
Abstract
SARS-CoV-2 surveillance by wastewater-based epidemiology is poised to provide a complementary approach to sequencing individual cases. However, robust quantification of variants and de novo detection of emerging variants remains challenging for existing strategies. We deep sequenced 3,413 wastewater samples representing 94 municipal catchments, covering >59% of the population of Austria, from December 2020 to February 2022. Our system of variant quantification in sewage pipeline designed for robustness (termed VaQuERo) enabled us to deduce the spatiotemporal abundance of predefined variants from complex wastewater samples. These results were validated against epidemiological records of >311,000 individual cases. Furthermore, we describe elevated viral genetic diversity during the Delta variant period, provide a framework to predict emerging variants and measure the reproductive advantage of variants of concern by calculating variant-specific reproduction numbers from wastewater. Together, this study demonstrates the power of national-scale WBE to support public health and promises particular value for countries without extensive individual monitoring.
Similar content being viewed by others
Main
The recent introduction of SARS-CoV-2 into the human population has led to a pandemic with immense health and socioeconomic impact worldwide1,2 The emergence of new variants with higher transmissibility and/or immune escape properties pose future challenges for containing the circulation of the virus3,4 Potential contributing factors like antiviral immunity by natural infections or vaccines as well as social behaviour, viral reservoirs, testing strategies and global transmission make it difficult to predict the dynamic nature of future evolutions of SARS-CoV-25,6,7,8. As previously shown for epidemic infectious diseases, such as influenza virus and ebolavirus, continuous pathogenomic surveillance of circulating virus is pivotal to understand outbreak origins and progressions and to inform public health interventions9,10,11,12,13. Comprehensive SARS-CoV-2 surveillance programmes allow monitoring of the epidemiological situation and link it to viral variants and clinical outcomes14,15. Results thereof are of importance to decision makers for assessing the current situation and preparing for imminent developments14. Nonetheless, such programmes do come with limitations in their applicability at a global scale. Certain population groups including asymptomatic and individuals with reduced access to healthcare may be inherently underrepresented depending on the national testing and surveillance strategy16. Neglecting such groups potentially introduces a bias to the detection of variants with altered clinical manifestation. Moreover, few countries can muster enough resources for comprehensive case monitoring, which requires adequately funded organisational structures, cross-disciplinary scientific expertise and the readiness to integrate these resources into public health operations17,18,19.
WBE can overcome sampling bias and some economic constraints of epidemiological surveillance20. SARS-CoV-2 RNA is excreted in faeces, urine and saliva from infected individuals, and what makes its way to the sewer can be potentially found in the wastewater (WW)21. Hence, municipal WW drainage systems can serve as point of use for representative sampling of circulating SARS-CoV-2 variants22,23. Initially, PCR-based approaches were applied to detect viral RNA in the sewage24,25,26,27,28,29. Deduced virus titres were shown to robustly reflect prevalence rates in the catchments30,31,32 and empower forecasts of infection dynamics for the near future33. The successful sequencing of SARS-CoV-2 genomes from WW was reported34,35 and used to detect regional occurrence of selected virus variants based on the presence of characteristic mutations25,26,29,34,35,36,37,38,39,40,41,42,43,44,45,46. Later, WW was used to construct and quantify haplotype signatures of variants of concern (VOC) and to deduce the reproductive number47,48. Yet, the suitability of WBE to survey the spatiotemporal viral dynamics and integrate variant-specific epidemiological parameters on a national scale has not been assessed so far.
Austria, a Central European country of around 9 million inhabitants with a high degree of international mobility owing to tourism, which detected 3.34 Mio SARS-CoV-2 positive cases as of 17 March 2022, responded with strict non-pharmaceutical interventions to the pandemic. Like other European countries, the pandemic was characterised by the sequential emergence of different VOC, namely, Alpha (pangolin nomenclature49: B.1.1.7), Delta (B.1.617.2) and Omicron (B.1.1.529). Notably, the province of Tyrol harboured one of the largest clusters of the Beta variant (B.1.351) outside of South Africa from January to March 202150,51 Such spatiotemporal dynamics of emerging viral variants coupled with a comprehensive national epidemiological surveillance system52 including routine genotyping of positive cases, and a high population share of 93% being connected to sewer infrastructure53 set the stage to develop and validate the necessary methodology for WW-based variant monitoring by sequencing.
In this study, we present a validated, robust framework to deduce public health-relevant epidemiological indicators such as relative abundance and reproduction numbers for predefined variants from WW-derived deep-sequenced SARS-CoV-2 genomes within the context of a national-scale WBE programme. Furthermore, we show the feasibility of de novo detection of emerging variants from WW sequencing data.
Results
National-scale WBE surveillance programme
For the presented study, 94 WW treatment plants (WWTPs), covering >59% of the Austrian population (Fig. 1a and Extended Data Fig. 1a), were selected. Thereby, the population of the nine different federal states were covered between 18 and 100%, with a median value of 53% (Extended Data Fig. 1b). The selected WWTPs varied in the population size of the catchment, with between 1,490 to 1,900,000 people (median: 22,725). The selection of monitored WWTPs was guided by the objective to cover as many people in as many parts of the country as possible, with a special emphasis on towns with larger school facilities, as well as access to samples and appropriate logistic chains. Samples collected between 1 December 2020 and 10 February 2022 were considered (Extended Data Fig. 1c). All samples were subjected to quantitative PCR with reverse transcription (RT-qPCR)-based measurements of SARS-CoV-2 genome quantity, using internal spike-in controls to deduce copy numbers per volume. WW collection, pre-processing and quantitative screening were performed at three different laboratories. In total 3,413 samples were analysed by amplicon-based whole-genome deep sequencing, using a pipeline described previously54. Of all samples, 10.2% failed during sequencing, mostly owing to low viral loads in the WW during the low-incidence summer period (from June to August 2021) as indicated by CT values >35. The remaining 89.8% produced a detection confidence of SARS-CoV-2, and 80.0% passed our quality criteria of at least 40% genome coverage, to be considered for follow-up analysis (Extended Data Fig. 1c).

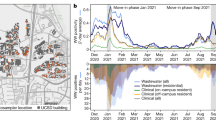
a, Location of Austrian WWTPs (grey) and those included in the surveillance programme (black). WWTPs labelled with names are used to showcase analysis details throughout the manuscript. These WWTPs were selected for their size and geographic position, namely, a small and a large WWTP from west, south and east. b, Results of the VaQuERo variant quantification analysis for the ten most abundant variants for all WWTPs with more than 17 time points. c, Spatiotemporal spread of selected variants. Time is projected on the vertical axis; position along the two main transportation axes (west-east and east-south mobility axis) is projected on the horizontal axis. The colour indicates relative variant abundance of B.1.160, B.1.258, Alpha (B.1.1.7), Delta (B.1.617.2) and Omicron (BA.1 and BA.2), from top left to bottom right, as deduced from the measured points and interpolated with B-splines. White crosses indicate measured data points.
The reproducibility of our sequencing pipeline to call low-frequency mutations down to 1% was shown previously for clinical samples54. To validate mutation calling in WW samples, we sequenced triplicate samples from two WWTP locations, representing varying WW characteristics (Extended Data Fig. 2a), namely, the WWTP Hofsteig with a high industrial share and the more rural WWTP Kuchl. Comparing the observed mutations qualitatively between the triplicates revealed that only 29% and 21% of the mutations in the WWTPs Hofsteig and Kuchl, respectively, were observed in all three replicates (Extended Data Fig. 2b). For mutations called by at least two of the three replicates, this proportion rose to 47% and 40%, respectively. Despite the high dropout rate, the overall correlation of observed mutation frequencies was encouraging with Pearson’s correlation coefficients in pairwise comparisons between 0.69 and 0.87, respectively (Extended Data Fig. 2c). Notably, the read coverage along the genome was not uniform, leaving substantial regions with few informative reads. The positioning of poorly covered regions did not show a strong reproducibility between the replicates, indicating that variable sample quality and stochasticity have a more pronounced effect than systematic technical causes such as amplicon primer affinities (Extended Data Fig. 2d). Consequently, mutations that were expected to be present, for example, defining mutations for a viral strain dominating at a certain time, often failed to manifest in the data (Extended Data Fig. 2e). Comparing the deduced mutation frequencies from the single samples to the median of all triplicates also revealed substantial deviation from the expected mutation frequency, especially up to a relative abundance of 50% (Extended Data Fig. 2f). Most missed mutations were found in regions of low read coverage, explaining their regular dropout. However, some of the missed mutations would have been covered by a substantial number of reads. In this case, the lack of detection may be explained with sampling errors during the amplification step due to low amounts of viral RNA in the complex WW matrix (Extended Data Fig. 2e,f).
Variant characterisation and quantification
Our technical assessments of WW sequencing determined the overall reliability and reproducibility of the approach, but also showed inherent shortcomings related to dropouts at individual sites. Consequently, we aimed to develop a robust and error-tolerant method for the detection and quantification of different viral variants, which we named VaQuERo (variant quantification in sewage designed for robustness). To increase the positive predictive value, a reduced set of relevant variants for targeted quantification was defined first. Thereby, detectable variants were restricted to variants of concern, of interest, or under monitoring as defined by the European Centre for Disease Prevention and Control (ECDC) and variants circulating in Austria according to GISAID database records55,56. Each mutation that occurred in more than 80% of all genomes associated with a particular variant deposited in the GISAID database, was considered a marker mutation for the respective variant. Mutations found in no other variants of relevance with more than 40% were denominated as unique markers. Subclades separated by less than three unique mutations were collapsed into an aggregated representative variant. In total, 33 variants of relevance were included in the analysis. On the basis of this comprehensive set of marker mutations, we developed a method to detect and quantify variants. To gain robustness against the anticipated dropouts and hence inflated number of mutations with an observed frequency of zero, we implemented a hurdle model inspired scheme57. In a first step, we removed all mutations not found in the set of marker mutations or that were observed at a mutation frequency below 2%58. Variants of relevance, for which at least two individual and at least 10% of all uniquely defining mutations were detected, were designated as detected and subjected to the subsequent quantification step. For quantification, both unique and non-unique marker mutations were used in a SIMPLEX regression to deduce the overall variant frequencies.
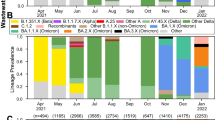
Applying VaQuERo to our data enabled us to quantify the temporal development of the variant composition in WW (Fig. 1b). Thereby, the VaQuERo approach converted the detailed but complex mutation patterns as observed in the WW into a format that was reminiscent of case-based epidemiological reports and made it more accessible for public health stakeholders in Austria. Moreover, as these data provides information on variant-specific relative abundances over time, it can serve as the basis for further in-depth analysis (Extended Data Fig. 3).
To visualise the regional patterns of expansion and decline of single variants, we reduced Austria to its main axis of mobility, as documented by mobile communications network records (Extended Data Fig. 4). Imposed by its geographical drawn-out shape and its topography with a large mountain range (the Alps) and main river (Danube), the two defined mobility axes follow a west-east and an east-south direction. This way, we used the second dimension to depict changes over time. Interpolation in this reduced space–time coordinate system allowed to visualise the spatiotemporal development of variant incidences (Fig. 1c). This was demonstrated by the steady retreat of B.1.160 and B.1.258 and their replacement by Alpha, ushering in the eastern provinces of Austria during January 2021 and being completed in the western provinces only around three months later. By contrast, the displacement of Alpha by the Delta variant during early summer 2021 as well as the subsequent change from Delta to Omicron during a high-prevalence phase during winter 2021/22, started almost concomitantly throughout Austria and were completed within a month (Fig. 1c).
Comparison between WBE and epidemiological case surveillance
In a next step, we sought to validate our WBE analysis results through a comprehensive integration with available surveillance data from individual cases in the catchment areas (>310,000) collected by the Austrian Agency of Health and Food Safety (AGES). Of note, during the period from 1 January 2021 to 10 February 2022, 50.6% of all positive cases in Austria were tested for VOC. Testing was performed by mutation-specific PCR assays against characteristic mutations, mutation hotspot sequencing and/or whole-genome sequencing. The testing regime was implemented according to the official guidelines of the Austrian health authorities but may have been affected by occasional technical and practical constraints, such as delays in the development and roll out of adapted diagnostic tests for newly emerged variants (for example, January 2021) or overloaded infrastructure during periods of high incidence (for example, November 2021) (Extended Data Fig. 5a). Nevertheless, together with the overall high testing rate (median of 37.4 tests per thousand inhabitants and day between January 2021 and February 2022) and the low positivity rate (median 0.7% between January 2021 and February 2022)52 the number of unreported cases is assumed to be comparatively low. The absolute case counts per detected variant from communities within the catchment areas were aggregated to proportions per total genotyped cases per week and per catchment area. Contrasting the two independent data sources indicates a high concordance of the WW data with the epidemiological trends and the time of introduction of emerging variants (Fig. 2a). WBE also reliably captured smaller epidemiologically confirmed outbreaks as shown, for example, for Beta (B.1.351) and B.1.1.318 in the municipal area discharging to the WWTP Bregenz (Fig. 2a). In some instances, even time points with a single confirmed infection case in the catchment area were reflected by a proportionate signal in the WW analysis. The size of the WWTP catchment in terms of people served (that is, people residing in the catchment and being connected to the sewer) influenced the sensitivity to detect individual infected cases. To examine the overall agreement between the case-based and WW-based variant surveillance, we applied Cohen’s κ coefficient to measure inter-rater reliability for each monitored variant separately. First, we tested the ability to detect a variant in a defined catchment. The test revealed robust agreement between the two approaches, with κ values significantly different from zero for all variants (Fig. 2b). Additionally, we tested our ability to identify the dominating variant with a relative frequency of >50%. There, the two approaches agreed for the variants that showed prolonged dominance across the country, that is, Alpha, Delta and Omicron. For variants which reached higher frequencies only at isolated instances, concordance between the approaches was less pronounced (Fig. 2c). Quantitative agreement between the two approaches was tested applying a rank correlation analysis. To avoid feigned confidence caused by the bimodal distribution, with a heavy overrepresentation of values close to 0 and close to 1 (Extended Data Fig. 5b), relative frequencies <0.1 and >0.9 were omitted. Collectively, the signal from the WW variant surveillance and case surveillance exhibited a Kendall correlation coefficient τ of 0.43 (P = 10−27; Fig. 2d). Independent analyses of different variants produced comparable results for Alpha, Delta and Omicron, but not for Beta likely owing to its low number of data points (Extended Data Fig. 5c). To estimate the sensitivity of the WW-based approach, reports from the epidemiological case surveillance were examined for regions and time points where no corresponding variant-specific signal in the WW was detected. These missed variant occurrences constituted between 1 and 117 epidemiologically assigned cases, with a median of just 2 cases. Typically, we found that the larger the catchment the higher the absolute number of missed cases, but at the same time the lower the relative proportion of missed signal (Extended Data Fig. 5d). In terms of relative frequencies, missed variant signals ranged from 0.005% to 100% with a median of 3.74%. This is close to the expected detection limit of our method, given that mutations with frequencies below 2% in WW samples are dismissed (Fig. 2c). Considering the median missed case count and the median missed variant frequency altogether, 77.5% of all missed data points were below either of them. Conversely, a variant linked to more than 2 cases and more than 3.74% of all cases in the catchment area can be expected to be reliably detected in the WW. With 50.6% of positive individual cases being genotyped, effective detection limit of absolute cases can be expected to be slightly higher. Overall, the comparative analyses revealed that WBE provides a detailed reflection of the epidemiological dynamics at regional resolution across Austria (Extended Data Fig. 6).

Comparison between relative variant abundance as deduced from WW variant surveillance and from the aggregated statistics of the epidemiological case surveillance. Single data points are aggregated over a week and catchment areas. a, Detailed comparison between relative variant frequencies deduced from WW and from all variant-typed individual cases in the catchment of selected WW treatment plants (WWTP) from west, central and eastern Austria with a small and a large population size, respectively. Black line represents the signal from the WW. The capped bars (lollipop plot) represent the case surveillance records, whereas the respective colour indicates the range of absolute case numbers per variant for the respective catchment area and week. The chosen breaks correspond to the 0, 0.2, 0.5 and 0.75 quantile of the observed zero-truncated absolute case count distribution. b, Enumeration of the agreement and disagreement between case-based and WW-based surveillance with respect to the detection of each variant in a catchment in the same week. Detection is defined by a relative frequency >0. Below the associated Cohen’s κ coefficient, testing a one-sided alternative hypothesis, its 95% confidence interval and corresponding multiple testing corrected P values are depicted. c, Like the inter-rater reliability test for detection presented in b but examining the agreement to identify dominant variant with a relative frequency >0.5. d, Rank correlation across all detected variants and all surveyed WWTPs. To reduce bias caused by the bimodal distribution of relative frequencies with many values close to 0 or 1, values <0.1 and >0.9 were removed before the analysis. e, Absolute case counts and the relative case counts for all data points of the case-based monitoring programme for which no corresponding variant-specific signal was detected in the WW. Black bars depict distribution of both variables, black lines the corresponding medians.
De novo recognition of mutation constellations
Beside the reliable detection and relative quantification of known variants, detecting novel emerging variants is the second purpose a variant surveillance programme should serve. Effectively, this is equivalent to a deconvolution of single haplotypes in the amalgamation of genotypes found in the sewage. The nature of amplicon-based sequencing of fragments of around 400 bases59 renders direct linkages of mutations across amplicons largely unfeasible. To overcome this obstacle, we conflate associated mutations by their corresponding frequency pattern in time and space. Thereby, it is possible to designate different mutation constellations by a hierarchical, unsupervised two-step clustering approach implemented in our DeViVa (deconvolution of virus variants) tool. Evaluation of this approach showed that single samples typically feature not enough supportive information to deduce revealing mutation constellations. Apparently, a sensible composition of the sample set is pivotal to produce reliable results. To this effect, DeViVa was applied to all samples from the same calendar week from the province Carinthia, chosen for its substantial amount of data before the emergence of the Alpha variant. Thereby, Alpha-related mutations were observed in a dedicated constellation deduced from six samples from 1 February to 6 February 2021. The respective constellation contained a total of 30 mutations, of which 19 are defining mutations for Alpha (Fig. 3a). Identified mutation constellations can again be used as input for VaQuERo to examine the timely trends and geographic spread of the de novo identified mutation constellations. Such an analysis immediately displays that the observed constellation was indeed novel and sharply rising in three out of four WWTPs (Extended Data Fig. 7).

a, Mutation constellations in tree representation as clustered by their mutation frequencies in weekly sample batches from all Carinthian samples in the period from December 2020 to February 2021. The first constellation enriched with Alpha mutations is showcased with the frequencies of the mutations as observed in the samples used for deconvolution (heat map, left) and the frequency of the respective mutation in all samples of a specific variant as deposited in GISAID (heat map, right). b, Comparison of the relative abundance of Alpha, Delta and Omicron variants and the observed nucleotide diversity π in the WW samples. WWTPs with more than three pre-Alpha data points are shown individually (bottom), all WWTP are shown cumulatively (top). To test for reduction of nucleotide diversity during emergence and dominance of the occurrent variants, a one-sided Mann-Whitney U test between variant introduction (dotted vertical line) and end of dominance (dot-dashed vertical line) was performed. Corresponding P values are indicated.
Dynamics of genetic diversity
One important concept in molecular population biology is the nucleotide diversity π, expressing the mean number of nucleotide differences across all loci between two genome sequences from all possible pairs60. A small π indicates a very homogeneous population. Emerging variants with a higher reproductive fitness, gradually outdistancing prevailing variants, are expected to concomitantly see a reduction in observed π-values. By contrast, repeated introduction of virus through infected individuals should manifest in elevated π-values. To test these hypotheses, we first calculated the nucleotide diversity for each sequenced sample and compared its progression with the relative amount of the Alpha, Delta and Omicron variants (Fig. 3b). The recurring pattern seen for Alpha and Omicron, was a decreasing nucleotide diversity soon after the emergence of the variant. These reductions demonstrated the ongoing selective sweep in the genomic diversity caused by the replacement of the previously prevalent variants and their accumulated nucleotide diversity. By contrast, upon the takeover of the Delta variant, no reduction of π was observed, rather a steady increase in nucleotide diversity during the dominance of Delta could be observed (Fig. 3b). As the effect of the selective sweep seems to dominate the nucleotide diversity in the measured period, further hypotheses were subsequently evaluated only on samples with at least a 95% relative frequency of dominant variants. Population size and number of active infections in the catchment area show a weak but highly significant positive correlation (Kendall τ of 0.15 and 0.19, respectively) with the observed nucleotide diversity (Extended Data Fig. 8a,b). To test the hypothesis that regional migration of people contributed to elevated π-values, we resorted to time-resolved mobile communication network records and official numbers of Austria’s Federal Statistical Office on registered commuters and registered tourist overnight stays. These mobility-based analyses showed only a weak correlation (Kendall τ of −0.06 to 0.03) between people’s regional movements and the diversity of the virus population measured in the WW (Extended Data Fig. 8c–f).
Variant-specific reproduction number from wastewater
Sequencing-based abundance estimation typically produces only relative ratios, but no absolute quantification of single variants. Yet, it can be combined with ex-ante measured RT-qPCR-based quantification of the total viral load of the same sample47. Thereby, it can be assumed that the SARS-CoV-2 load normalised per population equivalents λ reflects a proxy for the prevalence of COVID-19 within the population connected to a sewage system61. To estimate the prevalence for single variants, the total normalised virus load λ can be apportioned according to the relative variant frequencies ν by λν = νλ. Accordingly, its first derivative with respect to time dλν/dt is a surrogate for the effective reproduction number Reff62, which we denoted as reproduction number from WW Rww. Hence, our data allowed a time-, region- and variant-resolved tracing of the effective reproduction number (Extended Data Fig. 9). Variant-specific Rww values were calculated considering the total viral load according to RT-qPCR normalised to the ammonium nitrogen load as a population size marker61 and the variant-specific relative abundances derived by VaQuERo from the same WW sample (Fig. 4a). Rww for different variants deduced at the same time and the same location, hence being produced by the same well-defined population, allow a direct comparison of the fitness of the examined variants. For Alpha, the variant-specific Rww was increased by a factor of 1.38 as compared to concomitantly circulating variants, which is supported by epidemiological studies63,64,65. The takeover by Delta fell into a low-prevalence phase with little WW sequencing information and was too sudden to yield enough time points with simultaneous signals of Delta and its predecessor to quantify its growth advantage with the applied method. For the exchange from Delta to Omicron, our WW-based Rww measurements indicated a reproductive advantage of Omicron over Delta by a factor of 1.85, exhibiting the same trend albeit reduced magnitude compared to reported Reff from individual case epidemiology66 (Fig. 4b).

a, Distribution of WW deduced reproduction number Rww for Alpha (B.1.1.7), Delta (B.1.617.2), Omicron (BA.2) and all other variants as calculated by an integration of absolute case estimate by RT-qPCR and variant estimate by sequencing, showcased for six WWTPs across the time period from January 2021 to February 2022. Depicted distribution are deduced from between 38 to 242 data points. b, Ratios of the variant-specific Rww from the same time point and the same WWTP exhibit a systematic shift to the values >1, with a median of 1.38 between Alpha and pre-Alpha and of 1.85 between Omicron and Delta, indicating a 38% increased transmissibility of Alpha over other competing variants and of 85% of Omicron over Delta, when subjected to an equal environment. Depicted distribution are deduced from between 25 to 96 data points. Horizontal box plots indicate 25th, 50th and 75th percentile (boxes) and up to the 1.5 times interquartile range contiguous from there (whiskers).
Discussion
This study aimed to convey insights deduced from WBE into classical, established concepts of case-based SARS-CoV-2 epidemiology. This includes the detection and quantification of a broad range of variants as defined by pangolin and the calculation of a WW-derived reproduction number Rww. Thereby, we facilitated its direct use in decision-making processes by public health authorities. Results of deconvoluted variant information and associated quantitative viral loads were reported to provincial and federal public health authorities since mid 2021 and continue to support regular risk assessments in Austria. We demonstrated that our VaQuERo method allows robustly deduction of relative virus variant frequencies from WW. We show at large scale that variant characterisation from WW and from case surveillance agree qualitatively and quantitatively. Onset, duration and scale of variant prevalence are in good agreement. The robustness of this finding is warranted by the comprehensive testing strategy implemented in Austria and the longitudinal and transversal breadth of the WW sequencing data presented here.
With respect to detection sensitivity of single cases, we note that the accuracy of sequencing-based WBE depends on prevalence and population number in the catchment area. In a catchment area with too many positive cases, the signal of an individual case becomes undetectable. A too-low prevalence rate, on the other hand, leads to a high dilution of viral RNA in the WW, thus impeding their detection. On average, with the given relation of (time variable) prevalence and catchment size, we observed that an absolute prevalence of more than 2 documented cases or a relative share in all documented cases of above 3.74% constitute the lower bound to reliably detect individual variants in Austrian municipal WWTP catchments early upon emergence. We show that after its introduction, Alpha exhibited a consistently elevated growth by 38% in comparison to pre-Alpha variants, as expressed by a higher variant-specific reproduction number Rww deduced from WW. Similarly, Omicron presented an 85% increased growth as compared to Delta.
Methods for the quantification of previously described and/or de novo variants from the complex genotype mixtures in WW are being developed but not well established yet42,67,68,69. Our approach to assign mutations to mutation constellations based on their individual mutation frequencies across several samples using DeViVa serves as a proof of principle and can be integrated directly with the variant definition-guided VaQuERo approach to identify emerging mutation constellations and investigate their temporal and spatial development patterns. A robust confirmation that the deduced constellations are indeed novel haplotypes, and of clinical relevance remains to be shown on the basis of individual patient samples.
In contrast to epidemiological case samples, virus sequencing from WW remains a technical challenge owing to the low concentration of target molecules, nucleic acid degradation as well as numerous interfering factors in the WW matrix70. The results presented here were derived despite considerable deficiencies in the raw sequencing data quality caused by the complex WW matrix, making our analysis approach applicable to versatile WBE operating conditions. However, fine-grained sample designs71 and/or more customised extraction methods72 and long-read sequencing techniques are expected to promise an even higher resolution of the future designation, detection and quantification of SARS-CoV-2 variants and other viruses.
Our results indicate that WBE recapitulates surveillance of epidemiological cases at a high spatiotemporal resolution, at reduced sample number and logistical effort. WBE complements classical surveillance programmes, but it cannot replace individual case-based surveillance owing to inherent limitations such as the inability to identify positive individuals and correlate clinical outcomes of novel variants. Yet, comprehensive WBE represents an economic way, given the reduced number of assays needed to gain an overview of the pandemic situation, to inform traditional case-based epidemiology and to adjust the testing strategy on a regional level. Further, WBE allows to observe the emergence of variants on a global scale, including in countries with limited resources to maintain individual case-based surveillance programmes73. In this regard, it is critical to iterate the presented methodology to other modes of WW disposal in absence of a centralised sewerage system74,75 Once in place, WBE will also provide valuable synergies to survey the prevalence of a wider range of public health-relevant pathogens, such as influenza virus and enteric viruses76,77,78
It is well established in population genomics that certain applications benefit from a high sampling rate79 Here we aimed to synergise the two fields by connecting the concept of nucleotide diversity and epidemiological parameters, such as prevalence and mobility, as a surrogate for introductions. We observed that the absolute case number is indeed imprinted into the observed nucleotide diversity. By contrast, no effect of mobility was found. Further studies are needed to clarify if this is due to a limited role of regional mobility during viral spread, or if the available data does not provide enough sensitivity for its detection. The introduction of the Alpha and the Omicron variant led to a pronounced selective sweep, resulting in an overall reduced nucleotide diversity. Notably, no similar reduction could be observed after the takeover of the Delta variant, which was accompanied by an increase in nucleotide diversity of the circulating virus population. The circumstances around the establishment of Alpha, Delta and Omicron as dominant circulating variants differed in many aspects. Alpha and Omicron were introduced during winter amidst a fading seasonal wave, immediately causing a new rise in incidence numbers. By contrast, Delta emerged during a low-incidence period in early summer, leading to a prominent but only slowly rising epidemiological wave. The reduction in nucleotide diversity after the introduction of Omicron, observed in the first two months after introduction, is unexpected since the two related sub-variants BA.1 and BA.2 emerged time-shifted and circulated simultaneously. It is tempting to account the peculiarity of Delta to a higher genomic plasticity in contrast to other variants as discussed previously80,81. Generally, tracking the overall changes of nucleotide diversity, variant-specific reproduction numbers and emergence of novel mutation constellations in WW allow observation of evolutionary processes, potentially assisting understanding and anticipation of future shifts in circulating virus populations.
The field of WBE received a strong impetus driven by the needs of the current pandemic management22,72. Nonetheless, many technical, scientific and policy challenges must be resolved to realise its full potential. For a robust quantification and interpretation of the data, a better understanding of the amount and the duration of virus shedding via faeces is needed, especially considering the influence of patient immune status and possibly altered variant-specific properties82. The timely transfer of insights gained from WBE into actionable results for public health will be key83. Our current setup of sample logistics, experimental procedures and sequence analyses led to the sharing of results with stakeholders typically within 10–14 days after sampling, providing a valuable layer for risk assessment to the Austrian authorities. Future improvements are expected to lead to reduced turnaround times with additional value for the pandemic management. On an international level, WBE data sharing will be crucial to support global pathogen surveillance. This national-scale study demonstrates the accuracy and the information richness of sequencing-based WW surveillance for the current SARS-CoV-2 pandemic as well as its potential for future improved global surveillance of other infectious diseases.
Methods
Analysis of WW parameters
Chemical oxygen demand, total nitrogen and ammonium nitrogen were analysed on site by respective WWTP operators using cuvette tests and following standards specified by the International Organisation for Standardisation: ISO1570584, ISO1190585 and ISO1173286, respectively. Deposition and daily WW flow were also recorded on site.
Sample processing
Sampling was performed by the WWTP operator with onsite installed 24-h refrigerated automatic composite samplers. RNA extraction and RT-qPCR-based virus quantification was performed by the members of the Coron-A consortium (https://www.coron-a.at), at three different laboratories (TU Wien, Medical University of Innsbruck and University Innsbruck). The protocols for WW sample pre-treatment and PEG-NaCl-based precipitation of the virus fraction were based on the methods described by Ye et al. and Wu et al.87,88. The details of the implemented protocols varied in each of the executing laboratory. Generally, all workflows followed a common procedure. Twenty-four-hour volume-equivalent mixed samples were stored at 4 °C until processing. Before PEG-precipitation-based concentration, large particles were removed. Nucleic acid extracted from the pellet after concentration were analysed via one-step RT-qPCR for presence of the N-gene of SARS-CoV-2 and passed on for sequencing library preparation. Experimental details have been described previously for each laboratory: Medical University of Innsbruck30,TU Wien31, and University Innsbruck32.
To warrant comparability of different laboratory procedures applied, a ring trial was performed. To this end, raw WW from three different WWTPs in Carinthia in calendar week 45 of 2021 was collected from catchments selected according to their 7-day incidence within the respective population, to represent WW with low, medium and high viral loads. Each of three cooled (4 °C) samples was aliquoted and sent to the three laboratories. The samples were processed on the same day via laboratory-specific methods. After concentration and nucleic acid extraction, extracts were sent on dry ice to one laboratory (Department of Microbiology, University of Innsbruck) to quantify the absolute SARS-CoV-2 concentration in the extract. The analysis was conducted using a digital PCR system (Qiacuity One 5plex System, Qiagen) to impede possible bias from reference standards and calibration curves. Confidence intervals were deduced from single samples, assuming a Poisson-distributed absolute number of responding partitions of the digital PCR system and calculated by the Qiacuity Software Suite 1.2. The results from the ring trial (Extended Data Fig. 10) reflect that the different methods as described above yielded nucleic acid extracts with a comparable amount of SARS-CoV-2 gene copies across the concentration range.
Samples generated for sequencing reproducibility were prepared in triplicates as described above. Each of the triplicates were drawn from the same 24-h volume-equivalent sample. All subsequent analysis step were performed independently.
RNA library prep and sequencing
RNA from WW extracts was reverse transcribed with Superscript IV Reverse Transcriptase (Thermo Fisher Scientific). The resulting complementary DNA was used to amplify viral sequences with modified primer pools from the Artic Network initiative59 or VarSkip 1a primer pool (NEB). For amplicon cleanup AMPure XP beads (Beckman Coulter) were used at a 1:1 ratio and amplicon concentrations and size distributions were checked with the Qubit Fluorometric Quantitation (Life Technologies) and the 2100 Bioanalyzer system (Agilent), respectively. Amplicon concentrations were normalised, and sequencing libraries created with the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) according to the manufacturer’s instructions. As before library concentrations and size distribution were assessed again, the pools mixed at equimolar concentrations and sequencing carried out on the NovaSeq 6000 platform (Illumina) using an SP flow cell with a read length of 2 × 250 bp in paired-end mode.
Mutation calling
After demultiplexing, adapter sequences were trimmed with BBDUK, and the overlapping regions of paired reads were corrected with BBMERGE from the BBTools suite (v 38.90)89. BWA-MEM90 (v.0.7.17) was then used to map read pairs to the combined GRCh38 and SARS-CoV-2 genomes (RefSeq: NC_045512.2) with a minimal seed length of 17. Only read mappings uniquely and properly paired to the SARS-CoV-2 genome were kept and primer sequences were masked with iVar91 (v.1.2.3). LoFreq92 (v 2.1.2) was used for low-frequency variant calling, after first using its Viterbi method to realign reads around indels and adding indel qualities. Variants were then filtered with LoFreq and Bcftools93 (v.1.9) only considering variants with a minimum coverage of 75 reads, a minimum Phred scaled calling quality value of 90 and indels with a HRUN value of at less than four. Variant annotation was performed with SnpEff94 (v.4.3) and SnpSift95 (v.4.3).
Variant detection and quantification
Multiple genome alignments of 6.8 million global SARS-CoV-2 genomes provided by the global initiative on sharing avian influenza data (GISAID database, retrieved on 29 January 2022)55,56 and the associated pangolin lineage assignments were used to extract all nucleotide variants as compared to the reference genome. For each pangolin lineage, all mutations that could be observed in at least 80% of the analysed genomes were defined as sensitive marker mutations for the respective lineage. Mutations occurring only in samples of one lineage with a frequency greater 40% were considered specific or unique mutations for that lineage. To reduce complexity, the scope was focused on all lineages which were detected in Austria (at least ten sequenced genomes according to GISAID) and all de-escalated variants, variants of concern/interest and variants under monitoring as defined by the European Centre for Disease Prevention and Control (ECDC; https://www.ecdc.europa.eu/en/covid-19/variants-concern; accessed on 29 January 2022). A total of 33 different regionally relevant variants were included. Deduced marker mutation, together with inferred mutation frequencies, were further used to quantify all respective variants of relevance per WWTP in the available time course data according to the following scheme: (i) remove all samples with more than 60% of the genomic position covered with less than 10 reads; (ii) filter marker mutations with observed frequency >0.02 and >75 supportive reads; (iii) for each time point, denote all variants with at least two unique mutations and at least 10% of all unique mutations as detected; (iv) add mutation frequencies from samples from the preceding 8 days; (v) filter frequencies of marker mutation of detected variants; (vi) transform96 remaining mutation frequencies to avoid zeros and ones by f′ = (f(d − 1) + 0.5)/d, where d denotes the sequencing depth at the respective locus, f and f′ denote the observed and the transformed mutation frequency, respectively; (vii) infer expected frequency per variant applying a regression model Y = Xβ, where Y represents the mutation frequencies, X represents the n × m design matrix assigning the considered n mutations to one or more of the m variants and β estimated model coefficients. To this end the package gamlss97 (generalised additive model for location scale and shape; v.5.3.4) is used with a SIMPLEX98 linker function and weights w per mutation i: \(w_i = \left( {\frac{1}{{\Delta t + 1}}} \right){{{\mathrm{log}}}}10\left( {d_i} \right)\), with the time difference Δt to the current sample in focus in days and the sequencing depth di for mutation position i. If the SIMPLEX function does not converge, resort to the Beta function. If the sum of the estimated variant frequencies exceeds 1, the frequencies are scaled down proportionally. The model is then used to predict the frequencies of the unique markers, as an estimate of the variant frequencies. An implementation of the described software is available on GitHub (https://github.com/fabou-uobaf/VaQuERo). Spatiotemporal visualisation was performed from the deduced variant frequencies across Austria using the Multilevel B-Spline Approximation99 using the mba.surf function implemented in the R package MBA (v.0.0.9) with the following non-default parameters: no.X = no.Y = 30; m = range of longitude (°) divided by range of date (days); and extend set to FALSE.
Variant-specific reproduction number from WW
SARS-CoV-2 load was normalised to the ammonium nitrogen load as a population size marker, assuming an ammonium nitrogen load per capita of 8 g per day61. The normalisation results in the viral load parameter λ, representing viral copies per population equivalent per day, which is proportional to the absolute number of infected individuals47. In combination with the relative proportions from the sequencing-based variant quantification (derived from VaQuERo), the absolute quantity λν from a certain variant ν was estimated, by multiplication of the total virus load with the relative frequency of the variant. From there, the load time series is fitted and interpolated with a spline using the Matlab 2018a function FIT with option SmoothingSpline and smoothing parameter p of 0.001. The smoothing parameter p is defined between 0 and 1. p = 0 produces a least-squares straight-line fit to the data, while p = 1 produces a cubic spline interpolant. Next, to account for time delays in viral shedding of infected individuals, the smoothed time series is deconvoluted according to an experimentally inferred shedding distribution (using an iterative blind deconvolution algorithm) with a gamma distribution truncated after 25 days and normalised as previously described21. Finally, Rww is estimated from this time series using the Epi-Estim62,100 implementation provided in MATLAB script estimate_R (https://mathworks.com/matlabcentral/fileexchange/78760-estimate_r). We use variant-specific mean generation times of 5.5 (s.d. 4), 4.6 (s.d. 3.1), 3.3 (s.d. 3.5), 6 (s.d. 4.9) for the Alpha, Delta, Omicron and pre-Alpha variants, respectively101,102,103. Deduced Rww on the basis of viral load signal in the WW below 5 mega gene copies per population equivalent per day are rejected.
Evaluation by epidemiological case records
The national COVID-19 surveillance system, maintained by AGES, collects all confirmed SARS-CoV-2 cases, as identified, by routine tests of asymptomatic persons, suspected cases with symptoms and cases detected through contact tracing, amongst other methods. This data set extends to, if available, comprehensive, aggregated medical records including virus variant-related differential diagnosis. In the period of examination between 1 January 2021 and 10 February 2022, on average 50.6% of all identified SARS-CoV-2 cases in all municipalities connected to any examined WWTP were tested for SARS-CoV-2 mutations and variants. Sequencing-based tests were performed by either partial sequencing of the spike gene50, Sanger sequencing or whole-genome sequencing. However, most cases were analysed using RT-qPCRs for specific mutations, performed in the responsible diagnosis laboratory and reported to AGES. On the basis of the set of reported mutations, a case was classified as a particular variant of concern or as the wild type, accordingly. Definition of indicator mutations were adapted during the pandemic. Since initially BA.1 and BA.2 were not distinguished but identified as B.1.1.529. Omicron subclades were aggregated in WW and clinical data for comparative analyses. Individual cases are allocated to their place of residence. Data were extracted from the AGES records for all municipalities connected to any examined WWTP and aggregated for all municipalities within the same catchment area and from the same calendar week, to deduce absolute case counts and relative frequency per variant, per catchment area and per week. Municipalities which discharge into more than one WWTP were omitted. If omitting municipalities led to the loss of >40% of the people served by one WWTP, the entire WWTP was omitted. For plotting purposes and for direct comparison with WW-derived data, both epidemiological cases and data from WW samples were shifted to Wednesday of that respective week. One-sided κ statistics were calculated using the function epi.kappa with the Fleiss method from the epiR package (https://cran.r-project.org/web/packages/epiR). Deduced P values were corrected for multiple testing with the method of Benjamini and Hochberg.
Mutation deconvolution
For mutation deconvolution, a hierarchical, unsupervised two-step clustering approach was used. First, a silhouette analysis collected mutations with similar pattern of relative abundances within a specific set of samples. The method is implemented in a software tool named DeViVa (deconvolution of virus variants), written in Python (v.3.8) and available from GitHub (https://github.com/SebH87/DeViVa). The first clustering step used Ward’s method on the basis of Euclidian distance to segregate spurious mutations. To guarantee consistent results, only mutations with a relative abundance >5% were accepted. The relative abundance of the remaining mutations was transformed (centred log-ratio transformation; 0 was replaced by 10−7) and subjected to a second hierarchical clustering step. For this, distances between mutations were calculated by applying the complete-linkage algorithm, which is based on square Euclidian distance. To identify distinct mutation constellations a silhouette analysis was performed104. Thereby, a silhouette coefficient is calculated for each clustering produced by a critical distance threshold between 2 and 20. The clustering exhibiting the highest silhouette coefficient is considered optimal and is used. Finally, each cluster is variant-typed by introducing its mutations into the reference genome of SARS-CoV-2 and assigning this newly assembled genome with the pangolin lineage tool49
Nucleotide diversity
The LoFreq-filtered variant call format files of all WWTP for which more than 18 time points were available with at least 2/3 of the genome covered with more than ten reads were used to extract variants with an allele frequency above 1%. Multiallelic SNPs were joined using Bcftools93 (v.1.12). For the amplicon primer sets used (ARTIC version 3 and VarSkip version 1) a total of 69 and 40, respectively, runs of independently processed and sequenced, synthetic SARS-CoV-2 RNA (Twist BioScience) control samples were produced and used to define sites with increased false-positive variant callings. For that purpose, the controls were sequenced and analysed as described above. Mutations which exhibit a median observed allele frequency >2% or an allele frequency >5% in at least 10% of the samples were flagged as problematic mutations and omitted from further analysis. In total 39 mutations were identified as such. Thereof, 9 and 30 mutations were identified in ARTIC v3 and VarSkip v1 produced datasets, respectively. SNPgenie105 (v.2019.10.31) was used to calculate genome wide nucleotide diversity π. For samples with more than 20 time points, the data was integrated with abundances of variant of concern as deduced by VaQuERo. Significance of reduction of π between the introduction and the end of the dominance periods was evaluated using a one-sided Mann-Whitney U test between π-values from ±4 weeks around the first time the variant rose above 10% relative frequency and from ±4 weeks around the last time the variant was observed with a relative frequency above 90%.
Mobility data
Statistics on registered accommodation in the hospitality industry (Beherbergungsstatistik 2019/20, https://www.statistik.at/atlas/?mapid=them_tourismus_winter_uebernachtungen) and the statistic on registered commuting dynamics (Registerzählung 2011, Abgestimmte Erwerbsstatistik, https://www.statistik.at/atlas/?mapid=them_bevoelkerung_pendler) were gathered from publicly available databases. Of note, both registers do not represent the same period, and moreover, it is incalculable how social adaptation (work from home and circumvention of hospitality restrictions) has affected the registered behaviour. Therefore, time-resolved mobility data, which is based on the mobile communications network records from one undisclosed mobile provider, were used in addition. A movement from region A to B in the origin–destination matrix was measured if a mobile phone was first logged in to A for at least 15 min and afterwards logged in to B for at least 15 min. Detailed information about the origin–destination matrix calculation process have been described previously106. This movement data was used to define main axes of transportation for visualisation purpose and to deduce the proportion of population who at least once per day left the catchment area in the direction of another area or even another federal state. Therefore, the number of movements leaving the districts which are assigned to the WWTP were counted for each catchment. The retrieved statistics were normalised to the number of people connected to the sewage system and log10 transformed.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All raw sequencing data used during this study, with additional sequencing data produced for samples drawn from Austrian WWTP before December 2020, are available under the ENA accession number: PRJEB48985. Raw variant quantification of samples described in this contribution are visualised and can be browsed in a dedicated dashboard available at https://ww-dashboard.bergthalerlab.com107. Access to aggregated epidemiological data, collected by the Austrian Agency for Health and Food Safety (AGES; https://covid19-dashboard.ages.at/), can be granted for research purposes in compliance with local regulatory and legal frameworks, to exclude retraceability of individual cases, upon request via the corresponding author. SARS-CoV-2 genome sequences for marker mutation definition can be obtained from GISAID (https://www.gisaid.org/) from registered users agreeing to the effective terms of use (https://www.gisaid.org/registration/terms-of-use/).
Code availability
The source code for the developed and applied software for variant quantification (VaQuERo) and mutation deconvolution (DeViVa) is available on GitHub (https://github.com/fabou-uobaf/VaQuERo and https://github.com/SebH87/DeViVa).
References
Nicola, M. et al. The socio-economic implications of the coronavirus pandemic (COVID-19): a review. Int. J. Surg. 78, 185–193 (2020).
Josephson, A., Kilic, T. & Michler, J. D. Socioeconomic impacts of COVID-19 in low-income countries. Nat. Hum. Behav. 5, 557–565 (2021).
Harvey, W. T. et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 19, 409–424 (2021).
Callaway, E. Heavily mutated coronavirus variant puts scientists on alert. Nature 21, 600 (2021).
Truong, T. T. et al. Increased viral variants in children and young adults with impaired humoral immunity and persistent SARS-CoV-2 infection: a consecutive case series. EBioMedicine 67, 103355 (2021).
Lucas, C. et al. Impact of circulating SARS-CoV-2 variants on mRNA vaccine-induced immunity. Nature 600, 523–529 (2021).
Chandler, J.C. et al. SARS-CoV-2 exposure in wild white-tailed deer (Odocoileus virginianus). Proc. Natl Acad. Sci. USA 118, e2114828118 (2021).
Plante, J. A. et al. The variant gambit: COVID-19’s next move. Cell Host Microbe 29, 508–515 (2021).
Gardy, J. L. & Loman, N. J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 19, 9–20 (2018).
Grubaugh, N. D. et al. Tracking virus outbreaks in the twenty-first century. Nat Microbiol. 4, 10–19 (2019).
Smith, G. J. D. et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza a epidemic. Nature 459, 1122–1125 (2009).
Inzaule, S. C., Tessema, S. K., Kebede, Y., Ogwell Ouma, A. E. & Nkengasong, J. N. Genomic-informed pathogen surveillance in Africa: opportunities and challenges. Lancet Infect. Dis. 21, e281–e289 (2021).
Woolhouse, M. E. J., Rambaut, A. & Kellam, P. Lessons from Ebola: improving infectious disease surveillance to inform outbreak management. Sci. Transl. Med. 7, 307rv5 (2015).
Furuse, Y. Genomic sequencing effort for SARS-CoV-2 by country during the pandemic. Int. J. Infect. Dis. 103, 305–307 (2021).
The COVID-19 Genomics UK (COG-UK) consortium. An integrated national scale SARS-CoV-2 genomic surveillance network. Lancet Microbe 1, e99–e100 (2020).
Treibel, T. A. et al. COVID-19: PCR screening of asymptomatic health-care workers at London hospital. Lancet 395, 1608–1610 (2020).
Brito, A. F. et al. Global disparities in SARS-CoV-2 genomic surveillance. Preprint at medRxiv https://doi.org/10.1101/2021.08.21.21262393 (2021)..
Belman, S., Saha, S. & Beale, M. A. SARS-CoV-2 genomics as a springboard for future disease mitigation in LMICs. Nat. Rev. Microbiol. https://doi.org/10.1038/s41579-021-00664-y (2021).
Majid, F., Omer, S. B. & Khwaja, A. I. Optimising SARS-CoV-2 pooled testing for low-resource settings. Lancet Microbe 1, e101–e102 (2020).
Larsen, D. A., Green, H., Collins, M. B. & Kmush, B. L. Wastewater monitoring, surveillance and epidemiology: a review of terminology for a common understanding. FEMS Microbes 2, xtab011 (2021).
Cavany, S. et al. Inferring SARS-CoV-2 RNA shedding into wastewater relative to the time of infection. Epidemiol Infect. 150, e21 (2022).
Bonanno Ferraro, G. et al. A state-of-the-art scoping review on SARS-CoV-2 in sewage focusing on the potential of wastewater surveillance for the monitoring of the COVID-19 pandemic. Food Environ. Virol. https://doi.org/10.1007/s12560-021-09498-6 (2021).
Hassard, F., Lundy, L., Singer, A. C., Grimsley, J. & di Cesare, M. Innovation in wastewater near-source tracking for rapid identification of COVID-19 in schools. Lancet Microbe 2, e4–e5 (2021).
la Rosa, G. et al. SARS-CoV-2 has been circulating in northern Italy since December 2019: evidence from environmental monitoring. Sci. Total Environ. 750, 141711 (2021).
Martin, J. et al. Tracking SARS-CoV-2 in sewage: evidence of changes in virus variant predominance during COVID-19 pandemic. Viruses 12, 1144 (2020).
Nemudryi, A. et al. Temporal Detection and Phylogenetic Assessment of SARS-CoV-2 in Municipal Wastewater. Cell Rep. Med. 1, 100098 (2020).
Wurtzer, S. et al. Monitoring the propagation of SARS CoV2 variants by tracking identified mutation in wastewater using specific RT–qPCR. Preprint at medRxiv https://doi.org/10.1101/2021.03.10.21253291 (2021).
Peccia, J. et al. Measurement of SARS-CoV-2 RNA in wastewater tracks community infection dynamics. Nat. Biotechnol. 38, 1164–1167 (2020).
Agrawal, S., Orschler, L. & Lackner, S. Long-term monitoring of SARS-CoV-2 RNA in wastewater of the Frankfurt metropolitan area in Southern Germany. Sci Rep. 11, 5372 (2021).
Daleiden, B. et al. Wastewater surveillance of SARS-CoV-2 in Austria: development, implementation, and operation of the Tyrolean wastewater monitoring program. J. Water Health 20, 314–328 (2022).
Radu, E. et al. Emergence of SARS-CoV-2 Alpha lineage and its correlation with quantitative wastewater-based epidemiology data. Water Res. 215, 118257 (2022).
Markt, R. et al. Detection and abundance of SARS-CoV-2 in wastewater in Liechtenstein, and the estimation of prevalence and impact of the B.1.1.7 variant. J. Water Health 20, 114–125 (2021).
Karthikeyan, S. et al. High-throughput wastewater SARS-CoV-2 detection enables forecasting of community infection dynamics in San Diego County. mSystems 6, e00045-21 (2021).
Crits-Christoph, A. et al. Genome sequencing of sewage detects regionally prevalent SARS-CoV-2 variants. mBio 12, 02703-20 (2021).
Izquierdo-Lara, R. et al. Monitoring SARS-CoV-2 circulation and diversity through community wastewater sequencing, the Netherlands and Belgium. Emerg. Infect. Dis. 27, 1405–1415 (2021).
Agrawal, S. et al. A pan-European study of SARS-CoV-2 variants in wastewater 2 under the EU Sewage Sentinel System. Preprint at medRxiv https://doi.org/10.1101/2021.06.11.21258756 (2021).
Bar-Or, I. et al. Detection of SARS-CoV-2 variants by genomic analysis of wastewater samples in Israel. Sci. Total Environ. 789, 148002 (2021).
Fontenele, S. et al. High-throughput sequencing of SARS-CoV-2 in wastewater provides insights into circulating variants. Water Res. 205, 117710 (2021).
Fuqua, J. L. et al. The rapid assessment of aggregated wastewater samples for genomic surveillance of SARS-CoV-2 on a city-wide scale. Pathogens 10, 1271 (2021).
Jahn, K. et al. Detection of SARS-CoV-2 variants in Switzerland by genomic analysis of wastewater samples. Preprint at medRxiv https://doi.org/10.1101/2021.01.08.21249379 (2021).
Pechlivanis, N. et al. Detecting SARS-CoV-2 lineages and mutational load in municipal wastewater and a use-case in the metropolitan area of Thessaloniki, Greece. Sci. Rep. 12, 2659 (2021).
Smyth, D.S., Trujillo, M., Gregory, D.A. et al. Tracking cryptic SARS-CoV-2 lineages detected in NYC wastewater. Nat Commun 13, 635 (2022). https://doi.org/10.1038/s41467-022-28246-3
la Rosa, G. et al. Rapid screening for SARS-CoV-2 variants of concern in clinical and environmental samples using nested RT-PCR assays targeting key mutations of the spike protein. Water Res. 197, 117104 (2021).
Prado, T. et al. Wastewater-based epidemiology as a useful tool to track SARS-CoV-2 and support public health policies at municipal level in Brazil. Water Res. 191, 116810 (2021).
Rimoldi, S. G. et al. Presence and infectivity of SARS-CoV-2 virus in wastewaters and rivers. Sci. Total Environ. 744, 140911 (2020).
Agrawal, S., Orschler, L. & Lackner, S. Metatranscriptomic analysis reveals SARS-CoV-2 mutations in wastewater of the Frankfurt metropolitan area in Southern Germany. Microbiol. Resour. Announc. 10, e00280-21 (2021).
Huisman, J. S. et al. Wastewater-based estimation of the effective reproductive number of SARS-CoV-2. Environ. Health Perspect. 130, 057011 (2022).
Jahn, K. et al. Detection and surveillance of SARS-CoV-2 genomic variants in wastewater. Preprint at medRxiv https://doi.org/10.1101/2021.01.08.21249379 (2021)..
O’Toole, Á. et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 7, veab064 (2021).
Özkan, E. et al. High-throughput mutational surveillance of the SARS-CoV-2 spike gene. Preprint at medRxiv https://doi.org/10.1101/2021.07.22.21259587 (2021).
Paetzold, J. et al. Impacts of rapid mass vaccination against SARS-CoV2 in an early variant of concern hotspot. Nat. Commun. 13, 612 (2022).
Hasell, J. et al. A cross-country database of COVID-19 testing. Sci. Data 7, 345 (2020).
Progress on household drinking water, sanitation and hygiene 2000-2020: five years into the SDGs. (WHO and UNICEF, 2021).
Popa, A. et al. Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci. Transl. Med. 12, eabe2555 (2020).
Elbe, S. & Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Challenges 1, 33–46 (2017).
Shu, Y. & McCauley, J. GISAID: global initiative on sharing all influenza data—from vision to reality. Eurosurveillance 22, 30494 (2017).
Cragg, J. G. Some statistical models for limited dependent variables with application to the demand for durable goods. Econometrica 39, 829–844 (1971).
van Poelvoorde, L. A. et al. Strategy and performance evaluation of low-frequency variant calling for SARS-CoV-2 using targeted deep Illumina sequencing. Front. Microbiol. 12, 747458 (2021).
Itokawa, K., Sekizuka, T., Hashino, M., Tanaka, R. & Kuroda, M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS One 15, e0239403 (2020).
Nei, M. & Li, W.-H. Mathematical model for studying genetic variation in terms of restriction endonucleases (molecular evolution/mitochondrial DNA/nucleotide diversity). Genetics 76, 5269–5273 (1979).
Been, F. et al. Population normalization with ammonium in wastewater-based epidemiology: application to illicit drug monitoring. Environ. Sci. Technol. 48, 8162–8169 (2014).
Cori, A., Ferguson, N. M., Fraser, C. & Cauchemez, S. A new framework and software to estimate time-varying reproduction numbers during epidemics. Am. J. Epidemiology 178, 1505–1512 (2013).
Campbell, F. et al. Increased transmissibility and global spread of SARSCoV- 2 variants of concern as at June 2021. Eurosurveillance 26, 1–6 (2021).
Davies, N. G. et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 372, eabg3055 (2021).
Washington, N. L. et al. Emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States. Cell 184, 2587–2594.e7 (2021).
Ito, K., Piantham, C. & Nishiura, H. Relative instantaneous reproduction number of Omicron SARS-CoV-2 variant with respect to the Delta variant in Denmark. J. Med. Virol. 94, 2265–2268 (2021).
Baaijens, J. A. et al. Variant abundance estimation for SARS-CoV-2 in 1 wastewater using RNA-Seq quantification 2. Preprint at medRxiv https://doi.org/10.1101/2021.08.31.21262938 (2021).
Pipes, L., Chen, Z., Afanaseva, S. & Nielsen, R. Estimating the relative proportions of SARS-CoV-2 strains from 2 wastewater samples. Preprint at medRxiv https://doi.org/10.1101/2022.01.13.22269236 (2021).
Sapoval, N. et al. Enhanced Detection of Recently Emerged SARS-CoV-2 Variants of Concern in Wastewater. Preprint at medRxiv https://doi.org/10.1101/2021.09.08.21263279 (2021).
Wade, M. J. et al. Understanding and managing uncertainty and variability for wastewater monitoring beyond the pandemic: lessons learned from the United Kingdom national COVID-19 surveillance programmes. J. Hazard. Mater. 424, 127456 (2022).
Karthikeyan, S. et al. Rapid, large-scale wastewater surveillance and automated reporting system enable early detection of nearly 85% of COVID-19 cases on a university campus. mSystems 6, 793–814 (2021).
Calderón-Franco, D., Orschler, L., Lackner, S., Agrawal, S. & Weissbrodt, D. G. Monitoring SARS-CoV-2 in sewage: toward sentinels with analytical accuracy. Sci. Total Environ. 804, 150244 (2022).
Shrestha, S. et al. Wastewater-based epidemiology for cost-effective mass surveillance of covid-19 in low-and middle-income countries: challenges and opportunities. Water 13, 2897 (2021).
Hong, P. Y. et al. Estimating the minimum number of SARS-CoV-2 infected cases needed to detect viral RNA in wastewater: to what extent of the outbreak can surveillance of wastewater tell us? Environ. Res. 195, 110748 (2021).
Basu, P. et al. Surveillance of SARS-CoV-2 RNA in open-water sewage canals contaminated with untreated wastewater in resource-constrained regions. Access Microbiol. 4, 000318 (2022).
Chan, M. C. W. et al. Seasonal influenza a virus in feces of hospitalized adults. Emerg. Infect. Dis. 17, 2038–2042 (2011).
Pogka, V. et al. Laboratory surveillance of polio and other enteroviruses in high-risk populations and environmental samples. Appl. Environ. Microbiol. 83, e02872-16 (2017).
Wolfe, M. K. et al. Wastewater-based detection of an influenza outbreak. Preprint at medRxiv https://doi.org/10.1101/2022.02.15.22271027 (2022).
Lynch, M., Bost, D., Wilson, S., Maruki, T. & Harrison, S. Population-genetic inference from pooled-sequencing data. Genome Biol. Evol. 6, 1210–1218 (2014).
Suratekar, R. et al. High diversity in Delta variant across countries revealed by genome‐wide analysis of SARS‐CoV‐2 beyond the Spike protein. Mol. Syst. Biol. 18, e10673 (2022).
Stern, A. et al. The unique evolutionary dynamics of the SARS-CoV-2 Delta variant-2 sequencing. Preprint at medRxiv https://doi.org/10.1101/2021.08.05.21261642 (2021).
Yuan, S. et al. Pathogenicity, transmissibility, and fitness of SARS-CoV-2 Omicron in Syrian hamsters. Science. 0, eabn8939. 10.1126/science.abn8939 (2022).
Safford, H. R., Shapiro, K. & Bischel, H. N. Wastewater analysis can be a powerful public health tool—if it’s done sensibly. Proc. Natl Acad. Sci. USA 119, e2119600119 (2022).
Water quality—determination of the chemical oxygen demand index (ST-COD)—small-scale sealed-tube method. DS/ISO 15705:2002(E) (International Standards Organisation, 2002).
Water quality—determination of nitrogen—part 1: method using oxidative digestion with peroxodisulfate. ISO 11905-1:1997 (International Standards Organisation, 1997).
Water quality—determination of ammonium nitrogen—method by flow analysis (CFA and FIA) and spectrometric detection. ISO 11732:2005. (International Standards Organisation, 2005).
Ye, Y., Ellenberg, R. M., Graham, K. E. & Wigginton, K. R. Survivability, partitioning, and recovery of enveloped viruses in untreated municipal wastewater. Environ. Sci. Technol. 50, 5077–5085 (2016).
Wu, F. et al. SARS-CoV-2 titers in wastewater are higher than expected from clinically confirmed cases. mSystems 5, e00614-20 (2020).
Bushnell, B., Rood, J. & Singer, E. BBMerge – Accurate paired shotgun read merging via overlap. PLoS One 12, e0185056 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Grubaugh, N. D. et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 20, 8 (2019).
Wilm, A. et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201 (2012).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Cingolani, P. et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genet. 3, 35 (2012).
Smithson, M. & Verkuilen, J. A better lemon squeezer? Maximum-likelihood regression with beta-distributed dependent variables. Psychol. Methods 11, 54–71 (2006).
Rigby, R. A. & Stasinopoulos, D. M. Generalized additive models for location, scale and shape. J. R. Stat. Soc. C Appl. Stat. 54, 507–554 (2005).
Barndorff-Nielsen, E. & Jorgensen, B. Some Parametric Models on the Simplex. J.Multivar. Anal. 39, 106–116 (1991).
Lee, S., Wolberg, G. & Shin, S. Y. Scattered data interpolation with multilevel B-splines. IEEE Trans. Vis. Comput. Graph. 3, 228–244 (1997).
Thompson, R. N. et al. Improved inference of time-varying reproduction numbers during infectious disease outbreaks. Epidemics 29, 100356 (2019).
Hart, W. et al. Inference of the SARS-CoV-2 generation time using UK household data. eLife 11, e70767 (2022).
Hart, W. S. et al. Generation time of the alpha and delta SARS-CoV-2 variants: an epidemiological analysis. Lancet Infect. Dis. 22, 603–610 (2022).
Abbott, S., Sherratt, K., Moritz, G. & Funk, S. Estimation of the test to test distribution as a proxy for generation interval distribution for the Omicron variant in England. Preprint at medRxiv https://doi.org/10.1101/2022.01.08.22268920 (2022).
Rousseeuw, P. J. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65 (1987).
Nelson, C. W., Moncla, L. H. & Hughes, A. L. SNPGenie: estimating evolutionary parameters to detect natural selection using pooled next-generation sequencing data. Bioinformatics 31, 3709–3711 (2015).
Heiler, G. et al. Country-wide mobility changes observed using mobile phone data during COVID-19 pandemic. In 2020 IEEE International Conference on Big Data (Big Data) 3123–3132 (IEEE, 2020).
Triska, P., Amman, F., Endler, L. & Bergthaler, A. WAVES (Web-based tool for Analysis and Visualization of Environmental Samples) – a web application for visualization of wastewater pathogen sequencing results. Preprint at medRxiv https://doi.org/10.1101/2022.05.31.22275831 (2022).
Acknowledgements
We thank A. Kammerer, B. Urban, M. Chan, M. Schuster, C. Suete, P. Carey, A. Peisser and P. Pelz for their support. This project was funded in part by the Förderkreis 1669 der Universität Innsbruck, the FFG-Corona-emergency-call, the Austrian Agency for Health and Food Safety (AGES), the Vienna science and Technology Fund (WWTF) as part of the WWTF COVID-19 Rapid Response Funding 2020 (A.B.), the Austrian Science Fund (FWF1212P) and the Austrian Academy of Sciences. Z.K. was supported by a fellowship of the Marie Skłodowska-Curie Actions (MSCA) Innovative Training Network H2020-MSCA- ITN-2019 (grant agreement no. 813343). Access to WWTP and sample logistics was possible through the Coron-A Project (funded by the Austrian Federal Ministry of Education, Science and Research, the Austrian Federal Ministry of Agriculture, Regions and Tourism, the Provincial Governments of Lower Austria, Upper Austria, Salzburg, Carinthia, Tyrol, Styria, Burgenland, Vorarlberg and the Austrian Association of Cities and Towns) and the national WW surveillance programmes conducted by the Austrian Federal Ministry of Education, Science and Research and by the Austrian Federal Ministry of Social Affairs, Health, Care and Consumer Protection. Additional samples and background information were provided by the COVID-19 WW surveillance programmes of the federal states of Carinthia, Vorarlberg and Vienna. Sequencing was performed by the Biomedical Sequencing Facility (https://biomedical-sequencing.org/) at CeMM. We thank M. Wade for critically proofreading the manuscript. A waiver of ethical approval was received from the Ethics commission of the City of Vienna.
Author information
Authors and Affiliations
Contributions
F.Am., R.M., L.E., S.H., L.R., M.Z., P.T., N.P., D.S., C.B., P.K. provided and/or analysed data. R.M., B.A., A.S., M.Z., M.B., G.H., P.T., M.T., T.P., M.Se., J.L., Z.K., B.D., M.St., H.N., C.S., G.V., G.W., A.O.W., K.S., A.M., E.R., F.Al., H.O., N.K., H.I., F.N., P.H., W.R., M.M. provided processed WW samples, generated SARS-CoV-2 genome sequences and/or provided data. F.Am., R.M. and A.B. wrote the manuscript. F.Am. and A.B. conceived the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks Ana Maria de Roda Husman and David Larsen for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Data collection and WWTP selection.
a, Cumulative population curve for all Austrian WWTP and their served people. The positioning of the six showcase WWTP is highlighted. b, Proportion of population for each Austrian federal state being connected to a WWTP included in the surveillance programme. c, Graphic representation of all sequenced samples from WWTP with more than three timepoints. Sample date and location and sequencing success are indicated. Samples are classified into passed and detected if 40% and 5% of the genome, respectively, is covered with more than 10 reads. A coverage of more than 5% of the genome was considered as robust detection of the virus, since at least three independent amplicons responded, but too little information is provided to reliably characterise viral variants. Therefore, detected samples were not used further. If less than 5% of the genome was covered, the sequencing of the respective samples was categorised as failed. 80% of all samples were classified as passed. Another 10.2% and 9.8% were classified as detected and failed, respectively.
Extended Data Fig. 2 Sequencing reproducibility analysis.
Analysis of triplicate sample sets collected on the same day at two different WWTP and processed independently. a, Distribution of parameter characterising the WW of Austrian WWTP. The values of the two WWTP selected for reproducibility analysis are marked with solid (Hofsteig) and dashed (Kuchl) lines. Boxplots indicate 25%, 50% and 75% percentile (boxes) and up to the 1.5 times interquartile range contiguous from there (whiskers). b, Reproducibility analysis for detection of mutations. Venn diagram over the sets of mutations observed with a frequency greater zero in an individual sample for all mutations called in any of the triplicates with an allele frequency greater 2%. c, Analysis of triplicate samples with respect to the qualitative reproducibility of observed allele frequencies. Correlation between allele frequencies is modest with correlation factors between 0.69 and 0.87. Generally, mutation dropout in one sample contributes heavily to a reduced correlation factor. d, Reproducibility of normalised read coverage per amplicon between the triplicates. e, Deduced allele frequencies of marker mutations of the Alpha (B.1.1.7) variant (dominating variant according to WW and to epidemiological case surveillance data at the corresponding time) in the triplicates. Point colour indicates read depth at the respective loci. f, Scatter plot depicting the median mutation frequency and the individual mutation frequencies for each detected mutation (with median frequency greater than 2%) in the two replication sets. Samples at the baseline (y = 0) represent complete dropouts in one replicate. Point colour indicates read depth at the respective loci. As in panel d, some mutations with expected presence according to the replicates are absent even though the position is covered by a substantial number of reads.
Extended Data Fig. 3 Analysis workflow.
Illustration of the main analysis workflow steps from the WW sampling point, the wet lab procedures (orange panels), via the bioinformatics analysis steps (blue panels) to the analysis results (green panels). Major readouts of the analysis are nucleotide diversity π, cluster of correlated mutation constellations, variant quantification and determination of reproduction number in the catchment area. Analysis steps implemented in the software tool VaQuERo are highlighted by red shading.
Extended Data Fig. 4 Dimensionality reduction of Austrian geolocations.
Given the mountainous topography and the elongated shape of Austria two pronounced axes of transportation can be defined. a, Movements between municipals based on mobile communications, network records and the two main axes of movement following arterial roads defined thereof. b, Localisation of monitored WWTP and their association to the closest point on the main transportation axis. c, One-dimension representation of the WWTP along the two main axes of transportation and the distance to the defined axis.
Extended Data Fig. 5 Validation of WW-based variant quantification.
a, Compendium of the development of the case-based variant surveillance programme conducted by the Austrian health authorities, Austria hospital associations and registered by the Austrian Agency for Health and Food Safety. Based on guidelines issued by the authorities, the fraction of positive cases within the examined WWTP catchments which were subjected to variant screening changed over the course of the pandemic. Between January 2021 and January 2022, on average 50.6% of the positive cases residing within a monitored catchment were conveyed to variant screening. b, Visualisation of the bimodal distribution of relative variant frequency with an accumulation close to zero and close to one and the two-sided Kendall rank correlation analysis thereof. c, Two-sided Kendall rank correlation analysis between relative variant frequencies deduced from clinical surveillance and from WBE, omitting extreme relative variant frequencies close to zero and one. d, Linear regression between magnitude of variant signal observed in case-based variant surveillance and missed in WW-based variant surveillance, and the population size of the respective catchment. For each WWTP the minimal, median and maximal absolute and relative size of the signal is plotted against the number of people served by the WWTP. Linear regression lines are calculated and drawn including the 95% confidence interval for the predicted linear model.
Extended Data Fig. 6 Comparison between clinical and WBE variant surveillance.
The relative variant frequency of different variants for each WWTP catchment deduced with either of the two surveillance programmes is arranged along the two main transportation axes in Austria (x-axis) and the observation period (y-axis) and interpolated individually using B-splines. For each data point in this artificial plane, the two independently deduced frequency values are visualised by an additive colour encoding, as depicted in the right upper corner. Generally, the colour encode agreement on a low level (black, agreement on a higher level (green) is only observed in the WW programme (red) and only observed in the case-based programme (yellow).
Extended Data Fig. 7 Mutation constellation deconvolution.
Mutations which are grouped into related mutation constellation based on their mutation frequencies, as depicted in Fig. 3a, are used as input for VaQuERo, instead of the normal GISAID trained variant defining mutations. The graphic illustrates the geographic distribution and the dynamics of the de novo inferred mutation constellation enriched in Alpha (B.1.1.7) associated mutations in all monitored WWTP from the examined province Carinthia.
Extended Data Fig. 8 Virus population diversity and mobility.
Two-sided Kendall rank correlation analysis between nucleotide diversity measure π of all samples with dominated by one variant with a frequency above 95% and external data, describing (from left to right) the number of people in the catchment area a, people connected to the WWTP in the catchment area. b, the 14-day incidence rate per 100,000 people c, the proportion of in and out commuters (as officially registered) to the total number of people in the catchment area d, the number of registered over-night days in the hospitality industry in the catchment area e, the proportion of population leaving the commune f, or the federal state, as observed by mobile communications network records.
Extended Data Fig. 9 Reproduction number Rww deduced from WW.
Selected WWTP with the variant-specific reproduction number Rww deduced from changes in virus load observed in the wastewater and relative variant frequency observed in sequencing data thereof. The grey area indicates the estimated Reff per respective federal state as deduced from epidemiological surveillance according to official records (https://wissenaktuell.ages.at/fileadmin/AGES2015/Wissen-Aktuell/COVID19/R_eff_bundesland.csv).
Extended Data Fig. 10 Reproducibility of RNA extraction method between collaborating laboratories.
To validate comparability of RNA extracts produced by different participating laboratories, namely, Medical University Innsbruck (GMI), Technical University Vienna (TU-Wien) and University Innsbruck (UIBK), a ring trial was performed. Samples at three locations selected based on a high, medium and low COVID-19 incidence rate as observed in epidemiological case surveillance were collected with n=1 in each group and shipped to the participating laboratories and independently processed with the respective routine protocol. Produced RNA extracts were transported to a centralised laboratory and quantified with a digital PCR (dPCR) system. Observed virus concentrations (gene copies per microliter) are depicted including the 95% confidence interval as deduced from the absolute count of responding single dPCR partitions assuming a Poisson distribution.
Supplementary information
Rights and permissions
About this article

Cite this article
Amman, F., Markt, R., Endler, L. et al. Viral variant-resolved wastewater surveillance of SARS-CoV-2 at national scale. Nat Biotechnol 40, 1814–1822 (2022). https://doi.org/10.1038/s41587-022-01387-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-022-01387-y
This article is cited by
-
Integrating wastewater and randomised prevalence survey data for national COVID surveillance
Scientific Reports (2024)
-
Estimating actual SARS-CoV-2 infections from secondary data
Scientific Reports (2024)
-
Dynamic SARS-CoV-2 surveillance model combining seroprevalence and wastewater concentrations for post-vaccine disease burden estimates
Communications Medicine (2024)
-
Spatiotemporal genome diversity of SARS-CoV-2 in wastewater: a two-year global epidemiological study
Environmental Monitoring and Assessment (2024)
-
Detection of SARS-COV-2 variants and their proportions in wastewater samples using next-generation sequencing in Finland
Scientific Reports (2024)
