
Abstract
Nitrogen (N2) fixation in oligotrophic surface waters is the main source of new nitrogen to the ocean1 and has a key role in fuelling the biological carbon pump2. Oceanic N2 fixation has been attributed almost exclusively to cyanobacteria, even though genes encoding nitrogenase, the enzyme that fixes N2 into ammonia, are widespread among marine bacteria and archaea3,4,5. Little is known about these non-cyanobacterial N2 fixers, and direct proof that they can fix nitrogen in the ocean has so far been lacking. Here we report the discovery of a non-cyanobacterial N2-fixing symbiont, ‘Candidatus Tectiglobus diatomicola’, which provides its diatom host with fixed nitrogen in return for photosynthetic carbon. The N2-fixing symbiont belongs to the order Rhizobiales and its association with a unicellular diatom expands the known hosts for this order beyond the well-known N2-fixing rhizobia–legume symbioses on land6. Our results show that the rhizobia–diatom symbioses can contribute as much fixed nitrogen as can cyanobacterial N2 fixers in the tropical North Atlantic, and that they might be responsible for N2 fixation in the vast regions of the ocean in which cyanobacteria are too rare to account for the measured rates.
Similar content being viewed by others


Main
Nitrogen is an essential component of all living organisms and limits life in the ocean. Atmospheric N2 gas is the largest reservoir of freely accessible nitrogen, but it is biologically available only to microorganisms that carry the nitrogenase metalloenzyme and thus can fix N2 into ammonia7. Even though a wide diversity of marine bacteria and archaea encode nitrogenase, the bulk of nitrogen fixation in the ocean has been attributed to cyanobacteria (ref. 4 and references therein). These phototrophs are capable of both free-living and symbiotic lifestyles, and can directly or indirectly contribute to carbon fixation and export production in the regions where they are abundant, such as oligotrophic coastal waters and margins of subtropical gyres8. Notably, in vast regions of the ocean, such as the centres of subtropical gyres, cyanobacterial N2 fixers are too rare to account for the measured rates of N2 fixation. Instead, a role of non-cyanobacterial N2 fixers has been invoked, on the basis of the abundance of nitrogenase-encoding gene sequences (nifH), most of which belong to uncultured proteobacteria (for example, refs. 3,5,9,10,11). So far, the most frequently detected non-cyanobacterial N2 fixer is the so-called gamma-A, named after its nifH gene phylogeny that clusters within the Gammaproteobacteria12. This enigmatic microorganism has been shown to be distributed in most world oceans, and its potential activity has been inferred from in situ nifH transcription13,14. To date, however, there is no proof that gamma-A fixes N2 in situ, and essentially all aspects of its physiology remain unknown.
An N2-fixing rhizobial diatom endophyte
We investigated the role of non-cyanobacterial N2 fixation in the tropical North Atlantic during an expedition in January–February 2020. This region is responsible for around 20% of oceanic N2 fixation8, and cyanobacteria can only explain approximately half of the rates measured in the region10. We detected high N2 fixation rates of up to 40 nmol N l−1 d−1 in the surface waters (Extended Data Table 1), and the presence of both cyanobacterial and heterotrophic N2 fixers—specifically, gamma-A—was confirmed by metagenomic sequencing (Extended Data Fig. 1a). Gamma-A nifH sequences were retrieved only from the large size fraction (greater than 3 µm) suggesting particle attachment or an association with a host organism (Extended Data Fig. 1a). We recovered a near-complete metagenome-assembled genome (MAG; 1.7 Mb, 37.8% GC, 98% completion with 0% redundancy) containing the gamma-A nifH gene, as well as a complete cluster of rRNA genes (Supplementary Table 1). Although the retrieved nifH sequence clustered within the Gammaproteobacteria as previously reported3,14,15 (Extended Data Fig. 2), both 16S-rRNA-gene-based and whole-genome-based taxonomy16 firmly placed this MAG within the alphaproteobacterial family Hyphomicrobiaceae (Fig. 1a). This family belongs to the order Rhizobiales, which comprises the prominent rhizobial symbionts of nodule-forming terrestrial legumes6,17,18. In addition to nifH, most other genes of the nif regulon are of gammaproteobacterial origin, including nifD and nifK, which encode the catalytic component of the nitrogenase; nifE, nifN and nifB, which encode the iron-molybdenum cofactor assembly proteins; and nifS, which is involved in metallocluster biosynthesis (Extended Data Fig. 2a). Almost all other genes in the gamma-A MAG are of alphaproteobacterial origin (Supplementary Table 1). On the basis of these results, we conclude that the gamma-A N2 fixer is, in fact, an alphaproteobacterium that has acquired its nitrogenase genes through horizontal gene transfer from a gammaproteobacterial donor. Besides gamma-A, several other bacteria, including members of the order Rhizobiales, obtained their nitrogenase genes through horizontal gene transfer from a gammaproteobacterial donor (Extended Data Fig. 2b). Such horizontal gene transfer across classes, resulting in the acquisition of nitrogenase genes, has been reported previously for other N2 fixers19,20.

a, Maximum likelihood phylogenetic tree of concatenated bacterial marker genes from the order Rhizobiales, showing the placement of Ca. T. diatomicola within the Hyphomicrobiaceae family (see Methods). The novel genus Ca. Tectiglobus, comprising Ca. T. diatomicola and its closest relative Ca. T. profundi, is highlighted in pink. Families within the Rhizobiales that contain known N2-fixing legume symbionts and their exemplary host plants are shown. The order Parvibaculales was used as an outgroup. Black dots indicate more than 95% bootstrap support. Scale bar indicates amino acid substitutions per site. Plant icons were designed by Freepik (Neptunia oleracea) or created with BioRender.com. b,c, False coloured scanning electron microscopy (SEM) image (b) and confocal laser scanning microscopy image (c) of a Haslea diatom. Four Ca. T. diatomicola cells (pink, overlay of Hypho1147 and Hypho734 fluorescence in situ hybridization (FISH) probes; Extended Data Table 2) were detected next to the host nucleus (white; stained with DAPI). Scale bars, 5 µm.
We name the newly discovered species ‘Candidatus Tectiglobus diatomicola’ within a novel genus ‘Candidatus Tectiglobus’ (see Methods for etymology). One other marine MAG from the North Pacific, which we now name ‘Candidatus Tectiglobus profundi’, is affiliated with this novel genus, with 72% average amino acid identity with Ca. T. diatomicola (Supplementary Methods). Compared with their closest relative, a MAG from the Mediterranean Sea, both Ca. Tectiglobus species have a substantially reduced genome size (around 1.7 Mb versus around 5 Mb) and a strongly decreased GC content (around 38% versus around 54%) (Extended Data Fig. 3), which are features typical of endosymbionts21. Notably, a similar reduction in genome size and GC content is observed for the N2-fixing cyanobacterial endosymbiont Candidatus Atelocyanobacterium thalassa, or UCYN-A, which lives in symbiosis with a haptophyte alga22,23. Thus, the genome properties of Ca. T. diatomicola, together with its presence in the large size fraction, strongly indicate a host-associated lifestyle.
We designed specific 16S rRNA oligonucleotide probes to visualize Ca. T. diatomicola (Extended Data Table 2), and found hybridized cells (1–2 µm cocci) that were located exclusively inside diatom hosts (Fig. 1b,c and Extended Data Fig. 4). The hosts showed large variation in cell sizes (20–58 μm long and 3–8 μm wide), probably representing different diatom life stages24. Typically, four Ca. T. diatomicola symbionts were observed in the proximity of the centrally located host nucleus, with some dividing hosts containing up to eight symbionts (Fig. 1c and Extended Data Fig. 4). On the basis of scanning electron micrographs combined with fluorescence microscopy, the host was identified as a pennate diatom likely to belong to the genus Haslea within the Naviculaceae family (Extended Data Fig. 4; see also Supplementary Information). Indeed, sequences belonging to this genus were recovered from the metagenome with the highest abundance of Ca. T. diatomicola (Supplementary Table 2, see also Methods). Haslea are ubiquitous marine diatoms that are found in surface waters (Extended Data Fig. 5a) and coastal sediments throughout the world’s oceans25, but they have not previously been reported to contain N2-fixing symbionts. Most marine diatom species that form associations with N2-fixing cyanobacteria are centric diatoms such as Hemiaulus, Rhizosolenia and Chaetoceros26. The new Haslea hosts thus expand the range of diatoms that can associate with N2-fixing symbionts. More importantly, all so-far-known N2-fixing symbionts of diatoms belong exclusively to the cyanobacteria27,28. Our discovery represents the first example—to our knowledge—of a symbiosis between a diatom and a non-cyanobacterial N2-fixing microorganism.
Host–symbiont metabolic interactions
To gain insights into the metabolic interactions between Ca. T. diatomicola and its Haslea host, we studied the Ca. T. diatomicola genome together with its in situ transcriptome. The Ca. T. diatomicola genome encodes all genes necessary for N2 fixation to ammonia, most of which were highly transcribed (Fig. 2a,b and Supplementary Table 1). These include key genes encoding the nitrogenase (nifH, nifD and nifK) and the iron-molybdenum cofactor assembly proteins (nifE, nifN and nifB).

a, Circular representation of the Ca. T. diatomicola genome with 13 encoding contigs (grey), GC content (black) and the average transcription of protein-coding genes as transcripts per million (TPM) (blue; TPM values higher than 800 were cut off). Genes related to N2 fixation (orange), electron transport chain and ATP generation (blue) and the TCA cycle (red) are highlighted. CDS, coding sequence; comp., completeness; red., redundancy; tmRNA, transfer-messenger RNA. b, Schematic of the proposed metabolic potential of Ca. T. diatomicola (white) and its interactions with Haslea (grey and green), indicating the transfer of fixed nitrogen from the N2-fixing symbiont in return for diatom-derived C4-dicarboxylic acids, such as succinate. Proteins and corresponding gene names are: Complex I (NADH–quinone oxidoreductase, nuoBNEF), Complex II (succinate dehydrogenase, sdhABCD), Complex III (cytochrome b/c1, fbcH_1/2, fbcF), Complex IV (cbb3-type oxidase, ccoNOP), Complex V (ATP synthase, atpABDEGF); fumarate hydratase (fumC); aconitate hydratase (acnB); 2-oxoglutarate dehydrogenase (sucAB, lpd); succinyl-CoA synthetase (sucCD); malate dehydrogenase (mdh); isocitrate dehydrogenase (icd); citrate synthase (gltA); nitrogenase (nifHDK) and its ancillary proteins (nifAENBMQSTUVWXZ) and ferredoxins (fdxABN); rnf complex (rnfBCD); dicarboxylic acid transporter (dctPQM); pyruvate dehydrogenase (aceEF, lpd); pyruvate kinase (pyk); malic enzyme (maeB); and phosphoenolpyruvate carboxykinase (pckA). 2-OG, 2-oxoglutarate; PEP, phosphoenolpyruvate.
Ca. T. diatomicola has a strongly reduced genome size, but the genome still encodes core carbon-processing pathways such as glycolysis and the tricarboxylic acid (TCA) cycle, which are present in many heterotrophic bacteria. However, on the basis of the low transcription of glycolysis genes (Supplementary Table 1), Ca. T. diatomicola probably does not grow on sugars. Instead, many genes involved in the TCA cycle were highly transcribed, in particular malate (mdh) and succinate (sdh) using enzymes (Fig. 2a,b and Supplementary Table 1), indicating growth on dicarboxylic acids. The dicarboxylates can be converted via pyruvate to acetyl-CoA, driving the TCA cycle independent of the glycolysis pathway (Fig. 2b). This is supported by the high transcription of genes encoding a TRAP-type dicarboxylic acid transporter (dctP, dctQ and dctM) and enzymes that decarboxylate malate (maeB) and oxaloacetate (pckA and pyk) to phosphoenolpyruvate and pyruvate. On the basis of the combined genomic and transcriptomic data, it seems that the N2-fixing Ca. T. diatomicola provides ammonia to the Haslea diatom host in return for dicarboxylic acids (Fig. 2b). This metabolite exchange is strongly reminiscent of the metabolic interaction in rhizobia–legume symbioses29,30, in which N2-fixing rhizobia grow on host-provided dicarboxylic acids, such as succinate and malate, and in return provide fixed nitrogen to the host plant. By contrast, in symbioses between marine diatoms and N2-fixing cyanobacteria, both partners are photosynthetic and grow on inorganic carbon31.
Notably, Ca. T. diatomicola seems to have lost its low-affinity terminal oxidase (Supplementary Table 1), which is typically present in other members of the Hyphomicrobiaceae family, with the notable exception of Ca. T. profundi (Supplementary Table 3). Instead, Ca. T. diatomicola encodes and highly transcribes the high-affinity cytochrome cbb3-type (ccoN, ccoO and ccoP) terminal oxidase (Fig. 2a, Extended Data Fig. 6 and Supplementary Table 1), which is used for respiration under low-oxygen conditions, and is generally poorly transcribed in high-oxygen environments such as the oxic surface waters of the tropical North Atlantic32. Legume-associated N2-fixing rhizobia also rely on high-affinity terminal oxidases when growing symbiotically29, because the plant hosts restrict the oxygen supply to the symbionts to control their growth and optimize nitrogen fixation33. The legume hosts also suppress the activity of the AMT-type ammonium transporters of nodulating rhizobia, to prevent the uptake of ammonium by the bacteria and to enhance ammonium transfer to the plant30. The lack of AMT transporters in Ca. T. diatomicola would similarly maximize the transfer of ammonia to the Haslea host. Ca. T. diatomicola seems to lack the capacity for de novo biosynthesis of some essential amino acids (aromatic amino acids, histidine and proline) and vitamins (for example, biotin and thiamine; Supplementary Table 1), a trait also found in nodulating Rhizobiales that are dependent on their plant host for these essential compounds34,35. Together, these results indicate that, similarly to nodulating rhizobia in legume symbioses, growth and N2 fixation by Ca. T. diatomicola is tightly regulated by its host.
To confirm that Ca. T. diatomicola fixes N2, we measured the assimilation of 15N from 15N2 in individual Ca. T. diatomicola–Haslea symbioses using nanoscale secondary ion mass spectrometry (nanoSIMS). All investigated Ca. T. diatomicola cells fixed 15N2 and more than 99% of the fixed nitrogen was subsequently transferred to the diatom host, which is likely to have been facilitated by the lack of AMT transporters in Ca. T. diatomicola (Fig. 3a,c). As such, the symbiont fixed 100-fold more nitrogen than would be needed for its own growth, which is similar to previous reports for N2-fixing cyanobacteria–diatom symbioses26.

a,b, NanoSIMS images showing the enrichment in 15N from 15N2 fixation (a) and 13C from 13CO2 fixation (b). The inset shows the corresponding fluorescence image after hybridization of Ca. T. diatomicola cells (indicated by white arrowheads) with specific oligonucleotide probes (in pink, overlay of Hypho638–Hypho825 mix in blue and Hypho1147 in red, respectively) (Extended Data Table 2). Scale bars, 5 µm. c, Cellular CO2 and N2 fixation rates of Ca. T. diatomicola symbionts (pink triangles, n = 64) and their diatom hosts (blue circles, n = 16). d, Carbon-based growth rates of symbionts (pink triangles, n = 64) and hosts (blue circles, n = 16) (black lines indicate mean; see Methods).
Single-cell uptake of 13C carbon from 13CO2, measured simultaneously with N2 fixation, revealed that the photosynthetic diatom in return transferred around 1% of fixed carbon to the symbiont for growth (Fig. 3b,c). The carbon supplied by the diatom might also be stored as glycogen, lipids or Calvin–Benson–Bassham-cycle products, as indicated by the carbon-rich biomass of the symbiont relative to the diatom host (Extended Data Fig. 7). Similar to nodulating rhizobia, Ca. T. diatomicola might store reduced carbon compounds to regulate its carbon flux and act as reductant storage30. Furthermore, on the basis of the similar 13C enrichments, both Ca. T. diatomicola and the Haslea host have comparable carbon-based growth rates (0.6 ± 0.3 and 0.8 ± 0.1 divisions per day (mean ± s.d.), respectively; Fig. 3d). When considered together with microscopic observations of dividing Ca. T. diatomicola–Haslea symbioses, this indicates the coordinated division of the symbiotic partners and vertical transmission of the symbiont (Extended Data Fig. 4a–d). Such an intricate coordination between host and symbiont growth is required for the long-term persistence and stability of a symbiosis36. Moreover, the fast growth of the rhizobia–diatom symbioses (mean, around 0.8 d−1; Extended Data Table 1) relative to the cyanobacteria–diatom symbioses (mean, around 0.2 d−1; Extended Data Table 1) suggests that Ca. T. diatomicola might contribute substantially to the nitrogen input to the oligotrophic tropical North Atlantic.
Ecological and evolutionary implications
To assess the relative importance of the Ca. T. diatomicola–Haslea symbiosis for N2 fixation in surface waters of the tropical North Atlantic, we calculated their total N2 fixation activity on the basis of their cellular N2 fixation rates and abundance. Owing to the large transfer of nitrogen from the Ca. T. diatomicola symbiont to its host diatom (Fig. 3a,c), the biomass of both Ca. T. diatomicola and its Haslea host was considered for calculations (see Supplementary Methods). On average, N2 fixation rates for the Ca. T. diatomicola–Haslea symbiosis were around 650 fmol N d−1, which equates to around 1.5 nmol N l−1 d−1 on the basis of its in situ abundance (around 2,000 cells per litre; Extended Data Table 1). This is comparable to the combined contribution of the most abundant cyanobacteria–diatom symbioses that we observed in these waters: the cyanobacterium Richelia, which associates with the diatoms Hemiaulus and Guinardia (around 1.6 nmol N l−1 d−1; Extended Data Table 1). Moreover, N2 fixation by the Ca. T. diatomicola–Haslea symbiosis is in the same range of N2 fixation previously reported from this region for the most abundant cyanobacterial N2 fixers Trichodesmium and UCYN-A (up to 4 and 1.5 nmol N l−1 d−1, respectively)10. To our knowledge, our nanoSIMS measurements present the first direct quantitative results showing that non-cyanobacterial heterotrophic N2 fixers fix nitrogen in situ at rates that can account for a substantial part of the high N2 fixation in the tropical North Atlantic.
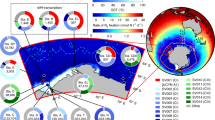
To investigate the global distribution of this symbiosis, we retrieved sequences related to Ca. T. diatomicola from our own and previously published metagenomes, as well as nifH abundances from compilations of quantitative PCR (qPCR) data8. These analyses revealed that Ca. T. diatomicola is widespread and present in all major oligotrophic ocean regions (Fig. 4a). Notably, our metagenomic data revealed the presence of the Ca. T. diatomicola symbiont in regions where gamma-A was previously not reported, such as the oligotrophic South Pacific, Indian and South Atlantic Oceans. In many of these oligotrophic regions, cyanobacterial N2 fixers are rare8 and thus cannot account for the measured N2 fixation. We hypothesize that part of this missing nitrogen is provided by the Ca. T. diatomicola symbiosis. Furthermore, genomic evidence suggests that the closest relative of Ca. T. diatomicola, Ca. T. profundi, is also a widespread heterotrophic N2-fixing symbiont (Fig. 4a, Extended Data Figs. 3 and 8 and Supplementary Methods). A global-scale metagenomic survey5 indicates that heterotrophic N2 fixers are more common than N2-fixing cyanobacteria in large parts of the surface ocean (Fig. 4b). Although the contribution of these heterotrophs to oceanic N2 fixation remains unclear, it is noteworthy that they were frequently retrieved from the large size fraction5 (greater than 3 µm; Extended Data Fig. 1a), suggesting possible host association. Hence, it might be common for N2-fixing heterotrophs to form obligate or facultative symbioses with diatoms or other unicellular algae. By living in symbiosis with photosynthetic hosts, heterotrophic N2 fixers would directly fuel CO2 drawdown and thus contribute to oceanic carbon sequestration.

a, Distribution of Ca. T. diatomicola (pink circles) and Ca. T. profundi (black circles) based on read detection in metagenome datasets from Tara Oceans and our own samples (see Methods and Supplementary Table 4). Black-and-pink circles are metagenomes in which both Ca. Tectiglobus species were detected. The abundance of Ca. T. diatomicola on the basis of gamma-A-specific nifH qPCR data is shown (circles in blue-to-yellow gradient; data from a previous study8). Sample locations in which gamma-A nifH qPCR counts were zero are shown in Extended Data Fig. 5b. b, Proportion of heterotrophic (orange) versus cyanobacterial (cyan) N2 fixers identified in a previous study5 (0.8–2,000 µm size fraction) in metagenome datasets from Tara Oceans.
Ca. T. diatomicola and probably Ca. T. profundi represent the first host-associated members of the family Hyphomicrobiaceae, as well as the first marine beneficial N2-fixing symbionts within the order Rhizobiales. Moreover, the finding that rhizobial N2 fixers can form tight symbioses with unicellular algae expands the known photosynthetic hosts for Rhizobiales beyond the well-described rhizobia–legume symbiosis17,18. Besides Ca. T. diatomicola and Ca. T. profundi, nine other members of the Hyphomicrobiaceae were found to have the genomic capacity to fix N2, eight of which contain nif genes that are of alphaproteobacterial origin (Extended Data Figs. 2 and 3). The alphaproteobacterial nifH, nifD, nifK, nifE, nifN, nifB and nifS gene sequences from these eight Hyphomicrobiaceae form deeply branching sister clades to two of the major nodulating Rhizobiales nif clusters (the Allorhizobium–Mesorhizobium–Rhizobium–Sinorhizobium and the Bradyrhizobium clusters; Extended Data Fig. 2b). The prevalence of nif genes throughout the Hyphomicrobiaceae family indicates that their last common ancestor was capable of N2 fixation. This trait was subsequently lost in some members and the ancestor of Ca. T. diatomicola and Ca. T. profundi is likely to have re-acquired the capacity to fix N2 through horizontal gene transfer from a gammaproteobacterial donor. Such loss and subsequent re-acquisition of N2 fixation capacity also occurred during the evolution of the nodulating Rhizobiales17,18.
Because the Hyphomicrobiaceae evolved more than 1,000 million years ago, well before nodulating Rhizobiales lineages began to form symbioses with legume plants around 100 million years ago17,18, we speculate that beneficial N2-fixing symbioses in the Rhizobiales order evolved independently in marine environments much earlier than the nodulating species on land. Although Ca. T. diatomicola and the nodulating Rhizobiales evolved from one common ancestor and have similar metabolic interactions with their hosts, different degrees of host dependency have resulted in different evolutionary genome adaptations. The terrestrial nodulating rhizobial lineages form facultative symbioses with their host, and have undergone genome expansion to accommodate both a free-living and an intracellular lifestyle18. By contrast, the marine Ca. T. diatomicola has strongly reduced its genome size, in line with its proposed obligate symbiotic lifestyle. As such, the evolutionary adaptations of Ca. T. diatomicola are similar to those of the endosymbiotic cyanobacterium UCYN-A, which functions as an early-stage N2-fixing organelle37. It is tempting to speculate that Ca. T. diatomicola, which fulfils the same function in diatoms as UCYN-A does in haptophyte algae, is also in the early stages of becoming an N2-fixing organelle. This raises the possibility that endosymbiosis-derived N2-fixing organelles have originated not only from the cyanobacteria, but also from the Rhizobiales.
Nitrogen-fixing symbiotic Rhizobiales are crucial players in terrestrial productivity; they enable legumes to produce biomass through photosynthesis and consequently provide 20% of the proteins in food production (ref. 7 and references therein). Our results show that symbiotic marine N2-fixing Rhizobiales, such as Ca. T. diatomicola, are major contributors to oceanic N2 fixation and have a crucial role in sustaining marine productivity and global CO2 sequestration.
Methods
Etymology of the Candidatus taxa
‘Candidatus Tectiglobus’: Tec.ti.glo’bus. L. past part. tectus, hidden; L. masc. n. globus, a sphere; N.L. masc. n. Tectiglobus, a hidden sphere.
‘Candidatus Tectiglobus diatomicola’: di.a.to.mi’co.la. N.L. fem. n. diatoma, a diatom; L. suff. -cola (from L. masc. or fem. n. incola), inhabitant; N.L. masc. n. diatomicola, an inhabitant of diatoms.
‘Candidatus Tectiglobus profundi’: pro.fun’di. L. gen. n. profundi, from the depth of the sea, referring to the recovery of its genome from a 4,000-m-deep sediment trap.
Sample collection and experimental set-up
Sampling was performed during two parallel cruises on board RV Maria S. Merian (cruise MSM89; Bridgetown, Barbados–Bridgetown, Barbados) and RV Meteor (cruise M161; Bridgetown, Barbados–Ponta Delgada, Azores, Portugal) in January–February 2020 in the western tropical North Atlantic. Samples were obtained from Niskin rosette samplers equipped with conductivity, temperature and depth (CTD) systems. At each station, CTD casts were performed to obtain surface water (around 10 m) for dawn-to-dawn incubation experiments of CO2 and N2 fixation rates using stable isotope tracers. At the beginning of the incubation experiments, subsamples for DNA and RNA sequencing and FISH were taken. Samples for DNA and RNA sequencing were taken by sequential filtration of 10 l of seawater through 10-µm and 3-µm polycarbonate filters (Isopore, 47 mm diameter) followed by two parallel 0.22-µm Sterivex filters (5 l was filtered through each of the two 0.22-µm filters; all filters from Merck). After filtration, filters were flash-frozen in liquid nitrogen and stored at −80 °C until processing. At the end of the approximately 24-h incubation experiments, subsamples for measurements of bulk rate and for FISH and single-cell analyses were taken. On the RV Meteor cruise, additional DNA and RNA samples were collected at the end of the approximately 24-h incubation period through sequential size filtration of around 3 l of incubated seawater. Samples for FISH and nanoSIMS were preserved with methanol-free paraformaldehyde solution (1% w/v final concentration) either for around 24 h at 4 °C or for a few hours at 4 °C followed by 0.5 h at room temperature. Preserved samples were subsequently filtered onto polycarbonate filters (Isopore, 0.2 µm pore size, 25 mm diameter); all samples intended for nanoSIMS analyses were filtered onto gold (Au)-coated polycarbonate filters. Filters were subsequently rinsed with ultrapure water (MilliQ), dried and stored at −20 °C until further analyses.
Metagenomic and metatranscriptomic sequencing
Samples from a total of eight stations were selected for DNA and RNA extractions and subsequent long- and short-read metagenomic and metatranscriptomic sequencing. All library preparation steps and sequencing were performed at the Max Planck Genome Centre (http://mpgc.mpipz.mpg.de/home/). See Supplementary Methods for details of samples, DNA and RNA extraction protocols, library preparation for short- and long-read sequencing and quality trimming.
Recovery and annotation of the Candidatus Tectiglobus diatomicola genome
The genome of Ca. T. diatomicola was reconstructed from a deeply sequenced short-read metagenomic sample (around 315 Gb, 3–10 µm size fraction from surface water after 24 h of incubation, station 4 from the M161 cruise) together with long-read metagenomes from six stations from the MSM89 cruise (all size fractions from surface water before incubations), as follows. Raw metagenomic short reads were trimmed using Trimmomatic38 v.0.39 (ILLUMINACLIP:TruSeq3-PE.fa:2:30:10, LEADING:3, TRAILING:3, SLIDINGWINDOW:4:15, MINLEN:36) and assembled using MEGAHIT39 v.1.2.9. To reduce the size of the assembly (33 million contigs, totalling around 17.7 Gb), it was filtered to retain only contigs with a length of more than 2 kb, 25–40% GC content and a coverage of 29–44; the latter two parameters were chosen on the basis of a preliminary reconstruction of the Ca. T. diatomicola genome (for further details, see Supplementary Methods). The remaining 8,218 contigs were visualized in anvi’o40 v.7.1, and those with high similarity to the previously reconstructed Ca. T. diatomicola genome were identified using blastn (BLAST+ (ref. 41) v.2.9.0). Using this approach, a tightly clustered group of 189 contigs, most of which matched with contigs of the previously reconstructed Ca. T. diatomicola, was identified in anvi’o40 v.7.1. The 189 contigs were then iteratively extended and refined, and one iteration included the following steps (see ‘Code availability’). Long and short metagenomic reads were mapped onto contigs using minimap2 (ref. 42) v.2.22-r1101 (‘-ax map-hifi’ for long reads and ‘-ax sr --score-N 2’ for short reads) and the sam mapping files were converted into bam format using SAMtools43 v.1.14 (‘samtools view’) and filtered to retain only reads that mapped with more than 98% identity (and more than 80% of the read length for short reads only) using CoverM v.0.6.1 (https://github.com/wwood/CoverM). The remaining mapped long and short reads were converted into fasta format using SAMtools43 v.1.14 (‘samtools fasta’). Short-read pairs in which only a single read was mapped were completed using seqkit44 v.2.3.0. Subsequently, mapped long and short reads were assembled together using SPAdes45 v.3.15.3 (‘--isolate -k 21,33,55,77,99,111’, mapped long reads were supplied using ‘-s’ and scaffolds of the previous iteration were supplied using ‘--trusted-contigs’) and the assembled scaffolds were filtered to retain only scaffolds of at least 1 kb. The filtered scaffolds were then used as input for the next iteration. After 23 iterations, the scaffolds were manually inspected and refined using anvi’o40 v.7.1, resulting in the current Ca. T. diatomicola genome. Completeness and redundancy were estimated using CheckM2 (ref. 46) v.0.1.2 and taxonomy was assigned using GTDB-TK47 v.2.1.0 and the Genome Taxonomy Database (GTDB)16 v.r214.
The Ca. T. diatomicola genome was at first annotated using Prokka48 v.1.14.6. Additional information about gene functions was sourced from the RAST web server49 (https://rast.nmpdr.org/) and through DIAMOND50 v.2.0.8 similarity searches against the KEGG51 v.58 and eggNOG52 v.4.5 databases using the utility script ‘sqm_annot.pl’ from the SqueezeMeta metagenomics pipeline53 v.1.6.2. The utility script ‘sqm_annot.pl’ was further used for taxonomic annotation of all coding sequences using a last common ancestor approach53. For genes of interest on the nif cluster encoding contig, the taxonomic origin was further investigated using phylogenetic analyses and/or by manual inspection of their best blast hits (see Supplementary Methods). Highly transcribed genes (top 20%) were further inspected using the InterPro web server54 v.95.0-97.0 (https://www.ebi.ac.uk/interpro/result/InterProScan) and searches against the NCBI-nr database55 using the NCBI BLAST web server (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Genome comparisons
The gene content of the two Ca. Tectiglobus genomes was compared to that of two closely related MAGs (GCA_905480435 and GCA_002689605). The genes for each genome were annotated with COG identifiers using anvi’o40 v.7.1 (https://merenlab.org/2016/10/25/cog-annotation/) and genes within each COG category were summed. Whole-genome alignment and identification of blocks of conserved regions within the two Ca. Tectiglobus genomes were performed using mauve56 (development snapshot 2015-02-26).
Phylogenetic analyses
A maximum likelihood phylogenetic tree of Ca. T. diatomicola, Ca. T. profundi and all Rhizobiales and Parvibaculales (outgroup) genomes from the GTDB16 (v.r214), as well as HBD_Alpha_05 (a marine MAG from the Hyphomicrobiaceae family also containing nif genes5) was calculated on the basis of 16 ribosomal proteins57, using muscle58 v.3.8.1551 for alignment and FastTree59 v.2.1.11 for tree calculation. The resulting tree was visualized in iToL60 v.6.8.1. For further details and the phylogeny of marker proteins (NifH, NifD, NifK, NifE, NifN, NifB, NifS and CcoN), see Supplementary Methods.
Candidatus Tectiglobus diatomicola transcriptome analysis
To obtain gene transcription information for Ca. T. diatomicola, all sequenced metatranscriptome reads were combined and mapped to the Ca. T. diatomicola genome using BWA-MEM61 v.0.7.17-r1188, and the resulting mapping files were filtered requiring at least 95% sequence identity and at least 80% of the read to align (mapping and filtering were done through CoverM v.0.6.1). Gene counts were generated using featureCounts62 v.2.0.1 and TPM values for protein-coding genes were calculated as previously described63. The genome plot (Fig. 2a) including TPM values was generated using BRIG64 v.0.95 and DNAPlotter65 v.18.1.0.
Global abundance of Candidatus Tectiglobus diatomicola and Candidatus Tectiglobus profundi
To determine the global distribution and abundance of Ca. T. diatomicola and Ca. T. profundi, we analysed their presence in metagenomes and in publicly available qPCR data. To this end, we used metagenomes from the Tara Oceans campaign (total of 1,241 metagenomes from projects PRJEB4352, PRJEB1787, PRJEB9691 and PRJEB9740) and metagenomes that we obtained from the tropical North Atlantic and the South Pacific gyre (see Supplementary Table 4). The contigs for each of the two genomes were concatenated (not including the regions encoding the rRNA gene clusters to reduce non-specific read recruitment) and metagenomic reads were mapped using bbmap v.38.70 (https://sourceforge.net/projects/bbmap/) with a minimum identity threshold of 90%. We only considered each genome to be present in a metagenome when the breadth of coverage (fraction of the genome covered by at least one read) was close to the expected breadth following a previously reported formula66:
Expected breadth = 1 – e(−0.883 × coverage) (Extended Data Fig. 5c,d).
In addition, we downloaded gamma-A (that is, Ca. T. diatomicola) nifH qPCR data67 from a previous study8 (referred to as NCD_gammaA_nifH_gene and NCD_g24774A11_nifH_gene, around 2,500 data points), added a pseudocount of 1 to the nifH copy numbers, log-transformed the counts and filtered out all samples with a count value of 0. We then plotted the coordinates of all metagenomes in which either of the two Ca. Tectiglobus genomes were detected together with the log-transformed Ca. T. diatomicola nifH qPCR counts on a world map using R68 (Fig. 4). qPCR samples in which Ca. T. diatomicola (gamma-A) nifH had a count value of 0 were plotted separately (Extended Data Fig. 5b).
Software for bioinformatics analyses
Further software that was used during the analysis of the sequencing data that is described in the Supplementary Information: hifiasm-meta69 v.0.2-r043 for the assembly of long-read metagenomes; CompareM v.0.1.2 (https://github.com/dparks1134/CompareM) for the calculation of average amino acid identity between Ca. T. diatomicola and closely related genomes; fastANI70 v.1.33 for the calculation of average nucleotide identity between preliminary MAGs; USEARCH71 v.11.0.667 for clustering sequences on the basis of similarity before phylogenetic tree constructions; MAFFT72 v.7.505 for calculating and trimAl73 v.1.4.1 for trimming multiple sequence alignments; ModelFinder74 for predicting best-fitting models; and UFBoot2 (ref. 75) for calculating ultrafast bootstraps during the construction of maximum likelihood trees with IQ-TREE76 v.2.2.0.3 and v.2.2.2.7. The following software was used as part of the SqueezeMeta metagenomics pipeline53 v.1.6.2: Barrnap 0.9-dev (https://github.com/tseemann/barrnap) for the prediction of ribosomal RNAs; the RDP classifier77 v.2.10.2 for the taxonomic classification of predicted 16S rRNA sequences; prodigal78 v.2.6.3 for gene prediction; HMMER v.3.1b2 (http://hmmer.org/) for HMM homology searches against the Pfam database79; Bowtie2 (ref. 80) v.2.3.4.1 for mapping short reads; MetaBAT 2 (ref. 81) v.2.12.1, MaxBin 2.0 (ref. 82) and CONCOCT83 v1.1.0 for the binning of contigs into MAGs; DAS Tool84 v.1.1.1 for integrating the results from the three binning tools; and bbduk v38.87 (https://sourceforge.net/projects/bbmap/) for trimming of metatranscriptomic reads.
Bulk rates of CO2 and N2 fixation
Rates of CO2 and N2 fixation were determined as previously described10,85 (with a detailed description in the Supplementary Methods). Stable isotope incubations (15N-N2 and 13C-DIC (dissolved inorganic carbon)) were performed in triplicate for 24 h (dawn to dawn). Bottles were incubated in on-deck incubators continuously flushed with surface seawater, with simulated light conditions86. After around 24 h, subsamples were taken for elemental and isotopic biomass analyses as well as FISH and nanoSIMS analyses. Fixation rates were calculated on the basis of the incorporation of 13C and 15N into biomass (that is, the change in isotopic composition) for both bulk and single-cell activities (Supplementary Methods).
Visualization and abundance of Candidatus Tectiglobus diatomicola
To visualize the newly identified Ca. T. diatomicola, we designed FISH probes targeting the 16S rRNA87,88 (Supplementary Methods). In total, four FISH probes were designed: two specifically targeting Ca. T. diatomicola (Hypho825 and Hypho638) and two with a broader coverage, targeting many members of the Hyphomicrobiaceae (Hypho1147) and several members of the Hyphomicrobiaceae genera Hyphomicrobium, Filomicrobium and Pedomicrobium (Hypho734) (see Extended Data Table 2 and Supplementary Methods). The optimal formamide concentrations for these new probes were tested using Clone-FISH89 (Supplementary Methods).
The Ca. T. diatomicola cells were visualized using catalysed reporter deposition-FISH (CARD-FISH) with the four new horseradish peroxidase (HRP)-labelled probes either alone or in combination (double hybridization) and together with helpers or competitors to increase signal intensity (Extended Data Table 2). CARD-FISH was performed as previously described90. Microscopy was performed using a Zeiss Axio Imager.M2 wide-field epifluorescence microscope equipped with a Zeiss Axiocam 506 mono camera and Zeiss ZEN 3.2 blue edition software, a Zeiss LSM 780 confocal laser scanning microscope equipped with Zeiss Elyra PS.1 super-resolution-structured illumination microscopy and a laser microdissection (LMD) microscope (LMD 7000, Leica). For better identification of the diatom host of Ca. T. diatomicola, host diatoms containing FISH-positive cells were visualized by SEM with a FEI Quanta 250 FEG ESEM (Thermo Fisher Scientific, FEI) (see Supplementary Methods). Abundances of the Ca. T. diatomicola–Haslea symbiosis were determined as the number of diatom hosts containing FISH-positive cells from filter pieces representing around 25 ml of sampled water (Extended Data Table 1).
Abundance of free-living and host-associated cyanobacterial N2 fixers
Free-living (Trichodesmium, Crocosphaera) and host-associated (diatom-associated Richelia) cyanobacterial N2 fixers were identified by morphology and chlorophyll a- and/or phycoerythrin autofluorescence using an LMD microscope (LMD 7000, Leica). Richelia were found associated with the diatoms Guinardia, sometimes also referred to as Rhizosolenia, and Hemiaulus. Abundances were determined as the number of Richelia-containing host diatoms. Abundances of Trichodesmium were determined by measuring the total (summed) length of all free trichomes, and by dividing the total trichome length by the average cell length. Abundances of Crocosphaera cells were determined by direct cell counts. Abundances were determined from whole filters representing around 300–360 ml of sampled water (Extended Data Table 1).
Single-cell N2-fixation activities using nanoSIMS
Au-sputtered filters from the end of the stable isotope incubations were used for nanoSIMS (nanoSIMS 50L, CAMECA) to measure the single-cell carbon and nitrogen isotopic composition and to determine the single-cell CO2 and N2 fixation activities of Ca. T. diatomicola and their Haslea hosts as well as Richelia and their diatom hosts (Hemiaulus and Guinardia) (Extended Data Table 1). Both target symbioses were visualized as described above and subsequently marked using an LMD microscope (LMD 7000, Leica). NanoSIMS analyses were performed as previously described90 and details are provided in the Supplementary Methods. The isotopic ratios (13C/12C and 12C15N/12C14N) of regions of interest were determined by overlaying epifluorescence images of FISH-positive cells and their host diatom (for Ca. T. diatomicola–Haslea symbiosis) or brightfield and autofluorescence images (diatom-associated Richelia) with the nanoSIMS images (secondary electrons). For the elemental imaging (Extended Data Fig. 7), carbon (12C), nitrogen (12C14N) and secondary electrons were overlaid; for this image only, background signals were removed. Cellular rates and abundances were combined to determine the absolute contributions of the Ca. T. diatomicola–Haslea symbiosis and diatom-associated Richelia to the bulk N2 fixation rate as previously described10. For both the Ca. T. diatomicola–Haslea symbiosis and the diatom-associated Richelia, the amount of nitrogen recovered in the diatom was also taken into account. Carbon-based growth rates for individual cells as well as whole symbioses were determined using the equation provided in a previous report10 and mass-balancing host and symbiont. Single-cell activities and contributions in our study can be considered conservative because 13C/12C and 15N/14N ratios can be diluted during sample preparation, which can lead to underestimation91,92,93.
Statistics and reproducibility
For Fig. 1b,c, the correlative SEM and fluorescence (confocal as well as epifluorescence) images are representative of a total of 11 diatoms from surface waters of 3 independent environmental samples. Additional fluorescence images (Fig. 1c) were obtained from a total of 27 diatoms from 6 independent environmental samples.
For Fig. 3a,b, the correlative nanoSIMS images are representative of a total of 16 diatoms (containing a total of 64 symbionts) from 3 independent environmental samples.
For Extended Data Fig. 4, the correlative fluorescence (confocal as well as epifluorescence) and SEM images (Extended Data Fig. 4a–d) are representative of a total of 13 diatoms, which contained more than 4 symbionts, from 5 independent environmental samples. The NON338-probe image (Extended Data Fig. 4e) is representative of a total of 32 diatoms from 3 independent environmental samples; however, chloroplasts were not always visible. Extended Data Fig. 4f, showing Ca. T. diatomicola cells after hybridization with a specific oligonucleotide probe in close vicinity to the H-shaped nucleus and bilobed chloroplasts, is representative of a total of 18 diatoms from 3 independent environmental samples. The SEM image showing a whole diatom (Extended Data Fig. 4g) is representative of a total of 19 diatoms from 2 independent environmental samples. Extended Data Fig. 4h–k show a selection of magnified images that helped with the identification of the diatom host.
For Extended Data Fig. 7, one diatom was analysed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Read data from metagenomic analyses pertaining to Ca. T. diatomicola have been deposited at the NCBI under BioProject accession number PRJNA1036431, including the MAGs of Ca. T. diatomicola and Ca. T. profundi under the accession numbers JAZDSJ000000000 and DAWWJP000000000, respectively. RNA-sequencing data can be found under the same BioProject number with the accession numbers SRR26695118, SRR26695119 and SRR26695121–SRR26695130. The publicly available sequences used for phylogenetic tree construction and genome comparison can be found at the GTDB (https://gtdb.ecogenomic.org/) under the accession numbers given in Supplementary Data 1–9 (tree file for each tree). Publicly available MAGs from a previous study5 can be found at https://figshare.com/articles/dataset/Marine_diazotrophs/14248283. Tara Oceans metagenomes used in this study can be found at https://www.ncbi.nlm.nih.gov/bioproject/173486 with the following BioProject accessions used in this study: PRJEB4352 (size fractions for protists), PRJEB1787 (size fractions for prokaryotes), PRJEB9691 (size fractions for protists from polar circle samples) and PRJEB9740 (size fractions for prokaryotes from polar circle samples). For the reconstruction of the MAG of Ca. T. profundi, the metagenomic data that we used can be found at the GTDB (https://gtdb.ecogenomic.org/genome?gid=GCA_013214245.1; original MAG) and at the Sequence Read Archive under https://www.ncbi.nlm.nih.gov/bioproject/PRJNA482655 with the accession numbers SRR7648332, SRR7648341, SRR7648350, SRR7632647 and SRR7648334 (metagenomes used for MAG reconstruction). Publicly available qPCR data from a previous study8 can be found at https://doi.org/10.6084/m9.figshare.21677687.v3. Additional databases used in this study can found at the following links: eggNOG: http://eggnog45.embl.de/download/eggnog_4.5/data/NOG/; ncbi-nr database: https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz; pfam database: https://www.ebi.ac.uk/interpro/download/pfam/; and kegg database: http://andes.cnb.csic.es/SqueezeMeta/kegg.db.gz. Source data are provided with this paper.
Code availability
Custom scripts for iterative bin refinements are deposited at https://github.com/bresyd/mag_refinement/tree/main.
References
Gruber, N. & Galloway, J. N. An Earth-system perspective of the global nitrogen cycle. Nature 451, 293–296 (2008).
Wang, W.-L., Moore, J. K., Martiny, A. C. & Primeau, F. W. Convergent estimates of marine nitrogen fixation. Nature 566, 205–211 (2019).
Farnelid, H. et al. Nitrogenase gene amplicons from global marine surface waters are dominated by genes of non-cyanobacteria. PLoS One 6, e19223 (2011).
Zehr, J. P. & Capone, D. G. Changing perspectives in marine nitrogen fixation. Science 368, eaay9514 (2020).
Delmont, T. O. et al. Heterotrophic bacterial diazotrophs are more abundant than their cyanobacterial counterparts in metagenomes covering most of the sunlit ocean. ISME J. 16, 927–936 (2022).
Poole, P., Ramachandran, V. & Terpolilli, J. Rhizobia: from saprophytes to endosymbionts. Nat. Rev. Microbiol. 16, 291–303 (2018).
Kuypers, M. M. M., Marchant, H. K. & Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 16, 263–276 (2018).
Shao, Z. et al. Global oceanic diazotroph database version 2 and elevated estimate of global oceanic N2 fixation. Earth Syst. Sci. Data 15, 3673–3709 (2023).
Halm, H. et al. Heterotrophic organisms dominate nitrogen fixation in the South Pacific Gyre. ISME J. 6, 1238–1249 (2012).
Martínez-Pérez, C. et al. The small unicellular diazotrophic symbiont, UCYN-A, is a key player in the marine nitrogen cycle. Nat. Microbiol. 1, 16163 (2016).
Shiozaki, T. et al. Basin scale variability of active diazotrophs and nitrogen fixation in the North Pacific, from the tropics to the subarctic Bering Sea. Global Biogeochem. Cycles 31, 996–1009 (2017).
Zehr, J. P., Mellon, M. T. & Zani, S. New nitrogen-fixing microorganisms detected in oligotrophic oceans by amplification of nitrogenase (nifH) genes. Appl. Environ. Microbiol. 64, 3444–3450 (1998).
Riemann, L., Farnelid, H. & Steward, G. Nitrogenase genes in non-cyanobacterial plankton: prevalence, diversity and regulation in marine waters. Aquat. Microb. Ecol. 61, 235–247 (2010).
Langlois, R., Großkopf, T., Mills, M., Takeda, S. & LaRoche, J. Widespread distribution and expression of Gamma A (UMB), an uncultured, diazotrophic, γ-proteobacterial nifH phylotype. PLoS One 10, e0128912 (2015).
Langlois, R. J., Hümmer, D. & LaRoche, J. Abundances and distributions of the dominant nifH phylotypes in the Northern Atlantic Ocean. Appl. Environ. Microbiol. 74, 1922–1931 (2008).
Parks, D. H. et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004 (2018).
Garrido-Oter, R. et al. Modular traits of the rhizobiales root microbiota and their evolutionary relationship with symbiotic rhizobia. Cell Host Microbe 24, 155–167 (2018).
Wang, S., Meade, A., Lam, H.-M. & Luo, H. Evolutionary timeline and genomic plasticity underlying the lifestyle diversity in Rhizobiales. mSystems 5, e00438-20 (2020).
Koirala, A. & Brözel, V. S. Phylogeny of nitrogenase structural and assembly components reveals new insights into the origin and distribution of nitrogen fixation across bacteria and archaea. Microorganisms 9, 1662 (2021).
Bolhuis, H., Severin, I., Confurius-Guns, V., Wollenzien, U. I. A. & Stal, L. J. Horizontal transfer of the nitrogen fixation gene cluster in the cyanobacterium Microcoleus chthonoplastes. ISME J. 4, 121–130 (2010).
McCutcheon, J. P. & Moran, N. A. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26 (2012).
Tripp, H. J. et al. Metabolic streamlining in an open-ocean nitrogen-fixing cyanobacterium. Nature 464, 90–94 (2010).
Thompson, A. W. et al. Unicellular cyanobacterium symbiotic with a single-celled eukaryotic alga. Science 337, 1546–1550 (2012).
Armbrust, E. V. The life of diatoms in the world’s oceans. Nature 459, 185–192 (2009).
Sterrenburg, F. A. S., Tiffany, M. A., Hinz, F., Herwig, W. E. & Hargraves, P. E. Seven new species expand the morphological spectrum of Haslea. A comparison with Gyrosigma and Pleurosigma (Bacillariophyta). Phytotaxa 207, 143–162 (2015).
Foster, R. A. et al. Nitrogen fixation and transfer in open ocean diatom–cyanobacterial symbioses. ISME J. 5, 1484–1493 (2011).
Caputo, A., Nylander, J. A. A. & Foster, R. A. The genetic diversity and evolution of diatom-diazotroph associations highlights traits favoring symbiont integration. FEMS Microbiol. Lett. 366, fny297 (2019).
Schvarcz, C. R. et al. Overlooked and widespread pennate diatom–diazotroph symbioses in the sea. Nat. Commun. 13, 799 (2022).
Dixon, R. & Kahn, D. Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2, 621–631 (2004).
Udvardi, M. & Poole, P. S. Transport and metabolism in legume–rhizobia symbioses. Annu. Rev. Plant Biol. 64, 781–805 (2013).
Nieves-Morión, M., Flores, E. & Foster, R. A. Predicting substrate exchange in marine diatom-heterocystous cyanobacteria symbioses. Environ. Microbiol. 22, 2027–2052 (2020).
Berg, J. S. et al. How low can they go? Aerobic respiration by microorganisms under apparent anoxia. FEMS Microbiol. Rev. 46, fuac006 (2022).
Kiers, E. T., Rousseau, R. A., West, S. A. & Denison, R. F. Host sanctions and the legume–rhizobium mutualism. Nature 425, 78–81 (2003).
Dunn, M. F. Key roles of microsymbiont amino acid metabolism in rhizobia–legume interactions. Crit. Rev. Microbiol. 41, 411–451 (2015).
Palacios, O. A., Bashan, Y. & de-Bashan, L. E. Proven and potential involvement of vitamins in interactions of plants with plant growth-promoting bacteria—an overview. Biol. Fertil. Soils 50, 415–432 (2014).
Davy, S. K., Allemand, D. & Weis, V. M. Cell biology of cnidarian–dinoflagellate symbiosis. Microbiol. Mol. Biol. Rev. 76, 229–261 (2012).
Coale, T. H. et al. Nitrogen-fixing organelle in a marine alga. Science 384, 217–222 (2024).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Eren, A. M. et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6 (2021).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11, e0163962 (2016).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinformatics 70, e102 (2020).
Chklovski, A., Parks, D. H., Woodcroft, B. J. & Tyson, G. W. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 20, 1203–1212 (2023).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316 (2022).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Aziz, R. K. et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9, 75 (2008).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Huerta-Cepas, J. et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293 (2016).
Tamames, J. & Puente-Sánchez, F. SqueezeMeta, a highly portable, fully automatic metagenomic analysis pipeline. Front. Microbiol. 9, 3349 (2019).
Paysan-Lafosse, T. et al. InterPro in 2022. Nucleic Acids Res. 51, D418–D427 (2023).
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., & Wheeler, D. L. GenBank. Nucleic Acids Res. 33, D34–D38 (2005).
Darling, A. C. E., Mau, B., Blattner, F. R. & Perna, N. T. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2004).
Hug, L. A. et al. A new view of the tree of life. Nat. Microbiol. 1, 16048 (2016).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv https://doi.org/10.48550/arXiv.1303.3997 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Zhao, S., Ye, Z. & Stanton, R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA 26, 903–909 (2020).
Alikhan, N.-F., Petty, N. K., Ben Zakour, N. L. & Beatson, S. A. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12, 402 (2011).
Carver, T., Thomson, N., Bleasby, A., Berriman, M. & Parkhill, J. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics 25, 119–120 (2009).
Olm, M. R. et al. inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nat. Biotechnol. 39, 727–736 (2021).
Shao, Z. et al. Version 2 of the global ocean diazotroph database. Figshare https://doi.org/10.6084/m9.figshare.21677687.v3 (2023).
R Core Team. R: A Language and Environment for Statistical Computing. http://www.R-project.org/ (R Foundation for Statistical Computing, 2020).
Feng, X., Cheng, H., Portik, D. & Li, H. Metagenome assembly of high-fidelity long reads with hifiasm-meta. Nat. Methods 19, 671–674 (2022).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 7, e7359 (2019).
Wu, Y.-W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Sieber, C. M. K. et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843 (2018).
Großkopf, T. et al. Doubling of marine dinitrogen-fixation rates based on direct measurements. Nature 488, 361–364 (2012).
Duerschlag, J. et al. Niche partitioning by photosynthetic plankton as a driver of CO2-fixation across the oligotrophic South Pacific Subtropical Ocean. ISME J. 16, 465–476 (2022).
Kitzinger, K. et al. Single cell analyses reveal contrasting life strategies of the two main nitrifiers in the ocean. Nat. Commun. 11, 767 (2020).
Graf, J. S. et al. Anaerobic endosymbiont generates energy for ciliate host by denitrification. Nature 591, 445–450 (2021).
Schramm, A., Fuchs, B. M., Nielsen, J. L., Tonolla, M. & Stahl, D. A. Fluorescence in situ hybridization of 16S rRNA gene clones (Clone-FISH) for probe validation and screening of clone libraries. Environ. Microbiol. 4, 713–720 (2002).
Kitzinger, K. et al. in Fluorescence In-Situ Hybridization (FISH) for Microbial Cells: Methods and Concepts (eds Azevedo, N. F. & Almeida, C.) 207–224 (Springer, 2021).
Musat, N. et al. The effect of FISH and CARD-FISH on the isotopic composition of 13C- and 15N-labeled Pseudomonas putida cells measured by nanoSIMS. Syst. Appl. Microbiol. 37, 267–276 (2014).
Woebken, D. et al. Revisiting N2 fixation in Guerrero Negro intertidal microbial mats with a functional single-cell approach. ISME J. 9, 485–496 (2015).
Meyer, N. R., Fortney, J. L. & Dekas, A. E. NanoSIMS sample preparation decreases isotope enrichment: magnitude, variability and implications for single-cell rates of microbial activity. Environ. Microbiol. 23, 81–98 (2021).
Sargent, E. C. et al. Evidence for polyploidy in the globally important diazotroph Trichodesmium. FEMS Microbiol. Lett. 363, fnw244 (2016).
Bench, S. R., Ilikchyan, I. N., Tripp, H. J. & Zehr, J. P. Two strains of Crocosphaera watsonii with highly conserved genomes are distinguished by strain-specific features. Front. Microbiol. 2, 261 (2011).
Vernette, C. et al. The Ocean barcode atlas: a web service to explore the biodiversity and biogeography of marine organisms. Mol. Ecol. Resour. 21, 1347–1358 (2021).
Acknowledgements
We thank the Leitstelle Deutsche Forschungsschiffe of the German National Research Foundation (DFG) and the captains, crews and chief scientists J. Karstensen, G. Lavik and S. Kinne of the RV Maria S. Merian MSM89 and RV Meteor M161 cruises for their support; the Ministry of Foreign Affairs and Foreign Trade in Barbados and the Ministry of Foreign and CARICOM Affairs of the Republic of Trinidad and Tobago for permissions to conduct marine scientific research in territorial waters; B. Stevens and S. Bony for coordinating the EUREC4A++ project; D. Baranowski and his CTD team for help with sampling; M. Knutzen, G. Klockgether, S. Lilienthal, K. Imhoff, S. Piosek and N. Rujanski for technical support; P. Kociolek and L. Nicolas-Asselineau for advice on diatom taxonomy and phylogenomics, respectively; and A. Oren for his help with the etymology of Ca. T. diatomicola and Ca. T. profundi. The MSM89 and M161 cruises were financially supported by the DFG (funding numbers: GPF18-2-50 and GPF18-1-69, EUREC4A++) and the Bundesministerium für Bildung und Forschung. This study was funded by the Max Planck Society.
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
B.T., D.R.S. and S. Li processed and analysed all metagenomic and metatranscriptomic data. B.T., M.P., A.T.K. and W.M. performed ship-board sampling and experiments and analysed bulk rate data. M.P. and K.K. designed the FISH probes. D.T. and M.E. performed FISH and microscopy. A.T.K. performed nanoSIMS analyses. S. Littmann performed SEM. A.K. identified and characterized the diatom host. B.T., W.M. and M.M.M.K. designed the study. B.T., H.K.M., B.K., J.M., W.M. and M.M.M.K. wrote the manuscript with contributions from all coauthors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Douglas Capone, Tom O. Delmont and Philip Poole for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Relative abundance of heterotrophic and cyanobacterial N2 fixers in the tropical North Atlantic.
a, Relative abundance of ‘Ca. T. diatomicola’ (gamma-A), ‘Ca. T. profundi’, as well as selected cosmopolitan heterotrophic and cyanobacterial N2 fixers5 based on read detection in metagenome data from different size fractions from the tropical North Atlantic. Note that only few known heterotrophic N2 fixers were detected in the dataset, and that ‘Ca. T. diatomicola’ had the highest relative abundance. b, Relative abundance of the most abundant known N2 fixers determined from direct microscopy counts (surface waters of stations 4 and 7 from cruise M161 in the tropical North Atlantic). Note that the metagenome based relative abundances of Trichodesmium in the large size fraction overestimate its microscopy-based abundance by an order of magnitude, most likely due to the polyploidy of this genus94. By contrast, metagenome based relative abundances of Crocosphaera in the large size fraction underestimate its microscopy-based abundance, most likely due to DNA extraction biases95.
Extended Data Fig. 2 Organization and phylogeny of nitrogen fixation (nif) genes in ‘Candidatus Tectiglobus diatomicola’.
a, Organization of the nif regulon showing the phylogenetic affiliation and GC content of each gene. All nif genes are affiliated to gammaproteobacteria (yellow), whereas almost all other genes in and around the nif regulon are affiliated to alphaproteobacteria (blue). The presence of nifV indicates that unlike most nodulating rhizobia, ‘Ca. T. diatomicola’ has the capacity to synthesize homocitrate, a ligand of the FeMo cofactor6. Genes affiliated to gamma-and alphaproteobacteria were found together on individual PacBio reads (solid black lines). Note that there is no substantial difference in GC content between genes of gamma- and alphaproteobacterial origin. b, Maximum likelihood phylogenetic trees of NifHDKENBS sequences of the Pseudomonadota. Phylogeny is based on the alignment of full-length amino acid sequences retrieved from the GTDB. Cyanobacterial Nif sequences were used as outgroups. All ‘Ca. Tectiglobus diatomicola’/‘Ca. Tectiglobus profundi’ (pink line; 1) Nif protein sequences cluster with gammaproteobacterial sequences, while Nif sequences from other members of the Hyphomicrobiaceae family (purple lines; 2–9; Extended Data Fig. 3) cluster with alphaproteobacteria. These alphaproteobacterial Nif protein sequences form deeply branching sister clades to two of the major nodulating Rhizobiales nif clusters; i.e. Bradyrhizobium (I, solid line) and Allorhizobium-Mesorhizobium-Rhizobium-Sinorhizobium (II, dashed line). Note that some other Rhizobiales (turquoise lines; members of the Rhodobiaceae, Rhizobiaceae, Cohaesibacteraceae and BM303 families) Nif protein sequences also cluster with the gammaproteobacterial sequences indicating that they were also obtained via horizontal gene transfer from a (common) gammaproteobacterial donor. Tree scales indicate amino acid substitutions per site. Note that in panel A, individual nif genes are indicated by capital letters; fd, ferredoxin; hr, hemerythrin; hyp, hypothetical protein; prx, peroxiredoxin; rnd, ribonuclease D.
Extended Data Fig. 3 Maximum likelihood phylogenetic tree of concatenated bacterial marker genes, highlighting genomic features of the Hyphomicrobiaceae family.
Columns from left to right represent genome size (Mb), GC content (%), presence and phylogeny of nifH genes (yellow, gammaproteobacteria; blue, alphaproteobacteria), and presence (green) of ammonium transporter genes (amt). The numbers on the right indicate the nif-containing members of the Hyphomicrobiaceae from Extended Data Fig. 2. ‘Ca. T. diatomicola’ and ‘Ca. T. profundi’ are highlighted in pink. Only genomes with >80% completion are shown. Black dots indicate >95% bootstrap support. Scale bar indicates amino acid substitutions per site.
Extended Data Fig. 4 Microscopic characterization of ‘Candidatus Tectiglobus diatomicola’–Haslea symbioses.
a–d, Confocal laser scanning microscopy images (a,b) and corresponding scanning electron micrographs (c,d) of dividing diatoms with six (a,c) and eight (b,d) ‘Ca. T. diatomicola’ cells, after hybridization with oligonucleotide probes (in pink, overlay of Hypho1147 and Hypho734 in red and blue, respectively; see Extended Data Table 2) and counterstaining with DAPI (white). Solid and dashed lines indicate assumed outline of the dividing diatoms. e, Confocal laser scanning microscopy image of a Haslea diatom after CARD-FISH with the NON338-probe showing no unspecific binding at the typical symbiont location (autofluorescence of the chloroplasts at 488 nm in green, NON338-probe background signal in light blue, DAPI in white). f, Epifluorescence image of the ‘Ca. T. diatomicola’–Haslea symbiosis, showing ‘Ca. T. diatomicola’ cells after hybridization with a specific oligonucleotide probe (Hypho638 in blue), autofluorescence of the chloroplasts in green and the H-shaped nucleus counterstained with DAPI (white). The bilobed chloroplasts are located on either side of the central valve area, and the lobes of the chloroplasts are connected along the cell’s transapical axis. g–k, Scanning electron micrographs of the diatom host. g, Whole frustule showing the internal raphe structure and an apparent hyaline area around the central raphe region (arrow) as well as sections of the hyaline girdle bands (arrowheads). h, Valve end showing the ridges bordering the central raphe. i, Central valve area. The valve’s normal surface structure of longitudinal ribs overlying the transapical striae appears modified with the longitudinal ribs damaged and only the transapical structures visible. j, Valve pole showing the shape of raphe end (arrow head). k, Central valve area with the central valve area collapsed. Arrowheads point to a chamber-like structure. Scale bars are 5 µm in a–g and 1 µm in h–k.
Extended Data Fig. 5 Detection of ‘Candidatus Tectiglobus’ and Haslea species in metagenomic and nifH qPCR datasets.
a, Relative abundance of Haslea in the 0.8 to 5 µm size fraction determined from V9-18S rRNA amplicon sequencing data from Tara Oceans96. b, qPCR datasets in which no ‘Ca. T. diatomicola’ nifH were detected (grey circles; data from Shao et al. 8,67). c,d, Identification of ‘Ca. Tectiglobus’ in metagenomes from Tara Oceans (blue), the South Pacific gyre (yellow) and the tropical North Atlantic (green) datasets. Coverage and breadth (fraction of the genome covered by at least one read) of the ‘Ca. T. diatomicola’ (c) and ‘Ca. T. profundi’ (d) genomes within the metagenomic samples. The black line indicates the expected breadth, see Methods. ‘Ca. Tectiglobus’ was only considered to be present in metagenomes where the actual breadth was close to the expected breadth (all metagenomes inside the pink-shaded ellipses).
Extended Data Fig. 6 Maximum likelihood phylogenetic tree of the ‘Candidatus Tectiglobus diatomicola’ and ‘Candidatus Tectiglobus profundi’ high-affinity cytochrome cbb3-type terminal oxidase.
The tree is constructed from closely related full-length CcoN amino acid sequences retrieved from the GTDB. The CcoN sequences of the two ‘Ca. Tectiglobus’ species are highlighted in pink. The coloured squares indicate the taxonomic affiliation of the genome. Black dots indicate > 95% bootstrap support. Scale bar indicates amino acid substitutions per site.
Extended Data Fig. 7 Single-cell elemental imaging of the ‘Candidatus Tectiglobus diatomicola’–Haslea symbiosis.
NanoSIMS image of the distribution of carbon (12C, red) and nitrogen (12C14N, green) showing the carbon-rich symbionts embedded in nitrogen-rich host biomass. The image shows the same ‘Ca. T. diatomicola’–Haslea symbiosis as in Fig. 3a,b. SE, secondary electrons. Scale bar, 5 µm.
Extended Data Fig. 8 Genome comparison of ‘Candidatus T. diatomicola’ and ‘Candidatus T. profundi’.
a, Whole-genome alignment of ‘Ca. T. diatomicola’ (top) and ‘Ca. T. profundi’ (bottom). Coloured boxes represent colinear regions present in both genomes. Grey vertical lines indicate contig boundaries. Numbers represent genomic positions in kilobase. b, Distribution of genes among the COG functional gene categories for the genomes of ‘Ca. T. diatomicola’, ‘Ca. T. profundi’ and their two closest relatives (GCA_905480435 and GCA_002689605).
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Supplementary Discussion and Supplementary References.
Supplementary Table 1
This file contains Supplementary Table 1, which lists the genome annotation and transcriptome counts of ‘Ca. T. diatomicola’.
Supplementary Table 2
This file contains Supplementary Table 2, which lists Haslea spp. contigs identified in the metagenome assembly.
Supplementary Table 3
This file contains Supplementary Table 3, which lists the presence/absence of KEGG modules in the two ‘Ca. Tectiglobus’ (diatomicola and profundi) genomes and genomes of other Hyphomicrobiaceae.
Supplementary Table 4
This file contains Supplementary Table 4, which lists metagenomes used for the global distribution analysis.
Supplementary Table 5
This file contains Supplementary Table 5, which lists the genome annotation of ‘Ca. T. profundi’.
Supplementary Data 1–9
These are tree files corresponding to the phylogenetic trees shown in Fig.1 and Extended Data Figs. 2 and 3.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tschitschko, B., Esti, M., Philippi, M. et al. Rhizobia–diatom symbiosis fixes missing nitrogen in the ocean. Nature 630, 899–904 (2024). https://doi.org/10.1038/s41586-024-07495-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07495-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
