
Abstract
Appropriate use and interpretation of serological tests for assessments of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) exposure, infection and potential immunity require accurate data on assay performance. We conducted a head-to-head evaluation of ten point-of-care-style lateral flow assays (LFAs) and two laboratory-based enzyme-linked immunosorbent assays to detect anti-SARS-CoV-2 IgM and IgG antibodies in 5-d time intervals from symptom onset and studied the specificity of each assay in pre-coronavirus disease 2019 specimens. The percent of seropositive individuals increased with time, peaking in the latest time interval tested (>20 d after symptom onset). Test specificity ranged from 84.3% to 100.0% and was predominantly affected by variability in IgM results. LFA specificity could be increased by considering weak bands as negative, but this decreased detection of antibodies (sensitivity) in a subset of SARS-CoV-2 real-time PCR-positive cases. Our results underline the importance of seropositivity threshold determination and reader training for reliable LFA deployment. Although there was no standout serological assay, four tests achieved more than 80% positivity at later time points tested and more than 95% specificity.
Similar content being viewed by others


Main
To date, hundreds of thousands of deaths have been attributed to coronavirus disease 2019 (COVID-19)1. Millions of infections by SARS-CoV-2, the virus responsible for COVID-19, have been reported, although its full extent has yet to be determined owing to limited testing2. Government interventions to slow viral spread have disrupted daily life and economic activity for billions of people. Strategies to ease restraints on human mobility and interaction without provoking a major resurgence of transmission and mortality will depend on accurate estimates of population levels of infection and immunity3. Current testing for the virus largely depends on labor-intensive molecular techniques4. Individuals with positive molecular tests represent only a small fraction of all infections, given limited deployment and the brief time window when real-time (RT)–PCR testing has the highest sensitivity5,6,7. The proportion of undocumented cases in the original epidemic focus was estimated to be as high as 86%8, and asymptomatic infections are suspected to play a substantial role in transmission9,10,11,12,13,14.
Widely available, reliable antibody detection assays would enable more accurate estimates of SARS-CoV-2 prevalence and incidence. On February 4, 2020, the Secretary of the US Department of Health and Human Services issued an emergency use authorization (EUA) for the diagnosis of SARS-CoV-215, allowing nucleic acid detection and immunoassay tests to be offered based on manufacturer-reported data without formal US Food and Drug Administration (FDA) clearance16. In response, dozens of companies began to market laboratory-based immunoassays and point-of-care (POC) tests. Rigorous, comparative performance data are crucial to inform clinical care and public health responses.
We conducted a head-to-head comparison of serology tests available to our group in early April, comprising ten immunochromatographic LFAs and two enzyme-linked immunosorbent assays (ELISAs) (for details, see Supplementary Table 1). Specimens were obtained from patients with SARS-CoV-2 that was confirmed by RT–PCR, contemporaneous patients with other respiratory pathogen testing and/or without SARS-CoV-2 by RT–PCR and blood donor specimens collected before 2019. We included analyses of performance by time from symptom onset and disease severity. Our goal was to provide well-controlled performance data to help guide the use of serology in the response to COVID-19.
Results
Study population
This study included 128 plasma or serum specimens from 79 individuals who tested positive for SARS-CoV-2 and who were diagnosed in the University of California, San Francisco (UCSF) hospital system and Zuckerberg San Francisco General (ZSFG) Hospital. Patients ranged from 22 to over 90 years of age (Table 1). Most patients were Hispanic/Latinx (68%), reflecting the ZSFG patient population and demographics of the epidemic in San Francisco17,18. Most presented with cough (91%) and fever (86%). Chronic medical conditions, such as hypertension, type 2 diabetes mellitus, obesity and chronic kidney disease, were frequent. Of the 79 individuals, 18% were not admitted, 46% were inpatients without intensive care unit (ICU) care and 37% required ICU care. There were no reported deaths at the time of chart review.
Test performance
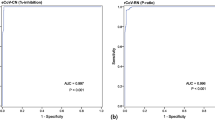
Because we lacked a gold standard against which to benchmark the 12 tests in our study, we assessed the positive percent agreement (positivity) compared with the RT–PCR assay. The percentage of specimens testing positive rose with increasing time from symptom onset (Table 2 and Fig. 1a), reaching the highest levels in the 16–20-d and >20-d time intervals. The highest detection rate was achieved by combining IgM and IgG results (Fig. 1b). However, 95% confidence intervals (CIs) for later time intervals showed substantial overlap with those for earlier intervals (Fig. 1b). Four assays (Bioperfectus, Premier, Wondfo and in-house ELISA) achieved more than 80% positivity in the later two time intervals (16–20 d and >20 d) while maintaining more than 95% specificity. Some tests were not performed on a subset of specimens owing to exhausted sample material, which might have affected reported percent positivity; the sample size tested is reflected in 95% CIs. IgM detection was less consistent than IgG for nearly all assays. Kappa agreement statistic ranged from 0.95 to 0.99 for IgG and from 0.81 to 1.00 for IgM for standardized intensity score (Supplementary Table 2 and Supplementary Fig. 2). Details on establishing intensity score values and reader training are available in the ‘Immunochromatographic LFAs’ section within Methods. Although mean band intensities varied among different assays, the approximate rate of sample positivity was generally consistent (Fig. 2). For ELISA tests, a normalized value of sample optical density at 450 nm (OD450) divided by calculated cutoff (signal-to-cutoff (S/CO)) was used to capture quantitative data about antibody levels for each specimen. S/CO values provide a quantitative value comparable between plates. Our ability to perform end-point dilutions was limited by specimen and assay availability.

a, The second reader’s score (0–6 based on band intensity) is reported for each assay, binned by time after patient-reported symptom onset. Biologically independent samples for each test are as follows: n = 126, Biomedomics; n = 126, Bioperfectus; n = 124, DecomBio; n = 128, DeepBlue; n = 114, Innovita; n = 127, Premier; n = 127, Sure; n = 128, UCP; n = 119, VivaChek; n = 124, Wondfo. The second reader’s score for pre-COVID-19 samples is also displayed (n = 107, Biomedomics; n = 104, Bioperfectus; n = 107, DecomBio; n = 108, DeepBlue; n = 108, Innovita; n = 108, Premier; n = 108, Sure; n = 107, UCP; n = 99, VivaChek; n = 106, Wondfo). For tests with separate IgG and IgM bands, the higher score is reported. Joint IgM/IgG signal is represented by a single band in Wondfo. The lower, dark gray line refers to the positivity threshold (score greater than or equal to 1) used in this study. The upper, light gray line refers to an alternative positivity threshold (score greater than or equal to 2) discussed in the text and Fig. 3. Box spans 25th to 75th percentiles with median indicated by the black bar; whiskers show maximum and minimum value within 1.5× the interquartile range from the box. b, Percent of SARS-CoV-2 RT–PCR-positive samples testing positive by each LFA and ELISA are plotted relative to time after patient-reported symptom onset (n = 126, Biomedomics; n = 126, Bioperfectus; n = 124, DecomBio; n = 128, DeepBlue; n = 114, Innovita; n = 127, Premier; n = 127, Sure; n = 128, UCP; n = 119, VivaChek; n = 124, Wondfo; n = 128, Epitope; n = 128, in-house). The ‘IgM or IgG’ category refers to positivity of either isotype. c, Specificity is plotted for each test using pre-COVID-19 negative control samples (n = 107, Biomedomics; n = 104, Bioperfectus; n = 107, DecomBio; n = 108, DeepBlue; n = 108, Innovita; n = 108, Premier; n = 108, Sure; n = 107, UCP; n = 99, VivaChek; n = 106, Wondfo; n = 108, Epitope; n = 108, in-house). For b and c, all nodes refer to the calculated percent positivity or specificity, respectively. Error bars signify 95% CIs.

a, LFA scores for each of two readers (blue) and mean ELISA S/CO (purple) for each specimen are grouped by binned time after patient-reported symptom onset and plotted by assay. White cells indicate samples not run with the corresponding assay. For ELISAs, gray indicates S/CO less than or equal to 1. The same legend applies to b and c. The F(ab′)2 specific secondary antibody used in our in-house ELISA preferentially binds the IgG light chain but per the manufacturer has some reactivity for other isotypes (IgM and IgA). b, LFA score and ELISA S/CO values are plotted for pre-COVID-19 historical control serum samples to determine assay specificity. c, LFA score and ELISA S/CO values are plotted for serum samples obtained from 51 individuals after the emergence of COVID-19 (post-COVID-19), some of which received BioFire FilmArray (BioFire Diagnostics) and/or SARS-CoV-2 RT–PCR testing (all negative) as indicated (black cells) in the appropriate columns. Arrows highlight specimens from five individuals with moderate to strong band intensity further discussed in the text. Specimens are grouped by positive testing for coronavirus HKU1 (CoV HKU1), coronavirus OC43 (CoV OC43), influenza A virus A/H3 (FluA H3), influenza A virus A/H1 2009 (FluA H1), parainfluenza type 1 virus (PIV-1), parainfluenza type 4 virus (PIV-4), human metapneumovirus (HMP), adenovirus (ADNV), respiratory syncytial virus (RSV), human rhinovirus/enterovirus (HRE) or negative testing for SARS-CoV-2 and other viruses (nco-).
We observed a trend toward higher percent positivity by LFA for patients admitted to the ICU compared to those having milder disease, but the specimen numbers per time interval were low, limiting statistical power (Supplementary Fig. 3).
Test specificity in 108 pre-COVID-19 blood donor plasma samples ranged from 84.3% to 100.0%, with 39 samples demonstrating false-positive results by at least one LFA (Table 2 and Fig. 2b). Of the false-positive results, 61.5% (24/39) had a weak intensity score of 1. Intensity scores of 2–3 were seen in 30.8% (12/39), and scores of 4–6 were seen in 7.7% (3/39).
We evaluated the tradeoff between percent positivity in samples from RT–PCR-positive individuals and specificity as a function of LFA reader score. RT–PCR measures the presence of viral nucleotides. Individuals with RT–PCR-proven SARS-CoV-2 infection are expected to seroconvert and develop anti-SARS-CoV-2 antibodies, although frequency and kinetics of seroconversion can vary5,6,19,20,21,22. We, therefore, assessed percent positivity at various time intervals after onset of symptoms. Changing the positive LFA threshold from 1 to 2 decreased the mean overall percent positivity across tests from 67.2% (range, 57.9–75.4%) to 57.8% (range, 44.7–65.6%) and increased the average specificity from 94.2% (range, 84.3–100.0%) to 98.1% (range, 94.4–100.0%) (Fig. 3).

a, The percent positivity of each assay tested on serum from patients who tested positive for SARS-CoV-2 by RT–PCR is plotted by time after patient-reported symptom onset. Biologically independent samples for each test are as follows: n = 126, Biomedomics; n = 126, Bioperfectus; n = 124, DecomBio; n = 128, DeepBlue; n = 114, Innovita; n = 127, Premier; n = 127, Sure; n = 128, UCP; n = 119, VivaChek; n = 124, Wondfo. Squares indicate percent positivity using reader score >0 (‘Weak bands positive’) as the positivity threshold. Triangles indicate percent positivity using reader score >1 (‘Weak bands negative’) as the positivity threshold. ‘IgM or IgG’ signifies detection of either isotype. Wondfo reports a single band for IgM and IgG together, and the results are plotted here as both ‘IgM’ and ‘IgG’ for horizontal comparison across assays. b, Comparison of percent positivity at each timepoint for BioMedomics assay at either the MGH (left, n = 48) or UCSF (right, n = 126) study site using low (square) or high (triangle) positivity thresholds. Note that a weak score at MGH is not directly equivalent to a 1 at UCSF owing to differences in reader training. c, The specificity of all assays on historical pre-COVID-19 serum using low (square) or high (triangle) positivity thresholds. UCSF BioMedomics data are plotted again in the right subpanel column for direct comparison to MGH BioMedomics data. All nodes refer to the calculated percent positivity or specificity (as designated), and all error bars indicate 95% CIs.
An independent study at Massachusetts General Hospital (MGH) compared three LFAs, of which BioMedomics was also assessed in the current study (Supplementary Table 3). Although study design and methods differed between sites, precluding direct comparison of results (see ‘Study design’ in Methods), test validation efforts at another site provided additional useful data. Overall, both studies showed a trend for increased detection of SARS-CoV-2-specific antibodies with increased time from symptom onset. However, the MGH study displayed increased specificity with lower percent positivity at early time points after symptom onset. MGH positivity thresholds were set higher to prioritize test specificity (Fig. 3b,c).
A set of specimens collected during the COVID-19 outbreak that had negative SARS-CoV-2 RT–PCR testing and/or alternative respiratory pathogen testing demonstrated higher numbers of positive results compared to the pre-COVID-19 sample set (Fig. 2c). Five specimens had positive results by more than three tests, all with respiratory symptoms and concurrent negative or un-performed SARS-CoV-2 RT–PCR testing (Fig. 2c, arrows). One patient was positive on eight different tests, including the in-house ELISA. In this limited panel, no consistent pattern of cross-reactivity was identified with non-SARS-CoV-2 respiratory viruses, including two strains of seasonal coronavirus (one coronavirus OC43 and three coronavirus HKU1).
Agreement among results of LFAs with those of IgG and IgM Epitope ELISAs ranged from 75.7% to 85.6%, whereas agreement with the in-house ELISA ranged from 83.5% to 94.8% (Fig. 4a). LFA band intensity scores showed a direct correlation with ELISA S/CO values (Fig. 4b).

a, Percent agreement is plotted across all assay combinations, and values signify the binomial regression of the two assays across all tests. Samples were labeled ‘positive’ if any antibody isotype was detected for each assay. b, IgM or IgG LFA scores for each assay are compared to S/CO from three different ELISAs for all SARS-CoV-2 RT–PCR-positive samples. Biologically independent samples for each test are as follows: n = 126, Biomedomics; n = 126, Bioperfectus; n = 124, DecomBio; n = 128, DeepBlue; n = 114, Innovita; n = 127, Premier; n = 127, Sure; n = 128, UCP; n = 119, VivaChek; n = 124, Wondfo. Joint IgM/IgG signal is represented by a single band in Wondfo, so data were plotted as IgM or IgG depending on ELISA comparison. The F(ab′)2-specific secondary antibody used in our in-house ELISA preferentially binds the IgG light chain but per the manufacturer contains some reactivity for other isotypes (IgM and IgA); it is compared in b to IgG band intensity. For b, the box spans the 25th to 75th percentiles with median indicated by the black bar; whiskers show maximum and minimum value within 1.5× the interquartile range from the box.
Discussion
This study describes test performance for 12 COVID-19 serology assays on a panel of 128 samples from 79 individuals with RT–PCR-confirmed SARS-CoV-2 infection and 108 pre-COVID-19 specimens. In April 2020, when we performed this analysis, there was no assay with sufficient performance data for use as a proven reference standard; only three serological assays had an FDA EUA23; and anti-SARS-CoV-2 IgM and IgG kinetics were poorly understood. We, therefore, chose a specimen set covering the first several weeks after illness onset in patients with SARS-CoV-2 proven by RT–PCR to avoid the potential bias of assuming superiority of one assay over the others. To date, no single assay or combination of assays has been accepted as a gold standard comparator for antibody testing. Additionally, we surveyed 51 specimens from individuals who were tested for other respiratory viral pathogens and/or had negative molecular testing for SARS-CoV-2 to evaluate potential cross-reactivity or infections detected only by serology. Our data are also available on a dedicated website (https://covidtestingproject.org). We hope these data will inform the use of serology by the medical and public health communities and provide feedback to test developers about areas of success and necessary improvement.
We focused on comparisons of percent positivity by time interval, rather than reporting the ‘sensitivity’ of each assay, both because of the lack of a gold standard to test against and our expectation that percent positivity would rise with increasing time after symptom onset5,6,19,20,21,22,24,25. Percent positivity above 80% was not reached until at least 2 weeks into clinical illness; diagnosis early in the course of illness remains dependent on viral detection methods. Our data are consistent with growing evidence that IgM and IgG tend to rise around the same time in COVID-195,19. The assays showed a trend to higher positive rates within time intervals for more severe disease, but this finding should be interpreted with caution, owing to the limited data from ambulatory cases. Most samples more than 20 d after symptom onset had detectable anti-SARS-CoV-2 antibodies, suggesting good to excellent sensitivity for all evaluated tests in hospitalized patients three or more weeks into their disease course. Additional studies assessing frozen versus fresh specimens and matrix effects between serum versus plasma will be useful in understanding potential limitations of our current test performance evaluations. Looking forward, well-powered studies testing ambulatory or asymptomatic individuals, including LFA performance with fresh capillary blood, will be essential to guide appropriate use of serology.
Our data demonstrate specificity of more than 95% for most tests evaluated and more than 99% for two LFAs (Wondfo and Sure Biotech) and the in-house ELISA (adapted from Amanat et al., 2020)26. We observed moderate to strong positive bands in several pre-COVID-19 blood donor specimens, some of them positive by multiple assays, suggesting the possibility of non-specific binding of plasma proteins, non-specific antibodies (potentially including auto-antibodies) or cross-reactivity with antibodies against other viruses. Three of the pre-COVID-19 specimens (2.8%) were scored positive by more than three assays. Intriguingly, the fraction of positive tests was higher in a set of recent specimens obtained during the COVID-19 outbreak from individuals undergoing respiratory infection workup, many with negative SARS-CoV-2 RT–PCR. Five of these (9.8%) had positive results by more than three assays, without relation to a specific viral pathogen, suggesting non-specific reactivity and/or missed COVID-19 diagnosis. Recent reports demonstrate that RT–PCR from nasopharyngeal swabs might yield false-negative results in over 20% of cases5,27, and co-infection with other respiratory pathogens might be significantly higher than previously anticipated28. One specimen was positive by 8 of 12 assays, including the in-house ELISA. The patient was over 90 years old and presented with altered mental status, fever and ground glass opacities on chest radiological imaging. SARS-CoV-2 RT–PCR was negative, and ancillary laboratory testing suggested a urinary tract infection. This case could represent COVID-19 not detected by RT–PCR, reinforcing the importance of caution in interpreting negative molecular results as ruling out the infection. Appropriate algorithms for serology testing, including confirmatory or reflexive testing, have yet to be determined. These algorithms will be affected by test performance characteristics and prevalence of disease, as well as pre-test probability of infection.
Importantly, we still do not know the extent to which positive results by serology reflect a protective immune response, nor how long such protection might last29. Neutralization assays measure the ability of blood-derived samples to prevent viral (most commonly pseudovirus) infection of cultured cells in vitro30,31. Although these assays provide information on the functional capabilities of an individual’s antibodies, their correlation with total IgG antibodies to serological test antigens (primarily spike and nucleocapsid proteins) is not well established. Additionally, most antibody neutralization assays are research laboratory based with limited test performance data and inter-lab standardization measures. Antibody neutralization assays should be harmonized across laboratories to establish the extent to which conventional serology assays correlate with neutralization. Further studies are needed to assess the relationships among positive serological testing, in vitro viral neutralization results and clinical protection from future SARS-CoV-2 infection and transmission. Epidemiological data and results from convalescent plasma treatment trials should help guide clinical and public health policies for use of serological testing.
High specificity testing is crucial in low-prevalence settings. One approach to increase specificity would employ confirmatory testing with an independent assay (perhaps recognizing a distinct epitope or antigen). Our comparison of UCSF and MGH data suggests that reclassifying faint bands as ‘negative’ or ‘inconclusive’ can change test performance characteristics by increasing specificity, albeit at the expense of sensitivity. However, the subjectivity of calling faint bands by individual readers might be difficult to standardize without specific control materials, operator training and/or objective methods of analyzing LFAs. In the clinical setting, these parameters and protocols should be independently assessed and validated by clinical laboratories for operation under the Clinical Laboratory Improvement Amendments32.
Objective methods to standardize LFA reading, such as digital image analysis, are potentially attractive. Image analysis tools can be benchtop or mobile (for example, smartphone applications). However, introduction of a separate device for reading LFAs will require specific validation. Variables, including lighting, camera quality, image compression and quantification algorithms, must all be assessed rigorously to ensure accuracy and precision.
A consensus has emerged that serological testing provides an essential tool in the pandemic response, but inadequate data on test performance characteristics in some early surveys and important gaps in immunological knowledge have impeded agreement on appropriate implementation strategies33,34. Our study highlights the need for rigorous assay validation using standardized sample sets with: 1) known positives from individuals with a range of clinical presentations at multiple time points after onset of symptoms; 2) pre-COVID-19 outbreak samples for specificity; and 3) samples from individuals with other viral and inflammatory illnesses as cross-reactivity controls. Coordinated efforts to ensure widespread availability of validated sample sets would facilitate data-driven decisions on the use of serology. The updated guidance released by the FDA in early May 202035 and the initiative recently launched by the FDA and the US National Cancer Institute/National Institutes of Health36 to systematize data generation for EUAs are substantive steps toward this goal and will help build the essential evidence base to guide serological testing during the COVID-19 pandemic.
Methods
Ethical approvals
This study was approved by institutional review boards at the UCSF/ZSFG and MGH.
Study design
The study population included individuals with symptomatic infection and positive SARS-CoV-2 RT–PCR testing of nasopharyngeal or oropharyngeal swabs who had remnant serum and plasma specimens in clinical laboratories serving the UCSF and ZSFG medical center networks. All samples were obtained from venous blood draws, with serum being collected in either uncoated or serum separator tubes and plasma from lithium heparin tubes depending on other ancillary testing orders. All samples were drawn in an outpatient or hospital setting, professionally couriered to the clinical laboratory and acquisitioned for routine testing within the clinical laboratory within the same day. Samples were stored at 4˚C and aliquoted for freezing at −20 °C within 1 week of the initial blood draw. Serum and plasma were used interchangeably. All but one assay (Epitope ELISA) noted that either specimen type could be used. We included multiple specimens per individual but no more than one sample per time interval (1–5, 6–10, 11–15, 16–20 and >20 d after symptom onset). If an individual had more than one specimen for a given time interval, only the later specimen was included. For specificity, we included 108 pre-COVID-19 plasma specimens from eligible blood donors collected before July 201837. We assessed detection of SARS-CoV-2 antibodies in 51 specimens from 2020: 49 with test results for detection of other respiratory viruses (BioFire FilmArray, BioFire Diagnostics) and 31 with negative results by SARS-CoV-2 RT–PCR. For these specimens, the median days from symptom onset was four with a range of 0–107 d, the latter end of the range owing to unresolving respiratory viral infection in the setting of HIV infection.
We based minimum sample size calculations on expected binomial exact 95% confidence limits. A total of 287 samples were included in the final analysis, including 128 from 79 individuals who tested positive for SARS-CoV-2 by RT–PCR. Some specimens were exhausted during the analysis and were not included in all tests. Data obtained from serial specimens that did not conform to our study design were excluded.
Clinical data were extracted from electronic health records and entered in a Health Insurance Portability and Accountability Act-secure REDCap database hosted by UCSF. Data included demographic information, major comorbidities, patient-reported symptom onset date, symptoms and indicators of severity.
Independent data from testing efforts at MGH, with slight deviations in methods, are included as Supplementary Data (Supplementary Fig. 3). Briefly, 48 heat-inactivated serum/plasma samples from 44 individuals who tested positive for SARS-CoV-2 by RT–PCR were included. For specificity, the MGH study included 60 heat-inactivated pre-COVID-19 samples from 30 asymptomatic adults and 30 individuals admitted with febrile and/or respiratory illness with a confirmed pathogen.
Sample preparation
Samples from UCSF and ZSFG were assigned a random well position in one of four 96-well plates. Samples were thawed at 37 °C, and up to 200 µl was transferred to the assigned well without heat inactivation. Samples were then sub-aliquoted (12.5 µl) to replica plates for testing. Replica plates were stored at −20 °C until needed and then thawed for 10 min at room temperature and briefly centrifuged before testing. All sample handling followed UCSF biosafety committee-approved practices.
For the MGH study, samples were heat inactivated at 56 °C for 60 min, aliquoted and stored at 4 °C and −20 °C. Samples stored at 4 °C were used within 7 d. Frozen aliquots were stored until needed with only a single freeze-thaw cycle for any sample. All samples were brought to room temperature and briefly centrifuged before adding the recommended volume to the LFA cartridge.
Immunochromatographic LFAs
Ten LFAs were evaluated (Supplementary Table 1). At the time of testing, cartridges were labeled by randomized sample location (plate and well). The appropriate sample volume was transferred from the plate to the indicated sample port, followed by provided diluent, following manufacturer instructions. The lateral flow cartridges were incubated for the recommended time at room temperature before readings. Each cartridge was assigned an integer score (0 for negative, 1–6 for positive) for test line intensity by two independent readers blinded to specimen status and to each other’s scores (Supplementary Fig. 1). Readers were trained to score intensity from images representative of each value from a previous LFA test performance evaluation37. Test line scoring was performed for research purposes to capture semi-quantitative data about the LFA readout and reproducibility of subjective interpretation, considering that these are the major analytical factors that affect test performance. These tests are prescribed to be interpreted qualitatively, and test performance characteristics in this report are derived from qualitative scoring of any interpreted band color. For some cartridges (DeepBlue, UCP and Bioperfectus), the positive control indicator failed to appear after addition of diluent in a significant fraction of tests. For these tests, two further drops of diluent were added to successfully recover control indicators in all affected tests. These results were included in analyses. During testing, two plates were transposed 180°, and assays were run in the opposite order from the wells documented on cartridges. These data were corrected, and accuracy was confirmed by empty well position and verification of a subset of results.
ELISAs
Epitope Diagnostics assays were carried out according to manufacturer instructions with minor deviations, including the mixed use of plasma and serum specimens (instead of serum only), use of frozen specimens (versus same day), blanking all specimens and controls instead of using raw OD450 values and performing samples in singlicate for three of four 96-well plates (instead of duplicate). Plate 4 was run in duplicate owing to availability of samples and assay wells. For IgM detection, 100 µl of control samples or 10 µl of patient serum and 100 µl of sample diluent were added to indicated wells. Plates were incubated for 30 min at 37 °C and manually washed five times in provided Wash Buffer. Each well received 100 µl of horseradish peroxidase (HRP)-labeled COVID-19 antigen, was incubated for 30 min at 37 °C and was manually washed five times in provided Wash Buffer. Each well then received 100 µl of colorimetric substrate, was incubated for 20 min and then received 100 µl of Stop Solution. The OD450 was measured using a Synergy H1 Microplate Reader (BioTek Instruments) within 10 min of adding Stop Solution. Positive cutoff for IgM detection was calculated as described in the Epitope Diagnostics protocol: IgM positive cutoff = 1.1 × ((average of negative control readings) + 0.10). Values less than or equal to the positive cutoff were interpreted as negative. For IgG detection, 1 µl of serum was diluted 1:100 in Sample Diluent and loaded into designated wells. Plates were incubated for 30 min at room temperature and manually washed five times in provided Wash Buffer. Each well received 100 µl of provided HRP-labeled COVID-19 Tracer Antibody; plates were incubated for 30 min at room temperature and manually washed five times in provided Wash Buffer. Then, each well received 100 µl of Substrate, was incubated for 20 min and then received 100 µl of Stop Solution. The absorbance at OD450 was measured using a Synergy H1 Microplate Reader (BioTek Instruments) within 10 min of adding Stop Solution. Positive cutoffs for IgG detection were calculated as described in the Epitope Diagnostics protocol: IgG positive cutoff = 1.1 × ((average of negative control readings) + 0.18). Values less than or equal to the positive cutoff were interpreted as negative.
An in-house receptor binding domain (RBD)-based ELISA was performed with minor deviations from a published protocol (Amanat et al.26, Krammer Lab, Mount Sinai School of Medicine). SARS-CoV-2 RBD protein was produced using the published construct (NR-52306, BEI Resources) by Aashish Manglik (UCSF). Next, 96-well plates (3855, Thermo Fisher Scientific) were coated with 2 µg ml−1 RBD protein and stored at 4 °C for up to 5 d before use. Specimen aliquots (12 µl) were diluted 1:5 in 1× phosphate-buffered saline (PBS) (10010-023, Gibco), mixed and heat inactivated at 56 °C for 1 h. RBD-treated plates were washed three times with PBS-Tween (PBST, BP337-500, Fisher Bioreagents) using a 405 TS Microplate Washer (BioTek Instruments) and blocked with PBST-Milk (3% wt/vol, AB10109-01000, AmericanBio) for 1 h at 20 °C. Samples were further diluted 1:10 (1:50 final) in PBST-Milk (1% wt/vol), and 100 µl was transferred to the blocked ELISA plates in duplicate plates. Samples were incubated for 2 h at 20 °C and washed three times with PBST. The peroxidase AffiniPure Goat Anti-human IgG (F(ab′)20-specific) secondary antibody (109-035-097, lot 146576, Jackson ImmunoResearch) used in this study binds the IgG light chain and has some reactivity for other isotypes (IgM and IgA). This secondary antibody was diluted 1:750 in PBST-Milk (1% wt/vol), 50 µl was added to each sample well and samples were incubated for 1 h at 20 °C. Plates were subsequently washed three times with PBST. We dispensed 100 µl of 1× SigmaFast OPD Solution (P9187, Sigma-Aldrich) to each sample well and incubated plates for 10 min at room temperature. We added 50 µl of 3M HCl (A144-212, Fisher Chemical) to stop the reaction and immediately read the optical density at 490 nm (OD490) using a Synergy H1 Microplate Reader (BioTek Instruments). OD490 values were corrected for each plate by subtracting the mean value of each plate’s blank wells. To determine a cutoff for positive values, we calculated the mean value of negative wells for each plate, plus three standard deviations.
Data analysis
For LFA testing, the second reader’s scores were used for performance calculations, and the first reader’s scores were used to calculate inter-reader agreement statistics. Percent seropositivity among RT–PCR-confirmed cases was calculated by time interval from symptom onset. Specificity was based on results in pre-COVID-2019 samples. Binomial exact 95% CIs were calculated for all estimates. Analyses were conducted in R (3.6.3) and SAS (9.4).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The source data for figures are provided as a supplementary file or can be viewed at https://covidtestingproject.org.
References
Johns Hopkins University. COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU), https://gisanddata.maps.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6 (2020).
Infectious Diseases Society of America. IDSA Statement on COVID-19 Testing, https://www.idsociety.org/globalassets/idsa/public-health/covid-19-idsa-testing-intro.pdf (2020).
Weitz, J. S. et al. Modeling shield immunity to reduce COVID-19 epidemic spread. Nat. Med. https://doi.org/10.1038/s41591-020-0895-3 (2020).
World Health Organization. Laboratory Testing for Coronavirus Disease 2019 (COVID-19) in Suspected Human Cases: Interim Guidance, 2 March 2020 (World Health Organization, 2020).
Zhao, J. et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin. Infect. Dis., https://doi.org/10.1093/cid/ciaa344 (2020).
Wolfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature https://doi.org/10.1038/s41586-020-2196-x (2020).
He, X. et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. https://doi.org/10.1038/s41591-020-0869-5 (2020).
Li, R. et al. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV2). Science https://doi.org/10.1126/science.abb3221 (2020).
Bai, Y. et al. Presumed asymptomatic carrier transmission of COVID-19. JAMA https://doi.org/10.1001/jama.2020.2565 (2020).
Du, Z. et al. Serial interval of COVID-19 among publicly reported confirmed cases. Emerg. Infect. Dis. https://doi.org/10.3201/eid2606.200357 (2020).
Kimball, A. et al. Asymptomatic and presymptomatic SARS-CoV-2 infections in residents of a long-term care skilled nursing facility - King County, Washington, March 2020. MMWR Morb. Mortal. Wkly. Rep. 69, 377–381 (2020).
Mizumoto, K., Kagaya, K., Zarebski, A. & Chowell, G. Estimating the asymptomatic proportion of coronavirus disease 2019 (COVID-19) cases on board the Diamond Princess cruise ship, Yokohama, Japan, 2020. Euro. Surveill. https://doi.org/10.2807/1560-7917.ES.2020.25.10.2000180 (2020).
Qiu, H. et al. Clinical and epidemiological features of 36 children with coronavirus disease 2019 (COVID-19) in Zhejiang, China: an observational cohort study. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(20)30198-5 (2020).
Tong, Z. D. et al. Potential presymptomatic transmission of SARS-CoV-2, Zhejiang Province, China, 2020. Emerg. Infect. Dis. 26, 1052–1054 (2020).
Secretary of Health and Human Services. Determination of a public health emergency and declaration that circumstances exist justifying authorizations pursuant to section 564(b) of the Federal Food, Drug, And Cosmetic Act, 21 U.S.C. § 360bbb-3. https://www.fda.gov/media/135010/download (2020).
Food and Drug Administration. Policy for diagnostic tests for coronavirus disease-2019 during the public health emergency. https://www.fda.gov/media/135659/download (2020).
San Francisco Department of Public Health. San Francisco Department of Public Health Annual Report 2017-2018, https://www.sfdph.org/dph/files/reports/PolicyProcOfc/DPHAnnualReportFY17-18.pdf (2018).
DataSF. San Francisco COVID-19 Data Tracker, https://data.sfgov.org/stories/s/San-Francisco-COVID-19-Data-Tracker/fjki-2fab/ (2020).
Long, Q. X. et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat. Med. https://doi.org/10.1038/s41591-020-0897-1 (2020).
Tan, W. et al. Viral kinetics and antibody responses in patients with COVID-19. Preprint at medRxiv https://doi.org/10.1101/2020.03.24.20042382 (2020).
Ng, D. et al. SARS-CoV-2 seroprevalence and neutralizing activity in donor and patient blood from the San Francisco Bay Area. Preprint at medRxiv https://doi.org/10.1101/2020.05.19.20107482 (2020).
Lynch, K. L. et al. Magnitude and kinetics of anti-SARS-CoV-2 antibody responses and their relationship to disease severity. Preprint at medRxiv https://doi.org/10.1101/2020.06.03.20121525 (2020).
U.S. Food and Drug Administration. Emergency Use Authorization, https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization (2020).
Lassaunière, R. et al. Evaluation of nine commercial SARS-CoV-2 immunoassays. Preprint at medRxiv https://doi.org/10.1101/2020.04.09.20056325 (2020).
To, K. K. et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(20)30196-1 (2020).
Amanat, F. et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. https://doi.org/10.1038/s41591-020-0913-5 (2020).
Wang, W. et al. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA https://doi.org/10.1001/jama.2020.3786 (2020).
Kim, D., Quinn, J., Pinsky, B., Shah, N. H. & Brown, I. Rates of co-infection between SARS-CoV-2 and other respiratory pathogens. JAMA https://doi.org/10.1001/jama.2020.6266 (2020).
Jiang, S., Hillyer, C. & Du, L. Neutralizing antibodies against SARS-CoV-2 and other human coronaviruses. Trends Immunol. https://doi.org/10.1016/j.it.2020.03.007 (2020).
Nie, J. et al. Establishment and validation of a pseudovirus neutralization assay for SARS-CoV-2. Emerg. Microbes Infect. 9, 680–686 (2020).
Crawford, K. H. D. et al. Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 spike protein for neutralization assays. Viruses https://doi.org/10.3390/v12050513 (2020).
Food and Drug Administration. Clinical Laboratory Improvement Amendments (CLIA), https://www.fda.gov/medical-devices/ivd-regulatory-assistance/clinical-laboratory-improvement-amendments-clia (2020).
Altmann, D. M., Douek, D. C. & Boyton, R. J. What policy makers need to know about COVID-19 protective immunity. Lancet 395, 1527–1529 (2020).
Weinstein, M. C., Freedberg, K. A., Hyle, E. P. & Paltiel, A. D. Waiting for certainty on COVID-19 antibody tests — at what cost? N. Engl. J. Med. https://doi.org/10.1056/NEJMp2017739 (2020).
U.S. Food and Drug Administration. Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency (Revised), https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-coronavirus-disease-2019-tests-during-public-health-emergency-revised (2020).
National Cancer Institute. NCI Part of Federal Effort to Evaluate Antibody Tests for Novel Coronavirus, https://www.cancer.gov/news-events/cancer-currents-blog/2020/covid-19-nci-antibody-testing-review (2020).
Whitman, J. D. et al. Chagas disease serological test performance in United States blood donor specimens. J. Clin. Microbiol. https://doi.org/10.1128/JCM.01217-19 (2019).
Acknowledgements
We thank all members of the Marson lab and the Hsu lab, P. Kim, S. Boyd, J. DeRisi, S. Quake, B. Greenhouse, C. Tato, J. Doudna, F. Urnov, D. Friedberg, D. Neeleman, J. Hering, C. Cheng, N. Khosla, M. Krisiloff, L. Groom, C. Xu, D. Fontenot, J. Karkanias, G. Worthington, B. Burkholder, C. Craik, XPrize Pandemic Alliance, W. Bokhari, Z. Joaquin, S. Sarlati, S. Nesbit, W. Poe, S. Broder, Verily, C. Kim, A. Kijac, M. Solit and the Coronavirus Standards Working Group, D. Havlir, J. Engel, P. Farley, J. MacGregor, K. Hou, B. Sanders, S. Yang and S. Parker. We thank Y. E. Castro-Sesquen for sharing her semi-quantitative LFA scale, which was adapted for use in our current study. This work was supported by gifts from Anthem Blue Cross Blue Shield, the Chan Zuckerberg Biohub and anonymous philanthropy. We thank the following sources for donation of test kits: the manufacturers of Bioperfectus, DecomBio, Sure Biotech, UCP Biosciences; D. Friedberg, J. Hering and H. Schein. The Wilson lab has received support from the Rachleff Family Foundation. The Hsu lab has received support from S. Altman, V. and N. Khosla, D. and S. Deb, the Curci Foundation and Emergent Ventures. P.D.H. holds a Deb Faculty Fellowship from the University of California, Berkeley College of Engineering and is the recipient of the Rainwater Foundation Prize for Innovative Early-Career Scientist. The Marson lab has received gifts from J. Aronov, G. Hoskin, K. Jordan, B. Bakar, the Caufield family and funds from the Innovative Genomics Institute, the Northern California JDRF Center of Excellence and the Parker Institute for Cancer Immunotherapy. We thank the National Institutes of Health for its support (to J.D.W., R38HL143581; to A.E.G., F30AI150061; to D.N.N., L40 AI140341; to S.P.B., NHLBI R38HL143581, to G.M.G., NHLBI R38HL143581; to T.A.M., 1F30HD093116; to D.W., 1F31NS106868-01; to J.G.C., R01 AI40098; to E.T.R. and R.C.C., CDC U01CK000490; MSTP students are supported by T32GM007618). R.Y. was supported by an AP Giannini Postdoctoral Fellowship. J.A.S. was supported by the Larry L. Hillblom Foundation (2019-D-006-FEL). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, is an investigator at the Chan Zuckerberg Biohub and is a recipient of the Cancer Research Institute Lloyd J. Old STAR grant.
Author information
Authors and Affiliations
Contributions
As co-first authors, J.D.W., J.H., C.T.M., B.R.S. and R.Y. were leaders of study design, data management and statistical analysis, test procurement, laboratory analysis, clinical correlation and manuscript drafting. A.M. and P.D.H. conceived this project and, with C.B., co-supervised this work and manuscript drafting. T.N.Y., U.R., C.W., J.M.W., A.E.G., S.A.B., A.M.L., S.L., A.W.W., E.M.G.B., Z.B.S., Y.L., R.A., M.J.L., J.A.S., T.Z., I.C.B., E.I., J.C., D.D.A., J.L., T.A.M., T.S.K., D.W., D.L.N., W.G., V.A.Y., H.A.E., P.C.C., L.W., M.E.M., J.L., L.T., L.F., R.L. K.K. and K.C.Z. were involved in test kit procurement, laboratory analysis and laboratory administration. G.M.G., A.G.L., D.N.N., S.P.B., J.B. and S.B. contributed to chart review and clinical correlation. S.M., C.Y.C., S.L.S., M.R.W., A.M., C.J.Y., N.J.K., M.S.A., J.G.C, J.D.E., A.H.B.W. and K.L.L. contributed to study design and supervision of UCSF co-authors. T.E.M., W.F.G.B., D.Y., M.G.A., B.E.B., J.A.G., E.T.R., R.C.C., A.J.I. and J.K.L. contributed to study design, laboratory analysis and data analysis at Massachusetts General Hospital. All authors contributed to manuscript review and editing.
Corresponding authors
Ethics declarations
Competing interests
This work was supported by gifts from Anthem Blue Cross Blue Shield, the Chan Zuckerberg Biohub and anonymous philanthropy. C.Y.C. is the director of the UCSF-Abbott Viral Diagnostics and Discovery Center, receives research support funding from Abbott Laboratories and is on the scientific advisory board of Mammoth Biosciences. C.J.Y. is co-founder of DropPrint Genomics and serves as an advisor to them. M.S.A. holds stock in Medtronic and Merck. P.D.H. is a co-founder of Spotlight Therapeutics and serves on the board of directors and scientific advisory board and is an advisor to Serotiny. P.D.H. holds stock in Spotlight Therapeutics and Editas Medicine. A.M. is a co-founder of Spotlight Therapeutics and Arsenal Biosciences and serves on their board of directors and scientific advisory board. A.M. has served as an advisor to Juno Therapeutics, was a member of the scientific advisory board at PACT Pharma and was an advisor to Trizell. A.M. owns stock in Arsenal Biosciences, Spotlight Therapeutics and PACT Pharma. R.Y. owns stock in AbbVie, Bluebird Bio, Bristol-Myers Squibb, Cara Therapeutics, Editas Medicine, Esperion and Gilead Sciences. Unrelated to this current work, the Marson lab has received sponsored research support from Juno Therapeutics, Epinomics, Sanofi and GlaxoSmithKline and a gift from Gilead.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Tables 1–3 and Figs. 1–3
Supplementary Data 1
Serology test performance comparison data set
Rights and permissions
About this article

Cite this article
Whitman, J.D., Hiatt, J., Mowery, C.T. et al. Evaluation of SARS-CoV-2 serology assays reveals a range of test performance. Nat Biotechnol 38, 1174–1183 (2020). https://doi.org/10.1038/s41587-020-0659-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-020-0659-0
This article is cited by
-
Overview of diagnostic tools and nano-based therapy of SARS-CoV-2 infection
Chemical Papers (2024)
-
A peek behind the curtain in the diagnosis and management of COVID‑19‑Associated Mucormycosis (CAM)
Journal of the Egyptian Public Health Association (2023)
-
Screening and confirmation tests for SARS-CoV-2: benefits and drawbacks
Beni-Suef University Journal of Basic and Applied Sciences (2023)
-
A bead-based multiplex assay covering all coronaviruses pathogenic for humans for sensitive and specific surveillance of SARS-CoV-2 humoral immunity
Scientific Reports (2023)
-
Comparison of six antibody assays and two combination assays for COVID-19
Virology Journal (2022)
