
Abstract
Characterization of microbiomes has been enabled by high-throughput metagenomic sequencing. However, existing methods are not designed to combine reads from short- and long-read technologies. We present a hybrid metagenomic assembler named OPERA-MS that integrates assembly-based metagenome clustering with repeat-aware, exact scaffolding to accurately assemble complex communities. Evaluation using defined in vitro and virtual gut microbiomes revealed that OPERA-MS assembles metagenomes with greater base pair accuracy than long-read (>5×; Canu), higher contiguity than short-read (~10× NGA50; MEGAHIT, IDBA-UD, metaSPAdes) and fewer assembly errors than non-metagenomic hybrid assemblers (2×; hybridSPAdes). OPERA-MS provides strain-resolved assembly in the presence of multiple genomes of the same species, high-quality reference genomes for rare species (<1%) with ~9× long-read coverage and near-complete genomes with higher coverage. We used OPERA-MS to assemble 28 gut metagenomes of antibiotic-treated patients, and showed that the inclusion of long nanopore reads produces more contiguous assemblies (200× improvement over short-read assemblies), including more than 80 closed plasmid or phage sequences and a new 263 kbp jumbo phage. High-quality hybrid assemblies enable an exquisitely detailed view of the gut resistome in human patients.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

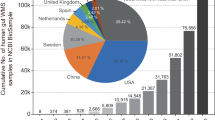
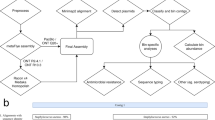
Data availability
GIS20 mock community sequencing data can be obtained from the European Nucleotide Archive (ENA) under project ID PRJEB29139 (Illumina, PacBio and ONT) and sequencing data for the 28 gut metagenomes can be found under project ID PRJEB29152 (Illumina and ONT).
Code availability
OPERA-MS is freely available under the MIT license at https://github.com/CSB5/OPERA-MS.
References
Zhu, B., Wang, X. & Li, L. Human gut microbiome: the second genome of human body. Protein Cell 1, 718–725 (2010).
Liu, L. et al. The human microbiome: a hot spot of microbial horizontal gene transfer. Genomics 100, 265–270 (2012).
Penders, J., Stobberingh, E. E., Savelkoul, P. H. M. & Wolffs, P. F. G. The human microbiome as a reservoir of antimicrobial resistance. Front. Microbiol. 4, 87 (2013).
Loman, N. J. et al. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA 309, 1502 (2013).
Forbes, J. D., Knox, N. C., Ronholm, J., Pagotto, F. & Reimer, A. Metagenomics: the next culture-independent game changer. Front. Microbiol. 8, 1069 (2017).
Sczyrba, A. et al. Critical assessment of metagenome interpretation—a benchmark of metagenomics software. Nat. Methods 14, 1063–1071 (2017).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Wu, Y.-W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Nielsen, H. B. et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32, 822–828 (2014).
Sangwan, N., Xia, F. & Gilbert, J. A. Recovering complete and draft population genomes from metagenome datasets. Microbiome 4, 8 (2016).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Brooks, B. et al. Strain-resolved analysis of hospital rooms and infants reveals overlap between the human and room microbiome. Nat. Commun. 8, 1814 (2017).
Frank, J. A. et al. Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Sci. Rep. 6, 25373 (2016).
Kuleshov, V. et al. Synthetic long-read sequencing reveals intraspecies diversity in the human microbiome. Nat. Biotechnol. 34, 64–9 (2016).
Beaulaurier, J. et al. Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat. Biotechnol. 36, 61–69 (2017).
Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018).
Juul, S. et al. What’s in my pot? Real-time species identification on the MinION. Preprint at bioRxiv https://doi.org/10.1101/030742 (2015).
Daims, H. et al. Complete nitrification by Nitrospira bacteria. Nature 528, 504–509 (2015).
Leggett, R. M. et al. Rapid MinION metagenomic profiling of the preterm infant gut microbiota to aid in pathogen diagnostics. Preprint at bioRxiv https://doi.org/10.1101/180406 (2017).
Koren, S. et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 30, 693–700 (2012).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput. Biol. 13, e1005595 (2017).
Yin, M. et al. Carriage duration of carbapenemase-producing Enterobacteriaceae in a hospital cohort - implications for infection control measures. Preprint at med Rxiv 2019/001479 (2019).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Peng, Y., Leung, H. C. M., Yiu, S. M. & Chin, F. Y. L. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Gao, S., Bertrand, D., Chia, B. K. H. & Nagarajan, N. OPERA-LG: efficient and exact scaffolding of large, repeat-rich eukaryotic genomes with performance guarantees. Genome Biol. 17, 102 (2016).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Antipov, D., Korobeynikov, A., McLean, J. S. & Pevzner, P. A. hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics 32, 1009–1015 (2016).
Hanson, N. W. et al. Metabolic pathways for the whole community. BMC Genomics 15, 619 (2014).
Nandi, T. et al. Gut microbiome recovery after antibiotic usage is mediated by specific bacterial species. Preprint at bioRxiv https://doi.org/10.1101/350470 (2018).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–55 (2015).
Pasolli, E. et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662.e20 (2019).
Orlek, A. et al. Plasmid classification in an era of whole-genome sequencing: application in studies of antibiotic resistance epidemiology. Front. Microbiol. 8, 182 (2017).
Yuan, Y. & Gao, M. Jumbo bacteriophages: an overview. Front. Microbiol. 8, 403 (2017).
Devoto, A. E. et al. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 4, 693–700 (2019).
Lin, D. M., Koskella, B. & Lin, H. C. Phage therapy: an alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 8, 162–173 (2017).
Morrill, H. J., Pogue, J. M., Kaye, K. S. & LaPlante, K. L. Treatment options for carbapenem-resistant Enterobacteriaceae infections. Open Forum Infect. Dis. 2, ofv050–ofv050 (2015).
Meletis, G., Chatzidimitriou, D. & Malisiovas, N. Double- and multi-carbapenemase-producers: the excessively armored bacilli of the current decade. Eur. J. Clin. Microbiol. Infect. Dis. 34, 1487–93 (2015).
Trecarichi, E. M. & Tumbarello, M. Therapeutic options for carbapenem-resistant Enterobacteriaceae infections. Virulence 8, 470–484 (2017).
Lee, C.-S. & Doi, Y. Therapy of infections due to carbapenem-resistant Gram-negative pathogens. Infect. Chemother. 46, 149–64 (2014).
Partridge, S. R. Analysis of antibiotic resistance regions in Gram-negative bacteria. FEMS Microbiol. Rev. 35, 820–55 (2011).
Press, M. O. et al. Hi-C deconvolution of a human gut microbiome yields high-quality draft genomes and reveals plasmid-genome interactions. Preprint at bioRxiv https://doi.org/10.1101/198713 (2017).
Bishara, A. et al. High-quality genome sequences of uncultured microbes by assembly of read clouds. Nat. Biotechnol. 36, 1067–1075 (2018).
Nayfach, S. & Pollard, K. S. Toward accurate and quantitative comparative metagenomics. Cell 166, 1103–1116 (2016).
Magnúsdóttir, S. et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35, 81–89 (2016).
Luo, C. et al. ConStrains identifies microbial strains in metagenomic datasets. Nat. Biotechnol. 33, 1045–52 (2015).
Quince, C. et al. DESMAN: a new tool for de novo extraction of strains from metagenomes. Genome Biol. 18, 181 (2017).
Mirzaei, M. K. & Maurice, C. F. Ménage à trois in the human gut: interactions between host, bacteria and phages. Nat. Rev. Microbiol. 15, 397–408 (2017).
Truong, D. T. et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903 (2015).
Loman, N. J. & Quinlan, A. R. Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics 30, 3399–3401 (2014).
Chaisson, M. J. & Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13, 238 (2012).
Sedlar, K., Kupkova, K. & Provaznik, I. Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics. Comput. Struct. Biotechnol. J. 15, 48–55 (2017).
RAFTERY, A. E. Bayes factors and BIC. Sociol. Methods Res. 27, 411–427 (1999).
Wasserman, L. Bayesian model selection and model averaging. J. Math. Psychol. 44, 92–107 (2000).
Navlakha, S., White, J., Nagarajan, N., Pop, M. & Kingsford, C. Finding biologically accurate clusterings in hierarchical tree decompositions using the variation of information. J. Comput. Biol. 17, 503–516 (2010).
Ondov, B. D. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Brown, C. T., Olm, M. R., Thomas, B. C. & Banfield, J. F. Measurement of bacterial replication rates in microbial communities. Nat. Biotechnol. 34, 1256–1263 (2016).
Rosenblatt, M. Remarks on some nonparametric estimates of a density function. Ann. Math. Stat. 27, 832–837 (1956).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at http://arxiv.org/abs/1303.3997 (2013).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Preprint at bioRxiv https://doi.org/10.1101/530972 (2019).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Sović, I. et al. Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nat. Commun. 7, 11307 (2016).
Mikheenko, A., Saveliev, V. & Gurevich, A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 32, 1088–90 (2016).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Gupta, S. K. et al. ARG-ANNOT, a New bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220 (2014).
Aziz, R. K. et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9, 75 (2008).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Marimuthu, K. et al. Clinical and molecular epidemiology of carbapenem-resistant enterobacteriaceae among adult inpatients in Singapore. Clin. Infect. Dis. 64, S68–S75 (2017).
Zerbino, D. R. & Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Gao, S., Bertrand, D. & Nagarajan, N. in Algorithms in Bioinformatics (eds Raphael, B. & Tang, J.) 314–325 (Springer, 2012).
Acknowledgments
This work was supported by funding from the National Healthcare Group (NHG-CSCS/12008 and SIDI/2013/008) to K.M. and O.T.N., BMRC IAF (IAF311018) to K.M., O.T.N. and N.N., HBMS IAF-PP (H18/01/a0/016) and A*STAR Singapore to N.N.
Author information
Authors and Affiliations
Contributions
D.B. and N.N. designed the algorithm with inputs from J.S. and M.S.K. D.B., J.S., M.K., M.S.K., M.D. and J.P.S. implemented OPERA-MS. D.B., M.K., C.L., J.Y.K., C.T. and K.R.C. conducted computational experiments and analysis with guidance from N.N. O.T.N., T.B., B.Y. and K.M. organized volunteer recruitment and sampling. A.H.Q.N. performed wet-lab experiments. D.B. and N.N. wrote the manuscript with inputs from M.K., K.R.C. and M.S. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–16, Supplementary Tables 1–5 and Supplementary Notes 1–3
Rights and permissions
About this article

Cite this article
Bertrand, D., Shaw, J., Kalathiyappan, M. et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat Biotechnol 37, 937–944 (2019). https://doi.org/10.1038/s41587-019-0191-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-019-0191-2
This article is cited by
-
Bacterial dynamics of the plastisphere microbiome exposed to sub-lethal antibiotic pollution
Microbiome (2024)
-
Distributions, interactions, and dynamics of prokaryotes and phages in a hybrid biological wastewater treatment system
Microbiome (2024)
-
MAGICIAN: MAG simulation for investigating criteria for bioinformatic analysis
BMC Genomics (2024)
-
VirRep: a hybrid language representation learning framework for identifying viruses from human gut metagenomes
Genome Biology (2024)
-
Comparative analysis of metagenomic classifiers for long-read sequencing datasets
BMC Bioinformatics (2024)
