greatly facilitate deployment in the developing world, where the supply chain necessary for cold transport may be inadequate. They would also allow for vaccines to be cost-efficiently stockpiled in preparation for a potential outbreak55. Deployment of COVID-19 vaccines in particular has been hampered by poor shelf life and reliance on cold chain storage and transport systems. Until now, to our knowledge, thermostable mRNA-LNP vaccines have not been developed33,56, perhaps because of their nanoparticulate nature (for example, very high surface area) and tendency to aggregate irreversibly, which makes them exceptionally difficult to stabilize in a dry formulation33,57,58,59.
We show that MNP vaccines printed by the MVP can be stored at room temperature and used for immunizations months after fabrication. In contrast, certain current mRNA vaccines must be held at −60 °C to −80 °C for long-term storage and 4 °C for short-term storage. Modeling data suggest that a human dose of mRNA-LNP COVID-19 vaccine can be formulated into a 2-cm-square microneedle patch (Fig. 3j). This assumes that needle-to-needle spacing is maintained as patch size increases, but, for eventual translation, skin penetration and needle dissolution of a larger patch must be evaluated. Additional engineering of the MVP will be required to develop an end-to-end microneedle fabrication process for application in humans, including an aseptic enclosure to reduce bioburden and an integrated packaging step60,61. We think that the device could be easily adapted to any mRNA vaccine for diseases of interest in specific regions and sized according to the desired scale of MNP vaccine production.
Methods
Design, assembly and automation of the MNP device
The MVP was designed with the requirement to integrate and automate the necessary functions to convert ingredients necessary for fabricating MNPs into finished MNPs with minimal operator skill level and in a form factor suitable for transport. The resulting design consisted of three main functions: loading, dispensing and drying. These three functions were organized into the MVP (Extended Data Fig. 3f) and were programmed to function simultaneously. All programming related to MVP was implemented in the EPSON RC 7.0 SPEL programming language. The operator is required to load molds, fill the reservoirs for liquid formulation and polymer backing and unload MNPs.
A key operating function was developed such that, during the dispensing step, the liquid formulation is first dispensed above the MNPʼs mold and then drawn inside the negative mold cavity using vacuum underneath (taking approximately 15 min), and then the loading stage moves to the drying station where its atmosphere is sealed to accelerate the drying time.
The dispensing stage consists of a syringe pump (Harvard PHD 2000) and a robotic arm (Epson T3) holding a pair of nozzles (18-gauge needles for dye solutions, 25-gauge for vaccine printing) connected via tubing to the syringe pump. The motion of the robotic arm was programmed to be synchronized with the syringe pump. The motion of the robotic arm/pump was optimized to maximize the coverage of the MNP mold by the liquid formulation. A dispensing time gap was imposed between each two adjacent MNPs to minimize inter-patch dripping and vaccine waste.
Both processes were based on different sequences of applying vacuum on PDMS molds and dispensing the polymer solution (loaded with dye or biological agents), followed by vacuum drying. In the first process (de-gassing), vacuum was initially applied on empty PDMS molds, followed by dispensing polymer solution within a specific time (~7 min) right after vacuum application, eventually drying the polymer solution with vacuum. Vacuum de-gassing facilitated diffusion of polymer solution into the microneedle cavities in the PDMS mold (Extended Data Fig. 1b and Supplementary Video 1). In the second approach, polymer solution was dispensed on the PDMS mold, and vacuum was subsequently applied both from underneath and on top of the sheets (Extended Data Fig. 1a and Supplementary Video 2) to induce diffusion of polymer but also accelerate the drying time without bubble formation. When drying the solution using vacuum, it is important to maintain the pressure gradient through the PDMS mold by keeping a higher negative pressure below the PDMS sheet. By doing so, the air dissolved in the polymer–vaccine solution is driven downwards (toward the vacuum chamber below the PDMS mold), which avoids bubble formation during drying. Due to simplicity of automation, the second approach was further implemented into the device.
The loading stage was a custom-made 10 × 10 array of MNP molds, which was used to apply vacuum (−1 bar) below the PDMS sheets. The loading stage was fixed to a single-axis motion stage (Thorlabs) allowing transfer between the loading station and the drying station. Before dispensing the formulation, the alignment of the loading station with the dispensing station was calibrated.
The drying stage was a custom-made vacuum chamber that moved vertically via a single-axis motion stage (Thorlabs). The headcover comprised a vacuum connection as well as a desiccant bag (Extended Data Fig. 3c). The vacuum was set to −0.6 bar to accelerate drying. A single vacuum pump was used for both the loading and drying stages. The vacuum pump was directly connected to the loading stage, and a pressure controller was used to decrease the vacuum to −0.6 bar in the headcover. The lower vacuum in the headcover compared to inside the loading stage maintained a negative pressure gradient in the sheet of PDMS molds, avoiding bubble formation in the MNP solutions. Moreover, the pressure controller was used to automatically bring back the drying stage to atmospheric pressure after drying and release the vacuum chamber from the loading stage.
Patch coverage quantification
Patches (n = 100) were dispensed and dried with the MVP to study the effect of liquid formulation on patch coverage caused by different drying patterns on individual MNPs. The liquid formulation included a polymer formulation (PVP, PVA, PVP:PVA 1:1) dissolved at 20% (w/w) in PBS and mixed with dye. Patch coverage was defined as the number of microneedles formed successfully after drying divided by the total number of patches fabricable (100). The patch coverage was quantified individually for each patch under an optical microscope for each formulation. Drop area and circularity were quantified using image analysis after imaging.
Dispensing vaccines and other biological cargos
A two-step dispensing approach was followed for loading vaccine and other biological cargos. In the first step, vaccine solution was aspirated into the dispensing tube through the dispensing needles (25-gauge, BD Biosciences). The liquid formulation in the first step was composed of a certain concentration of the biological agent mixed with PVP:PVA 1:1, 4% (w/w). Approximately 36 ± 6 µl of vaccine solution (or other cargos) was then dispensed using a pair of 1-ml syringes (BD Biosciences) on the center of MNP molds on the dispensing tray. A time gap of 9 s was imposed between dispensing each two adjacent MNPs to minimize vaccine waste during the transfer of the dispensing robot from mold to mold. The backing solution (PVP:PVA 1:1, 20% (w/w)) was aspirated directly to a new dispensing syringe (10 ml-sized, BD Biosciences). An amount 48 ± 6 µl of the backing solution was dispensed in the second stage right on top of the previously dispensed (dried) vaccine spots. Before and after each dispensing round, the needles and the tubes were washed with ethanol (70%) followed by sterile water. In some experiments, a pair of cargos was co-dispensed such that each syringe/needle dispensed a different cargo. The same loading/dispensing procedure as in mono-dispensing was followed in this case, except that each syringe was loaded with a different cargo.
Computer-aided design and illustration
SolidWorks 2021 (Dassault Systèmes) was used for 3D illustrations of the MVP, robotic arm, dispensing station, vacuum devices, negative pressure chamber, sterile environment container, drying rack, conveyer belt and automatic microneedle patch demolding station. The Epson T3 robotic arm drawing was directly downloaded from the official Epson website. The XYZ plotter, drying rack and conveyer belt models were downloaded from GrabCAD. All other parts were custom designed in reference to the actual dimensions of the prototype parts.
Dispensing optimization
A design of experiments (DOE) approach was followed to systemically study the effect of various design parameters on the collective size of the drops, as the output response, dispensed on an individual PDMS array from the robotic nozzle (needle). To this end, an L18 orthogonal array design of experiment (Taguchi DOE) was constructed in software (Minitab). The effect of the following design parameters was studied: (1) attachment of a 0.2-µm filter to the nozzle, (2) needle gauge, (3) dispensing height, (4) dispensing flow rate and (5) polymer viscosity as a function of polymer composition. The projected drop area of total drops at the end of each dispensing was quantified using ImageJ software. Results (n = 3–5) were analyzed in Minitab to find the mean response of drop area as a function of change in these parameters. A schematic illustration of the experimental setup is shown in Extended Data Fig. 3g. The total dispensed volume was considered constant and equal to 200 µl. Different flowrates were achieved by keeping the total dispensed volume constant (200 µl) but varying the dispensing duration.
Microneedle formation quantification
MNPs were printed using the automated MVP and imaged with an optical microscope to determine if microneedles were the full length and sharp (n = 30). Image analysis was used to estimate the needle height.
Scanning electron microscopy
Scanning electron microscopy (SEM) was used to image fabricated microneedles. Samples were initially coated by a thin layer of Au/Pd using a Hummer 6.2 Sputtering System (ANATECH) and then imaged using a JSM-5600LV SEM (JEOL) with an acceleration voltage of 5–10 kV.
PDMS molds fabrication
Steel MNP positives were used to generate negative molds made of PDMS (Sylgard 184, Dow Corning), which was mixed according to the manufacturer’s instructions and cured overnight at 60 °C. To create additional MNP positives, UV-curable Norland Optical Adhesive 61 was filled into the PDMS negative molds using a centrifuge at 3,234g for 1 min, placed in a UV-curing oven at room temperature for 20 min and manually removed.
To create a tray of negative molds, MNP positives were first aligned using a laser-cut 5 × 5 acrylic grid. Then, Norland Optical Adhesive 61 was poured between the individual resin positives replicates, UV cured at room temperature for 20 min and then held at 60 °C overnight. The resulting tray of evenly spaced 5 × 5 positive MNPs (Extended Data Fig. 3d) was used to fabricate a 5 × 5 PDMS sheet of negative molds (Extended Data Fig. 3e). PDMS was used to cover the aligned resin positives and create an additional 1-mm layer of PDMS above the positives. The sheet was leveled, cured overnight at 60 °C and removed using isopropanol to help separate the negative from the positive.
Dissolvable microneedle fabrication
MNPs were fabricated by loading and drying 200 µl of a 20% w/w PVP, PVA or PVP:PVA solution in a PDMS mold. When dye, LNPs, DNA or protein were loaded in the MNP, a two-step loading procedure was used. First, 200 µl of a solution in deionized (DI) water or PBS containing 0.8 mg, 1.6 mg, 2 mg or 8 mg of PVP:PVA and various amounts of LNPs (expressed as encapsulated mRNA mass), protein or DNA were loaded and dried. Then, a variable volume of PVP:PVA 20% w/w in DI water or PBS was used to bring the total MNP mass to 40 mg of PVP:PVA. That polymer solution was loaded and dried to create a thin polymer backing. All MNPs used in in vivo studies were fabricated using manual dispensing unless otherwise specified. All MNPs loaded with LNPs, DNA or protein were fabricated by applying vacuum through the mold using the pattern with lines at −1 bar gauge pressure and applying vacuum in the drying chamber at −0.6 bar gauge pressure.
For the one-step fabrication method, the 40 mg of PVP:PVA was mixed with various amounts of protein and added as one step.
Measurement of loading time
The time needed to load ink into the PDMS negative mold was evaluated by recording the loading with an electronic microscope (n = 3). The microscope was placed on the side of the PMDS mold and focused on the microneedles through the transparent PDMS.
Vacuum de-gassing loading method
Polymer solution was loaded in PDMS molds that were de-gassed under various vacuum values from 0 to −0.5 bar gauge pressure for various amounts of time. The amount of time between de-gassing and loading was varied from 5–10 min to 1 h. MNPs were then dried and removed from the PDMS mold. Optical images of the polymer–dye solution moving into the PDMS mold were acquired 0 min, 2 min, 5 min and 10 min after adding the solution to the mold. From these images, the needle height was estimated using image analysis.
Measurement of viscosity
Polymer solutions (n = 1) were characterized using a TA Instruments Discovery HR-2 Rheometer using a 40-mm parallel plate and a shear rate ramp from 0.01 s−1 to 5,000 s−1.
Protein quantification
Needles of an MNP loaded with 50 µg, 100 µg or 200 µg of BSA and 0.8 mg, 4 mg or 8 mg of PVP:PVA (for two-step tip loading) were cut and dissolved in DI water (n = 6). A bicinchoninic acid assay (BCA) (Thermo Fisher Scientific) was used to quantify the protein loading using the standard procedure for a 96-well plate. An internal standard of PVP and PVA was added to the calibration curve to account for their interference. The amount of PVP and PVA added to the calibration curve was calculated based on the needle volume (0.08 mm3), a density of 1.2 g/cm3 and the number of needles used for BSA quantification (adjusted to the dilution).
DNA quantification
For the loading efficiency comparison between manual and automated fabrication, needles of an MNP loaded with 100 µg of DNA and 1.6 mg of PVP:PVA (two-step tip loading) were cut and dissolved in Tris-EDTA (TE) buffer (n = 8). DNA was quantified using UV absorption at 260 nm and 280 nm using a NanoDrop. Because PVP also absorbs in UV range, the same amount of PVP was added to the calibration curve to account for its contribution. The amount of PVP to add was calculated the same way as described in protein quantification and assuming a 1:1 PVP-to-PVA ratio. This was repeated for the tray position heat map and loading efficiency calculation with DNA, but 10 µg of DNA and 2 mg of PVP:PVA were used per patch.
LNPs synthesis
Purified mRNAs were obtained with CleanCap AG cap, full N1-methyl-pseudouridine substitution and polyadenylated tail (120 A) (TriLink BioTechnologies). Various high-purity lipids were used for LNP synthesis, including 3,6-bis({4-[bis(2-hydroxydodecyl)amino]butyl})piperazine-2,5-dione (cKK-E12, Organix), heptadecane-9-yl 8-((2-hydroxyethyl)(8-nonyloxy)-8-oxoctyl)amino)octanoate (Lipid 5, Organix), 1-2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, Avanti), cholesterol (Sigma-Aldrich) and 1-2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (C14-PEG2000, Avanti). LNPs were prepared using procedures previously described34,35,36. Lipids were dissolved in ethanol at a molar ratio of 35:16:46.5:2.5 cKK-E12:DOPE:cholesterol:C14-PEG2000 or 38.4:12.3:47.4:1.9 Lipid 5:DOPE:cholestorol:C14-PEG2000 when using cKK-E12 or Lipid 5 as the ionizable lipid, respectively. To prepare the LNPs, the ethanoic solution was rapidly added to and mixed with an mRNA solution buffered with citrate at pH 3 at volume ratio 3:1 (aqueous:ethanol). When using cKKe12, the ionizable lipid-to-mRNA weight ratio was set to 10, and the final mRNA concentration was 0.1 mg ml−1. When using Lipid 5, the ionizable lipid-to-mRNA weight ratio was set to 5, and the final mRNA concentration was 0.135 mg ml−1. All nucleic acids were stored at −80 °C and allowed to thaw on ice before use. The LNPs were then dialyzed for at least 2 h in PBS at 4 °C in a 20,000 molecular weight cutoff (MWCO) cassette34. For LNPs in DI water, the solution was dialyzed against DI water for an additional minimum of 2 h at 4 °C. When needed, the LNPs were concentrated on an Amicon filter by centrifuging at 3,000g42. All solutions were kept at 4 °C and used within 1 week.
mRNA concentration and encapsulation efficiency in the LNPs was estimated with a Quant-iT RiboGreen assay (Thermo Fisher Scientific) using a modified procedure described elsewhere (n = 3 per batch)34. In brief, LNPs were diluted in either TE or TE mixed with Triton X-100 buffer (TX). Then, the Quant-iT RiboGreen assay was used to quantify the mRNA that is not encapsulated (when diluted with TE) and the total mRNA concentration (when diluted with TX). For size, LNPs were diluted 200 times in PBS and characterized using a Zetasizer Nano-NS (Malvern Instruments). When measuring the mRNA loading in MNPs, microneedles were cut and dissolved in TE and TX. When measuring the mRNA loaded in MNP backing, the microneedles were cut, and the remaining MNP was dissolved in TE and TX. Subtracting the un-encapsulated mRNA from the total mRNA yielded the encapsulated mRNA concentration.
Testing various polymer–vaccine inks for stabilizing dry LNPs
Polymers were dissolved in PBS or DI water at a concentration ranging from 10% to 30% w/w depending on their solubility (n = 4). These solutions were then weighed and mixed with LNP suspension to reach the appropriate polymer-to-mRNA mass ratio. The ink mixture was immediately dried in a LoBind Eppendorf tube in a desiccator under −0.5 bar vacuum. After 24 h drying, the ink pellet was redissolved in PBS and incubated for 10 min at 37 °C. PBS was used to adjust the volume so that 15 µl of dissolved ink contains 50 ng of encapsulated mRNA. This mixture was used to transfect cells. The ionizable lipid used was cKK-E12.
For the in vitro evaluation of MNPs loaded with LNPs, four MNPs were fabricated using the two-step loading, 10 µg of mRNA and 8 mg PVP:PVA for the tip loading. Two MNPs were used to quantify LNP tip loading, and two MNPs were used to transfect HeLa cells. Needles were cut and dissolved in 1 ml of TE buffer for the RNA quantification and in 1 ml of DMEM for the cell transfection. The procedure described in LNP synthesis was used to quantify mRNA, and HeLa cells were transfected as described hereunder. The ionizable lipid used was Lipid 5.
HeLa cells were cultured in high-glucose DMEM with phenol red (Invitrogen) supplemented with 10% FBS (Invitrogen) and 1% antibiotic (Invitrogen). Then, 10,000 cells were seeded in wells of a white 96-well plate in full growth medium. Twenty hours after seeding, 15 µl of fresh LNPs or dissolved formulation, or 50 µl of fresh LNPs or dissolved MNPs, was added to the growth medium. In all cases, 50 ng of encapsulated mRNA was added to each well. Twenty-four hours after transfection, 100 µl of Bright-Glo Luciferase Assay Kit (Promega) reagent was added, and luminescence was measured in the following 2 min using a Tecan Infinite 200 PRO plate reader.
HeLa cell viability was measured in presence of 0.6 mg of polymer formulation and 50 ng of mRNA encapsulated in LNPs made with cKK-e12 as the ionizable lipid. Fifteen microliters of ink were used in each well. Cell viability was measured in a 96-well plate using a CKK8 assay as described by the manufacturer (ab228554).
mRNA spatial distribution in MNP
The MNP was fabricated using the two-step loading, using 10 µg of mRNA and 8 mg PVP:PVA in the first step. Each of the 100 needles of the MNP was manually cut at their base under a microscope and sorted based on their position on the array. RNA was quantified the same way as described in the LNP synthesis section. The ionizable lipid used was Lipid 5.
Particle size analysis of mRNA-LNPs from dissolved MNPs
FLuc mRNA-LNPs were used for all samples. LNPs were analyzed (1) after dialysis to PBS, (2) after concentrating to 320 μg ml−1, (3) after diluting to 200 μg ml−1 in 4% w/w polymer solution to form vaccine ink and (4) after drying to form MNPs. Samples were dissolved in 20 µl, vortexed for 2 s, diluted with an additional 200 µl and mixed. Then, 10 µl of reconstituted mRNA-LNPs was diluted in 490 µl of water, vortexed for 2 s and then analyzed using dynamic light scattering (DLS) on a Zetasizer Nano-NS (Malvern Instruments). Samples (n = 5 per group) were equilibrated for 60 s before measurement. Three technical replicates were performed for each sample.
TEM and cryogenic-TEM
FLuc mRNA-LNPs were used for all samples. LNPs were analyzed (1) after dialysis to PBS (2), after concentrating to 320 μg ml−1 (3), after diluting to 200 μg ml−1 in 4% w/w polymer solution to form vaccine ink and (4) after drying to form MNPs. Dissolved samples were added onto the carbon-coated copper TEM grids and blotted to remove the excess solution. Next, samples were stained using 1% phosphotungstic acid aqueous solution; the excess stain solution was removed; and samples were dried at room temperature before TEM imaging. For cryogenic-TEM imaging, samples were plunge-frozen using a 930 Gatan Cryoplunge 3. All the samples were imaged using a JEOL 2100 FEG microscope at 200-kV acceleration voltage.
Capillary electrophoresis
FLuc mRNA-LNPs were used for all samples. mRNA was analyzed (1) as provided by the manufacturer, (2) after mRNA-LNP synthesis and dialysis to PBS, (3) after diluting to 200 μg ml−1 in 4% w/w polymer solution to form vaccine ink and (4) after drying to form MNPs. Then, 2 µl of dissolved samples was analyzed using an Agilent Femto Pulse using the Ultra Sensitivity RNA Kit (FP-1201). Samples were prepared using TE or TX buffer.
Water content analysis
The water content of the fabricated MNPs at different timepoints and stages of drying was quantified by thermogravimetric analysis (TGA) using a Pyris 1 Thermogravimetric analyzer (PerkinElmer) with a heating rate of 20 °C per minute from 50 °C to 600 °C under nitrogen flow (20 ml min−1) (n = 3). The water content was evaluated by analyzing only the needles of the MNPs placed in ceramic pans and not the backing. The drying rate was estimated using a linear regression of water content measured over time throughout the drying process. All analyses were conducted in triplicate.
Compressive mechanical testing
For compression testing, a single MNP was mounted between compression platens (Instron 2501 Series) and compressed at a rate of 1 mm min−1 using an Instron 5943 with a 500 N load cell (n = 10). The peak force before microneedle failure was reported and the slope of the linear region of initial compression. Microneedle failure mode was determined by imaging the patches after testing.
Microneedle skin penetration and dissolution assessment
MNPs were applied ex vivo on excised pig skin using a mini spring-loaded applicator (Micropoint Technologies) for 2 min, 5 min, 10 min or 30 min (n = 3). Subsequently, the skin samples were fixed into formalin for 48 h and then transferred to 70% ethanol and embedded in paraffin wax. Samples were sectioned and stained with hematoxylin and eosin.
For quantifying microneedle dissolution as a function of application time, microneedle patches were imaged before and after application on excised pig skin using a Leica DFC450. The patches were placed in a transverse manner for imaging using LAS version 4.7 software. Microneedle length was calculated using ImageJ for at least 10 microneedles from each patch, and these measurements were performed on three patches per timepoint.
Luciferase mRNA expression in mice
All animal procedures were approved and performed under the guidelines of the Massachusetts Institute of Technology Committee on Animal Care (1019-061-22). Six- to eight-week-old female C57BL/6 mice (Charles River Laboratories) were used and monitored for safety (n = 5). MNPs were fabricated with mRNA-LNPs encoding for FLuc (L-7010, TriLink BioTechnologies) using the two-step loading method described above. LNP dose and ionizable lipid chemistry were varied, maintaining at least a 100:1 polymer:mRNA mass ratio. Two ionizable lipids that were previously selected for IV or IM mRNA administration methods were studied, respectively: cKK-E12 (ref. 34) and Lipid 5 (ref. 35). MNPs were applied to either footpad with anesthesia. To more fully dissolve the full dose of LNPs carried in an MNP, when a 10 × 10 array was applied, MNPs were divided into two halves and applied consecutively to the same footpad, for 10 min per half. Hand pressure was applied for the first minute, and then the MNP was secured to the footpad using tape for the remaining 9 min. As a positive control, LNPs were administered to the caudal thigh muscle as a 40-µl suspension in PBS at matching doses. The footpad was selected because it has been previously used for IVIS imaging of luminescence after microneedle application37 and because it is a well-studied site for ID administration of vaccines, allowing for isolation of the draining lymph node62,63.
Twenty-four hours after MNP application, mice were imaged for bioluminescence in an IVIS kinetic imaging system (PerkinElmer). Then, 15 min before imaging, mice were injected intraperitoneally with RediJect D-Luciferin Ultra (PerkinElmer) at 150 mg kg−1. Luminescence was quantified using LivingImage software (PerkinElmer). Comparison between luminescence results must be carefully considered because injection site light absorption, diffusion and depth of delivery can affect the signal measured using IVIS. For the kinetics experiment, imaging was performed at 2 h, 6 h, 24 h, 48 h and 72 h after administration.
RBD expression in HEK293
In total, 89,000 HEK293 cells were seeded per well of a BioCoat Poly-D-Lysine four-well culture slide (354577). The same growth medium as HeLa cells was used. Twenty-four hours after seeding, cells were transfected with 265 ng of mRNA per well from an LNP suspension made with Lipid 5 and encapsulating mRNA encoding for RBD. LNPs dissolved from an MNP were also used to demonstrate bioactivity after loading into MNPs. The same MNP as the ones used in the SARS-CoV-2 vaccination study was used. Needles were cut, dissolved in DMEM and used to transfect cells using the same mRNA mass as the control LNP suspension. The mRNA was quantified by dissolving needles of an independent replicate of the MNP in TE buffer and using the procedure described in LNP synthesis (PVP and PVA were also added as internal standards to the RNA calibration curve of the RiboGreen). Forty-eight hours after seeding, cells were washed two times with PBS at 37 °C. Cells were washed with PBS between all subsequent steps. Cells were left to equilibrate to room temperature for 15 min. Then, 1 ml of Image-iT Fixative Solution (Invitrogen) was added per well (FB002) and left for 15 min. Next, 1 mL of eBioscience Intracellular Fixation and Permeabilization Buffer (Invitrogen) was added and left to incubate for 10 min. Then, 1 ml of BSA 1% (w:v) in PBS was added for 1 h at room temperature. Then, 300 µl of SARS-CoV-2 Spike Protein (RBD) Recombinant Human Monoclonal Antibody (T01KHu, Thermo Fisher Scientific, 703958) diluted 100-fold in PBS was added to each well and left to incubate for 1 h. PBS-Tween was used to wash cells. Next, 300 µl of goat anti-human IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Thermo Fisher Scientific, A-11013), diluted 400-fold in PBS was used as the secondary antibody and incubated for 1 h. Then, 300 µl of Rhodamine-Phalloidin (Invitrogen R415) staining diluted 400-fold in BSA 1% was used for cellular cytoskeleton imaging and incubated for 30 min. Cells were finally fixed using one drop of DAPI ProLong (Invitrogen, P36982). Cells were imaged on a confocal microscope (Olympus FV1000).
SARS-CoV-2 MNP vaccination with mRNA
All animal procedures were approved and performed under the guidelines of the Massachusetts Institute of Technology Committee on Animal Care (1019-061-22). Six- to eight-week-old female C57BL/6 mice (Charles River Laboratories) were used and monitored for safety (n = 5). MNPs were fabricated with human codon-optimized mRNA-LNPs encoding for SARS-CoV-2 RBD and a T4 trimerization motif, using the two-step loading method described above. Smaller MNP arrays were fabricated containing 1.5 µg of encapsulated mRNA, which was verified by mRNA quantification. Four MNPs with approximately 12 microneedles were applied to the left and right footpad of mice with anesthesia, with a 10-min application time for each, for a total encapsulated mRNA dose of 6.0 µg per mouse. Hand pressure was applied for the first minute, and then the MNP was secured to the footpad using tape for the remaining 9 min. As a positive control, LNPs were administered to the right caudal thigh muscle as a 40-µl suspension in PBS at 10.0 µg of encapsulated mRNA per mouse.
mRNA stability evaluation in mice
All animal procedures were approved and performed under the guidelines of the Massachusetts Institute of Technology Committee on Animal Care (1019-061-22). MNPs were fabricated with FLuc mRNA LNPs using the two-step loading method described above, at an mRNA dose of 1.0 µg per patch with a 500:1 polymer:mRNA mass ratio. Lipid 5 was used as the ionizable lipid. MNP size and application are identical to the above studies of fLuc expression (n = 5). MNPs were stored in a container with silica desiccant at various temperatures. As a positive control, sealed vials of suspension containing the same amount of fLuc mRNA in LNPs made with Lipid 5 were stored at various temperatures alongside MNPs.
SARS-CoV-2 anti-RBD binding titers
A SARS-CoV-2 Spike S1-RBD ELISA detection kit was used (L00845, GenScript). Plates were coated with purified recombinant SARS-CoV-2 Spike S1-RBD antigen. Plates were incubated with serial dilutions of heat-inactivated sera and incubated at 37 °C for 30 min. A 1:10,000 dilution of rabbit anti-mouse IgG-horseradish peroxidase conjugate (Abcam, ab6728) was used as a secondary antibody, and 3,5,3′,5′-tetramethylbenzidine (TMB) was used as a substrate. Interpolated endpoint titers were calculated as the dilution that emitted an optical density exceeding 3× background produced by serum from naive mice.
Electrochemiluminescence assay
ECLA plates (Meso Scale Discovery SARS-CoV-2 IgG, N05CA-1, Panel 7) were designed and produced with four antigen spots in each well. Antigens included were WA1/2020, B.1.1.7, P.1 and B.1.351 S1-RBD. Plates were blocked with 50 µl of 1% BSA solution for at least 30 min at room temperature with shaking at 700 r.p.m. During blocking, the serum was diluted 1:5,000 with Diluent 100 (Meso Scale Discovery). The plates were washed with 150 µl of wash buffer and blotted dry, and 50 µl of diluted samples was added in duplicate to the plates. The samples were incubated at room temperature with shaking at 700 r.p.m. for 2 h. Secondary antibody was prepared using Jackson ImmunoResearch rabbit anti-mouse IgG detection antibody (315-005-045) conjugated to the MSD GOLD SULFO-TAG by NHS-Ester chemistry per the manufacturer’s guidelines (R91AO-1). Plates were again washed three times, and 50 µl of SULFO-tagged anti-mouse IgG detection antibody diluted to 1× in Diluent 100 was added to each well and incubated for 1 h with 700 r.p.m. shaking. Plates were washed three times; 150 µl of MSD GOLD Read Buffer B was added to each well; and the plates were read on a MESO Quick-Plex SQ 120. MSD titers for each sample were reported as relative light units (RLU), which were calculated as sample minus blank for each spot and sample. The limit of detection was defined as 500 RLU for all assays.
Shelf-life study with SARS-CoV-2 mRNA MNPs
MNPs containing 1.5 μg of encapsulated SARS-CoV-2 mRNA were stored in a container with silica desiccant at various temperatures. As a positive control, sealed vials of suspension containing SARS-CoV-2 mRNA-LNPs at 200 µg ml−1 were stored at various temperatures alongside MNPs. Six- to eight-week-old female BALB/c mice (Charles River Laboratories) were used and monitored for safety (n = 5). All mice received a prime dose of 10 µg of encapsulated mRNA in mRNA-LNPs administered to the right caudal thigh muscle as a 40-µl suspension in PBS. Mice were then boosted with various materials, including fresh controls and patches or suspension stored for 1 month or 3 months. For MNP groups, as in previous experiments, four MNPs with 12 microneedles were applied to the left and right footpad of mice with anesthesia, with a 10-min application time for each, for an estimated total mRNA dose of 6.0 µg per mouse. For IM groups, 4 μg of encapsulated mRNA in mRNA-LNPs was administered to the right caudal thigh muscle as a 40-µl suspension in PBS.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software. P values are represented by: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets generated and analyzed during these studies are available from the corresponding author upon reasonable request. The RBD sequence used for this study is available at GenBank (OP839194).
Code availability
The codes used for throughput modeling and device programming are available at a public Zenodo repository (https://doi.org/10.5281/zenodo.7735167).
References
Khan, M. I., Ikram, A. & Hamza, H. B. Vaccine manufacturing capacity in low- and middle-income countries. Bull. World Health Organ. 99, 479–479A (2021).
Koff, W. C. et al. Development and deployment of COVID-19 vaccines for those most vulnerable. Sci. Transl. Med. 13, eabd1525 (2021).
Wells, C. R. & Galvani, A. P. The global impact of disproportionate vaccination coverage on COVID-19 mortality. Lancet Infect. Dis. 22, 1254–1255 (2022).
Kakule, B., Lubukayi, N., Muhindo, E., Janoch E. & Prather A. At the last mile: COVID-19 vaccines in DRC (CARE International, 2022); https://careevaluations.org/wp-content/uploads/COVID-19-vaccines-in-Democratic-Republic-of-the-Congo-April-2022.pdf
O’Shea, J., Prausnitz, M. R. & Rouphael, N. Dissolvable microneedle patches to enable increased access to vaccines against SARS-CoV-2 and future pandemic outbreaks. Vaccines (Basel) 9, 320 (2021).
Sullivan, S. P., Murthy, N. & Prausnitz, M. R. Minimally invasive protein delivery with rapidly dissolving polymer microneedles. Adv. Mater. 20, 933–938 (2008).
Sullivan, S. P. et al. Dissolving polymer microneedle patches for influenza vaccination. Nat. Med. 16, 915–920 (2010).
Kim, Y. C., Park, J. H. & Prausnitz, M. R. Microneedles for drug and vaccine delivery. Adv. Drug Deliv. Rev. 64, 1547–1568 (2012).
Mistilis, M. J. et al. Long-term stability of influenza vaccine in a dissolving microneedle patch. Drug Deliv. Transl. Res. 7, 195–205 (2017).
Gill, H. S., Denson, D. D., Burris, B. A. & Prausnitz, M. R. Effect of microneedle design on pain in human subjects. Clin. J. Pain 24, 585–594 (2008).
Rodgers, A. M., Cordeiro, A. S. & Donnelly, R. F. Technology update: dissolvable microneedle patches for vaccine delivery. Med. Devices (Auckl). 12, 379–398 (2019).
Noh, I., Lee, K. & Rhee, Y. S. Microneedle systems for delivering nucleic acid drugs. J. Pharm. Investig. 52, 273–292 (2022).
Koh, K. J. et al. Formulation, characterization and evaluation of mRNA-loaded dissolvable polymeric microneedles (RNApatch). Sci Rep. 8, 11842 (2018).
Golombek, S. et al. Intradermal delivery of synthetic mRNA using hollow microneedles for efficient and rapid production of exogenous proteins in skin. Mol. Ther. Nucleic Acids. 11, 382–392 (2018).
Creelman, B., Frivold, C., Jessup, S., Saxon, G. & Jarrahian, C. Manufacturing readiness assessment for evaluation of the microneedle array patch industry: an exploration of barriers to full-scale manufacturing. Drug Deliv. Transl. Res. 12, 368–375 (2021).
Moga, K. A. et al. Rapidly-dissolvable microneedle patches via a highly scalable and reproducible soft lithography approach. Adv. Mater. 25, 5060–5066 (2013).
Fakhraei Lahiji, S. et al. Effects of dissolving microneedle fabrication parameters on the activity of encapsulated lysozyme. Eur. J. Pharm. Sci. 117, 290–296 (2018).
Markarian, J. Manufacturing microneedle array patches for vaccine delivery. Pharm. Technol. 44, 31–32 (2020).
Dalvi, M. et al. Panorama of dissolving microneedles for transdermal drug delivery. Life Sci. 284, 119877 (2021).
Du, G. & Sun, X. Current advances in sustained release microneedles. Pharmaceut. Fronts 2, e11–e22 (2020).
Chen, W. et al. Microneedle-array patches loaded with dual mineralized protein/peptide particles for type 2 diabetes therapy. Nat. Commun. 8, 1777 (2017).
Zhao, H. et al. High-fidelity replica molding of glassy liquid crystalline polymer microstructures. ACS Appl. Mater. Interfaces 8, 8110–8117 (2016).
Indermun, S. et al. Current advances in the fabrication of microneedles for transdermal delivery. J. Control. Release 185, 130–138 (2014).
Daly, R., Harrington, T. S., Martin, G. D. & Hutchings, I. M. Inkjet printing for pharmaceutics—a review of research and manufacturing. Int. J. Pharm. 494, 554–567 (2015).
Boehm, R. D., Miller, P. R., Schell, W. A., Perfect, J. R. & Narayan, R. J. Inkjet printing of amphotericin B onto biodegradable microneedles using piezoelectric inkjet printing. JOM 65, 525–533 (2013).
Economidou, S. N. et al. 3D printed microneedle patches using stereolithography (SLA) for intradermal insulin delivery. Mater. Sci. Eng. C Biol. Appl. 102, 743–755 (2019).
Caudill, C. et al. Transdermal vaccination via 3D-printed microneedles induces potent humoral and cellular immunity. Proc. Natl Acad. Sci. USA 118, e2102595118 (2021).
Elahpour, N. et al. 3D printed microneedles for transdermal drug delivery: a brief review of two decades. Int. J. Pharm. 597, 120301 (2021).
Xu, L., Lee, H., Jetta, D. & Oh, K. W. Vacuum-driven power-free microfluidics utilizing the gas solubility or permeability of polydimethylsiloxane (PDMS). Lab Chip 15, 3962–3979 (2015).
Yang, S. et al. A scalable fabrication process of polymer microneedles. Int. J. Nanomedicine. 7, 1415–1422 (2012).
Nejad, H. R., Sadeqi, A., Kiaee, G. & Sonkusale, S. Low-cost and cleanroom-free fabrication of microneedles. Microsyst. Nanoeng. 4, 17073 (2018).
Fukushima, K. et al. Two-layered dissolving microneedles for percutaneous delivery of peptide/protein drugs in rats. Pharm. Res. 28, 7–21 (2011).
Schoenmaker, L. et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int. J. Pharm. 601, 120586 (2021).
Fenton, O. S. et al. Customizable lipid nanoparticle materials for the delivery of siRNAs and mRNAs. Angew. Chem. Int. Ed. Engl. 57, 13582–13586 (2018).
Hassett, K. J. et al. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. Nucleic Acids. 15, 1–11 (2019).
Sabnis, S. et al. Amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 26, 1509–1519 (2018).
Gonzalez-Gonzalez, E. et al. Silencing of reporter gene expression in skin using siRNAs and expression of plasmid DNA delivered by a soluble Protrusion Array Device (PAD). Mol. Ther. 18, 1667–1674 (2010).
Li, W., Li, S., Fan, X. & Prausnitz, M. R. Microneedle patch designs to increase dose administered to human subjects. J. Control. Release 339, 350–360 (2021).
Davis, S. P., Landis, B. J., Adams, Z. H., Allen, M. G. & Prausnitz, M. R. Insertion of microneedles into skin: measurement and prediction of insertion force and needle fracture force. J. Biomech. 37, 1155–1163 (2004).
Aoyagi, S., Izumi, H. & Fukuda, M. Biodegradable polymer needle with various tip angles and consideration on insertion mechanism of mosquito’s proboscis. Sens. Actuators A Phys. 143, 20–28 (2008).
Walsh, E. E. et al. Safety and immunogenicity of two RNA-based Covid-19 vaccine candidates. N. Engl. J. Med. 383, 2439–2450 (2020).
Mihaila, R. et al. Lipid nanoparticle purification by Spin Centrifugation–Dialysis (SCD): a facile and high-throughput approach for small scale preparation of siRNA–lipid complexes. Int. J. Pharm. 420, 118–121 (2011).
Corbett, K. S. et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 586, 567–571 (2020).
Gebre, M. S. et al. mRNA vaccines induce rapid antibody responses in mice. NPJ Vaccines 7, 88 (2022).
Yu, J. et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science 369, 806–811 (2020).
Fenton, O. S. et al. Synthesis and biological evaluation of ionizable lipid materials for the in vivo delivery of messenger RNA to B lymphocytes. Adv. Mater. 29, https://doi.org/10.1002/adma.201606944 (2017).
Zhang, H., Leal, J., Soto, M. R., Smyth, H. D. C. & Ghosh, D. Aerosolizable lipid nanoparticles for pulmonary delivery of mRNA through design of experiments. Pharmaceutics. 12, 1042 (2020).
Chen, X. et al. Rapid kinetics to peak serum antibodies is achieved following influenza vaccination by dry-coated densely packed microprojections to skin. J. Control. Release 158, 78–84 (2012).
Quan, F.-S. et al. Intradermal vaccination with influenza virus-like particles by using microneedles induces protection superior to that with intramuscular immunization. J. Virol. 84, 7760–7769 (2010).
Stewart-Jones, G. B. E. et al. Development of SARS-CoV-2 mRNA vaccines encoding spike N-terminal and receptor binding domains. Preprint at bioRxiv https://doi.org/10.1101/2022.10.07.511319 (2022).
Beals, C. R. et al. Immune response and reactogenicity of intradermal administration versus subcutaneous administration of varicella-zoster virus vaccine: an exploratory, randomised, partly blinded trial. Lancet Infect. Dis. 16, 915–922 (2016).
Levin, Y., Kochba, E., Hung, I. & Kenney, R. Intradermal vaccination using the novel microneedle device MicronJet600: past, present, and future. Hum. Vaccines Immunother. 11, 991–997 (2015).
World Health Organization. Meeting of the Strategic Advisory Group of Experts on immunization, April 2012: conclusions and recommendations. Wkly Epidemiol. Rec. 87, 201–216 (2012).
Dey, A. et al. Immunogenic potential of DNA vaccine candidate, ZyCoV-D against SARS-CoV-2 in animal models. Vaccine. 39, 4108–4116 (2021).
Massinga Loembé, M. & Nkengasong, J. N. COVID-19 vaccine access in Africa: global distribution, vaccine platforms, and challenges ahead. Immunity 54, 1353–1362 (2021).
Crommelin, D. J. A., Anchordoquy, T. J., Volkin, D. B., Jiskoot, W. & Mastrobattista, E. Addressing the cold reality of mRNA vaccine stability. J. Pharm. Sci. 110, 997–1001 (2021).
Kuwentrai, C. et al. Intradermal delivery of receptor-binding domain of SARS-CoV-2 spike protein with dissolvable microneedles to induce humoral and cellular responses in mice. Bioeng. Transl. Med. 6, e10202 (2021).
Zhao, P. et al. Long-term storage of lipid-like nanoparticles for mRNA delivery. Bioact. Mater. 5, 358–363 (2020).
Ball, R., Bajaj, P. & Whitehead, K. Achieving long-term stability of lipid nanoparticles: examining the effect of pH, temperature, and lyophilization. Int. J. Nanomedicine. 12, 305–315 (2016).
The Lancet Infectious Diseases. Time for Africa to future-proof, starting with COVID-19. Lancet. Infect. Dis. 22, 151 (2022).
mRNA made in Africa. Nat. Biotechnol. 40, 284 (2022).
Maloy, K. J., Donachie, A. M., O'Hagan, D. T. & Mowat, A. M. Induction of mucosal and systemic immune responses by immunization with ovalbumin entrapped in poly(lactide-co-glycolide) microparticles. Immunology 81, 661–667 (1994).
Zhang, Y. N. et al. Nanoparticle size influences antigen retention and presentation in lymph node follicles for humoral immunity. Nano Lett. 19, 7226–7235 (2019).
Acknowledgements
We thank Meso Scale Discovery for generous advice, assistance and reagents. We thank the Koch Institute’s Robert A. Swanson (1969) Biotechnology Center (RRID: SCR_018674) for technical support, specifically the Histology core, the Nanotechnology Materials core, the BioMicroCenter and the Animal Imaging and Preclinical Testing facilities. We acknowledge the Lab for Engineering Materials at the MIT Department of Materials Science and Engineering for technical support. This work was supported, in part, by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute. This work was also supported by Biomedical Advanced Research and Development Authority (BARDA) grant DHHS/OS/ASPR/BARDA/DRIVe EZ-BAA-18-100-SOL-00018. A.V.S. is a fellowship recipient and would like to thank the Belgian American Educational Foundation (BAEF) and Wallonia-Brussels International for the Excellence Scholarship (WBI.World). M.K. is a fellowship recipient and would like to acknowledge the Bodossaki Foundation and the Onassis Foundation. We acknowledge support from the National Institutes of Health (T32 AI0073787) to L.H.T.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.V.S., M.S., J.D., M.K., J.C., A.J. and R.F. Methodology: A.V.S., M.S., J.D., M.K., L.T., J.C., A.P., D.V., B.E., J.G., T.F., G.L., N.M., C.J.D., S.L., L.Z., O.P., K.H., S.A. and M.W. Investigation: A.V.S., M.S., J.D., M.K., L.T., J.C., A.P., D.V., B.E., J.G., T.F., G.L., N.M., S.P., C.J.D., S.L., L.Z., O.P., K.H. and M.W. Visualization: A.V.S., M.S., J.D. and J.H. Funding acquisition: A.J., R.L., A.V.S. and M.K. Project administration: A.V.S., J.D., J.C. and A.J. Supervision: A.J., R.L., D.B. and M.T. Writing—original draft: A.V.S., M.S. and J.D. Writing—review and editing: A.V.S., M.S., J.D., L.T., J.H., R.F., D.B., R.L. and A.J.
Corresponding authors
Ethics declarations
Competing interests
For a list of entities with which R.L. is involved, compensated or uncompensated, see www.dropbox.com/s/yc3xqb5s8s94v7x/Rev Langer COI.pdf?dl=0. For a list of entities with which A.J. is involved, compensated or uncompensated, see https://www.dropbox.com/s/wre16lh7jr7bvoi/230410%20Jaklenec%20COI.pdf?dl=0. A.V.S., M.S., J.D., M.K., J.C., R.L. and A.J. are named as inventors on a pending patent application (17/903586) related to the described technology. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Independent consultant: Robert Farra.
Extended data
Extended Data Fig. 1 Vacuum loading microneedle molds.
(a) Loading in the negative microneedle mold based on PDMS air permeability followed by drying of the polymer solution. Vacuum is applied below the PDMS sheet to remove air from beneath the vaccine ink through the air permeable PDMS mold. Drying is accelerated by also applying vacuum above the mold. (b) Loading in the negative microneedle mold based on PDMS air solubility followed by drying of the polymer solution. (c) Vacuum device configurations for applying vacuum to the bottom of the PDMS mold. (d) Polymer solution loading time for PDMS made from different polymer to cross-linker ratios (n = 3 independent samples). An ordinary one-way ANOVA (Sidak’s multiple comparisons test) was used. (e) Images of MNPs from PDMS molds that were degassed under various vacuum values and loaded with polymer solution. (f) Images of MNPs where the polymer solution was added to the degassed PDMS mold after 5–10 min or 1 hour. (g–h) Progression of needle height over time after dispensing polymer solution (n = 13–18 individual needles). (i–j) Normalized needle height from images after various degassing conditions (n = 10–18 individual needles). (k) Viscosity measured for various polymer solutions used in this study (n = 1). Data represent mean ± s.d. P values are represented by: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. MPa, megapascal; NS, not significant.
Extended Data Fig. 2 Minimizing vaccine waste.
(a) Images of MNPs made from various volumes of 20% w/w PVP:PVA solution, which minimize vaccine waste with an ultrathin backing. The 20% w/w PVP:PVA solutions were colored with crystal violet. (b–c) Dispensing volume optimization images and circularity measurement of dried backing resulted from various MNP matrix formulations (n = 1).
Extended Data Fig. 3 Vacuum loading device utilizing PDMS air permeability.
(a) CAD design of the acrylic pieces that were cut and mounted together to form the loading station. Pictures of (b) the loading station, (c) the headcover of the drying station, (d) the 5×5 MNP positive and (e) the 5×5 PDMS sheet of negative MNP molds. (f) A photograph of the MVP system with features annotated. (g) Schematic depicting the dispensing system and dispensing height. Effect of dispensing parameters flow rate (h), filtration (i), needle size (j), and dispensing height (k) on resulting drop size. All differences are significant at (P < 0.01) unless otherwise noted. An ordinary one-way ANOVA (Sidak’s multiple comparisons test) was used (n = 3 MNPs per group). (l) An image of 100 MNPs dispensed and dried onto a printing tray. Data represent mean ± s.d. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. NS, not significant.
Extended Data Fig. 4 Vaccine loading methods and MNP throughput.
(a) Schematic of the processes used for (a) one-step loading and (b) two-step tip-loading. A solution containing the vaccine and polymer is first dispensed using a robotic arm above the PDMS mold. Simultaneously, a vacuum of –1 bar is applied below to load the solution into the negative mold. Then, then solution is dried under desiccant atmosphere and –0.6 bar vacuum. For one-step loading (a), vaccine and enough polymer to form tips and backing are deposited in one step. For two-step tip-loading (b), vaccine and polymer are first deposited in the tips only, and then the same procedure is repeated using only polymer to form the backing of MNPs. (c) Characterization of correctly-formed full length, sharp microneedles fabricated by the automated MVP, as a percentage of total needles produced (n = 30 patches). MNPs fabricated had on average 37 ± 8 needles per patch. (d) Dispensing throughput is a function of dispensed drop height and patch size, while (e) drying throughput is a function of drying time and number of needles per patch. Both throughputs are functions of (f) drying time and (g) patch size. The step with the lowest throughput determines the overall throughput of the microneedle vaccine printer.
Extended Data Fig. 5 Alternate printer configurations and MNP demolding.
(a) Throughput of automated MNP fabrication can be increased by modular moving trays of MNP molds to a drying rack to expedite the vaccine drying step. Vacuum-through application to fill the microneedle mold can occur either on the mold transport device (middle) or in the tray rack (right). (b) Drying throughput is presented as a function of tray length and the number of trays in the vertical drying rack. Size (h) of the drying rack was estimated assuming a single tray height of 50 mm. Like the single tray device, drying time is assumed to be 48 h. (c) PDMS molds can be pretreated with a degassing technique using negative pressure prior to vaccine dispensing for an expedited MNP fabrication process. With degassed PDMS molds, a robotic arm can dispense vaccine solution in a continuous manner. Fully dispensed PDMS molds can then be moved to a drying rack using a conveyer belt. This entire process will be contained within an aseptic enclosure to maintain sterility during processing. (d–i) Design for automated patch demolding. (d) After vaccine microneedle patches are fully dried in a PDMS mold, (e) a robotic arm brings an acrylic backing with double-sided tape. (f) The robotic arm aligns and attaches the acrylic adhesive backing to a microneedle patch, (g) and the MNP is removed from PDMS mold as the robotic arm is raised vertically. (h) The tip of the robotic arm enters between a metal slit designed for MNP removal. As the robotic arm raises vertically, the MNP detaches from the robot arm tip and falls into its packaging (i), where the MNP is hovering to prevent any direct contact with the needle tips. Demolded MNPs are packaged and stored in a sterile and dry environment until use.
Extended Data Fig. 6 Effect of multiple variables on loading efficiency.
Comparison of tip-loading (a) and tip-loading efficiency (b) using the one-step or two-step fabrication process as a function of protein mass used for dispensing. (c) Comparison of polymer (PVP:PVA) mass used with the protein (BSA) during the first step of the two-step fabrication process. Low polymer mass is 0.8 mg PVP:PVA while high polymer mass is 4 mg and 8 mg PVP:PVA for 100 µg and 200 µg BSA respectively. An ordinary two-way ANOVA (Sidak’s multiple comparisons test) was used (n = 6 independent samples). (d) Relative mass of bovine serum albumin (BSA) loaded for three different large batches of MNPs, normalized to the average. Bartlett’s test (p = 0.006) revealed that the standard deviations could be significantly different, and two-sided t-tests between the groups revealed that patches dispensed at 30 mm had a significantly different standard deviation from the other two groups, which were comparable to each other (n = 50 patches for handmade; n = 40 patches for either of dispensing heights studied). (e) Loading efficiency per patch for 10 μg of a DNA vaccine for n = 100 MNPs fabricated using the MVP. Data represent mean ± s.d. P values are represented by: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. NS, not significant.
Extended Data Fig. 7 mRNA-LNP characterization.
(a) mRNA encapsulated concentration, (b) mRNA encapsulation efficiency, and (c) diameter of LNPs made with two different ionizable lipids (cKK-e12 or Lipid 5) and encapsulating mRNA encoding for fLuc or RBD (n = 3). (d) HeLa cells viability measured in presence of 0.6 mg of formulation and 50 ng of mRNA encapsulated in LNPs made with cKK-e12 (n = 4 technical replicates). (e) Total drying time for MNPs made of different polymer formulations using polyvinyl alcohol (PVA) or polyvinylpyrrolidone (PVP10 or PVP60, 10 kDa or 60 kDa molecular weight, respectively). Two different drying approaches were studied: drying under nitrogen flow for 10 h prior to vacuum or drying with desiccant for 24 h prior to vacuum (n = 1). 100% PVP patches were too brittle to remove from the mold without fracturing. (f) Stiffness and (g) peak force measured in compression testing of mRNA-LNP loaded MNPs made from different polymers. An ordinary one-way ANOVA (Sidak’s multiple comparisons test) was used (n = 5 per group). (h) mrna-lnp diameter measured by dynamic light scattering (dls) immediately after synthesis, after concentration, after formulating into vaccine ink, and after drying to form MNP matrix. (n = 3 per group). Representative transmission electron microscopy (TEM) and cryo-TEM images for (i) mRNA-LNPs after synthesis, (j) mRNA-LNPs in vaccine ink, and (k) mRNA-LNPs in microneedle patches. (l) mRNA fragment analysis from FLuc mRNA-LNPs, mRNA from vaccine ink, and mRNA from MNPs, characterized using capillary electrophoresis. (m) Encapsulated mRNA distribution in the needles of one microneedle patch. (n) Histogram of encapsulated mRNA loading per needle. (o) Encapsulated mRNA loading and (p) loading efficiency per patch (n = 2 independent samples). (q) Protein expression after HeLa transfection with LNPs (6 technical replicates) or the same LNPs redissolved from the microneedle patch (n = 2 independent samples, 6 technical replicates per MNP). Data represent mean ± s.d.
Extended Data Fig. 8 Microneedle dissolution.
(a) Dimensions of an individual microneedle used in pyramid or conical microneedle patches. Spacing between two adjacent needles (pitch) is 1000 µm. (b) Representative optical imaging of needle dissolution upon application ex vivo on pig skin as a function of time for MNPs of pyramid and conical shape. (c–e) Comparison between conical and pyramid microneedles made from PVP:PVA 2:1 and 1:1, in terms of dissolved volume in ex vivo pig skin. Unpaired, two-tailed t-tests (n = 23–59 needles per group). Data represent mean ± s.d. P values are represented by: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. NS, not significant.
Extended Data Fig. 9 Characterization of SARS-CoV-2 MNP.
(a) Schematic of SARS-CoV-2 RBD sequence of the mRNA constructs used in this paper. The signal sequence (SS) is colored orange, RBD domain is colored blue, and the T4 trimerization motif is colored green. (b-c) LNPs encapsulating mRNA that encodes for fLuc were mixed with various water-soluble polymers and dried. LNPs were either dialyzed in PBS or DI water before their mixing with the polymer matrix. Protein expression in HeLa cells was measured after transfection with re-dissolved polymer to screen for a combination of polymers that produces protein expression comparable to LNPs in suspension (n = 4 technical replicates). (d-E) Droplet size is critical for high vaccine loading efficiency. Due to the Marangoni effect, a larger droplet size (d) has lower loading efficiency than a small droplet (e) dispensed in the center of a mold. (f) HEK cells transfected with LNP suspension made with Lipid 5 and encapsulating mRNA encoding for RBD. LNPs encapsulating mRNA encoding for RBD were dissolved from a MNP and added to demonstrate bioactivity after loading into MNPs. RBD is shown in green (Alexa Fluor 488 used as secondary antibody), actin is shown in red (rhodamine phalloidin) and nucleus in blue (DAPI). (g) MNP tip-loading (h) and loading efficiency of different mRNA-LNP constructs used for in vivo studies (n = 3 independent samples). Four MNPs were administered for in vivo studies at 1.5 μg per patch to yield a theoretical 6 μg dose. (i) Of the 6 μg mRNA used for MNP fabrication, we assessed the percent mRNA recovered in various regions of the MNP. Approximately 25% were found in the microneedles, matching the loading efficiency measurement, and the remaining mRNA was found in the backing. The total reported is the sum of needles and backing, showing that all mRNA was accounted for in the MNP (n = 5 independent samples). Data represent mean ± s.d.
Extended Data Fig. 10 In vivo evaluation of mRNA-LNP-loaded MNPs.
(a) Immunization regimen for in vivo experiment in C57BL/6 mice. (b) Kinetics of FLuc mRNA expression as measured by luminescence. 1 μg of mRNA in mRNA-LNPs was administered to C57BL/6 mice either by intramuscular (IM) injection or microneedle patch (MNP) application (n = 5 biologically independent samples). mRNA expressions peaks later for MNPs compared to suspension of mRNA-LNPs. (c) FLuc expression after 1 month storage at 37 °C in PVP:PVA 1:1 MNP mRNA-LNPs. An ordinary two-way ANOVA (Sidak’s multiple comparisons test) was used (n = 5 biologically independent samples). (d) Immunogenicity of microneedle patches stored for either 1 month (1 mo) or 3 months (3 mo) using an electrochemiluminescence assay that measures the strength of binding responses SARS-CoV-2 S-protein receptor binding domain. All BALB/c mice were primed with a 10 μg dose of SARS-CoV-2 RBD mRNA via intramuscular injection (IM). At week 4, a booster dose was applied which varied depending on storage time and condition, except for a group which received no booster for comparison. Serum was collected at 3 weeks (pre-boost) and 9 weeks (post-boost) (n = 5 biologically independent samples). Data represent mean ± s.d. NS, not significant.
Supplementary information
Supplementary Information
Supplementary Notes 1–4 and Supplementary Table 1.
Supplementary Video 1
Movie S1.
Supplementary Video 2
Movie S2.
Supplementary Video 3
Movie S3.
Supplementary Video 4
Movie S4.
Supplementary Video 5
Movie S5.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
vander Straeten, A., Sarmadi, M., Daristotle, J.L. et al. A microneedle vaccine printer for thermostable COVID-19 mRNA vaccines. Nat Biotechnol 42, 510–517 (2024). https://doi.org/10.1038/s41587-023-01774-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-023-01774-z
This article is cited by
-
Long-term monitoring of ultratrace nucleic acids using tetrahedral nanostructure-based NgAgo on wearable microneedles
Nature Communications (2024)
-
Cyclosporin A-loaded dissolving microneedles for dermatitis therapy: Development, characterisation and efficacy in a delayed-type hypersensitivity in vivo model
Drug Delivery and Translational Research (2024)
-
Biomimetic microneedles: exploring the recent advances on a microfabricated system for precision delivery of drugs, peptides, and proteins
Future Journal of Pharmaceutical Sciences (2023)
-
Enhancing the immunogenicity of lipid-nanoparticle mRNA vaccines by adjuvanting the ionizable lipid and the mRNA
Nature Biomedical Engineering (2023)
-
Nanotechnology-based mRNA vaccines
Nature Reviews Methods Primers (2023)
