
Abstract
The gut microbiota produce hundreds of small molecules, many of which modulate host physiology. Although efforts have been made to identify biosynthetic genes for secondary metabolites, the chemical output of the gut microbiome consists predominantly of primary metabolites. Here we introduce the gutSMASH algorithm for identification of primary metabolic gene clusters, and we used it to systematically profile gut microbiome metabolism, identifying 19,890 gene clusters in 4,240 high-quality microbial genomes. We found marked differences in pathway distribution among phyla, reflecting distinct strategies for energy capture. These data explain taxonomic differences in short-chain fatty acid production and suggest a characteristic metabolic niche for each taxon. Analysis of 1,135 individuals from a Dutch population-based cohort shows that the level of microbiome-derived metabolites in plasma and feces is almost completely uncorrelated with the metagenomic abundance of corresponding metabolic genes, indicating a crucial role for pathway-specific gene regulation and metabolite flux. This work is a starting point for understanding differences in how bacterial taxa contribute to the chemistry of the microbiome.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
The LifeLines DEEP cohort raw metagenomic sequencing data, metabolome data and human phenotypes (that is, age and sex) used for the analysis presented in this study are available at the European Genome-phenome Archive under accession EGAS00001001704. Taxonomic assignments of bacteria were performed according to Genome Taxonomy Database release 95 (https://gtdb.ecogenomic.org/). Lists of accessions of genome assemblies used are available in Supplementary Tables 3 and 4. iHMP multi-omics data were downloaded from https://ibdmdb.org. Raw sequence data of the iHMP are available from the National Center for Biotechnology Informationʼs Sequence Read Archive via BioProject PRJNA398089; metatranscriptome data are available through Gene Expression Omnibus series accession number GSE111889; and metabolomics data are available at the Metabolomics Workbench (http://www.metabolomicsworkbench.org; Project ID PR000639). Source data are provided with this paper.
Code availability
The gutSMASH source code is available freely under an open-source AGPL-3.0 license from https://github.com/victoriapascal/gutsmash/.
References
Blin, K. et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87 (2019).
Karp, P. D. et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 20, 1085–1093 (2019).
Abubucker, S. et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 8, e1002358 (2012).
Medema, M. H. & Fischbach, M. A. Computational approaches to natural product discovery. Nat. Chem. Biol. 11, 639–648 (2015).
Ziemert, N., Alanjary, M. & Weber, T. The evolution of genome mining in microbes—a review. Nat. Prod. Rep. 33, 988–1005 (2016).
Medema, M. H., de Rond, T. & Moore, B. S. Mining genomes to illuminate the specialized chemistry of life. Nat. Rev. Genet. 22, 553–571 (2021).
Navarro-Muñoz, J. C. et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 16, 60–68 (2020).
Kitamoto, S. et al. Dietary l-serine confers a competitive fitness advantage to Enterobacteriaceae in the inflamed gut. Nat. Microbiol 5, 116–125 (2020).
Zou, Y. et al. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 37, 179–185 (2019).
Lloyd-Price, J. et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66 (2017).
Tracy, B. P., Jones, S. W., Fast, A. G., Indurthi, D. C. & Papoutsakis, E. T. Clostridia: the importance of their exceptional substrate and metabolite diversity for biofuel and biorefinery applications. Curr. Opin. Biotechnol. 23, 364–381 (2012).
Maguire, F. et al. Metagenome-assembled genome binning methods with short reads disproportionately fail for plasmids and genomic islands. Microb. Genom. 6, mgen000436 (2020).
Vieira-Silva, S. et al. Species–function relationships shape ecological properties of the human gut microbiome. Nat. Microbiol 1, 16088 (2016).
Rautio, M et al. Reclassification of Bacteroides putredinis (Weinberg et al., 1937) in a new genus Alistipes gen. nov., as Alistipes putredinis comb. Nov., and description of Alistipes finegoldii sp. Nov., from human sources. Syst. Appl. Microbiol. 26, 182–188 (2003).
Cummings, J. H., Pomare, E. W., Branch, W. J., Naylor, C. P. & Macfarlane, G. T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28, 1221–1227 (1987).
Jones, S. A. et al. Anaerobic respiration of Escherichia coli in the mouse intestine. Infect. Immun. 79, 4218–4226 (2011).
Tremblay, P. L., Zhang, T., Dar, S. A., Leang, C. & Lovley, D. R. The Rnf complex of Clostridium ljungdahlii is a proton-translocating ferredoxin:NAD+ oxidoreductase essential for autotrophic growth. mBio 4, e00406–e00412 (2012).
Liu, Y. et al. Clostridium sporogenes uses reductive Stickland metabolism in the gut to generate ATP and produce circulating metabolites. Nat. Microbiol 7, 695–706 (2022).
Andreu, V. P. et al. BiG-MAP: an automated pipeline to profile metabolic gene cluster abundance and expression in microbiomes. mSystems 6, e0093721 (2021).
Tigchelaar, E. F. et al. Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open 5, e006772 (2015).
Faber, F. et al. Respiration of microbiota-derived 1,2-propanediol drives Salmonella expansion during colitis. PLoS Pathog. 13, e1006129 (2017).
Andriamihaja, M. et al. The deleterious metabolic and genotoxic effects of the bacterial metabolite p-cresol on colonic epithelial cells. Free Radic. Biol. Med. 85, 219–227 (2015).
Funabashi, M. et al. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 582, 566–570 (2020).
Mallick, H. et al. Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat. Commun. 10, 3136 (2019).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688 (2020).
Vandeputte, D. et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511 (2017).
Karp, P. D. et al. Pathway Tools version 23.0 update: software for pathway/genome informatics and systems biology. Brief. Bioform. 22, 109–126 (2021).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes—a 2019 update. Nucleic Acids Res. 48, D445–D453 (2020).
Richardson, L. J. et al. Genome properties in 2019: a new companion database to InterPro for the inference of complete functional attributes. Nucleic Acids Res. 47, D564–D572 (2019).
Franzosa, E. A. et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968 (2018).
Pascal Andreu, V., Fischbach, M. A. & Medema, M. H. Computational genomic discovery of diverse gene clusters harbouring Fe-S flavoenzymes in anaerobic gut microbiota. Microb. Genom. 6, e000373 (2020).
Parks, D. H. et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004 (2018).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Chen, C. et al. Representative proteomes: a stable, scalable and unbiased proteome set for sequence analysis and functional annotation. PLoS ONE 6, e18910 (2011).
Eddy, S. R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M. & Barton, G. J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
de los Santos, E. L. C. & Challis, G. L. clusterTools: proximity searches for functional elements to identify putative biosynthetic gene clusters. Preprint at https://www.biorxiv.org/content/10.1101/119214v2 (2017).
Medema, M. H., Takano, E. & Breitling, R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol. Biol. Evol. 30, 1218–1223 (2013).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Zhernakova, A. et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569 (2016).
Ondov, B. D. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016).
Lloyd-Price, J. et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019).
Chen, L. et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat. Med. 28, 2333–2343 (2022).
Acknowledgements
This work was supported by the Chan Zuckerberg Biohub (M.A.F.); DARPA awards HR0011-15-C-0084 and HR0112020030 (M.A.F.); National Institutes of Health (NIH) awards R01 DK101674, DP1 DK113598 and P01 HL147823 (to M.A.F.); the Leducq Foundation; and a European Research Council (ERC) Starting Grant (948770-DECIPHER to M.H.M.). A.Z. is supported by ERC Starting Grant 715772; Netherlands Organization for Scientific Research NWO-VIDI grant 016.178.056; Netherlands Heart Foundation CVON grant 2018-27; and NWO Gravitation grant ExposomeNL 024.004.017. J.F. is supported by the ERC Consolidator Grant (grant agreement no. 101001678); NWO-VICI grant VI.C.202.022; Dutch Heart Foundation IN-CONTROL (CVON2018-27); the Netherlands Organ-on-Chip Initiative; and the NWO Gravitation Project (024.003.001), funded by the Ministry of Education, Culture and Science of the government of The Netherlands. L.C. is supported by a Foundation de Cock-Hadders grant (20:20-13) and a joint fellowship from the University Medical Centre Groningen and the China Scholarship Council (CSC201708320268). D.D. was supported by NIH awards K08 DK110335, R35 GM142873 and R01 AT011396.
Author information
Authors and Affiliations
Contributions
M.A.F. and M.H.M. initially conceived the project, with modifications and extensions introduced on the advice of V.P.A., A.Z., J.F. and D.D. The gutSMASH software was developed and used to analyze genomic data by V.P.A., with input from M.H.M., D.D. and M.A.F. Analysis of metagenomic and metatranscriptomics data was performed by H.E.A., V.P.A. and L.C. Correlations with metabolomic data were performed by L.C. M.H.M., D.D. and M.A.F. coordinated and supervised the study as a whole, and A.Z. and J.F. coordinated and supervised analysis of cohort data. All authors contributed to data interpretation. V.P.A., M.A.F., D.D. and M.H.M. drafted the initial manuscript, with input from the other authors. All authors read and contributed to the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
M.A.F. is a co-founder and director of Federation Bio, a co-founder of Revolution Medicines and a member of the scientific advisory board of NGM Biopharmaceuticals. D.D. is a co-founder of Federation Bio. M.H.M. is a co-founder of Design Pharmaceuticals and a member of the scientific advisory board of Hexagon Bio. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
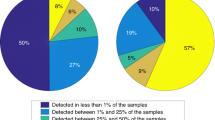
Extended Data Fig. 1 Pathway prevalence using different core coverage thresholds.
Pathway prevalence was computed by assessing the number of reads (per sample) mapping to known gene clusters at a certain core coverage cutoff. The figure illustrates how the pathway prevalence gradually changes when increasing the core coverage cutoff from 10 to 80%.
Extended Data Fig. 2 Limited correlation of genetic pathway abundance with metabolites abundance in blood plasma.
This figure shows correlation plots for additional metabolites not shown in Fig. 4a. Spearman correlation (two sided with rho and empirical P value are reported) is used to check the relationship between pathway abundances and metabolite levels after adjusting for age, sex and read depth. n = 1054 biologically independent samples.
Extended Data Fig. 3 Network of putative non-redundant MGCs predicted by gutSMASH.
From all the unknown predicted MGCs, a redundancy filtering of 0.9 sequence similarity was applied using MMseqs2. From each cluster, two representatives were picked, and all representatives were used as input for BiG-SCAPE using the default cutoffs. The network contains 2,921 nodes and 7,474 edges. The MGCs have been classified into four different categories based on the key enzyme classes they code for. The GR (glycyl-radical) category is composed of MGCs that include pyruvate formate-lyase (PFL-like) and/or glycyl radical (Gly_radical), OD (oxidative decarboxylation) involves MGCs with at least one of the following Pfam domains: pyruvate ferredoxin/flavodoxin oxidoreductase (POR), pyruvate flavodoxin/ferredoxin oxidoreductase, thiamine diP-bdg (POR_N), pyruvate:ferredoxin oxidoreductase core domain II (PFOR_II) and thiamine pyrophosphate enzyme, C-terminal TPP binding domain (TPP_enzyme_C). The flavoenzymes category is a combination of MGCs harbouring at least one of the custom-made BaiCD and BaiH pHMMs. HGD-D-related MGCs, as the name states, include enzymes matching any of the 2-hydroxyglutaryl-CoA dehydratase, D-component (HGD-D)-related pHMM domains.
Extended Data Fig. 4 Subset of unknown MGCs predicted by gutSMASH manually picked.
The network/nodes present in the left side of the figure represent the subnetwork extracted from the complete network in Extended Data Fig. 3. The arrows have been coloured-coded based on the Pfam domains found in the protein-coding sequences and the functional annotations of these proteins.
Supplementary information
Supplementary Information
Supplementary Results, Supplementary Figs. 1–3 and legends for Supplementary Tables 1–18
Supplementary Tables
Supplementary Tables 1–18
Source data
Source Data Fig. 2
Raw pathway abundance across most representative genera of bacteria in the human gut, absolute counts of genomes harboring genes and MGCs corresponding to the main acetate-producing pathways, summarized at phylum level (also found in Supplementary Tables 5 and 6).
Source Data Fig. 3
Pathway prevalence and abundance counts across 1,135 human microbiome samples (also found in Supplementary Tables 7 and 8).
Source Data Fig. 4
Multi-omics correlation data.
Source Data Extended Data Fig. 1
Pathway prevalence values of the 41 pathways across 1,135 human microbiomes using different BiG-MAP mapping coverage threshold values (also found in Supplementary Table 7).
Source Data Extended Data Fig. 2
Correlations between different metabolites and the MGC abundance of those pathways that have the same end product.
Source Data Extended Data Figs. 3 and 4
Sequence similarity network (raw data, can be imported into Cytoscape) of putatively novel types of MGCs identified by gutSMASH general rules.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pascal Andreu, V., Augustijn, H.E., Chen, L. et al. gutSMASH predicts specialized primary metabolic pathways from the human gut microbiota. Nat Biotechnol 41, 1416–1423 (2023). https://doi.org/10.1038/s41587-023-01675-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-023-01675-1
This article is cited by
-
Williams–Beuren syndrome shapes the gut microbiota metaproteome
Scientific Reports (2023)
