
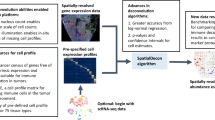
Abstract
Many spatially resolved transcriptomic technologies do not have single-cell resolution but measure the average gene expression for each spot from a mixture of cells of potentially heterogeneous cell types. Here, we introduce a deconvolution method, conditional autoregressive-based deconvolution (CARD), that combines cell-type-specific expression information from single-cell RNA sequencing (scRNA-seq) with correlation in cell-type composition across tissue locations. Modeling spatial correlation allows us to borrow the cell-type composition information across locations, improving accuracy of deconvolution even with a mismatched scRNA-seq reference. CARD can also impute cell-type compositions and gene expression levels at unmeasured tissue locations to enable the construction of a refined spatial tissue map with a resolution arbitrarily higher than that measured in the original study and can perform deconvolution without an scRNA-seq reference. Applications to four datasets, including a pancreatic cancer dataset, identified multiple cell types and molecular markers with distinct spatial localization that define the progression, heterogeneity and compartmentalization of pancreatic cancer.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

Data availability
This study made use of publicly available datasets. These include the MOB dataset (http://www.spatialtranscriptomicsresearch.org), human PDAC dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE111672), mouse hippocampus Slide-seqV2 dataset (https://singlecell.broadinstitute.org/single_cell/study/SCP948/robust-decomposition-of-cell-type-mixtures-in-spatial-transcriptomics) and mouse brain (coronal section) 10x Visium dataset (https://www.10xgenomics.com/resources/datasets/). For the scRNA-seq references used in this study, all are publicly available, with details provided in Supplementary Tables 2 and 3.
Code availability
The CARD software package and source code have been deposited at www.xzlab.org/software.html. All scripts used to reproduce all the analyses are also available at the same website.
References
Burgess, D. J. Spatial transcriptomics coming of age. Nat. Rev. Genet. 20, 317 (2019).
Soldatov, R. et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 364, eaas9536 (2019).
Prinz, M., Priller, J., Sisodia, S. S. & Ransohoff, R. M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 14, 1227–1235 (2011).
Svensson, V., Teichmann, S. A. & Stegle, O. SpatialDE: Identification of spatially variable genes. Nat. Methods 15, 343–346 (2018).
Dries, R. et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 78 (2021).
Pham, D. et al. stLearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell–cell interactions and spatial trajectories within undissociated tissues. Preprint at bioRxiv https://doi.org/10.1101/2020.05.31.125658 (2020).
Biancalani, T. et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 18, 1352–1362 (2021).
Fu, H. et al. Unsupervised spatially embedded deep representation of spatial transcriptomics. Preprint at bioRxiv https://doi.org/10.1101/2021.06.15.448542 (2021).
Fischl, A. M., Heron, P. M., Stromberg, A. J. & McClintock, T. S. Activity-dependent genes in mouse olfactory sensory neurons. Chem. Senses 39, 439–449 (2014).
Moses, L. & Pachter, L. Museum of spatial transcriptomics. Nat. Methods https://doi.org/10.1038/s41592-022-01409-2 (2022).
Asp, M., Bergenstråhle, J. & Lundeberg, J. Spatially resolved transcriptomes—next generation tools for tissue exploration. Bioessays 42, e1900221 (2020).
Genomics, 10x. 10x Genomics Visium. https://www.10xgenomics.com/spatial-transcriptomics/
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Liao, J., Lu, X., Shao, X., Zhu, L. & Fan, X. Uncovering an organ’s molecular architecture at single-cell resolution by spatially resolved transcriptomics. Trends Biotechnol. 39, 43–58 (2020).
Rao, A., Barkley, D., França, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220 (2021).
Hwang, B., Lee, J. H. & Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 50, 1–14 (2018).
Cobos, F. A., Alquicira-Hernandez, J., Powell, J. E., Mestdagh, P. & De Preter, K. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat. Commun. 11, 5650 (2020).
Wang, X., Park, J., Susztak, K., Zhang, N. R. & Li, M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 10, 380 (2019).
Dong, M. et al. SCDC: bulk gene expression deconvolution by multiple single-cell RNA sequencing references. Brief. Bioinform. 22, 416–427 (2020).
Jew, B. et al. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat. Commun. 11, 1971 (2020).
Elosua-Bayes, M., Nieto, P., Mereu, E., Gut, I. & Heyn, H. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 49, e50 (2021).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-00830-w (2021).
Song, Q. & Su, J. DSTG: deconvoluting spatial transcriptomics data through graph-based artificial intelligence. Brief. Bioinform. 22, bbaa414 (2021).
Lopez, R. et al. Multi-resolution deconvolution of spatial transcriptomics data reveals continuous patterns of inflammation. Preprint at bioRxiv https://doi.org/10.1101/2021.05.10.443517 (2021).
Danaher, P. et al. Advances in mixed cell deconvolution enable quantification of cell types in spatial transcriptomic data. Nat. Commun. 13, 385 (2022).
Gayoso, A. et al. scvi-tools: a library for deep probabilistic analysis of single-cell omics data. Preprint at bioRxiv https://doi.org/10.1101/2021.04.28.441833 (2021).
Andersson, A. et al. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun. Biol. 3, 565 (2020).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-01139-4 (2022).
Dong, R. & Yuan, G.-C. SpatialDWLS: accurate deconvolution of spatial transcriptomic data. Genome Biol. 22, 145 (2021).
Stoltzfus, C. R. et al. CytoMAP: a spatial analysis toolbox reveals features of myeloid cell organization in lymphoid tissues. Cell Rep. 31, 107523 (2020).
Dudas, M., Wysocki, A., Gelpi, B. & Tuan, T.-L. Memory encoded throughout our bodies: molecular and cellular basis of tissue regeneration. Pediatr. Res. 63, 502–512 (2008).
Bove, A. et al. Local cellular neighborhood controls proliferation in cell competition. Mol. Biol. Cell 28, 3215–3228 (2017).
Van Vliet, S. et al. Spatially correlated gene expression in bacterial groups: the role of lineage history, spatial gradients, and cell–cell interactions. Cell Syst. 6, 496–507 (2018).
Phillips, D. et al. Immune cell topography predicts response to PD-1 blockade in cutaneous T cell lymphoma. Nat. Commun. 12, 6726 (2021).
Schürch, C. M. et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182, 1341–1359 (2020).
Xia, C., Fan, J., Emanuel, G., Hao, J. & Zhuang, X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl Acad. Sci. USA 116, 19490–19499 (2019).
Eng, C. H. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568, 235–239 (2019).
Banerjee, S., Carlin, B. P. & Gelfand, A. E. Hierarchical Modeling and Analysis for Spatial Data 2nd edn (Chapman and Hall/CRC, 2014).
Lee, D. A comparison of conditional autoregressive models used in Bayesian disease mapping. Spat. Spatiotemporal Epidemiol. 2, 79–89 (2011).
Zhao, E. et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 39, 1375–1384 (2021).
Zeisel, A. et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014 (2018).
Yang, S. & Zhou, X. Accurate and scalable construction of polygenic scores in large biobank data sets. Am. J. Hum. Genet. 106, 679–693 (2020).
Zhou, X., Carbonetto, P. & Stephens, M. Polygenic modeling with Bayesian sparse linear mixed models. PLoS Genet. 9, e1003264 (2013).
Tepe, B. et al. Single-cell RNA-seq of mouse olfactory bulb reveals cellular heterogeneity and activity-dependent molecular census of adult-born neurons. Cell Rep. 25, 2689–2703 (2018).
Nagayama, S., Homma, R. & Imamura, F. Neuronal organization of olfactory bulb circuits. Front. Neural Circuits 8, 98 (2014).
Moncada, R. et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 38, 333–342 (2020).
Zheng, B. et al. TM4SF1 as a prognostic marker of pancreatic ductal adenocarcinoma is involved in migration and invasion of cancer cells. Int. J. Oncol. 47, 490–498 (2015).
Fu, F. et al. Role of transmembrane 4 L six family 1 in the development and progression of cancer. Front. Mol. Biosci. 7, 202 (2020).
Xu, D. et al. Lost miR-141 and upregulated TM4SF1 expressions associate with poor prognosis of pancreatic cancer: regulation of EMT and angiogenesis by miR-141 and TM4SF1 via AKT. Cancer Biol. Ther. 21, 354–363 (2020).
Zhang, X. et al. Expression pattern of cancer-associated fibroblast and its clinical relevance in intrahepatic cholangiocarcinoma. Hum. Pathol. 65, 92–100 (2017).
Morvaridi, S., Dhall, D., Greene, M. I., Pandol, S. J. & Wang, Q. Role of YAP and TAZ in pancreatic ductal adenocarcinoma and in stellate cells associated with cancer and chronic pancreatitis. Sci. Rep. 5, 16759 (2015).
Nielsen, M. F. B., Mortensen, M. B. & Detlefsen, S. Key players in pancreatic cancer-stroma interaction: cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 22, 2678–2700 (2016).
Zheng, C. et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356 (2017).
Comito, G., Ippolito, L., Chiarugi, P. & Cirri, P. Nutritional exchanges within tumor microenvironment: impact for cancer aggressiveness. Front. Oncol. 10, 396 (2020).
Lambrechts, D. et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24, 1277–1289 (2018).
Peng, J. et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 29, 725–738 (2019).
Stickels, R. R. et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 39, 313–319 (2021).
Saunders, A. et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030 (2018).
Del Bigio, M. R. Ependymal cells: biology and pathology. Acta Neuropathol. 119, 55–73 (2010).
Ramachandran, V. S. (ed.) Encyclopedia of the Human Brain, 1st edn (Elsevier, 2003).
Meyer, G. Building a human cortex: the evolutionary differentiation of Cajal–Retzius cells and the cortical hem. J. Anat. 217, 334–343 (2010).
Hawrylycz, M. et al. in Springer Handbook of Bio-/Neuroinformatics (ed. Kasabov N.) 1111–1126 (Springer, 2014).
Wozny, C. et al. VGLUT2 functions as a differential marker for hippocampal output neurons. Front. Cell. Neurosci. 12, 337 (2018).
Wälchli, T. et al. Quantitative assessment of angiogenesis, perfused blood vessels and endothelial tip cells in the postnatal mouse brain. Nat. Protoc. 10, 53–74 (2015).
Lubeck, E., Coskun, A. F., Zhiyentayev, T., Ahmad, M. & Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
Moffitt, J. R. et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018).
He, Y. et al. ClusterMap for multi-scale clustering analysis of spatial gene expression. Nat. Commun. 12, 5909 (2021).
Moen, E. et al. Deep learning for cellular image analysis. Nat. Methods 16, 1233–1246 (2019).
Bergenstråhle, J., Larsson, L. & Lundeberg, J. Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genomics 21, 482 (2020).
Sun, S. et al. Differential expression analysis for RNAseq using Poisson mixed models. Nucleic Acids Res. 45, e106 (2017).
Sun, S. et al. Heritability estimation and differential analysis of count data with generalized linear mixed models in genomic sequencing studies. Bioinformatics 35, 487–496 (2019).
Sun, S., Zhu, J. & Zhou, X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat. Methods 17, 193–200 (2020).
Hu, J. et al. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods. 18, 1342–1351 (2021).
Soneson, C. & Robinson, M. D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 15, 255–261 (2018).
Duò, A., Robinson, M. D. & Soneson, C. A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000Res. 7, 1141 (2018).
De Oliveira, V. Bayesian analysis of conditional autoregressive models. Ann. Inst. Stat. Math. 64, 107–133 (2012).
Besag, J. Spatial interaction and the statistical analysis of lattice systems. J. R. Stat. Soc. Ser. B 36, 192–225 (1974).
Vanhatalo, J., Pietiläinen, V. & Vehtari, A. Approximate inference for disease mapping with sparse Gaussian processes. Stat. Med. 29, 1580–1607 (2010).
Rousset, F. & Ferdy, J. B. Testing environmental and genetic effects in the presence of spatial autocorrelation. Ecography 37, 781–790 (2014).
Cressie, N. Statistics for spatial data. Terra Nova 4, 613–617 (1992).
Rue, H. & Held, L. Gaussian Markov Random Fields: Theory and Applications 1st edn (Chapman and Hall/CRC, 2005).
Brook, D. On the distinction between the conditional probability and the joint probability approaches in the specification of nearest-neighbour systems. Biometrika 51, 481–483 (1964).
Lee, D. D. & Seung, H. S. in Advances in Neural Information Processing Systems (eds Leen T., Dietterich T. & Tresp V.) 556–562 (MIT Press, 2001).
Janecek, A. & Tan, Y. Iterative improvement of the multiplicative update nmf algorithm using nature-inspired optimization. In 2011 Seventh International Conference on Natural Computation Vol. 3 1668–1672 (IEEE, 2011).
Park, J.-S. & Oh, S.-J. A new concave hull algorithm and concaveness measure for n-dimensional datasets. J. Inf. Sci. Eng. 28, 587–600 (2012).
Ralston, A. & Shaw, K. Gene expression regulates cell differentiation. Nat. Educ. 1, 127–131 (2008).
Li, H., Calder, C. A. & Cressie, N. Beyond Moran’s I: testing for spatial dependence based on the spatial autoregressive model. Geogr. Anal. 39, 357–375 (2007).
Radeloff, V. C., Miller, T. F., He, H. S. & Mladenoff, D. J. Periodicity in spatial data and geostatistical models: autocorrelation between patches. Ecography 23, 81–91 (2000).
Bivand, R. et al. spdep: Spatial Dependence: Weighting Schemes, Statistics and Models. R package version 0.5-37 (2011).
Teschendorff, A. E., Zhu, T., Breeze, C. E. & Beck, S. EPISCORE: cell type deconvolution of bulk tissue DNA methylomes from single-cell RNA-seq data. Genome Biol. 21, 221 (2020).
Acknowledgements
This study was supported by the National Institutes of Health (NIH) grants R01GM126553, R01HG011883 and R01GM144960 (all to X.Z.).
Author information
Authors and Affiliations
Contributions
X.Z. conceived the idea and provided funding support. Y.M. and X.Z. designed the experiments. Y.M. developed the method, implemented the software, performed simulations and analyzed real data. Y.M. and X.Z. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–94, Tables 1–6 and Notes 1–9.
Rights and permissions
About this article

Cite this article
Ma, Y., Zhou, X. Spatially informed cell-type deconvolution for spatial transcriptomics. Nat Biotechnol 40, 1349–1359 (2022). https://doi.org/10.1038/s41587-022-01273-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-022-01273-7
This article is cited by
-
SRT-Server: powering the analysis of spatial transcriptomic data
Genome Medicine (2024)
-
Mapping cancer biology in space: applications and perspectives on spatial omics for oncology
Molecular Cancer (2024)
-
DANCE: a deep learning library and benchmark platform for single-cell analysis
Genome Biology (2024)
-
SPADE: spatial deconvolution for domain specific cell-type estimation
Communications Biology (2024)
-
Unsupervised and supervised discovery of tissue cellular neighborhoods from cell phenotypes
Nature Methods (2024)
