
Abstract
Blight-resistant rice lines are the most effective solution for bacterial blight, caused by Xanthomonas oryzae pv. oryzae (Xoo). Key resistance mechanisms involve SWEET genes as susceptibility factors. Bacterial transcription activator-like (TAL) effectors bind to effector-binding elements (EBEs) in SWEET gene promoters and induce SWEET genes. EBE variants that cannot be recognized by TAL effectors abrogate induction, causing resistance. Here we describe a diagnostic kit to enable analysis of bacterial blight in the field and identification of suitable resistant lines. Specifically, we include a SWEET promoter database, RT–PCR primers for detecting SWEET induction, engineered reporter rice lines to visualize SWEET protein accumulation and knock-out rice lines to identify virulence mechanisms in bacterial isolates. We also developed CRISPR–Cas9 genome-edited Kitaake rice to evaluate the efficacy of EBE mutations in resistance, software to predict the optimal resistance gene set for a specific geographic region, and two resistant ‘mega’ rice lines that will empower farmers to plant lines that are most likely to resist rice blight.
Similar content being viewed by others


Main
Rice is the most important food crop in the world, but biotic and abiotic stresses can reduce yields. Xoo causes the rice disease bacterial blight, which results in substantial yield losses across Asia1. Bacterial blight has been classed as the most serious bacterial disease of rice2, with the highest social impact. Epidemics severely affect smallholders: about 70% of farms in India are ~ 0.39 ha (ref. 3), roughly similar to 80% of farms in sub-Saharan Africa ( < 2 ha). Bacterial blight is a major problem in India that has increased in severity year on year since 2000 (ref. 4). Increased severity has been attributed in part to climate change (increased rainfall and higher cyclone frequency)4. Modeling indicates that climate change might result in an increased effect of Xoo-mediated disease in Africa, where rice production is rising, and researchers have suggested that losses due to rice blight would eventually exceed those caused by rice blast5. Moreover, the United States is concerned about the potential for introduction of Asian or African bacterial blight strains; currently, only low-virulence strains that lack TAL effector genes are present in the United States6. Xoo is on the US Select Agent list as a potential bioterrorism agent7.
Genetic resistance to disease reduces the need for pesticides8. The best known plant resistance (R) genes encode proteins that interact with specific pathogen effectors and endow plants with dominant resistance to pathogens. More than 40 R genes for bacterial blight have been identified; a few of these have been cloned, and several have been associated with modular TAL effectors8. Complete genome sequences of Xoo enable identification of TAL effectors, information that can then be used together with the TAL effector recognition code9,10 to identify the respective TAL EBEs throughout the rice genome. In some cases, however, resistance is recessive and caused by effectors that target susceptibility factors, as in the case of host sucrose transporter (SWEET) genes. Xoo produces TAL effectors that ectopically induce the SWEET genes that make rice susceptible to infection and disease1,12.
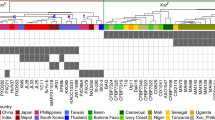
DNA polymorphisms in EBEs can prevent TAL effectors from binding to target promoters, and the respective rice lines with altered EBEs in a SWEET promoter are resistant to bacterial blight11,12. The first identified SWEET resistance variant was xa13, a naturally occurring promoter variant in SWEET11 (refs. 13,14). xa13 resistance is recessive and is used in rice breeding programs, as the xa13 promoter variants do not negatively affect yield4,15. The TAL effector PthXo1, which is present in several Xoo strains (PXO99 and PXO71), binds an EBE in the SWEET11 promoter11. The resistant rice line IRBB13 (xa13) carries a 38-bp deletion and a 252-bp insertion in the SWEET11 promoter, which abrogate binding of the EBE by PthXo1. Other resistant varieties carrying xa13 include Chinsurah Boro2, Tepa1, Aus274, AC-19-1-1, Long Grain (35023), Kalimekri77-5, Long Grain (64950) and BJ1 (ref. 13). xa13 resistance can be overcome by Xoo strains that produce alternative TAL effectors that bind to the promoter of a different SWEET paralog. For example, SWEET14 promoter elements can be bound by PthXo3 or AvrXa7 (ref. 16; Fig. 1a). Naturally occurring recessive resistance has also been identified for SWEET13 (xa25)17,18 and SWEET14 (xa41)19. Altogether, six EBEs in three SWEET genes that are targeted by naturally occurring TAL effectors (PthXo1 targeting SWEET11; PthXo2 and variants targeting SWEET13; PthXo3, AvrXa7, TalC and TalF targeting SWEET14) have been characterized. The TAL effectors target three of the five clade III SWEET genes; the other two SWEET genes can function as susceptibility (S) genes when artificially induced, but no Xoo strains targeting these genes have been identified20. SWEET genes in other clades do not function as S genes20.

a, Arrows indicate which TAL effectors can overcome a particular resistance mechanism by activating any of the other SWEET genes or by activating the same SWEET gene via targeting another EBE in the same promoter. For example, xa13-based resistance (resulting from a variant in the SWEET11 promoter that is not recognized by PthXo1) can be overcome by other TAL effectors (e.g., PthXo2 and PthXo3) that target the EBEs in another SWEET gene promoter or, in the case of SWEET14, target different EBEs in the same promoter. b, Example of SWEET gene induction as detected by RT–PCR using the SWEETup primer set. RT–PCR products are shown for the SWEET11, SWEET13 and SWEET14 genes in Kitaake infected by Xoo strain ME2 lacking a SWEET-targeting TAL effector and ME2 transformed with plasmids encoding PthXo1 (targeting SWEET11), PthXo2 or PthXo2B (both targeting SWEET13) and PthXo3, TalC, TalF or AvrXa7 (all targeting SWEET14). Actin served as a control. Leaves were infected using leaf-clipping assays; scissors dipped in water served as an additional negative control. The experiment was repeated twice independently with similar results.
The pool of naturally occurring EBE variants is small, meaning that extensive breeding efforts are needed to identify and introduce such resistance variants into elite rice varieties. TALEN- or CRISPR-based genome editing has been used to engineer EBE variants of SWEET11 and SWEET14 in the japonica variety Kitaake18,19,21. Additionally, five EBEs have been targeted, alone or in combination, in Kitaake, IR64 and Ciherang-Sub1 (refs. 18,21,22,23,24), generating resistance to a collection of 94 Xoo strains of Asian origin24. These R-gene-containing lines can now be deployed, individually or in combination, for broad resistance. However, Xoo will likely evolve TAL effector variants that can bind to other promoter sequences to enable plant infection.
To enable the strategic deployment of R genes to block novel virulent strains of Xoo when they emerge, we developed the SWEETR kit 1.0, a multicomponent tool set that integrates pathogen diagnostics, genetic resistance and customizable resistance line deployment to overcome bacterial blight.
Results
Content of the SWEETR kit 1.0
The SWEETR kit 1.0 (Supplementary Table 1) contains a SWEET promoter database (SWEETpDB), SWEETup primers for detecting mRNA accumulation for each SWEET gene, three SWEETacc-rice tester lines that report spatial SWEET protein accumulation during infection using translational reporter fusions26, single- and combined-knockout SWEETko mutants to identify which SWEET genes are required for susceptibility to individual Xoo strains, SWEETpR tester lines in the R-gene-free Kitaake background to evaluate whether particular EBE variants (or combinations thereof) are sufficient for resistance against an Xoo isolate, SWEET PathoTracer, a decision tool26 based on disease diagnostic surveys and population information to develop the most effective and most durable resistance for a specific region, and a total of 32 transgene-free EBE-edited lines of two mega varieties: IR64 and Ciherang-Sub1 (ref. 24).
SWEET promoter database
To predict resistance against, or susceptibility to, any Xoo strain with TAL effector(s) that target SWEET promoters, the EBE sequences in SWEET promoters must be characterized. We analyzed EBE variations for SWEET11, SWEET13 and SWEET14 in 4,726 accessions (in the first 400 bp of the SWEET promoters)27,28 as well as 5 rice lines grown in India, Southeast Asia and Africa, and identified 15 sequence variants. One A/G variant in the EBE for PthXo1 occurred at a frequency of 0.2% (Supplementary Fig. 1 and Supplementary Table 2)13,29. Seven variations were found in the PthXo2 EBE at frequencies between 1.3% and 20.8% (Supplementary Fig. 2 and Supplementary Table 2)29. Notably, five variations were found at the TATA box of SWEET13 (TATATAAA, TATTTAAA, TATATATA, TATATAA and TATATAAAA), which overlap with the EBE. The TATATAAA variant is known to occur in japonica varieties resistant to Xoo strains that harbor the TAL effector PthXo2 (ref. 18). In the PthXo3/AvrXa7 EBE, an insertion of one adenosine was found at a frequency of 7.7% (Supplementary Fig. 3 and Supplementary Table 2)29. Lines CX371 and CX372 (or NERICA1 and NERICA2) carried a G/T variation in the TalC EBE and an 18-bp deletion spanning the PthXo3/AvrXa7 and TalF EBEs. These variations could lead to broad-spectrum resistance against Xoo strains harboring TalC, PthXo3/AvrXa7 and TalF. To confirm the information mined from genomic sequences, 2 of the 4,726 accessions for each variation type and 5 lines grown in India, Southeast Asia and Africa (MTU1010, Samba Mahsuri, Komboka, BRRI Dhan28 and BRRI Dhan 29) (Supplementary Table 3) were chosen for validation. The first 400 bp of the three SWEET promoters were sequenced and included in a representative promoter sequence database (sequences in Supplementary Table 4).
SWEETup primers to detect SWEET mRNA levels
To enable rapid assessment of SWEET mRNA accumulation upon Xoo infection, we synthesized specific primer pairs for SWEET11, SWEET13 and SWEET14 (named SWEETup) and established robust RT–PCR protocols (Supplementary Table 5 and Supplementary Note 1). SWEET mRNA levels were detected in uninfected leaves (Supplementary Fig. 4) and in infected leaves in a strain-specific manner (Fig. 1b). Quantitative RT–PCR (qRT–PCR) showed that SWEET13 mRNA levels were highest among the five clade III SWEET genes in uninfected leaves, with lower levels of SWEET14. SWEET13 may have a role in phloem loading, as shown for its maize homologs30. SWEET11 mRNA levels were very low in leaves, consistent with its roles in seed filling26. Validated primer pairs are available in the kit for testing new Xoo isolates.
SWEETacc-rice tester lines for SWEET protein accumulation
First, we engineered transcriptional reporter lines to visualize SWEET RNA accumulation by histochemical β-glucuronidase (GUS) staining. However, reporter lines for all three promoters had nonspecific reporter activity (Supplementary Fig. 5). Previously, similar observations were reported for AtSWEET11 and AtSWEET12 in Arabidopsis; only translational SWEET reporters had cell-specific reporter activity12. To monitor SWEET protein accumulation, translational promoter reporter lines were engineered. The constructs included a 2-kb fragment from the SWEET promoter, the coding region of the SWEET gene including introns and a GUSPlus reporter (Supplementary Figs. 6–8). Consistent with the absolute mRNA levels measured by qRT–PCR, SWEET13 and SWEET14 translational promoter reporter activities were detected in uninfected leaves, whereas SWEET11 translational promoter fusion lines showed no detectable GUS activity (Fig. 2a–d). SWEET13 and SWEET14 translational reporter lines showed vein-specific protein accumulation, consistent with the roles of SWEET13 and SWEET14 in phloem loading (Fig. 2b–d). Three reporter lines for SWEET11, SWEET13 and SWEET14, named SWEETacc-rice tester lines, were each infected with five Xoo strains (PXO61, PXO71, PXO86, PXO99 and PXO112), which are known to induce specific SWEET genes. The Xoo strain ME2, lacking TAL effectors for SWEET induction14, was used as a control and did not trigger SWEETacc reporter activity. Induction of SWEET11 was detected upon infection with PXO71 and PXO99 (both of which harbor PthXo1) but not with the other strains. SWEET13 was induced only upon infection with PXO61 (harboring PthXo2B), whereas SWEET14 was induced upon infection with PXO61 (PthXo3), PXO86 (AvrXa7) and PXO112 (PthXo3; Fig. 2e). Infection with ME2 strains harboring PthXo1 (targeting SWEET11), PthXo2B (targeting SWEET13) or PthXo3, TalC, AvrXa7 and TalF (targeting SWEET14) further confirmed specific SWEET isoform induction by this set of reporter lines (Supplementary Fig. 9).

a–c, GUS activity in flag leaf blades of rice for pSWEET11:SWEET11-GUS (event 10) (a), pSWEET13:SWEET13-GUS (event 22) (b) and pSWEET14:SWEET14-GUS (event 3) (c). d, A cross-section of the leaf blade from pSWEET13:SWEET13-GUS in b. e, SWEET protein accumulation upon infection with Xoo strains (pSWEET11:SWEET11-GUS event 10, pSWEET13:SWEET13-GUS event 15 and pSWEET14:SWEET14-GUS event 3). Scale bars: 20 µm (a–d); 1 mm (e). The experiment was repeated at least three times independently with similar results.
SWEETko knock-out lines to detect Xoo SWEET targets
Tools that could identify the SWEET genes targeted by a specific Xoo strain, without previous knowledge of the targeted EBE, would inform efforts to engineer resistance. This is especially important because variant Xoo strains can target either different promoter elements or other clade III SWEET paralogs. Moreover, it is conceivable that promoter-edited lines or variants could impair yield, if the promoter variations affect normal gene function in uninfected plants. Knowledge of the specific defects is thus important to judge possible negative effects of such mutations on plant performance. SWEET-mutant lines could also provide insight into the role(s) of SWEET genes in resistance and yield. To diagnose which SWEET was targeted by any Xoo strain and to predict the yield effects of alterations in SWEET genes, knockout mutants were created for four of the five described clade III SWEET genes (SWEET11, SWEET13, SWEET14 and SWEET15) using CRISPR–Cas9 (Supplementary Fig. 10 and ref. 26). In all cases, lines containing frameshift mutations in the sequence corresponding to transmembrane domain I (TM I) were identified (Supplementary Table 6). Frameshifts that result in early termination should create nonfunctional transporters. For example, premature termination in the last transmembrane domain, TM VII, of OsSWEET11 results in defective transporters12. sweet13 mutant lines had reduced SWEET13 mRNA levels, which often occur concurrently with early termination (Fig. 3b). Knockout mutants for SWEET11 and SWEET15 were reported to have defects in seed filling26. Although SWEET11 and SWEET15 have important roles in seed filling in rice, widely used promoter variants, such as the resistance-conferring xa13 variant (SWEET11), do not seem to affect yield4,15.

a, Phenotypes of sweet13-1 and sweet13-2 knockout mutants relative to Kitaake controls at the mature stage. Scale bar, 10 cm. b, Relative mRNA levels of SWEET13 in flag leaf blades. Samples were harvested at 12:00 (mean ± s.e.m., n = 3 biological replicates with mRNA levels normalized to rice Ubiquitin1 levels; repeated independently three times with similar results). Center lines show medians; box limits indicate the 25th and 75th percentiles as determined by R software; and whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles. c, One-thousand-grain weight of greenhouse-grown Kitaake, sweet13-1 and sweet13-2 (mean ± s.e.m.; n = 4 biological replicates). The experiment was repeated at least three independent times with similar results. Center lines show medians; box limits indicate the 25th and 75th percentiles as determined by R software; and whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles. No significant differences (P = 0.051 for sweet13-1 and P = 0.758 for sweet13-2) were identified by Student’s t-test. d, Phenotypes of wild type and the sweet13;sweet14 double knockout grown in the greenhouse. No significant differences were identified. e, Length of lesions at 14 days after inoculation (DAI) caused by ME2 (negative control), PXO99 (positive control) and AXO1947 on single-, double- and triple-knockout (sweet11, sweet13 and sweet14) mutants relative to Kitaake wild type (mean ± s.e.m.; n = 10 inoculated leaves). The experiment was independently repeated twice with similar results. The difference observed for AXO1947 virulence between sweet14 and sweet11;14 in a single experiment was not significant when compared over a larger number of experiments (Supplementary Fig. 14).
Because maize ZmSWEET13 paralogs have roles in phloem loading30, the role of rice SWEET13 was investigated. SWEET13 is the most highly expressed SWEET gene in rice leaves, and the encoded protein localizes to the plasma membrane and, in common with SWEET14, transports sucrose (Supplementary Figs. 4 and 11)12,18. Analysis of GUS reporter fusions showed that SWEET13 accumulates in the phloem (Fig. 2b,d). SWEET14 also accumulated in the phloem but had substantially lower mRNA levels in leaves than SWEET13 (Fig. 2c; transcript levels not shown). Nevertheless, CRISPR–Cas9 sweet13 and sweet14 knockout mutant lines did not show detectable growth or yield defects under greenhouse conditions (Fig. 3a,c and Supplementary Fig. 12), nor were obvious differences observed in a single-season field experiment (based on visual inspection during the growth period and after harvest). To identify potential compensatory activity from other SWEET genes in the knockout mutants, the expression levels of other sucrose-transporting SWEET genes were analyzed. Only the weakly expressed SWEET14 and SWEET15, and none of the other clade III SWEET genes, showed substantial increases in mRNA accumulation in the leaf blade of sweet13 knockout lines (Supplementary Fig. 13). To test whether upregulation of SWEET14 could compensate for the loss of SWEET13 and thereby restore apoplasmic phloem loading, sweet14 single-knockout and sweet13;sweet14 double-knockout lines were generated. Double mutants did not show obvious growth differences relative to controls in the greenhouse (Fig. 3d). Because mutant lines had no clear defects in plant growth or yield, EBE-edited lines in which the normal promoter function of SWEET13 and SWEET14 is affected are not hypothesized to have yield penalties. Further, our data indicate that apoplasmic phloem loading in rice, in contrast to maize and Arabidopsis, either is not crucial to plant performance or does not entirely depend on SWEET function12,30.
Knockout lines can serve as diagnostic tools for testing whether Xoo strains require specific SWEET genes. The knockout mutants proved to be valuable tools for characterizing the virulence of a collection of 95 different Xoo strains with diverse geographic origins24. In this analysis, we observed that an African strain, AXO1947, which contains the effector TalC and can induce SWEET14, but does not induce SWEET13, was still able to infect a Kitaake mutant line edited in the TalC EBE present in the SWEET14 promoter24. Although technically weakly virulent on lines carrying TalC EBE variants, AXO1947 showed substantially reduced virulence in the quintuple-mutant promoter lines, which are likely sufficiently resistant in field conditions24. The quintuple-mutant line was moderately resistant, with lesion lengths of 5–7 cm upon infection by AXO1947, as compared to 18–25 cm in controls. A systematic screen for resistance using sweet13 and sweet14 single-knockout and sweet13;sweet14 double-knockout mutants showed that AXO1947 lost some virulence in sweet14-knockout lines but was unable to infect sweet13;sweet14 double mutants (Fig. 3e and Supplementary Fig. 14). These data demonstrate the utility of knockout lines for testing resistance (Fig. 3e, Table 1 and Supplementary Table 7). Co-dependence of strain AXO1947 on both SWEET13 and SWEET14 function is under investigation. Further characterization is needed, as dependence on SWEET13 is not understood, given that SWEET13 induction by AXO1947 was not detected. Notably, whereas in leaves basal SWEET11 mRNA levels are low and induction is easily detectable, SWEET13 and SWEET14 are expressed in leaves; thus, a further increase in expression in a few cells in the xylem is difficult to detect against this background. SWEETacc and SWEETko rice tester lines are thus better suited to detect SWEET dependence.
SWEETpR, genome-edited EBE tester lines for Xoo genotyping
Rice varieties have different numbers and types of R genes. The only known R gene for bacterial blight in the japonica rice variety Kitaake is a cryptic resistance gene similar to the recessive xa25, which interacts with the major TAL effector PthXo2 (ref. 18). Thus, Kitaake is a useful reference line for testing Xoo compatibility. In a parallel study24, a series of EBE variants of SWEET11, SWEET13 and SWEET14 in Kitaake were engineered by genome editing, and resistance/susceptibility to Xoo strains was validated24. These 20 genome-edited Kitaake tester lines (named SWEETpR)24 are available for genotyping Xoo isolates and function similarly to R-gene line panels for race characterization31 (e.g., Kitaake line 11.1-45 was resistant to strains containing the TAL effectors PthXo1 and AvrXa7, and line 12.2-12 was resistant to strains containing PthXo2B, PthXo3 and AvrXa7 (ref. 24)) (Supplementary Tables 8 and 9).
SWEET PathoTracer visualization
Geographic Information System (GIS)-based platforms that incorporate pathogen monitoring and resistance profiles of rice varieties are useful for management of local disease outbreaks26. We integrated the near-isogenic IR64 and Ciherang-Sub1 lines into the PathoTracer platform (http://webapps.irri.org/pathotracer/index.html). PathoTracer displays the predicted involvement of SWEET11, SWEET13 and SWEET14 on the basis of the Xoo population that is present in geographic regions and suggests effective edited variants for planting in the next season. For example, a dataset containing analyses about the ability of Xoo strains to infect the R-gene near-isogenic IRBB lines32 was compared to the proportion of endemic strains from an area of the Philippines that might activate SWEET14 (Fig. 4 and Supplementary Fig. 15). On the basis of this information, 47% of the strains in the Xoo population are predicted to be controlled by one or more of the SWEET14 EBE variants. By planting the recommended varieties, farmers can minimize the risk of infection and the resulting yield losses in the next season.

PathoTracer (http://webapps.irri.org/pathotracer/index.html) is an online repository that integrates genotypic and phenotypic pathogen data with the resistance profiles of rice accessions to support strategic deployment of varieties in the region. Tester- strain-based prediction of SWEET targets is provided here for SWEET14, using Xoo populations collected from 1972 to 2012 in Laguna, a disease-endemic area in the Philippines (n = 1,294 isolates). A screenshot of the full interface with the same map is shown in Supplementary Fig. 15.
Genome-edited Xoo-resistant mega variety lines
Mega rice varieties are defined as varieties planted on more than 1 million hectares. Although genome-edited bacterial-blight-resistant SWEETpR Kitaake lines can be used by breeders, SWEETR mega variety lines would reduce the need for further breeding efforts. This is of particular relevance because breeding efforts are more extensive in this context, owing to the recessive nature of SWEET-based resistance. CRISPR–Cas9 was used to edit five of the six EBE sites in the three SWEET promoters in widely used indica rice mega variety IR64 and in Ciherang-Sub1, a new flooding-tolerant elite line24,33,34. We generated edited lines with alterations in single or multiple EBEs. Together, 32 Cas9-free lines were produced, encompassing 35 single variations in the three SWEET promoters. Agronomic assessment and pathogenicity trials validated resistance against single or multiple Xoo strains (Fig. 5 and Supplementary Table 10)24.

Reactions of IR64 SWEET-promoter-edited lines to three representative Xoo strains (data from ref. 24). Lesion lengths were measured at 14 DAI with strains PXO99A, PXO339 and PXO86. Infections were carried out at the maximum tillering stage by inoculating 3–6 leaf samples using a leaf clipping. Four replicate experiments with two plants each were performed per strain (four replicates per strain, two plants per replicate (n = 8)) and scored for 3–6 inoculated leaf samples per plant. The experiment was repeated three times independently.
Discussion
Genetically narrow germplasm and extensive mono-cropping are two hallmarks of modern agriculture that favor disease and its spread35. Genome editing could provide efficient tools for rapid engineering of pathogen resistance in cropping lines. However, detection of pathogen strains, their virulence factors and cropping line susceptibilities will be crucial if we are to effectively reduce the impact of plant diseases. Here we present a diagnostic kit to enable breeders, crop management teams and farmers to reduce the effect of bacterial blight on rice yields worldwide.
Genome editing can create large pools of R-gene variants, providing new ways to implement and manage long-term genetic resistance. Diseases that use TAL effectors are excellent candidates for reducing yield loss by engineering resistant plant lines, because TAL effector binding depends on highly conserved and short cognate EBE sequences present in host S-gene promoters. One crucial finding in the fight against bacterial blight is that Xoo strains use different TAL effectors (eight effectors have been characterized to date) to directly target five EBEs in three SWEET gene promoters24. Pathogen-mediated induction of one of three SWEET genes is sufficient to cause disease. Extant Xoo strains harbor just eight SWEET-targeting TAL effectors, indicating that effectors with novel EBE recognition motifs may not evolve quickly. Therefore, genome editing could provide durable as well as broad bacterial blight resistance.
The success of genome-edited rice lines resistant to bacterial blight will be threatened by the emergence of pathogenic strains that have adapted to recessive resistance. The resistance endowed by SWEET promoter variants that prevent TAL effector binding can be overcome by the emergence of pathogenic Xoo strains. For example, resistance based on the natural promoter variant xa13 can be overcome by Xoo strains that produce alternative TAL effectors, such as PthXo3 or AvrXa7, to induce either the same or different SWEET genes16. SWEET promoter variants will likely need to be deployed in combination with other R genes, and Xoo strain emergence may, therefore, depend on multiple genetic alterations.
The underlying key to durable resistance may be preventing bacteria from multiplying in monocultures36. Genome editing enabled us to establish a portfolio of recessive resistance variants with different promoter sequences that are available as R-gene variants to use when a novel pathogen emerges. The portfolio allows us to swap between R-gene variants. Tracking pathogens, using diagnostics and GIS, will likely also help. Because the edited lines are isogenic, they can be deployed as mixtures to reduce pathogen spread37,38, using mosaic crop plotting in which different R genes are deployed in adjacent fields or R genes are rotated over different seasons38. Here we provide a diagnostic tool set to accompany a suite of resistant rice lines24.
The SWEETR kit comprises a SWEETpDB and SWEETup primers to detect which SWEET is targeted by an Xoo isolate. If suitable expertise is not available locally, leaf samples can be preserved and analyzed in central test labs39. We also include four SWEETacc-rice tester lines containing full-gene translational reporters that detect a particular isolate that induces a SWEET gene20,25; this set includes a line for analysis of SWEET15 induction, although so far no naturally occurring Xoo strains induce this SWEET. The kit further includes single- and combined-knockout SWEETko mutants to identify which SWEET genes are required for susceptibility and SWEETpR tester lines in Kitaake. We added a component to a web-based decision tool26 named PathoTracer, which uses population information, to aid development of the most effective and most durable R-gene combination for a specific region through breeding. We also include 31 transgene-free EBE-edited lines in the two mega varieties IR64 and Ciherang-Sub1 (Supplementary Table 10)24. Figure 6 provides a roadmap for using the kit for customized deployment of the SWEETR lines.

Farmers with Xoo-infected rice fields will send samples to local breeders or pathologists, who will isolate respective Xoo strains. Pathologists will then identify induced and critically important SWEET genes using SWEETup primers for mRNA accumulation and SWEET knock-out (SWEETKO) mutants. After validation with SWEET EBE-edited Kitaake lines (SWEETpR), pathologists will identify the optimal SWEETR line, which is then provided to local breeders. In parallel, labs will isolate Xoo DNA from infected leaves, identify TAL effectors (the TALeome) and predict SWEET targets using SWEETpDB. PathoTracer provides additional region-specific recommendations for deployment of SWEETR variants.
Recently, a new Gibson cloning technology for TAL effectors was developed that enables determination of the full TAL effector complement of a new isolate followed by bioinformatic target prediction, a tool that will be integrated into the kit40.
In the future, we will use our diagnostic kit to track newly emerging Xoo strains, use them to challenge R-gene variants and engineer promoter variants if TAL effectors evolve. Although only three of five clade III SWEET genes have been targeted24, we have also developed sweet15 knockout mutants that have no obvious growth defects in greenhouse and field conditions24. We plan to improve our kit by adding sweet knockout lines to cover all clade III SWEET genes. There is one report of an Xoo strain that does not induce SWEET genes41, but it is challenging to prove a lack of induction, as exemplified here for AXO1947.
SWEET-based resistance also occurs for bacterial blights of cotton and cassava, so our approach of providing a disease diagnosis and management kit together with a suite of genome-edited lines may prove useful for other pathogens that decimate crops such as cassava and cotton42,43.
Methods
Promoter variation analysis
Rice varieties having nucleotide variations in six EBEs (for the TAL effectors PthXo1, PthXo2, PthXo3, TalC, AvrXa7 and TalF) were found using the ‘Search for Variations in a Region’ and ‘Search for Genotype With Variation ID’ functions in RiceVarMap v.2 (http://ricevarmap.ncpgr.cn/v2/)27. Two varieties were selected for each variation type as representative. Sequences of the first 400 bp of SWEET11, SWEET13 and SWEET14 promoters of the selected varieties were subtracted from the 3K database (http://snp-seek.irri.org/). Alignment was performed using ClustalW2.1 in Geneious 11.1.5 (https://www.geneious.com).
Genotyping of rice plants
Rice genomic DNA was extracted using CTAB (http://gsl.irri.org/services/dna-extraction-king-fisher/met). PCR was performed using ExTaq DNA polymerase (Clontech) with a melting temperature of 56 °C for SWEET11, SWEET13 and SWEET14 (primers listed in Supplementary Table 5). The PCR amplicons from the mutant alleles were validated by Sanger sequencing. Chromatograms were analyzed and aligned using Sequencher (https://www.genecodes.com/).
RNA isolation and transcript analyses
Total RNA was isolated using Spectrum Plant Total RNA kits (Sigma) or TRIzol (Invitrogen), and first-strand cDNA was synthesized using a QuantiTect Reverse Transcription kit (Qiagen). qRT–PCR was performed using a LightCycler 480 (Roche), with the 2−ΔCt method for relative quantification44. Primers for SWEET11, SWEET13, SWEET14 and UBI1 are listed in Supplementary Table 5.
DNA constructs and plant transformation
Generation of GUS reporter constructs
The method for constructing pSWEET11:gSWEET11-GUSplus was described26. In short, the promoter and coding region were fused in frame to the GUSplus coding sequence with the NOS terminator, and the resulting constructs were PCR amplified and inserted into pC1300intC (GenBank AF294978.1)45. For tissue specificity analysis, a 4,354-bp genomic clone of SWEET13 containing 1,919 bp of the 5′ region upstream of the translational start codon (ATG) and 2,435 bp of the entire coding region without a stop codon and a 4,365-bp genomic clone of SWEET14 containing 2,176 bp of the 5′ upstream region and 2,189 bp of the entire coding region without a stop codon were amplified by PCR using Kitaake genomic DNA as a template (primers are listed in Supplementary Table 5). The PCR amplicons were subcloned into pJET2.1/blunt (Thermo Fisher), and resulting inserts were confirmed by DNA sequencing. Sequences are shown in Supplementary Table 11. The cloned fragments digested with XbaI and KpnI for SWEET13 or AvrII and XmaI for SWEET14 were subsequently inserted in front of the GUSplus coding sequence of a promoterless GUSplus coding vector26 restricted with XbaI and KpnI for SWEET13 and XbaI and XmaI for SWEET14. The resulting pSWEET13:gSWEET13-GUSplus and pSWEET14:gSWEET14-GUSplus constructs were used to transform Oryza sativa L. ssp. japonica Kitaake. Nine independent events were obtained for pSWEET13:gSWEET13-GUSplus and pSWEET14:gSWEET14-GUSplus. Whereas GUS activity levels were different in the independent lines, the GUS patterns were similar.
Kitaake was also used for CRISPR–Cas9- and TALEN-mediated genome editing of SWEET11, SWEET13 and SWEET14. The methods for the CRISPR–Cas9-induced mutant (sweet11-1) and the TALEN-induced mutant (sweet11-2) were described previously18,25. The knockout mutants sweet13-1, sweet13-2, sweet14-1 and sweet14-2 were obtained with a CRISPR–Cas9 construct targeting coding sequences 5′-GCCTGTCCCTGCAGCATCCCTGG-3′ of SWEET13 and 5′-GCATGTCTCTTCAGCATCCCTGG-3′ of SWEET14 (where underlining indicates the position of the protospacer-adjacent motif (PAM)) common to the first exon as previously described18. Double mutants (sweet13-2;sweet14-1) were created by crossing. SWEET13 RNA levels were analyzed in the sweet13 mutant and shown to be reduced (Fig. 3b).
Plant materials and growth conditions
Kitaake wild-type and mutant plants were grown either in field conditions (20 individual plants per genotype, paddy field in summer 2016, Carnegie) or in greenhouses under long-day conditions of 14-h day/10-h night at 28–30 °C and 50% relative humidity, with a light intensity of 500–1,000 µmol m–2 s–1. Independent transformants for the GUS fusions were characterized (transcriptional GUS fusions: SWEET11, n = 8; SWEET13, n = 8; SWEET14, n = 15; translational GUS fusions: SWEET11, n = 18; SWEET13, n = 9; SWEET14, n = 29). All independent transformants showed similar tissue specificity by histochemical staining. Analyses were performed for a minimum of six plants per infection of each Xoo strain. Three independent experiments were performed for specific induction in a growth chamber (12-h day at 28 °C/12-h night at 25 °C, 80% relative humidity). IR64 lines were grown in a small-scale field environment in a screenhouse (28 ± 7 °C during the day/23 ± 4 °C at night; 80–85% relative humidity) at IRRI.
Histochemical GUS analyses
Samples were collected in cold 90% acetone for fixation, vacuum infiltrated for 10 min and incubated for 30 min at room temperature. Leaf samples were vacuum infiltrated in GUS washing buffer (staining solution without 5-bromo-4-chloro-3-indole-b-glucuronide (X-Gluc)) on ice for 10 min. The solution was changed to GUS staining solution (50 mM sodium phosphate pH 7.0, 10 mM EDTA, 20% (vol/vol) methanol, 0.1% (vol/vol) Triton X-100, 1 mM potassium ferrocyanide, 1 mM potassium ferricyanide and 2 mM X-Gluc dissolved in DMSO). Samples were incubated at 37 °C. After 2 h of incubation, samples were cleared in an ethanol series (20%, 35% and 50%) at room temperature for 30 min. Samples from Xoo-inoculated leaves were incubated in 70% ethanol to remove the chlorophyll. Specimens were observed with a SteREO Discovery.V12 stereoscope (Zeiss). For paraffin sections, samples were fixed using FAA for 30 min (50% (vol/vol) ethanol, 3.7% (vol/vol) formaldehyde and 5% (vol/vol) acetic acid). Dehydration was performed with an ethanol series (70%, 80%, 90% and 100%, 30 min each) and 100% tert-butanol. Samples were transferred and embedded in Histosec pastilles (Millipore). Sections (10 μm) were obtained with a rotary microtome (Jung RM 2025). Specimens were observed with an Eclipse e600 microscope (Nikon). GUS histochemistry experiments were performed at least two times with 12 individual plants, with similar results.
Xoo strains and infection protocols
The Xoo strains collected from different geographic regions were reported24. Plasmid-containing Xoo strains were obtained through electroporation of competent cells46 with respective pHM1-derived plasmids (e.g., pHM1/ZWpthXo1 for the pthXo1 gene)47. For infection experiments, bacterial inocula were prepared by growing bacterial cells on tryptone sucrose plates with appropriate antibiotics. Cells were scraped from the plates and resuspended in sterile distilled water at OD600 ~ 0.5. (i) Leaf clipping48: the two youngest fully expanded leaves of 4- to 5-week-old rice plants were clipped about 1–2 cm from the tip with scissor blades that were immersed in bacteria immediately before clipping. Five plants were used for inoculation of each strain (ten leaves in total). Lesion length (distance from the cut to the leading edge of (gray) symptoms) was measured for each inoculated leaf at 12–14 DAI. The mean lesion length of ten leaves was used for each treatment. The Tukey test for analysis after ANOVA was used for statistical analyses. Leaf tissues were mounted in laminating film and photographed under white light. (ii) Syringe infiltration: bacterial suspensions were infiltrated into leaves from the bottom by pressing the opening of a needleless syringe to the leaf. Leaf fragments with inoculated spots were cut off 48 h after inoculation for RNA extraction and GUS staining analysis.
Statistical analysis
Data were plotted using BoxPlotR (http://shiny.chemgrid.org/boxplotr/) or are presented as mean ± s.e.m as specified in respective tables or figures. One-sided ANOVA was conducted on measurements. The Tukey honestly significant difference test was used after ANOVA pairwise tests for significance, which was set at P t-test was used. Exact P values, the statistical test used and sample number n can be found in figure legends or graphs.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Materials will be made available for nonprofit research under a material transfer agreement (Supplementary Notes 2 and 3). We aim at obtaining freedom to operate for use by low-income farmers and will work with breeders to make the materials available to subsistence farmers. For commercial applications, accessibility will be negotiated from appropriate patent holders, and profits will be used to support dissemination to subsistence farmers. Edited IR64- and Ciherang-Sub1-based materials can be obtained from R.O.; edited Kitaake lines can be obtained from B.Y.; and translational reporter lines can be obtained from W.B.F. Distribution of Xoo strains may be restricted because of regulations of Xanthomonas oryzae as a Select Agent by the US government, because of the Nagoya protocol, or because some strains were donated from other groups and thus these groups should be contacted directly (for details, see ref. 24).
References
Mew, T. W. Current status and future prospects of research on bacterial blight of rice. Annu. Rev. Phytopath. 25, 359–382 (1987).
Ke, Y., Deng, H. & Wang, S. Advances in understanding broad-spectrum resistance to pathogens in rice. Plant J. 90, 738–748 (2017).
Government of India. State of Indian Agriculture 2015–16 (Ministry of Agriculture & Farmers Welfare, Cooperation & Farmers Welfare Directorate of Economics & Statistics Department of Agriculture, 2016).
Laha, G. S. et al. Changes in Rice Disease Scenario in India: An Analysis from Production Oriented Survey (ICAR–Indian Institute of Rice Research, 2016).
Duku, C., Sparks, A. H. & Zwart, S. J. Spatial modelling of rice yield losses in Tanzania due to bacterial leaf blight and leaf blast in a changing climate. Clim. Change 135, 569–583 (2016).
Triplett, L. R. et al. Genomic analysis of Xanthomonas oryzae isolates from rice grown in the United States reveals substantial divergence from known X. oryzae pathovars. Appl. Environ. Microbiol. 77, 3930–3937 (2011).
Nature Editors. Rice pathogen is added to list of bioterror agents. Nature 455, 1163 (2008).
Vikal, Y. & Bhatia, D. in Advances in International Rice Research (ed. Li, J. Q.) 175–213 (Intech Open, 2017).
Boch, J. et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512 (2009).
Moscou, M. J. & Bogdanove, A. J. A simple cipher governs DNA recognition by TAL effectors. Science 326, 1501 (2009).
Chen, L. Q. et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532 (2010).
Chen, L. Q. et al. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335, 207–211 (2012).
Chu, Z. et al. Promoter mutations of an essential gene for pollen development result in disease resistance in rice. Genes Dev. 20, 1250–1255 (2006).
Yang, B., Sugio, A. & White, F. F. Os8N3 is a host disease-susceptibility gene for bacterial blight of rice. Proc. Natl Acad. Sci. USA 103, 10503–10508 (2006).
Sakthivel, K. et al. The host background of rice influences the resistance expression of a three genes pyramid (xa5 + xa13 + xa21) to bacterial blight (Xanthomonas oryzae pv. oryzae) pathotypes of Indian mainland and Bay islands. Plant Breed. 136, 357–364 (2017).
Antony, G. et al. Rice xa13 recessive resistance to bacterial blight is defeated by induction of the disease susceptibility gene Os-11N3. Plant Cell 22, 3864–3876 (2010).
Cheng, Q. et al. Characterization of a disease susceptibility locus for exploring an efficient way to improve rice resistance against bacterial blight. Sci. China Life Sci. 60, 298–306 (2017).
Zhou, J. et al. Gene targeting by the TAL effector PthXo2 reveals cryptic resistance gene for bacterial blight of rice. Plant J. 82, 632–643 (2015).
Hutin, M., Sabot, F., Ghesquière, A., Koebnik, R. & Szurek, B. A knowledge-based molecular screen uncovers a broad-spectrum OsSWEET14 resistance allele to bacterial blight from wild rice. Plant J. 84, 694–703 (2015).
Streubel, J. et al. Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. oryzae. New Phytol. 200, 808–819 (2013).
Li, T., Liu, B., Spalding, M. H., Weeks, D. P. & Yang, B. High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 30, 390–392 (2012).
Bi, H. & Yang, B. Gene editing with TALEN and CRISPR/Cas in rice. Prog. Mol. Biol. Transl. Sci. 149, 81–98 (2017).
Septiningsih, E. M. et al. Accelerating the development of new submergence tolerant rice varieties: the case of Ciherang-Sub1 and PSB Rc18-Sub1. Euphytica 202, 259–268 (2015).
Oliva, R. et al. Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat. Biotechnol. https://doi.org/10.1038/s41587-019-0267-z (2019).
Yang, J., Luo, D., Yang, B., Frommer, W. B. & Eom, J.-S. SWEET11 and 15 as key players in seed filling in rice. New Phytol. 218, 604–615 (2018).
Dossa, G. S., Sparks, A., Cruz, C. V. & Oliva, R. Decision tools for bacterial blight resistance gene deployment in rice-based agricultural ecosystems. Front. Plant Sci. 6, 305 (2015).
3,000 Rice Genomes Project. The 3,000 Rice Genomes Project. Gigascience 3, 7 (2014).
Zhao, H. et al. RiceVarMap: a comprehensive database of rice genomic variations. Nucleic Acids Res 43, D1018–D1022 (2015).
Zaka, A. et al. Natural variations in the promoter of OsSWEET13 and OsSWEET14 expand the range of resistance against Xanthomonas oryzae pv. oryzae. PLoS ONE 13, e0203711 (2018).
Bezrutczyk, M. et al. Impaired phloem loading in zmsweet13a,b,c sucrose transporter triple knock-out mutants in Zea mays. New Phytol. 218, 594–603 (2018).
Ogawa, T., Yamamoto, T., Khush, G. S. & Mew, T.-W. Breeding of near-isogenic lines of rice with single genes for resistance to bacterial blight pathogen (Xanthomonas campestris pv. oryzae). Jpn. J. Breed. 41, 523–529 (1991).
Quibod, I. L. et al. Effector diversification contributes to Xanthomonas oryzae pv. oryzae phenotypic adaptation in a semi-isolated environment. Sci. Rep. 6, 34137 (2016).
Toledo, A. M. U. et al. Development of improved Ciherang-Sub1 having tolerance to anaerobic germination conditions. Plant Breed. Biotech. 3, 77–87 (2015).
Mackill, D. J. & Khush, G. S. IR64: a high-quality and high-yielding mega variety. Rice 11, 18 (2018).
Webb, K. M. et al. A benefit of high temperature: increased effectiveness of a rice bacterial blight disease resistance gene. New Phytol. 185, 568–576 (2010).
Vera Cruz, C. M. et al. Predicting durability of a disease resistance gene based on an assessment of the fitness loss and epidemiological consequences of avirulence gene mutation. Proc. Natl Acad. Sci. USA 97, 13500–13505 (2000).
Mikaberidze, A., McDonald, B. A. & Bonhoeffer, S. Developing smarter host mixtures to control plant disease. Plant Pathol. 64, 996–1004 (2015).
Rimbaud, L., Papaïx, J., Rey, J.-F., Barrett, L. G. & Thrall, P. H. Assessing the durability and efficiency of landscape-based strategies to deploy plant resistance to pathogens. PLoS Comp. Biol. 14, e1006067 (2018).
Camacho-Sanchez, M., Burraco, P., Gomez-Mestre, I. & Leonard, J. A. Preservation of RNA and DNA from mammal samples under field conditions. Mol. Ecol. Resour. 13, 663–673 (2013).
Li, C. et al. An efficient method to clone TAL effector genes from Xanthomonas oryzae using Gibson assembly. Mol. Plant Pathol. https://doi.org/10.1111/mpp.12820 (2019).
Carpenter, S. C. D. et al. A strain of an emerging Indian Xanthomonas oryzae pv. oryzae pathotype defeats the rice bacterial blight resistance gene xa13 without inducing a clade III SWEET gene and is nearly identical to a recent Thai isolate. Front. Microbiol. 9, 2703 (2018).
Cohn, M. et al. Xanthomonas axonopodis virulence is promoted by a transcription activator-like effector-mediated induction of a SWEET sugar transporter in cassava. Mol. Plant Microbe Interact. 27, 1186–1198 (2014).
Cox, K. L. et al. TAL effector driven induction of a SWEET gene confers susceptibility to bacterial blight of cotton. Nat. Comm. 8, 15588 (2017).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-∆∆ CT method. Methods 25, 402–408 (2001).
Ouwerkerk, P. B., de Kam, R. J., Hoge, H. J. & Meijer, A. H. Glucocorticoid-inducible gene expression in rice. Planta 213, 370–378 (2001).
Yang, B. & Bogdanove, A. in Rice Protocols (ed. Yang, Y.) 249–255 (Humana Press, 2013).
Yang, B. & White, F. F. Diverse members of the AvrBs3/PthA family of type III effectors are major virulence determinants in bacterial blight disease of rice. Mol. Plant Microbe Interact. 17, 1192–1200 (2004).
Kauffman, H. E., Reddy, A. P. K., Hsieh, S. P. Y. & Merca, S. D. An improved technique for evaluating resistance of rice varieties to Xanthomonas oryzae. Plant Dis. Rep. 57, 537–541 (1973).
Acknowledgements
We are grateful to M. Stiebner, A. Crepcia-Pevzner, M. Bezrutczyk and P. Maldonado (Heinrich Heine University) for technical assistance and B.-H. Hou and D. Sosso (Carnegie) for transport assays in HEK293T cells. We gratefully acknowledge funding from the Bill and Melinda Gates Foundation to Heinrich Heine University, with subawards to Iowa State University/University of Missouri, University of Florida and International Rice Rsearch Institute. Characterization of SWEET promoters was supported by a joint grant from the National Science Foundation to B.Y., F.F.W. and W.B.F. (IOS-1258018 and IOS-1258028). W.B.F. was supported by an Alexander-von-Humboldt Professorship.
Author information
Authors and Affiliations
Contributions
J.-S.E., J.Y., R.O., C.V.C., B.S., F.F.W., B.Y. and W.B.F. conceived and designed experiments. C.J., J.-S.E., J.Y., V.T.L., S.N.C. and D.L. performed experiments. B.L. transformed Kitaake. J.-S.E., C.J., J.C.H.-T., G.A.-G., V.T.L., J.Y., B.Y. and W.B.F. analyzed the data. H.N. did informatics for PathoTracer. J.-S.E., S.M.S., B.Y. and W.B.F. wrote the manuscript, and J.-S.E., J.Y., R.O., S.M.S. and F.F.W. helped with revisions.
Corresponding authors
Ethics declarations
Competing interests
W.B.F., J.S.E., F.W., B.Y. and R.O. are inventors on US provisional patent application 62832300 that covers Kitaake, IR64 and Ciherang-Sub1 EBE-edited lines and kit components described here.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Alignment of the SWEET11 promoter sequences from selected rice varieties.
Rice varieties having nucleotide variations in the PthXo1 EBE were identified using RiceVarMap v.2 (http://ricevarmap.ncpgr.cn/v2/). Two varieties were selected for each variation type as representative. Sequences of the first 400 bp of SWEET11 promoters of the selected varieties were extracted from the 3K database (http://snp-seek.irri.org/). Alignment was done using ClustalW (v 2.1) in Geneious 11.1.5 (https://www.geneious.com). One A/G variation was found in the PthXo1 EBE. Variation was observed with a frequency of 0.2% in 4,726 rice varieties.
Supplementary Figure 2 Alignment of the SWEET13 promoter sequences from selected rice varieties.
Rice varieties having nucleotide variations in the PthXo2 EBE were identified using RiceVarMap v.2 (http://ricevarmap.ncpgr.cn/v2/). Two varieties were selected for each variation type as representative. Sequences of the first 400 bp of SWEET13 promoters of the selected varieties were extracted from the 3K database (http://snp-seek.irri.org/). Alignment was done using ClustalW in Geneious 11.1.5 (https://www.geneious.com). Nine variations were found in the PthXo2 EBE with frequencies ranging from 1.3% to 20.8%.
Supplementary Figure 3 Alignment of the SWEET14 promoter sequences from selected rice varieties.
Rice varieties having nucleotide variations in the PthXo3, TalC, AvrXa7 and TalF EBEs were identified using RiceVarMap v.2 (http://ricevarmap.ncpgr.cn/v2/). Two varieties were selected for each variation type as representative. Sequences of the first 400 bp of SWEET14 promoters of the selected varieties were extracted from the 3K database (http://snp-seek.irri.org/). Alignment was done using ClustalW in Geneious 11.1.5 (https://www.geneious.com). In the PthXo3/AvrXa7 EBEs, there was one A insertion with a frequency of 7.7%. CX371 and CX372 had one G/T variation in the TalC EBE and an 18-bp deletion in the PthXo3/AvrXa7 and TalF EBEs.
Supplementary Figure 4 SWEET mRNA levels in uninfected rice leaves.
a, Relative mRNA levels (qRT-PCR) of SWEET11, SWEET13, SWEET14 and SWEET15 in different regions of rice flag leaves. Samples were harvested at 12 pm (mean ± s.e.m., n = 3 leaf samples from siblings grown in parallel) with expression normalized to rice Ubiquitin1 levels. This experiment was repeated independently three times with similar results). b, Tissue-specific expression pattern of SWEET13 from public microarray data (http://ricexpro.dna.affrc.go.jp).
Supplementary Figure 5 Transcriptional fusion reporter lines for SWEET11, 13 and 14.
GUS staining patterns of SWEET11, 13 and 14 transcriptional GUS fusion lines. Transcriptional GUS fusion lines show a non-specific expression pattern in leaf tissues. a, SWEET11 transcriptional GUS fusion lines. b, SWEET13 transcriptional GUS fusion lines. c, SWEET14 transcriptional GUS fusion lines. Scale bar, 1 mm. This experiment was repeated independently at least three times (n = 3 leaf samples from siblings grown in parallel) with similar results.
Supplementary Figure 6
Map of the SWEET11 translational reporter fusion constructs.
Supplementary Figure 7
Map of the SWEET13 translational reporter fusion constructs.
Supplementary Figure 8
Map of the SWEET14 translational reporter fusion constructs.
Supplementary Figure 9 SWEET protein accumulation in rice leaves infected with Xoo strains expressing a specific TALe.
SWEET protein accumulation upon infection with Xoo-containing specific TAL effectors. Translational GUS fusion lines were infected with an ME2 strain harboring a specific effector. SWEET11 was induced upon inoculation with ME2 expressing the PthXo1 effector. SWEET13 was induced by ME2 with the PthXo2B effector. SWEET14 was induced by ME2 with PthXo3, AvrXa7, TalC or TalF. This experiment was repeated independently twice with similar results.
Supplementary Figure 10 CRISPR-Cas9 editing of SWEET13 and SWEET14 for knock-out lines and predicted truncated form of transporters.
Mutagenesis of SWEET13 and SWEET14 using CRISPR/Cas9 genome editing. The guide RNA-targeting site is marked with an underline, and the PAM is marked in green. a, Mutagenesis scheme of SWEET13 and SWEET14. The dashed line denotes a deleted nucleotide in sweet13-1 (10 nt), sweet13-2 (4 nt) and sweet14-1 (1 nt), respectively. 1nt insertion in sweet14-2 is marked in blue. Both deletion and frameshift of amino acids occurred in the 1st exon and caused early termination. b, Predicted amino acid sequence of sweet13-1, sweet13-2, sweet14-1 and sweet14-2, respectively. In sweet13-1 and sweet13-2, frameshifts occured at the position of codons 8 and 7 of the original open reading frame, respectively, leading to polypeptides with altered sequence and length due to premature stop codons. c, If we assume that the second ATG (codon 58 in wild-type SWEET13) was used for protein production, only truncated proteins could be formed. In both mutants, the mutations will lead to loss of the first two transmembrane spanning domains, most likely leading to non-functional transporters. d, Predicted topology of the truncated SWEET14 protein in the sweet14-1 and sweet14-2 mutants in case codon 23 would serve a start codon. In both mutants, the mutations will lead to loss of the first transmembrane-spanning domain, most likely leading to non-functional transporters. Typically, premature stop codons affect RNA stability. Moreover, typically only the first ATG is used; thus, it is likely that all four lines completely lost the transport functions for the respective SWEETs.
Supplementary Figure 11 Sucrose transport activity and subcellular localization of SWEET13 from rice.
a, Sucrose transport activity by SWEET13 in HEK293T cells co-expressing the FLIPsuc90μ∆1V sucrose sensor. Cells expressing the sensor without SWEET13 were used as negative controls. SWEET14 served as a positive control (mean ± s.e.m.). b, Confocal Z-stack of Agrobacterium-infiltrated N. benthamiana epidermal leaf cells. ZmSWEET13a-eGFP fluorescence indicated localization at the plasma membrane. The eGFP signal (green) was merged with fluorescence derived from chloroplasts (667–773 nm) (blue). Both experiments were repeated at least three times independently. Scale bar, 50 µm.
Supplementary Figure 12 Molecular and phenotypic characterization of alleles of sweet13 mutants.
a, Number of seeds per panicle of greenhouse-grown wild-type sweet13-1 and -2 lines grown side by side (n = 4). No significant differences (P = 0.131 for sweet13-1 and P = 0.054 for sweet13-2) were observed with Student’s t-test. b, Total soluble sugars in wild-type and sweet13-1 and -2 flag leaves. Both mutants showed similar sugar concentrations compared to wild type. Samples were harvested at dusk (8 pm; n = 4 leaf samples from siblings grown in parallel and repeated independently at least three times, with similar results). Data were plotted using BoxPlotR (http://shiny.chemgrid.org/boxplotr/). Center lines show medians; box limits indicate the 25th and 75th percentiles as determined by R software; and data points were plotted as open circles. No significant differences were observed with Student’s t-test (sweet13-1: P = 0.318 for sucrose, P = 0.170 for glucose and P = 0.242 for fructose; sweet13-2: P = 0.824 for sucrose, P = 0.573 for glucose and P = 0.882 for fructose).
Supplementary Figure 13 mRNA levels of clade III SWEETs in sweet13 mutants.
a–c, Relative mRNA levels (2–ΔΔCt) of SWEET11, SWEET14 and SWEET15 in the sweet13-1 mutant (wild-type control: Kitaake). SWEET14 was the only SWEET clade III that showed significant upregulation in the mutant (mean ± s.e.m., n = 2 leaf samples from siblings grown in parallel, with mRNA levels normalized to the rice Ubiquitin1 levels, and repeated independently three times, with similar results). d, SWEET14 mRNA levels in the region around the laminar joint of the flag leaf (~1 cm of flag leaf blade base region plus laminar joint plus ~1 cm of flag leaf sheath top region) of the second allele sweet13-2 (in blue; wild-type control in white: Kitaake; center lines show the medians; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles; outliers are represented by dots; and data points are plotted as open circles; n = 4 sample points).
Supplementary Figure 14 SWEETko knock-out mutants as diagnostic tools.
Lesion length caused by ME2 (negative control, black circles, grey box outline), PXO99 (positive control, blue circles, blue box outline) and the African strain AXO1947 (red circles, faint blue boxes) on single, double and triple (sweet11, sweet13 and sweet14) knock-out mutants relative to Kitaake wild type. Lesion lengths measured at 14 DAI were plotted using BoxPlotR (http://shiny.chemgrid.org/boxplotr/). Center lines show medians; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles; and data points are plotted as open circles. Number of tests n is indicated below each box on the x-axis (numbers between 8 and 23). The same lower letters above the graph bars indicate no significant different at P < 0.05 by one-way ANOVA.
Supplementary Figure 15 PathoTracer visualization under the SWEETR kit 1.0 showing prevalence of Xoo strains with putative SWEET14 induction in the Philippines.
PathoTracer (http://webapps.irri.org/pathotracer/site2/) is an online repository that integrates genotypic and phenotypic pathogen data with resistance profiles of rice accessions to support the strategic deployment of varieties in the region. Highlighted in this figure is the population of Xoo collected from 1972 to 2012 in Laguna, a BB-disease endemic area in the Philippines (n = 1,294 isolates). A screenshot of the same map is shown in Fig. 6. The righthand side panel displays detailed information on the population structure of Xoo in that region in relation to putative induction of SWEET14 (n = 1,294 isolates) and any recommended varieties.
Supplementary Information
Supplementary Information
Supplementary Figs. 1–15, Supplementary Tables 1–11 and Supplementary Notes 1–3
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eom, JS., Luo, D., Atienza-Grande, G. et al. Diagnostic kit for rice blight resistance. Nat Biotechnol 37, 1372–1379 (2019). https://doi.org/10.1038/s41587-019-0268-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-019-0268-y
This article is cited by
-
Integrative genomic and transcriptomic analysis of Xanthomonas oryzae pv. oryzae pathotype IV, V, and IX in China reveals rice defense-responsive genes
Phytopathology Research (2024)
-
Constructed Rice Tracers Identify the Major Virulent Transcription Activator-Like Effectors of the Bacterial Leaf Blight Pathogen
Rice (2024)
-
Rice Promoter Editing: An Efficient Genetic Improvement Strategy
Rice (2024)
-
Engineering disease-resistant plants with alternative translation efficiency by switching uORF types through CRISPR
Science China Life Sciences (2024)
-
OsWRKY9 is Involved in Transcriptional Regulatory Cascade Enhancing Broad-Spectrum Disease Resistance
Journal of Plant Biology (2024)
