
Abstract
Late Pliocene and Early Pleistocene epochs 3.6 to 0.8 million years ago1 had climates resembling those forecasted under future warming2. Palaeoclimatic records show strong polar amplification with mean annual temperatures of 11–19 °C above contemporary values3,4. The biological communities inhabiting the Arctic during this time remain poorly known because fossils are rare5. Here we report an ancient environmental DNA6 (eDNA) record describing the rich plant and animal assemblages of the Kap København Formation in North Greenland, dated to around two million years ago. The record shows an open boreal forest ecosystem with mixed vegetation of poplar, birch and thuja trees, as well as a variety of Arctic and boreal shrubs and herbs, many of which had not previously been detected at the site from macrofossil and pollen records. The DNA record confirms the presence of hare and mitochondrial DNA from animals including mastodons, reindeer, rodents and geese, all ancestral to their present-day and late Pleistocene relatives. The presence of marine species including horseshoe crab and green algae support a warmer climate than today. The reconstructed ecosystem has no modern analogue. The survival of such ancient eDNA probably relates to its binding to mineral surfaces. Our findings open new areas of genetic research, demonstrating that it is possible to track the ecology and evolution of biological communities from two million years ago using ancient eDNA.
Similar content being viewed by others
Main
The Kap København Formation is located in Peary Land, North Greenland (82° 24′ N 22° 12′ W) in what is now a polar desert. The upper depositional sequence contains well-preserved terrestrial animal and plant remains washed into an estuary during a warmer Early Pleistocene interglacial cycle7 (Fig. 1). Nearly 40 years of palaeoenvironmental and climate research at the site provide a unique perspective into a period when the site was situated at the boreal Arctic ecotone with reconstructed summer and winter average minimum temperatures of 10 °C and −17 °C respectively—more than 10 °C warmer than the present7,8,9,10,11. These conditions must have driven substantial ablation of the Greenland Ice Sheet, possibly producing one of the last ice-free intervals7 in the last 2.4 million years (Myr). Although the Kap København Formation is known to yield well-preserved macrofossils from a coniferous boreal forest and a rich insect fauna, few traces of vertebrates have been found. To date, these comprise remains from lagomorph genera, their coprolites and Aphodius beetles, which live in and on mammalian dung10,11. However, the approximately 3.4 Myr old Fyles Leaf bed and Beaver Pond on Ellesmere Island in Arctic Canada preserve fossils of mammals that potentially could have colonized Greenland, such as the extinct bear (Protarctos abstrusus), extinct beavers (Dipoides sp.), the small canine Eucyon and Arctic giant camelines4,12,13 (similar to Paracamelus). Whether the Nares Strait was a sufficient barrier to isolate northern Greenland from colonization by this fauna remains an open question.

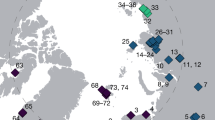
a. Location of Kap København Formation in North Greenland at the entrance to the Independence Fjord (82° 24′ N 22° 12′ W) and locations of other Arctic Plio-Pleistocene fossil-bearing sites (red dots). b, Spatial distribution of the erosional remnants of the 100-m thick succession of shallow marine near-shore sediments between Mudderbugt and the low mountains towards the north (a + b refers to location 74a and 74b). c, Glacial–interglacial division of the depositional succession of clay Member A and units B1, B2 and B3 constituting sandy Member B. Sampling intervals for all sites are projected onto the sedimentary succession of locality 50. Sedimentological log modified after ref. 7. Circled numbers on the map mark sample sites for environmental DNA analyses, absolute burial dating and palaeomagnetism. Numbered sites refer to previous publications7,10,11,14,61.
The Kap København Formation is formally subdivided into two members7 (Fig. 1). The lower Member A consists of up to 50 m of laminated mud with an Arctic ostracod, foraminifera and mollusc fauna deposited in an offshore glaciomarine environment14. The overlying Member B consists of 40–50 m of sandy (units B1 and B3) and silty (unit B2) deposits (Extended Data Fig. 1), including thin organic-rich beds with an interglacial macrofossil fauna that were deposited closer to the shore in a shallow marine or estuarine environment represented by upper and lower shoreface sedimentary facies7.
The specific depositional environments are also reflected in the mineralogy of the units, where the proximal B3 locality has the lowest clay and highest quartz contents (Sample compositions in Supplementary Tables 4.2.1 and 4.2.2 and unit averages in Supplementary Tables 4.2.3 and 4.2.4). The architecture of the basin infill suggests that Member B units thicken towards the present coast—that is, distal to the sediment source in the low mountains in the north (Fig. 1). Abundant organic detritus horizons are recorded in units B1 and B3, which also contain beds rich in Arctic and boreal plant and invertebrate macrofossils, as well as terrestrial mosses10,15. Therefore, the taphonomy of the DNA most probably reflects the biological communities eroded from a range of habitats, fluvially transported to the foreshore and concentrated as organic detritus mixed into sandy near-shore sediments within units B1 and B3. Conversely, the deeper water facies from Member A and unit B2 have a stronger marine signal. This scenario is supported by the similarities in the mineralogic composition between Kap København Formation sediments and Kim Fjelde sediments (Supplementary Tables 4.2.1 and 4.2.5).
Geological age
A series of complementary studies has successively narrowed the depositional age bracket of the Kap København Formation from 4.0–0.7 Myr to a 20,000-year-long age bracket around 2.4 Myr (see Supplementary Information, sections 1–3). This was achieved by a combination of palaeomagnetism, biostratigraphy and allostratigraphy7,14,16,17,18. Notably, the last appearance data of the mammals, foraminifera and molluscs in the stratigraphic record show an age close to 2.4 Myr (see Supplementary Information, section 2). Within this overall framework, we add new palaeomagnetic data showing that Member A has reversed magnetic polarity and the main part of the overlying unit B2 has normal magnetic polarity. In the context of previous work, this is consistent with three magnetostratigraphic intervals in the Early Pleistocene where there is a reversal: 1.93 Myr (scenario 1), 2.14 Myr (scenario 2) or 2.58 Myr (scenario 3) (Supplementary Information, section 1). Furthermore, we constrain the age using cosmogenic 26Al:10Be burial dating of Member B at four sites in this study (Supplementary Information, section 3). The recommended maximum burial age for the Kap København Formation is 2.70 ± 0.46 Myr (Fig. 2; Methods). However, we discard the older scenario 3 as it contradicts the evidence for a continuous sedimentation across Members A and B during a single glacial–interglacial depositional cycle7,14,16,18,19. This leaves two possible scenarios (scenarios 1 and 2), in which scenario 1 supports an age of 1.9 Myr and scenario 2 supports an age of 2.1 Myr.

a, Revised palaeomagnetic analysis shows unit B2 to have normal polarity and unlocks three possible age scenarios (S1–S3) including Members A (blue) and B (brown). Normal polarity is coloured black and reverse polarity is shown in white. Ja, Jaramillo; Co, Cobb Mountain; Ol, Olduvai; Fe, Feni; Ka, Kaena; Ma, Mammoth. b, Presence and last appearance datum (LAD) for marine foraminifera Cibicides grossus, rabbit-genus Hypolagus and the mollusc Arctica islandica in the High Arctic, Northern Hemisphere and North Greenland, respectively. The blue band on the far right indicates the age range for Member A estimated from amino acid ratios on shells7. c, Convolved probability distribution functions for cosmogenic burial ages calculated for two different production ratios (7.42 (black) and 6.75 (blue)). The dashed line and the solid line show the distributions for steady erosion and zero erosion, respectively. These distributions are all maximum ages. d, Molecular dating of Betula sp. yielding a median age of the DNA in the sediment of 1.323 Myr, with whiskers confining the 95% height posterior density (HPD) of 0.68 to 2.02 Myr (blue density plot), running Markov chain Monte Carlo estimation for 100 million iterations. The red dot is the median molecular age estimate found using the Mastodon mitochondrial genome restricting to radiocarbon-dated specimens, whereas the green area includes molecular clock estimated specimens in BEAST, running Markov chain Monte Carlo estimation for 400 million iterations. Whiskers confine the 95% HPD.
DNA preservation
DNA degrades with time owing to microbial enzymatic activity, mechanical shearing and spontaneous chemical reactions such as hydrolysis and oxidation20. The oldest known DNA obtained to date has been recovered from a permafrost-preserved mammoth molar dated to 1.2–1.1 Myr using geological methods and 1.7 Myr (95% highest posterior density, 2.1–1.3 Myr) using molecular clock dating21. To explore the likelihood of recovering DNA from sediments at the Kap København formation, we calculated the thermal age of the DNA and its expected degree of depurination at the Kap København Formation. Using the mean average temperature22 (MAT) of −17 °C, we found a thermal age of 2.7 thousand years for DNA at a constant 10 °C, which is 741 times less than the age of 2.0 Myr (Supplementary Information, section 4 and Supplementary Table 4.4.1). Using the rate of depurination from Moa bird fossils23, we found it plausible that DNA with an average size of 50 base pairs (bp) could survive at the Kap København Formation, assuming that the site remained frozen (Supplementary Information, section 4 and Supplementary Table 4.4.2). Mechanisms that preserve DNA in sediments are likely to be different from that of bone. Adsorption at mineral surfaces modifies the DNA conformation, probably impeding molecular recognition by enzymes, which effectively hinders enzymatic degradation24,25,26,27. To investigate whether the minerals found in Kap København Formation could have retained DNA during the deposition and preserved it, we determined the mineralogic composition of the sediments using X-ray diffraction and measured their adsorption capacities. Our findings highlight that the marine depositional environment favours adsorption of extracellular DNA on the mineral surfaces (Supplementary Information, section 4 and Supplementary Table 4.3.1.1). Specifically, the clay minerals (9.6–5.5 wt%) and particularly smectite (1.2–3.7 wt%), have higher adsorption capacity compared to the non-clay minerals (59–75 wt%). At a DNA concentration representative of the natural environments28 (4.9 ng ml−1 DNA), the DNA adsorption capacity of smectite is 200 times greater than for quartz. We applied a sedimentary eDNA extraction protocol29 on our mineral-adsorbed DNA samples, and retrieved only 5% of the adsorbed DNA from smectite and around 10% from the other clay minerals (Methods and Supplementary Information, section 4). By contrast, we retrieved around 40% of the DNA adsorbed to quartz. The difference in adsorption capacity and extraction yield from the different minerals demonstrates that mineral composition may have an important role in ancient eDNA preservation and retrieval.
Kap København metagenomes
We extracted DNA29 from 41 organic-rich sediment samples at five different sites within the Kap København Formation (Supplementary Information, section 6 and Source Data 1), which were converted into 65 dual-indexed Illumina sequencing libraries30. First, we tested 34 of the 65 libraries for plant plastid DNA by screening for the conserved photosystem II D2 (psbD) gene using droplet digital PCR (ddPCR) with a gene-targeting primer and probe spanning a 39-bp region and a P7 index primer. Further, we screened for the psbA gene using a similar assay targeting the Poaceae (Methods and Supplementary Fig. 6.12.1). A clear signal in 31 out of 34 samples tested confirmed the presence of plant plastid DNA in these libraries (Source Data 1, sheets 5 and 6). Additionally, we subjected 34 of the 65 libraries to mammalian mtDNA capture enrichment using the Arctic PaleoChip 1.031 and shotgun sequenced all libraries (initial and captured) using the Illumina HiSeq 4000 and NovaSeq 6000. A total of 16,882,114,068 reads were sequenced, which after adaptor trimming, filtering for ≥30 bp and a minimum phred quality of 30 and duplicate removal resulted in 2,873,998,429 reads. These were analysed for k-mer comparisons using simka32 (Supplementary Information, section 6) and then parsed for taxonomic classification using competitive mapping with HOLI (https://github.com/miwipe/KapCopenhagen.git), which includes a recently published dataset of more than 1,500 genome skims of Arctic and boreal plant taxa33,34 (Methods and Supplementary Information, section 6). Considering the age of the samples and thus the potential genetic distance to recent reference genomes, we allowed each read to have a similarity between 95–100% for it to be taxonomically classified using ngsLCA35. The metaDMG (v.0.14.0) program36 was subsequently used to quantify and filter each taxonomic node for postmortem DNA damage for all the metagenomic samples (Methods). This method estimates the average damage at the termini position (D-max) and a likelihood ratio (λ-LR) that quantifies how much better the damage model (that is, more damage at the beginning of the read) fits the data compared with a null model (that is, a constant amount of damage; see Supplementary Information, section 6). We found the DNA damage to be highly increased, especially for eukaryotes (mean D-max = 40.7%, see Supplementary Information, section 6). From this we set D-max ≥25% as a filtering threshold for a taxonomic node to be parsed for further downstream analysis as well as a λ-LR higher or equal to 1.5. We furthermore set a threshold requiring that the minimum number of reads per taxon exceeded the median of reads assigned across all taxa divided by two to filter for taxa in low abundance. Similarly, for a sample to be considered, the total number of reads for a sample had to exceed the median number of reads per sample divided by two, to filter for samples with fewest reads. Lastly, we filtered out taxa with fewer than three replicates and subsequently reads were normalized by conversion to proportions (Figs. 3 and 4a).

Taxonomic profiles of the plant assemblage found in the metagenomes. Taxa in bold are genera only found as DNA and not as macrofossil or pollen. Asterisks indicate those that are found at other Pliocene Arctic sites. Extinct species as identified by either macrofossils or phylogenetic placements are marked with a dagger. Reads classified as Pyrus and Malus are marked with a pound symbol, and are probably over-classified DNA sequences belonging to another species within Rosaceae that are not present as a reference genome.

a, Taxonomic profiles of the animal assemblage from units B1, B2 and B3. Taxa in bold are genera only found as DNA. b, Phylogenetic placement and pathPhynder62 results of mitochondrial reads uniquely classified to Elephantidae or lower (Source Data 1). Extinct species as identified by either macrofossils or phylogenetic placements are marked with a dagger.
DNA, pollen and macrofossils comparison
Greenland’s coasts extend from around 60° to 83° N and include bioclimatic zones from the subarctic to the northern polar desert37,38. There are 175 vascular plant genera native to Greenland, excluding historically introduced species39,40,41. Of these, 70 (40%) were detected by the metagenomic analysis (Fig. 3); the majority of these genera are today confined to bioclimatic zones well to the south of Kap København’s polar desert (see ref. 42 and references therein), for example, all aquatic macrophytes. Reads assigned to Salix, Dryas, Vaccinium, Betula, Carex and Equisetum dominate the assemblage, and of these genera, Equisetum, Dryas, Salix arctica and two species of Carex (Carex nardina and Carex stans) grow there currently, whereas only a few records of Vaccinium uliginosum are found above 80º N, and Betula nana are found above 74º N (ref. 43). Out of the 102 genera detected in the Kap København ancient eDNA assemblage, 39% no longer grow in Greenland but do occur in the North American boreal (for example, Picea and Populus) and northern deciduous and maritime forests (for example, Crataegus, Taxus, Thuja and Filipendula). Many of the plant genera in this diverse assemblage do not occur on permafrost substrates and require higher temperatures than those at any latitude on Greenland today.
In addition to the DNA, we counted pollen in six samples from locality 119, unit B3 (Methods and Supplementary Fig. 5.1.1). Percentages were calculated for 4 of the samples with pollen sums ranging from 71–225 terrestrial grains (mean = 170.25). Upland herbs, including taxa in the Cyperaceae, Ericales and Rosaceae comprised around 40% of sample 4. Samples 5 and 6 were dominated by arboreal taxa, particularly Betula. The Polypodiopsida (for example, Equisetum, Asplenium and Athyrium filix-mas) and Lycopodiopsida (Lycopodium annotinum and Selaginella rupestris) were also well represented and comprised over 30% of the assemblage in samples 1, 4 and 6.
A total of 39 plant genera out of the 102 identified by DNA also occurred as macrofossils or pollen at the genus level. A further 39 taxa were potentially identified as macrofossil or pollen but not to the same taxonomic level10,15 (Source Data 1, sheets 1 and 2). For example, 12 genera of Poaceae were identified by DNA (Alopecurus, Anthoxanthum, Arctagrostis, Arctophila, Calamagrostis, Cinna, Dupontia, Hordelymus, Leymus, Milium, Phippsia and Poa), of these only Hordelymus is not found in the Arctic today (http://panarcticflora.org/), but these were only distinguished to family level in the pollen analysis and only one Poaceae macrofossil was found. There were 24 taxa that were recorded only as DNA. These included the boreal tree Populus and a few shrubs and dwarf shrubs, but mainly herbaceous plants. Of the 73 plant genera recovered as macrofossils10,15, only 24 were not detected in the DNA analysis. Because macrofossils and DNA have similar taphonomies—as both are deposited locally—more overlap is expected between them than between DNA and pollen, which is typically dispersed regionally44. Nine of the taxa absent in DNA were bryophytes, probably owing to poor representation of this group within the genomic reference databases. Furthermore, the extinct taxon Araceae is not present in the reference databases. The remaining undetected genera were vascular plants, and all except two (Oxyria and Cornus) were rare in the macrofossil record. Because the detection of rare taxa is challenging in both macrofossil and DNA records45, we argue that this overlap between the DNA and macrofossil records is as high as can be expected on the basis of the limitations of both methods.
An additional 19 taxa were recorded in the pollen record presented here and in that of Bennike46 including four trees or shrubs, five ferns, three club mosses, and one each of algae, fungi and liverwort. We also find pollen from anemophilous trees, particularly gymnosperms, which can be distributed far north of the region where the plants actually grow10. Bennike46 also notes a high proportion of club mosses and ferns and suggests they may be overrepresented owing to their spore wall being resistant to degradation. Furthermore, if these taxa were preferentially distributed along streams flowing into the estuary, their spores could be relatively more concentrated in the alluvium than the pollen of more generally distributed taxa. Thus, both decay resistance and alluvial deposition could contribute to the relative frequencies we observe. This same alluvial dynamic might also have contributed to the very large read counts for Salix, Betula, Populus, Carex and Equisetum in the metagenomic record, implying that neither the proportion of these taxa in the pollen records nor read counts necessarily correlate with their actual abundance in the regional vegetation in terms of biomass or coverage.
Finally, we sought to date the age of the plant DNA by phylogenetic placement of the chloroplast DNA. We examined data for the genera Betula, Populus and Salix, because these had both sufficiently high chloroplast genome coverage (with mean depth 24.16×, 57.06× and 27.04×, respectively) and sufficient present-day whole chloroplast reference sequences (Methods). Owing to their age and hence potential genetic distance from the modern reference genomes, we lowered the similarity threshold of uniquely classified reads to 90% and merged these by unit to increase coverage. Both Betula and Salix placed basally to most of the represented species in the respective genera, and the Populus placement results showed support for a mixture of different species related to P. trichocarpa and P. balsamifera (Extended Data Figs. 7–9).
We used the Betula chloroplast reads for a molecular dating analysis, because they were placed confidently on a single edge of the phylogenetic tree (that is, not a mixture as in Populus), had a large number of reference sequences, and had high coverage in the ancient sample. We used BEAST47 v1.10.4 to obtain a molecular clock date estimate for our ancient Betula chloroplast sample (see Methods, ‘Molecular dating methods’ for details). We included 31 modern Betula and one Alnus chloroplast reference sequences, used only sites that had a depth of at least 20 in the ancient sample, and included a previously estimated Betula–Alnus chloroplast divergence time48 of 61.1 Myr for calibration of the root node. Our BEAST analysis was robust to both different priors on the age of the ancient sample, and to different nucleotide substitution models (Extended Data Fig. 10). This yielded a median age estimate of 1.323 Myr, with a 95% HPD of (0.6786, 2.0172) Myr (Fig. 2).
Animal DNA results
The metazoan mitochondrial and nuclear DNA record was much less diverse than that of the plants but contained one extinct family, one that is absent from Greenland today, and four vertebrate genera native to Greenland as well as representatives of four invertebrate families (Fig. 4a). Assignments were based on incomplete and variable representation of reference genomes, so we identified reads to family level, and only where sufficient mitochondrial reads were present, we refined the assignment to genus level by matching these into mitochondrial phylogenies based on more complete present-day mitochondrial sequences (Supplementary Information, section 6). As for the plant reads, uniquely classified animal reads with more than 90% similarity were parsed and merged by unit to increase coverage for phylogenetic placement.
Most notably, we found reads in unit B2 and B3 assigned to the family Elephantidae, which includes elephants and mammoths, but taxonomically not mastodon (Mammut sp.)—which are, however, in the NCBI taxonomy, and therefore our analysis reads classified to Elephantidae or below therefore include Mammut sp. A consensus genome of our Elephantidae mitochondrial reads falls on the Mammut sp. branch (Fig. 4b) and is placed basal to all clades of mastodons. However, we note that this placement within the mastodons depends on only two transition single-nucleotide polymorphisms (SNPs), with the first one supported by a read depth of three and the second by only one (Extended Data Fig. 4, Methods and Supplementary Information, section 6). Furthermore, we attempted dating the recovered mastodon mitochondrial genome using BEAST49. We implemented two dating approaches, one was based on using radiocarbon-dated specimens alone, while the other used radiocarbon- and molecular-dated mastodons. The first analysis yielded a median age estimate for our mastodon mitogenome of 1.2 Myr (95% HPD: 191,000 yr–3.27 Myr), the second approach resulted in a median age estimate of 5.2 Myr (95% HPD: 1.64–10.1 Myr) (Supplementary Fig. 6.8.5 and Supplementary Information, section 6).
Similarly, reads assigned to the Cervidae support a basal placement on the Rangifer (reindeer and caribou) branch (Extended Data Fig. 3). Mitochondrial reads mapping to Leporidae (hares and rabbits) place near the base to the Eurasian hare clade (Extended Data Fig. 2), which is the only mammal found in the fossil record7. Lepus, specifically Lepus arcticus, is also the only genus in the Leporidae living in Greenland today. Mitochondrial reads assigned to Cricetidae cover only one informative transversion SNP, which places them as deriving from the subfamily Arvicolinae (voles, lemmings and muskrats) (Extended Data Fig. 6). For the only avian taxon represented in our dataset—Anatidae, the family of geese and swans—we found a robust basal placement to the genus Branta of black geese, supported by three transversion SNPs with read depths ranging between two and four (Extended Data Fig. 5). The refined vertebrate assignments based on mitochondrial references are more biogeographically conserved than for plants. Dicrostonyx—specifically Dicrostonyx groenlandicus (the Nearctic collared lemming)—is the only genus of the Cricetidae native to Greenland today, just as Rangifer—specifically Rangifer tarandus groenlandicus (the barren-ground caribou)—is the only member of the Cervidae. The mastodon is the exception, as no member of the Elephantidae lives in present-day Greenland.
Ancient DNA from marine organisms
The other metazoan taxa identified in the DNA record were a single reef-building coral (Merulinidae) and several arthropods, with matches to two insects—Formicidae (ants) and Pulicidae (fleas)—and one marine family—Limulidae (horseshoe crabs). This is somewhat unexpected, given the rich insect macrofossil record from the Kap København Formation, which comprises more than 200 species, including Formica sp. The marine taxa are less abundant than the terrestrial taxa, and no mitochondrial DNA was identified from marine metazoans. The read lengths, DNA damage and the fact that the reads assigned distribute evenly across the reference genomes suggests that these are not artefacts but may be over-matched DNA sequences of closely related, potentially extinct species within the families that are currently absent from our reference databases owing to poor taxonomic representation. By contrast, Limulidae, in the subphylum Chelicerata, is unlikely to be misidentified as this distinct genus is the only surviving member within its order and thus deeply diverged from other extant organisms.
The probable source of these reads is a population of Limulus polyphemus, the only Atlantic member of the genus, which would have spawned directly onto the sediment as it accumulated. Today this genus does not spawn north of the Bay of Fundy (about 45° N), suggesting warmer surface water conditions in the Early Pleistocene at Kap København consistent with the +8 °C annual sea surface temperature anomaly reconstructed for the Pleistocene of the coast of northeast Greenland50. By aligning our reads against the Tara Oceans eukaryotic metagenomic assembled genomes (SMAGs) data (Methods), we further reveal the presence of 24 marine planktonic taxa in 14 samples, covering both zooplankton and phytoplankton (Fig. 5). These detected SMAGs belong to the supergroups Opisthokonta (6), Stramenopila (15) and Archaeplastida (3). The majority of these signals are from SMAGs associated with cold regions in the modern ocean (that is, the Arctic Ocean and Southern Ocean), such as diatoms (Bacillariophyta), Chrysophyceae and the MAST-4 group (Supplementary Table 6.11.1), as we expected. However, a few are cosmopolitan, whereas others, such as Archaeplastida (green microalgae), have an oceanic signal that is today confined to more temperate waters in the Pacific Ocean (Fig. 5). Although we do not know whether modern day ecologies can be extrapolated to ancient ecosystems, the abundance of green microalgae is believed to be increasing in Arctic regions, which tends to be associated with warming surface waters.

a, Detection of SMAGs and average damage (D-max) of a SMAG within a member unit. Top, the SMAG distribution in contemporary oceans based on the data of Delmont et al.63. The SMAGs are ordered on the basis of phylogenomic inference from Delmont et al.63. b–d, Distribution of DNA damage among the taxonomic supergroup Opisthokonta (b), Stramenopila (c) and Archaeplastida (d) (Source Data 1).
Discussion
The Kap København ancient eDNA record is extraordinary for several reasons; the upper limit of the 95% highest posterior density of the estimated molecular age is 2.0 Myr and independently supports a geological age of approximately 2 Myr (Fig. 2). This implies that the DNA is considerably older than any previously sequenced DNA21. Our DNA results detected five times as many plant genera as previous studies using shotgun sequencing of ancient sediments29,34,51,52, which is well within the range of the richest northern boreal metabarcoding records53. The accuracy of the assignments is strengthened by the observation that 76% of the taxa identified to the level of genus or family also occurred in macrofossil and/or pollen assemblages from the same units. Our results demonstrate the potential of ancient environmental metagenomics to reconstruct ancient environments, phylogenetically place and date ancient lineages from diverse taxa from around 2 Ma (Supplementary Information, section 6). Finally, the DNA identified a set of additional plant genera, which occur as macrofossils at other Arctic Late Pliocene and Early Pleistocene sites (Figs. 1 and 3 and Supplementary Information, section 5) but not as fossils at Kap København, thereby expanding the spatiotemporal distribution of these ancient floras.
Of note, the detection of both Rangifer (reindeer and caribou) and Mammut (mastodon) forces a revision of earlier palaeoenvironmental reconstructions based on the site’s relatively impoverished faunal record, entailing both higher productivity and habitat diversity for much of the deposition period. Because all the vertebrate taxa identified by DNA are herbivores, their representation may be a function of relative biomass (see discussion on taphonomy in Supplementary Information, section 6). Caribou, geese, hares and rodents can all be abundant, at least seasonally, in boreal environments. Additionally, the excrement of large herbivores (such as caribou and particularly mastodons) can be a significant component of sediments34. By contrast, carnivores are not represented, consistent with their smaller total biomass. This dynamic also explains the dominance of plant reads over metazoans and to some extent differences in representation of various plant genera (Supplementary Information, section 6). In the general absence of fossils, DNA may prove the most effective tool for reconstructing the biogeography of vertebrates through the Early Pleistocene. DNA from mastodon must imply a viable population of this large browsing megaherbivore, which would require a more productive boreal habitat than that inferred in earlier reconstructions based primarily on plant macrofossils7. Mastodon dung from a site in central Nova Scotia from around 75,000 years ago contained macrofossils from sedges, cattail, bulrush, bryophytes and even charophytes, but was dominated by spruce needles and birch samaras54. The Kap København units with mastodon DNA yielded macrofossils and DNA from Betula as well as more thermophilic arboreal taxa including Thuja, Taxus, Cornus and Viburnum, none of which range into Greenland’s hydric Arctic tundra or polar deserts today. The co-occurrence of these taxa in multiple units compels a revision of previous temperature estimates as well as the presence of permafrost.
No single modern plant community or habitat includes the range of taxa represented in many of the macrofossil and DNA samples from Kap København. The community assemblage represents a mixture of modern boreal and Arctic taxa, which has no analogue in modern vegetation10,15. To some degree, this is expected, as the ecological amplitudes of modern members of these genera have been modified by evolution55. Furthermore, the combination of the High Arctic photoperiod with warmer conditions and lower atmospheric CO2 concentrations56 made the Early Pleistocene climate of North Greenland very different from today. The mixed character of the terrestrial assemblage is also reflected in the marine record, where Arctic and more cosmopolitan SMAGs of Opistokonta and Stramenopila are found together with horseshoe crabs, corals and green microalgae (Archaeplastida), which today inhabit warmer waters at more southern latitudes.
Megaherbivores, particularly mastodons, could have had a significant impact on an interglacial taiga environment, even providing a top-down trophic control on vegetation structure and composition at this high latitude. The presence of mastodons57,58 coupled with the absence of anthropogenic fire, which has had a role in some Holocene boreal habitats59, are important differences. Another important factor is the proximity and biotic richness of the refugia from which pioneer species were able to disperse into North Greenland when conditions became favourable at the beginning of interglacials. The shorter duration of Early Pleistocene glaciations produced less extensive ice sheets allowing colonization from relatively species-rich coniferous-deciduous woodlands in northeastern Canada12,60. More extensive glaciation later in the Pleistocene increasingly isolated North Greenland and later re-colonizations were from increasingly distant and/or less diverse refugia.
In summary, we show the power of ancient eDNA to add substantial detail to our knowledge of this unique, ancient open boreal forest community intermixed with Arctic species, a community composition that has no modern analogues and included mastodons and reindeer, among others. Similar detailed flora and vertebrate DNA records may survive at other localities. If recovered, these would advance our understanding of the variability of climate and biotic interactions during the warmer Early Pleistocene epochs across the High Arctic.
Methods
Sampling
Sediment samples were obtained from the Kap København Formation in North Greenland (82° 24′ 00″ N 22° 12′ 00″ W) in the summers of 2006, 2012 and 2016 (see Supplementary Table 3.1.1). Sampled material consisted of organic-rich permafrost and dry permafrost. Prior to sampling, profiles were cleaned to expose fresh material. Samples were hereafter collected vertically from the slope of the hills either using a 10 cm diameter diamond headed drill bit or cutting out ~40 × 40 × 40 cm blocks. Sediments were kept frozen in the field and during transportation to the lab facility in Copenhagen. Disposable gloves and scalpels were used and changed between each sample to avoid cross-contamination. In a controlled laboratory environment, the cores and blocks were further sub-sampled for material taking only the inner part of sediment cores, leaving 1.5–2 cm between the inner core and the surface that provided a subsample of approximately 6–10 g. Subsequently, all samples were stored at temperatures below −22 °C.
We sampled organic-rich sediment by taking samples and biological replicates across the three stratigraphic units B1, B2 and B3, spanning 5 different sites, site: 50 (B3), 69 (B2), 74a (B1), 74b (B1) and 119 (B3). Each biological replicate from each unit at each site was further sampled in different sublayers (numbered L0–L4, Source Data 1, sheet 1).
Absolute age dating
In 2014, Be and Al oxide targets from 8× 1 kg quartz-rich sand samples collected at modern depths ranging from 3 to 21 m below stream cut terraces were analysed by accelerator mass spectrometry and the cosmogenic isotope concentrations interpreted as maximum ages using a simple burial dating approach1 (26Al:10Be versus normalized 10Be). The 26Al and 10Be isotopes were produced by cosmic ray interactions with exposed quartz in regolith and bedrock surfaces in the mountains above Kap København prior to deposition. We assume that the 26Al:10Be was uniform and steady for long time periods in the upper few metres of these gradually eroding palaeo-surfaces. Once eroded by streams and hillslope processes, the quartz sand was deposited in sandy braided stream sediment, deltaic distributary systems, or the near-shore environment and remained effectively shielded from cosmic ray nucleons buried (many tens of metres) under sediment, intermittent ice shelf or ice sheet cover, and—at least during interglacials—the marine water column until final emergence. The simple burial dating approach assumes that the sand grains experienced only one burial event. If multiple burial events separated by periods of re-exposure occurred, then the starting 26Al:10Be before the last burial event would be less than the initial production ratio (6.75 to 7.42, see discussion below) owing to the relatively faster decay of 26Al during burial, and therefore the calculated burial age would be a maximum limiting age. Multiple burial events can be caused by shielding by thick glacier ice in the source area, or by sediment storage in the catchment prior to final deposition. These shielding events mean that the 26Al:10Be is lower, and therefore a calculated burial age assuming the initial production ratio would overestimate the final burial duration. We also consider that once buried, the sand grains may have been exposed to secondary cosmogenic muons (their depth would be too great for submarine nucleonic production). As sedimentation rates in these glaciated near-shore environments are relatively rapid, we show that even the muonic production would be negligible (see Supplemental Information). However, once the marine sediments emerged above sea level, in-situ production by both nucleogenic and muogenic production could alter the 26Al:10Be. The 26Al versus 10Be isochron plot reveals this complex burial history (Supplementary Information, section 3) and the concentration versus depth composite profiles for both 26Al and 10Be reveal that the shallowest samples may have been exposed during a period of time (~15,000 years ago) that is consistent with deglaciation in the area (Supplemental Information). While we interpret the individual simple burial age of all samples as a maximum limiting age of deposition of the Kap København Formation Member B, we recommend using the three most deeply shielded samples in a single depth profile to minimize the effect of post-depositional production. We then calculate a convolved probability distribution age for these three samples (KK06A, B and C). However, this calculation depends on the 26Al:10Be production ratio we use (that is, between 6.75 and 7.42) and on whether we adjust for erosion in the catchment. So, we repeat the convolved probability distribution function age for the lowest and highest production ratio and zero to maximum possible erosion rate, to obtain the minimum and maximum limiting age range at 1σ confidence (Supplementary Information, section 3). Taking the midpoint between the negative and positive 3σ confidence limits, we obtain a maximum burial age of 2.70 ± 0.46 Myr. This age is also supported by the position of those three samples on the isochron plot, which suggests the true age may not be significantly different that this maximum limiting age.
Thermal age
The extent of thermal degradation of the Kap København DNA was compared to the DNA from the Krestovka Mammoth molar. Published kinetic parameters for DNA degradation64 were used to calculate the relative rate difference over a given interval of the long-term temperature record and to quantify the offset from the reference temperature of 10 °C, thus estimating the thermal age in years at 10 °C for each sample (Supplementary Information, section 4). The mean annual air temperature (MAT) for the the Kap København sediment was taken from Funder et al. (2001)6 and for the Krestovka Mammoth the MAT was calculated using temperature data from the Cerskij Weather Station (WMO no. 251230) 68.80° N 161.28° E, 32 m from the International Research Institute Data Library (https://iri.columbia.edu/) (Supplementary Table 4.4.1).
We did not correct for seasonal fluctuation for the thermal age calculation of the Kap København sediments or from the Krestovka Mammoth. We do provide theoretical average fragment length for four different thermal scenarios for the DNA in the Kap København sediments (Supplementary Table 4.4.2). A correction in the thermal age calculation was applied for altitude using the environmental lapse rate (6.49 °C km−1). We scaled the long-term temperature model of Hansen et al. (2013)65 to local estimates of current MATs by a scaling factor sufficient to account for the estimates of the local temperature decline at the last glacial maximum and then estimated the integrated rate using an activation energy (Ea) of 127 kJ mol−1 (ref. 64).
Mineralogic composition
The minerals in each of the Kap København sediment samples were identified using X-ray diffraction and their proportions were quantified using Rietveld refinement. The samples were homogenized by grinding ~1 g of sediment with ethanol for 10 min in a McCrone Mill. The samples were dried at 60 °C and added corundum (CR-1, Baikowski) as the internal standard to a final concentration of 20.0 wt%. Diffractograms were collected using a Bruker D8 Advance (Θ–Θ geometry) and the LynxEye detector (opening 2.71°), with Cu Kα1,2 radiation (1.54 Å; 40 kV, 40 mA) using a Ni-filter with thickness of 0.2 mm on the diffracted beam and a beam knife set at 3 mm. We scanned from 5–90° 2θ with a step size of 0.1° and a step time of 4 s while the sample was spun at 20 rpm. The opening of the divergence slit was 0.3° and of the antiscatter slit 3°. Primary and secondary Soller slits had an opening of 2.5° and the opening of the detector window was 2.71°. For the Rietveld analysis, we used the Profex interface for the BGMN software66,67. The instrumental parameters and peak broadening were determined by the fundamental parameters ray-tracing procedure68. A detailed description of identification of clay minerals can be found in the supporting information.
Adsorption
We used pure or purified minerals for adsorption studies. The minerals used and treatments for purifying them are listed in Supplementary Table 4.2.6. The purity of minerals was checked using X-ray diffraction with the same instrumental parameters and procedures as listed in the above section i.e., mineralogical composition. Notes on the origin, purification and impurities can be found in the Supplementary Information section 4. We used artificial seawater69 and salmon sperm DNA (low molecular weight, lyophilized powder, Sigma Aldrich) as a model for eDNA adsorption. A known amount of mineral powder was mixed with seawater and sonicated in an ultrasonic bath for 15 min. The DNA stock was then added to the suspension to reach a final concentration between 20–800 μg ml−1. The suspensions were equilibrated on a rotary shaker for 4 h. The samples were then centrifuged and the DNA concentration in the supernatant determined with UV spectrometry (Biophotometer, Eppendorf), with both positive and negative controls. All measurements were done in triplicates, and we made five to eight DNA concentrations per mineral. We used Langmuir and Freundlich equations to fit the model to the experimental isotherm and to obtain adsorption capacity of a mineral at a given equilibrium concentration.
Pollen
The pollen samples were extracted using the modified Grischuk protocol adopted in the Geological Institute of the Russian Academy of Science which utilizes sodium pyrophosphate and hydrofluoric acid70. Slides prepared from 6 samples were scanned at 400× magnification with a Motic BA 400 compound microscope and photographed using a Moticam 2300 camera. Pollen percentages were calculated as a proportion of the total palynomorphs including the unidentified grains. Only 4 of the 6 samples yielded terrestrial pollen counts ≥50. In these, the total palynomorphs identified ranged from 225 to 71 (mean = 170.25; median = 192.5). Identifications were made using several published keys71,72. The pollen diagram was initially compiled using Tilia version 1.5.1273 but replotted for this study using Psimpoll 4.1074.
DNA recovery
For recovery calculation, we saturated mineral surfaces with DNA. For this, we used the same protocol as for the determination of adsorption isotherms with an added step to remove DNA not adsorbed but only trapped in the interstitial pores of wet paste. This step was important because interstitial DNA would increase the amount of apparently adsorbed DNA and overestimate the recovery. To remove trapped DNA after adsorption, we redispersed the minerals in seawater. The process of redispersing the wet paste in seawater, ultracentrifugation and removal of supernatant lasted less than 2.5 min. After the second centrifugation, the wet pastes were kept frozen until extraction. We used the same extraction protocol as for the Kap København sediments. After the extraction, the DNA concentration was again determined using UV spectrometry.
Metagenomes
A total of 41 samples were extracted for DNA75 and converted to 65 dual-indexed Illumina sequencing libraries (including 13 negative extraction- and library controls)30. 34 libraries were thereafter subjected to ddPCR using a QX200 AutoDG Droplet Digital PCR System (Bio-Rad) following manufacturer’s protocol. Assays for ddPCR include a P7 index primer (5′-AGCAGAAGACGGCATAC-3′) (900nM), gene-targeting primer (900 nM), and a gene-targeting probe (250nM). We screened for Viridiplantae psbD (primer: 5′-TCATAATTGGACGTTGAACC-3′, probe: 5′-(FAM)ACTCCCATCATATGAAA(BHQ1)-3′) and Poaceae psbA (primer: 5′-CTCACAACTTCCCTCTAGAC-3′, probe 5′-(HEX)AGCTGCTGTTGAAGTTC(BHQ1)-3′). Additionally, 34 of the 65 libraries were enriched using targeted capture enrichment, for mammalian mitochondrial DNA using the PaleoChip Arctic1.0 bait-set31 and all libraries were hereafter sequenced on an Illumina HiSeq 4000 80 bp PE or a NovaSeq 6000 100 bp PE. We sequenced a total of 16,882,114,068 reads which, after low complexity filtering (Dust = 1), quality trimming (q ≥ 25), duplicate removal and filtering for reads longer than 29 bp (only paired read mates for NovaSeq data) resulted in 2,873,998,429 reads that were parsed for further downstream analysis. We next estimated kmer similarity between all samples using simka32 (setting heuristic count for max number of reads (-max-reads 0) and a kmer size of 31 (-kmer-size 31)), and performed a principal component analysis (PCA) on the obtained distance matrix (see Supplementary Information, ‘DNA’). We hereafter parsed all QC reads through HOLI33 for taxonomic assignment. To increase resolution and sensitivity of our taxonomic assignment, we supplemented the RefSeq (92 excluding bacteria) and the nucleotide database (NCBI) with a recently published Arctic-boreal plant database (PhyloNorway) and Arctic animal database34 as well as searched the NCBI SRA for 139 genomes of boreal animal taxa (March 2020) of which 16 partial-full genomes were found and added (Source Data 1, sheet 4) and used the GTDB microbial database version 95 as decoy. All alignments were hereafter merged using samtools and sorted using gz-sort (v. 1). Cytosine deamination frequencies were then estimated using the newly developed metaDMG, by first finding the lowest common ancestor across all possible alignments for each read and then calculating damage patterns for each taxonomic level36 (Supplementary Information, section 6). In parallel, we computed the mean read length as well as number of reads per taxonomic node (Supplementary Information, section 6). Our analysis of the DNA damage across all taxonomic levels pointed to a minimum filter for all samples at all taxonomic levels with a D-max ≥ 25% and a likelihood ratio (λ-LR) ≥ 1.5. This ensured that only taxa showing ancient DNA characteristics were parsed for downstream profiling and analysis and resulted in no taxa within any controls being found (Supplementary Information, section 6).
Marine eukaryotic metagenome
We sought to identify marine eukaryotes by first taxonomically labelling all quality-controlled reads as Eukaryota, Archaea, Bacteria or Virus using Kraken 276 with the parameters ‘--confidence 0.5 --minimum-hit-groups 3’ combined with an extra filtering step that only kept those reads with root-to-leaf score >0.25. For the initial Kraken 2 search, we used a coarse database created by the taxdb-integration workflow (https://github.com/aMG-tk/taxdb-integration) covering all domains of life and including a genomic database of marine planktonic eukaryotes63 that contain 683 metagenome-assembled genomes (MAGs) and 30 single-cell genomes (SAGs) from Tara Oceans77, following the naming convention in Delmont et al.63, we will refer to them as SMAGs. Reads labelled as root, unclassified, archaea, bacteria and virus were refined through a second Kraken 2 labelling step using a high-resolution database containing archaea, bacteria and virus created by the taxdb-integration workflow. We used the same Kraken 2 parameters and filtering thresholds as the initial search. Both Kraken 2 databases were built with parameters optimized for the study read length (--kmer-len 25 --minimizer-len 23 --minimizer-spaces 4).
Reads labelled as eukaryota, root and unclassified were hereafter mapped with Bowtie278 against the SMAGs. We used MarkDuplicates from Picard (https://github.com/broadinstitute/picard) to remove duplicates and then we calculated the mapping statistics for each SMAG in the BAM files with the filterBAM program (https://github.com/aMG-tk/bam-filter). We furthermore estimated the postmortem damage of the filtered BAM files with the Bayesian methods in metaDMG and selected those SMAGs with a D-max ≥ 0.25 and a fit quality (λ-LR) higher than 1.5. The SMAGs with fewer than 500 reads mapped, a mean read average nucleotide identity (ANI) of less than than 93% and a breadth of coverage ratio and coverage evenness of less than 0.75 were removed. We followed a data-driven approach to select the mean read ANI threshold, where we explored the variation of mapped reads as a function of the mean read ANI values from 90% to 100% and identified the elbow point in the curve (Supplementary Fig. 6.11.1). We used anvi’o79 in manual mode to plot the mapping and damage results using the SMAGs phylogenomic tree inferred by Delmont et al. as reference. We used the oceanic signal of Delmont et al. as a proxy to the contemporary distribution of the SMAGs in each ocean and sea (Fig. 5 and Supplementary Information, section 6).
Comparison of DNA, macrofossil and pollen
To allow comparison between records in DNA, macrofossil and pollen, the taxonomy was harmonized following the Pan Arctic Flora checklist43 and NCBI. For example, since Bennike (1990)18, Potamogeton has been split into Potamogeton and Stuckenia, Polygonym has been split to Polygonum and Bistorta, and Saxifraga was split to Saxifraga and Micranthes, whereas others have been merged, such as Melandrium with Silene40. Plant families have changed names—for instance, Gramineae is now called Poaceae and Scrophulariaceae has been re-circumscribed to exclude Plantaginaceae and Orobancheae80. We then classified the taxa into the following: category 1 all identical genus recorded by DNA and macrofossils or pollen, category 2 genera recorded by DNA also found by macrofossils or pollen including genus contained within family level classifications, category 3 taxa only recorded by DNA, category 4 taxa only recorded by macrofossils or pollen (Source Data 1).
Phylogenetic placement
We sought to phylogenetically place the set of ancient taxa with the most abundant number of reads assigned, and with a sufficient number of reference sequences to build a phylogeny. These taxa include reads mapped to the chloroplast genomes of the flora genera Salix, Populus and Betula, and to the mitochondrial genomes of the fauna families Elephantidae, Cricetidae, Leporidae, as well as the subfamilies Capreolinae and Anserinae. Although the evolution of the chloroplast genome is somewhat less stable than that of the plant mitochondrial genome, it has a faster rate of evolution, and is non-recombining, and hence is more likely to contain more informative sites for our analysis than the plant mitochondria81. Like the mitochondrial genome, the chloroplast genome also has a high copy number, so that we would expect a high number of sedimentary reads mapping to it.
For each of these taxa, we downloaded a representative set of either whole chloroplast or whole mitochondrial genome fasta sequences from NCBI Genbank82, including a single representative sequence from a recently diverged outgroup. For the Betula genus, we also included three chloroplast genomes from the PhyloNorway database34,83. We changed all ambiguous bases in the fasta files to N. We used MAFFT84 to align each of these sets of reference sequences, and inspected multiple sequence alignments in NCBI MSAViewer to confirm quality85. We trimmed mitochondrial alignments with insufficient quality due to highly variable control regions for Leporidae, Cricetidae and Anserinae by removing the d-loop in MegaX86.
The BEAST suite49 was used with default parameters to create ultrametric phylogenetic trees for each of the five sets of taxa from the multiple sequence alignments (MSAs) of reference sequences, which were converted from Nexus to Newick format in Figtree (https://github.com/rambaut/figtree). We then passed the multiple sequence alignments to the Python module AlignIO from BioPython87 to create a reference consensus fasta sequence for each set of taxa. Furthermore, we used SNPSites88 to create a vcf file from each of the MSAs. Since SNPSites outputs a slightly different format for missing data than needed for downstream analysis, we used a custom R script to modify the vcf format appropriately. We also filtered out non-biallelic SNPs.
From the damage filtered ngsLCA output, we extracted all readIDs uniquely classified to reference sequences within these respective taxa or assigned to any common ancestor inside the taxonomic group and converted these back to fastq files using seqtk (https://github.com/lh3/seqtk). We merged reads from all sites and layers to create a single read set for each respective taxon. Next, since these extracted reads were mapped against a reference database including multiple sequences from each taxon, the output files were not on the same coordinate system. To circumvent this issue and avoid mapping bias, we re-mapped each read set to the consensus sequence generated above for that taxon using bwa89 with ancient DNA parameters (bwa aln -n 0.001). We converted these reads to bam files, removed unmapped reads, and filtered for mapping quality > 25 using samtools90. This produced 103,042, 39,306, 91,272, 182 and 129 reads for Salix, Populus, Betula, Elephantidae and Capreolinae, respectively.
We next used pathPhynder62, a phylogenetic placement algorithm that identifies informative markers on a phylogeny from a reference panel, evaluates SNPs in the ancient sample overlapping these markers, and traverses the tree to place the ancient sample according to its derived and ancestral SNPs on each branch. We used the transversions-only filter to avoid errors due to deamination, except for Betula, Salix and Populus in which we used no filter due to sufficiently high coverage. Last, we investigated the pathPhynder output in each taxon set to determine the phylogenetic placement of our ancient samples (see Supplementary Information for discussion on phylogenetic placement).
Based on the analysis described above we further investigated the phylogenetic placement within the genus Mammut, or mastodons. To avoid mapping reference biases in the downstream results, we first built a consensus sequence from all comparative mitochondrial genomes used in said analysis and mapped the reads identified in ngsLCA as Elephantidae to the consensus sequence. Consensus sequences were constructed by first aligning all sequences of interest using MAFFT84 and taking a majority rule consensus base in Geneious v2020.0.5 (https://www.geneious.com). We performed three analyses for phylogenetic placement of our sequence: (1) Comparison against a single representative from each Elephantidae species including the sea cow (Dugong dugon) as outgroup, (2) Comparison against a single representative from each Elephantidae species, and (3) Comparison against all published mastodon mitochondrial genomes including the Asian elephant as outgroup.
For each of these analyses we first built a new reference tree using BEAST v1.10.4 (ref. 47) and repeated the previously described pathPhynder steps, with the exception that the pathPhynder tree path analysis for the Mammut SNPs was based on transitions and transversions, not restricting to only transversions due to low coverage.
Mammut americanum
We confirmed the phylogenetic placement of our sequence using a selection of Elephantidae mitochondrial reference sequences, GTR+G, strict clock, a birth-death substitution model, and ran the MCMC chain for 20,000,000 runs, sampling every 20,000 steps. Convergence was assessed using Tracer91 v1.7.2 and an effective sample size (ESS) > 200. To determine the approximate age of our recovered mastodon mitogenome we performed a molecular dating analysis with BEAST47 v1.10.4. We used two separate approaches when dating our mastodon mitogenome, as demonstrated in a recent publication92. First, we determined the age of our sequence by comparing against a dataset of radiocarbon-dated specimens (n = 13) only. Secondly, we estimated the age of our sequence including both molecularly (n = 22) and radiocarbon-dated (n = 13) specimens using the molecular dates previously determined92. We utilized the same BEAST parameters as Karpinski et al.92 and set the age of our sample with a gamma distribution (5% quantile: 8.72 × 104, Median: 1.178 × 106; 95% quantile: 5.093 × 106; initial value: 74,900; shape: 1; scale: 1,700,000). In short, we specified a substitution model of GTR+G4, a strict clock, constant population size, and ran the Markov Chain Monte Carlo chain for 50,000,000 runs, sampling every 50,000 steps. Convergence of the run was again determined using Tracer.
Molecular dating methods
In this section, we describe molecular dating of the ancient birch (Betula) chloroplast genome using BEAST v1.10.4 (ref. 47). In principle, the genera Betula, Populus and Salix had both sufficiently high chloroplast genome coverage (with mean depth 24.16×, 57.06× and 27.04×, respectively, although this coverage is highly uneven across the chloroplast genome) and enough reference sequences to attempt molecular dating on these samples. Notably, this is one of the reasons we included a recently diverged outgroup with a divergence time estimate in each of these phylogenetic trees. However, our Populus sample clearly contained a mixture of different species, as seen from its inconsistent placement in the pathPhynder output. In particular, there were multiple supporting SNPs to both Populus balsamifera and Populus trichocarpa, and both supporting and conflicting SNPs on branches above. Furthermore, upon inspection, our Salix sample contained a surprisingly high number of private SNPs which is inconsistent with any ancient or even modern age, especially considering the number of SNPs assigned to the edges of the phylogenetic tree leading to other Salix sequences. We are unsure what causes this inconsistency but hypothesize that our Salix sample is also a mixed sample, containing multiple Salix species that diverged from the same placement branch on the phylogenetic tree at different time periods. This is supported by looking at all the reads that cover these private SNP sites, which generally appear to be from a mixed sample, with reads containing both alternate and reference alleles present at a high proportion in many cases. Alternatively, or potentially jointly in parallel, this could be a consequence of the high number of nuclear plastid DNA sequences (NUPTs) in Salix93. Because of this, we continued with only Betula.
First, we downloaded 27 complete reference Betula chloroplast genome sequences and a single Alnus chloroplast genome sequence to use as an outgroup from the NCBI Genbank repository, and supplemented this with three Betula chloroplast sequences from the PhyloNorway database generated in a recent study29, for a total of 31 reference sequences. Since chloroplast sequences are circular, downloaded sequences may not always be in the same orientation or at the same starting point as is necessary for alignment, so we used custom code (https://github.com/miwipe/KapCopenhagen) that uses an anchor string to rotate the reference sequences to the same orientation and start them all from the same point. We created a MSA of these transformed reference sequences with Mafft84 and checked the quality of our alignment by eye in Seqotron94 and NCBI MsaViewer. Next, we called a consensus sequence from this MSA using the BioAlign consensus function87 in Python, which is a majority rule consensus caller. We will use this consensus sequence to map the ancient Betula reads to, both to avoid reference bias and to get the ancient Betula sample on the same coordinates as the reference MSA.
From the last common ancestor output in metaDMG36, we extracted read sets for all units, sites and levels that were uniquely classified to the taxonomic level of Betula or lower, with at a minimum sequence similarity of 90% or higher to any Betula sequence, using Seqtk95. We mapped these read sets against the consensus Betula chloroplast genome using BWA89 with ancient DNA parameters (-o 2 -n 0.001 -t 20), then removed unmapped reads, quality filtered for read quality ≥25, and sorted the resulting bam files using samtools89. For the purpose of molecular dating, it is appropriate to consider these read sets as a single sample, and so we merged the resulting bam files into one sample using samtools. We used bcftools89 to make an mpileup and call a vcf file, using options for haploidy and disabling the default calling algorithm, which can slightly biases the calls towards the reference sequence, in favour of a majority call on bases that passed the default base quality cut-off of 13. We included the default option using base alignment qualities96, which we found greatly reduced the read depths of some bases and removed spurious SNPs around indel regions. Lastly, we filtered the vcf file to include only single nucleotide variants, because we do not believe other variants such as insertions or deletions in an ancient environmental sample of this type to be of sufficiently high confidence to include in molecular dating.
We downloaded the gff3 annotation file for the longest Betula reference sequence, MG386368.1, from NCBI. Using custom R code97, we parsed this file and the associated fasta to label individual sites as protein-coding regions (in which we labelled the base with its position in the codon according to the phase and strand noted in the gff3 file), RNA, or neither coding nor RNA. We extracted the coding regions and checked in Seqotron94 and R that they translated to a protein alignment well (for example, no premature stop codons), both in the reference sequence and the associated positions in the ancient sequence. Though the modern reference sequence’s coding regions translated to a high-quality protein alignment, translating the associated positions in the ancient sequence with no depth cut-off leads to premature stop codons and an overall poor quality protein alignment. On the other hand, when using a depth cut-off of 20 and replacing sites in the ancient sequence which did not meet this filter with N, we see a high-quality protein alignment (except for the N sites). We also interrogated any positions in the ancient sequence which differed from the consensus, and found that any suspicious regions (for example, with multiple SNPs clustered closely together spatially in the genome) were removed with a depth cut-off of 20. Because of this, we moved forward only with sites in both the ancient and modern samples which met a depth cut-off of at least 20 in the ancient sample, which consisted of about 30% of the total sites.
Next, we parsed this annotation through the multiple sequence alignment to create partitions for BEAST47. After checking how many polymorphic and total sites were in each, we decided to use four partitions: (1) sites belonging to protein-coding positions 1 and 2, (2) coding position 3, (3) RNA, or (4) non-coding and non-RNA. To ensure that these were high confidence sites, each partition also only included those positions which had at least depth 20 in the ancient sequence and had less than 3 total gaps in the multiple sequence alignment. This gave partitions which had 11,668, 5,828, 2,690 and 29,538 sites, respectively. We used these four partitions to run BEAST47 v1.10.4, with unlinked substitution models for each partition and a strict clock, with a different relative rate for each partition. (There was insufficient information in these data to infer between-lineage rate variation from a single calibration). We assigned an age of 0 to all of the reference sequences, and used a normal distribution prior with mean 61.1 Myr and standard deviation 1.633 Myr for the root height48; standard deviation was obtained by conservatively converting the 95% HPD to z-scores. For the overall tree prior, we selected the coalescent model. The age of the ancient sequence was estimated following the overall procedures of Shapiro et al. (2011)98. To assess sensitivity to prior choice for this unknown date, we used two different priors, namely a gamma distribution metric towards a younger age (shape = 1, scale = 1.7); and a uniform prior on the range (0, 10 Myr). We also compared two different models of rate variation among sites and substitution types within each partition, namely a GTR+G with four rate categories, and base frequencies estimated from the data, and the much simpler Jukes Cantor model, which assumed no variation between substitution types nor sites within each partition. All other priors were set at their defaults. Neither rate model nor prior choice had a qualitative effect on results (Extended Data Fig. 10). We also ran the coding regions alone, since they translated correctly and are therefore highly reliable sites and found that they gave the same median and a much larger confidence interval, as expected when using fewer sites (Extended Data Fig. 10). We ran each Markov chain Monte Carlo for a total of 100 million iterations. After removing a burn-in of the first 10%, we verified convergence in Tracer91 v1.7.2 (apparent stationarity of traces, and all parameters having an Effective Sample Size > 100). We also verified that the resulting MCC tree from TreeAnnotator47 had placed the ancient sequence phylogenetically identically to pathPhynder62 placement, which is shown in Extended Data Fig. 9. For our major results, we report the uniform ancient age prior, and the GTR+G4 model applied to each of the four partitions. The associated XML is given in Source Data 3. The 95% HPD was (2.0172,0.6786) for the age of the ancient Betula chloroplast sequence, with a median estimate of 1.323 Myr, as shown in Fig. 2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequence data (13,135,646,556 reads following adapter trimming) are available through the ENA project accession PRJEB55522. Pollen counts are available through https://github.com/miwipe/KapCopenhagen.git. Source data are provided with this paper.
Code availability
All code used is available at https://github.com/miwipe/KapCopenhagen.git.
References
Salzmann, N. et al. Glacier changes and climate trends derived from multiple sources in the data scarce Cordillera Vilcanota region, southern Peruvian Andes. Cryosphere 7, 103–118 (2013).
IPCC Climate Change 2013: The Physical Science Basis (eds Stocker, T. F. et al.) (Cambridge Univ. Press, 2013).
Brigham-Grette, J. et al. Pliocene warmth, polar amplification, and stepped Pleistocene cooling recorded in NE Arctic Russia. Science 340, 1421–1427 (2013).
Gosse, J. C. et al. PoLAR-FIT: Pliocene Landscapes and Arctic Remains—Frozen in Time. Geosci. Can. 44, 47–54 (2017).
Matthews, J. V., Telka, A. Jr & Kuzmina, S. A. Late Neogene insect and other invertebrate fossils from Alaska and Arctic/Subarctic Canada. Zool. Bespozvon. 16, 126–153 (2019).
Willerslev, E. et al. Diverse plant and animal genetic records from Holocene and Pleistocene sediments. Science 300, 791–795 (2003).
Funder, S. et al. Late Pliocene Greenland—the Kap København formation in North Greenland. Bull. Geol. Soc. Den. 48, 117–134 (2001).
Funder, S. & Hjort, C. A reconnaissance of the Quaternary geology of eastern North Greenland. Rapp. Grønlands Geol. Unders. 99, 99–105 (1980).
Fredskild, B. & Røen, U. Macrofossils in an interglacial peat deposit at Kap København, North Greenland. Boreas 11, 181–185 (2008).
Bennike, O. & Böcher, J. Forest-tundra neighbouring the North Pole: plant and insect remains from the Plio-Pleistocene Kap København Formation, North Greenland. Arctic 43, 331–338 (1990).
Böcher, J. Palaeoentomology of the Kap Kobenhavn Formation, a Pilo-Pleistocene sequence in Peary Land, North Greenland (Museum Tusculanum Press, 1995).
Rybczynski, N. et al. Mid-Pliocene warm-period deposits in the High Arctic yield insight into camel evolution. Nat. Commun. 4, 1550 (2013).
Wang, X., Rybczynski, N., Harington, C. R., White, S. C. & Tedford, R. H. A basal ursine bear (Protarctos abstrusus) from the Pliocene High Arctic reveals Eurasian affinities and a diet rich in fermentable sugars. Sci Rep. 7, 17722 (2017).
Simonarson, L. A., Petersen, K. S. & Funder, S. Molluscan palaeontology of the Pliocene-Pleistocene Kap København Formation, North Greenland. Vol. 36 (Museum Tusculanum Press, University of Copenhagen, 1998).
Mogensen, G. S. Pliocene or Early Pleistocene mosses from Kap København, North Greenland. Lindbergia 10, 19–26 (1984).
Funder, S., Abrahamsen, N., Bennike, O. & Feyling-Hanssen, R. W. Forested Arctic: evidence from North Greenland. Geology 13, 542–546 (1985).
Abrahamsen, N. & Marcussen, C. Magnetostratigraphy of the Plio-Pleistocene Kap København Formation, eastern North Greenland. Phys. Earth Planet. Inter. 44, 53–61 (1986).
Bennike, O. The Kap København Formation: Stratigraphy and Palaeobotany of a Plio- Pleistocene Sequence in Peary Land, North Greenland. Meddelelser om Grønland, Geoscience Vol. 23 (Kommissionen for Videnskabelige Umdersøgelser i Grønland, 1990).
Feyling-Hanssen, R. W. Foraminiferal Stratigraphy in the Plio-Pleistocene Kap Kobenhavn Formation, North Greenland (Museum Tusculanum Press, 1990).
Lindahl, T. Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993).
van der Valk, T. et al. Million-year-old DNA sheds light on the genomic history of mammoths. Nature 591, 265–269 (2021).
Klimaet i Grønland. https://www.dmi.dk/klima/temaforside-klimaet-frem-til-i-dag/klimaet-i-groenland/ (DMI, 2021).
Allentoft, M. E. et al. The half-life of DNA in bone: measuring decay kinetics in 158 dated fossils. Proc. Biol. Sci. 279, 4724–4733 (2012).
Nguyen, T. H. & Elimelech, M. Plasmid DNA adsorption on silica: kinetics and conformational changes in monovalent and divalent salts. Biomacromolecules 8, 24–32 (2007).
Melzak, K. A., Sherwood, C. S., Turner, R. F. B. & Haynes, C. A. Driving forces for DNA adsorption to silica in perchlorate solutions. J. Colloid Interface Sci. 181, 635–644 (1996).
Cai, P., Huang, Q.-Y. & Zhang, X.-W. Interactions of DNA with clay minerals and soil colloidal particles and protection against degradation by DNase. Environ. Sci. Technol. 40, 2971–2976 (2006).
Fang, Y. & Hoh, J. H. Early intermediates in spermidine-induced DNA condensation on the surface of mica. J. Am. Chem. Soc. 120, 8903–8909 (1998).
Karl, D. M. & Bailiff, M. D. The measurement and distribution of dissolved nucleic acids in aquatic environments. Limnol. Oceanogr. 34, 543–558 (1989).
Pedersen, M. W. et al. Postglacial viability and colonization in North America’s ice-free corridor. Nature 537, 45–49 (2016).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, db.prot5448 (2010).
Murchie, T. J. et al. Optimizing extraction and targeted capture of ancient environmental DNA for reconstructing past environments using the PalaeoChip Arctic-1.0 bait-set. Quat. Res. 99, 305–328 (2021).
Benoit, G. et al. Multiple comparative metagenomics using multiset k-mer counting. Preprint at https://arxiv.org/abs/1604.02412 (2016).
Pedersen, M. W. et al. Environmental genomics of Late Pleistocene black bears and giant short-faced bears. Curr. Biol. 31, 2728–2736.e8 (2021).
Wang, Y. et al. Late Quaternary dynamics of Arctic Biota from ancient environmental genomics. Nature 600, 86–92 (2021).
Wang, Y. et al. ngsLCA—A toolkit for fast and flexible lowest common ancestor inference and taxonomic profiling of metagenomic data. Methods Ecol. Evol. https://doi.org/10.1111/2041-210X.14006 (2022).
Michelsen, C. et al. metaDMG: an ancient DNA damage toolkit. Zenodo https://doi.org/10.5281/zenodo.7284374 (2022).
Raynolds, M. K. et al. A raster version of the Circumpolar Arctic Vegetation Map (CAVM). Remote Sens. Environ. 232, 111297 (2019).
Bay, C. Floristical and ecological characterization of the polar desert zone of Greenland. J. Veg. Sci. 8, 685–696 (1997).
Boertmann, D. & Bay, C. Grønlands Rødliste 2018: Fortegnelse over Grønlandske Dyr og Planters Trusselstatus (Grønlands Naturinstitut, Aarhus Universitet, 2018).
Böcher, T. W., Holman, K. & Jakobson, K. Grønlands Flora 3rd edn (Forlaget Haase & Søn, 1978).
Elven, R., Murray, D. F., Razzhivin, V. Y. & Yurtsev, B. A. Annotated Checklist of the Panarctic Flora (PAF). http://panarcticflora.org (2022).
Bay, C. Four decades of new vascular plant records for Greenland. PhytoKeys 145, 63–92 (2020).
Bay, C. A Phytogeographical Study of the Vascular Plants of Northern Greenland—North of 74 Northern Latitude Vol. 36 (Kommissionen for Videnskabelige Undersøgelser i Grønland, 1992).
Parducci, L. et al. Ancient plant DNA in lake sediments. New Phytol. 214, 924–942 (2017).
Alsos, I. G. et al. Plant DNA metabarcoding of lake sediments: How does it represent the contemporary vegetation. PLoS ONE 13, e0195403 (2018).
Bennike, O. The Kap København Formation: Stratigraphy and Palaeobotany of a Plio-Pleistocene Sequence in Peary Land, North Greenland (Kommissionen for Videnskabelige Undersøgelser i Grønland, 1990).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Yang, X.-Y. et al. Plastomes of Betulaceae and phylogenetic implications. J. Syst. Evol. 57, 508–518 (2019).
Bouckaert, R. et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10, e1003537 (2014).
Dowsett, H. J., Chandler, M. A., Cronin, T. M. & Dwyer, G. S. Middle Pliocene sea surface temperature variability. Paleoceanography https://doi.org/10.1029/2005PA001133 (2005).
Graham, R. W. et al. Timing and causes of mid-Holocene mammoth extinction on St Paul Island, Alaska. Proc. Natl Acad. Sci. USA 113, 9310–9314 (2016).
Parducci, L. et al. Shotgun environmental DNA, pollen, and macrofossil analysis of lateglacial lake sediments from southern Sweden. Front. Ecol. Evol. 7, 189 (2019).
Rijal, D. P. et al. Sedimentary ancient DNA shows terrestrial plant richness continuously increased over the Holocene in northern Fennoscandia. Sci. Adv. 7, eabf9557 (2021).
Cocker, S. L. et al. Dung analysis of the East Milford mastodons: dietary and environmental reconstructions from central Nova Scotia at ~75 ka yr bp. Can. J. Earth Sci. https://doi.org/10.1139/cjes-2020-0164 (2021).
Fletcher, T. L., Telka, A., Rybczynski, N. & Matthews, J. V. Jr. Neogene and early Pleistocene flora from Alaska, USA and Arctic/Subarctic Canada: new data, intercontinental comparisons and correlations. Palaeontol. Electronica https://doi.org/10.26879/1121 (2021).
Feng, R. et al. Amplified Late Pliocene terrestrial warmth in northern high latitudes from greater radiative forcing and closed Arctic Ocean gateways. Earth Planet. Sci. Lett. 466, 129–138 (2017).
Galetti, M. et al. Ecological and evolutionary legacy of megafauna extinctions. Biol. Rev. Camb. Philos. Soc. 93, 845–862 (2018).
Malhi, Y. et al. Megafauna and ecosystem function from the Pleistocene to the Anthropocene. Proc. Natl Acad. Sci. USA 113, 838–846 (2016).
Rolstad, J., Blanck, Y.-L. & Storaunet, K. O. Fire history in a western Fennoscandian boreal forest as influenced by human land use and climate. Ecol. Monogr. 87, 219–245 (2017).
Elias, S. A. & Matthews, J. V. Jr Arctic North American seasonal temperatures from the latest Miocene to the Early Pleistocene, based on mutual climatic range analysis of fossil beetle assemblages. Can. J. Earth Sci. 39, 911–920 (2002).
Feyling-Hanssen, R. W. A remarkable foraminiferal assemblage from the Quaternary of northeast Greenland. Bull. Geol. Soc. Denmark 38, 101–107 (1989).
Martiniano, R., De Sanctis, B., Hallast, P. & Durbin, R. Placing ancient DNA sequences into reference phylogenies. Mol. Biol. Evol. 39, msac017 (2022).
Delmont, T. O. et al. Functional repertoire convergence of distantly related eukaryotic plankton lineages abundant in the sunlit ocean. Cell Genom. 2, 100123 (2022).
Lindahl, T. & Nyberg, B. Rate of depurination of native deoxyribonucleic acid. Biochemistry 11, 3610–3618 (1972).
Hansen, J., Sato, M., Russell, G. & Kharecha, P. Climate sensitivity, sea level and atmospheric carbon dioxide. Philos. Trans. A 371, 20120294 (2013).
Taut, T., Kleeberg, R. & Bergmann, J. The new Seifert Rietveld program BGMN and its application to quantitative phase analysis. Mater. Struct. 5, 57–66 (1998).
Doebelin, N. & Kleeberg, R. Profex: a graphical user interface for the Rietveld refinement program BGMN. J. Appl. Crystallogr. 48, 1573–1580 (2015).
Cheary, R. W. & Coelho, A. A fundamental parameters approach to X-ray line-profile fitting. J. Appl. Crystallogr. 25, 109–121 (1992).
Kester, D. R., Duedall, I. W., Connors, D. N. & Pytkowicz, R. M. Preparation of artificial seawater1. Limnol. Oceanogr. 12, 176–179 (1967).
Grichuk, E. D. & Zaklinskaya, V. P. The Analysis of Fossil Pollen and Spore and Using these Data in Paleogeography (GeographGIZ Press, 1948).
Kupriyanova, L. A. & Aleshina, L. A. Pollen and Spores of the European USSR Flora (Nauka, 1972).
Moore, P. D., Webb, J. A. & Collinson, M. E. Pollen Analysis. (Blackwell Scientific, 1991).
Grimm, E. C. Tilia and Tiliagraph (Illinois State Museum, 1991).
Bennett, K. D. Manual for psimpoll and pscomb. http://www.chrono.qub.ac.uk/psimpoll/psimpoll.html (2002).
Ardelean, C. F. et al. Evidence of human occupation in Mexico around the Last Glacial Maximum. Nature 584, 87–92 (2020).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Karsenti, E. et al. A holistic approach to marine eco-systems biology. PLoS Biol. 9, e1001177 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Eren, A. M. et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6 (2021).
The Angiosperm Phylogeny Group.An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot. J. Linn. Soc. 141, 399–436 (2003).
Chevigny, N., Schatz-Daas, D., Lotfi, F. & Gualberto, J. M. DNA repair and the stability of the plant mitochondrial genome. Int. J. Mol. Sci. 21, 328 (2020).
Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J. & Sayers, E. W. GenBank. Nucleic Acids Res. 44, D67–D72 (2016).
Alsos, I. G. et al. Last Glacial Maximum environmental conditions at Andøya, northern Norway; evidence for a northern ice-edge ecological ‘hotspot’. Quat. Sci. Rev. 239, 106364 (2020).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Yachdav, G. et al. MSAViewer: interactive JavaScript visualization of multiple sequence alignments. Bioinformatics 32, 3501–3503 (2016).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Cock, P. J. A. et al. BioPython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 (2009).
Page, A. J. et al. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2, e000056 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Karpinski, E. et al. American mastodon mitochondrial genomes suggest multiple dispersal events in response to Pleistocene climate oscillations. Nat. Commun. 11, 4048 (2020).
Huang, Y., Wang, J., Yang, Y., Fan, C. & Chen, J. Phylogenomic analysis and dynamic evolution of chloroplast genomes in Salicaceae. Front. Plant Sci. 8, 1050 (2017).
Fourment, M. & Holmes, E. C. Seqotron: a user-friendly sequence editor for Mac OS X. BMC Res. Notes 9, 106 (2016).
Li, H. et al. Seqtk: a fast and lightweight tool for processing FASTA or FASTQ sequences. https://github.com/lh3/seqtk (2018).
Li, H. Improving SNP discovery by base alignment quality. Bioinformatics 27, 1157–1158 (2011).
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2018).
Shapiro, B. et al. A Bayesian phylogenetic method to estimate unknown sequence ages. Mol. Biol. Evol. 28, 879–887 (2011).
Huang, D. I., Hefer, C. A., Kolosova, N., Douglas, C. J. & Cronk, Q. C. B. Whole plastome sequencing reveals deep plastid divergence and cytonuclear discordance between closely related balsam poplars, Populus balsamifera and P. trichocarpa (Salicaceae). New Phytol. 204, 693–703 (2014).
Levsen, N. D., Tiffin, P. & Olson, M. S. Pleistocene speciation in the genus Populus (salicaceae). Syst. Biol. 61, 401–412 (2012).
Zhang, L., Xi, Z., Wang, M., Guo, X. & Ma, T. Plastome phylogeny and lineage diversification of Salicaceae with focus on poplars and willows. Ecol. Evol. 8, 7817–7823 (2018).
Acknowledgements
We acknowledge support from the Carlsberg Foundation for logistics to carry out two expeditions to Kap København in 2006 and 2012 (S. Funder, principal investigator for Carlsberg foundation grant to LongTerm and Kap København—the age). The fieldwork in 2016 was supported by a grant to N.K.L. from the Villum Foundation. We highly appreciate the collaborative support by Illumina Inc. that was crucial for the success of the project. E.W. and K.H.K. thank the Danish National Research Foundation (DNRF) and the Lundbeck Foundation (R302-2018-2155) for providing long-term funds to develop the necessary DNA technology that eventually made it possible to retrieve environmental DNA from these ancient deposits in the Kap København Formation. E.W. also acknowledges the Wellcome Trust (UNS69906), the Carlsberg Foundation (CF18-0024), Novo Foundation (NNF18SA0035006), Leverhume (RPG-2016-235) and GRF EXC CRS Chair - Cluster of Excellence (44113220) for their support. M.W.P. acknowledges support from the Carlsberg Foundation (CF17-0275). K.K.S. and S.J. acknowledge support from VILLUM FONDEN (00025352). I.G.A. and E.C. have received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 819192). B.D.S. acknowledges support from the Wellcome Trust programme in Mathematical Genomics and Medicine (WT220023). J.Å.K. was supported by the Carlsberg Foundation (CF20-0238). C.B. acknowledges ERC Advanced Award Diatomic (grant agreement no. 835067). J.C.G. was supported by Natural Science and Engineering Research Council of Canada–Discovery Grant 06785 and Canada Foundation for Innovation grant 21305. M.J.C. acknowledges support from the Danish National Research Foundation DNRF128. We thank G. Yang for cosmogenic isotope AMS target chemistry; S. Funder for introducing us to the Kap København Formation and generating much of the platform that enabled us to conduct our research; T. O. Delmont for providing data and guidance on the SMAGs analysis; Minik Rosing for providing talc minerals; T. B. Zunic for providing tremolite, orthoclase and chlorite; Z. Vardanyan for help with the DNA extractions and library build; and L. B. Levy and D. Skov for their help collecting samples in 2016. This work was prepared in part by Lawrence Livermore National Laboratory under contract DE-AC52-07NA27344; LLNL-JRNL-830653. E.W. thanks St Johns College, Cambridge for providing him with a stimulating environment for scientific thoughts and discussion.
Author information
Authors and Affiliations
Consortia
Contributions
K.H.K. and E.W. conceived the idea. K.H.K., M.W.P. and E.W. designed the study. K.H.K., A.M.A.S., A.S.T., N.K.L. and E.W. provided samples, context and carried out fieldwork. M.W.P. undertook the DNA laboratory analysis and taxonomic profiling. M.W.P., B.D.S. and B.D.C. performed the phylogenetic placement with the supervision of M.S. and R.D. B.D.S., M.W.P. and B.D.C. performed the genetic dating with the supervision of R.D. and J.J.W. M.W.P., T.S.K. and C.S.M. conceived, designed and performed the DNA damage estimates. K.K.S. and S.J. conceived and designed the DNA–mineral aspects of the study, interpreted, and wrote about the DNA–mineral data, and participated in the thermal age calculations. K.H.K., M.W.P., A.H.R., A.R., I.G.A. and E.W. undertook the floristic analysis and interpretations. K.K.K. performed cartography and GIS analysis. I.S. designed and carried out palaeomagnetic analysis and interpreted the results. J.C.G. prepared and analysed eight samples for cosmogenic 26Al and 10Be and, with A.J.H., interpreted their burial age. I.G.A., E.C. and Y.W. provided access to the PhyloNorway reference database, and gave input to the phylogenetic placement of the chloroplasts. A.S.T. counted pollen from the six additional samples. J.Å.K. supported sediment provenance evaluation. M.B. provided mineralogical data from North Greenland. C.D., M.R., M.E.J. and B.S. designed and carried out ddPCR based assays to detect and identify ancient plant DNA in samples. A.F.-G. contributed to the bioinformatic analysis of SMAGs and C.B. contributed to interpretation of marine metagenomic signals. M.J.C. contributed to the thermal age and DNA modelling. M.E.A. contributed to the DNA decay rate estimates. K.H.K., M.W.P., A.H.R. and E.W. interpreted the results and wrote the manuscript with contributions from K.K.S., S.J., A.R., B.D.S., B.D.C., I.G.A., J.C.G., I.S. and N.K.L., with inputs from the other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Setting A.
Type locality 50 indicating units in formation b. Overview locality 74a+b with a complete sediment sequence. C. Overview of locality 69. D. Detail of organic rich sediment in unit B3 before excavation and cleaning for ancient eDNA samples. E. Sampling in the permafrost within unit B3 at locality 50. F. Organic rich sediment at the base of mega-scale cross-bedding within unit B2 at locality 74a+b. White circles mark persons for scale.
Extended Data Fig. 2 Phylogenetic placement results of Leporidae mitochondrial reads, using transversion SNPs only.
Reads have been merged from all layers and sites. The green numbers on each edge represent the number of supporting (+) SNPs, whereas the red numbers indicate conflicting (−) SNPs in the ancient Leporidae environmental mitochondrial genome overlapping the reference SNPs assigned to the respective edge. There is a clear placement for the ancient Leporidae environmental mitochondrial genome on the edge marked +2, basal to the extant Lepus lineage.
Extended Data Fig. 3 Phylogenetic placement results for representatives of the Capreolinae mitochondrial reads, using transversion SNPs only.
Reads have been merged from all layers and sites. The green numbers on each edge represent the number of supporting (+) SNPs, whereas the red numbers indicate conflicting (−) SNPs in the ancient Capreolinae environmental mitochondrial genome overlapping the reference SNPs assigned to the respective edge. There is a clear placement for the ancient Capreolinae environmental mitochondrial genome on the edge marked +8/−3, basal to the Rangifer genus.
Extended Data Fig. 4 Phylogenetic placement of Elephantidae mitochondrial reads within mastodons (Mammut americanum), using Elephas maximus as outgroup, including transitions and transversion SNPs.
(Please note that the NCBI taxonomy includes the Mammut genus within Elephantidae). The reference dataset consisted of mitochondria from mastodons (Mammut americanum) only and one Elephas maximus as an outgroup. Reads have been merged from all layers and sites. The green numbers on each edge represent the number of supporting (+) SNPs, whereas the red numbers indicate conflicting (−) SNPs in the ancient Elephantidae environmental mitochondrial genome overlapping the reference SNPs assigned to the respective edge. There is a placement for the ancient Elephantidae environmental mitochondrial genome on the edge marked +2/−1, identifying it as basal to the mastodon (Mammut americanum) clade, which contains most of all mastodon reference mitochondrial genomes. Please note that this placement is based on two transition SNPs with a read depth of three reads per SNP.
Extended Data Fig. 5 Phylogenetic placement of mitochondrial reads assigned within Anatidae and placed with representatives of the Anatidae, using transversion SNPs only.
Reads have been merged from all layers and sites. The green numbers on each edge represent the number of supporting (+) SNPs, whereas the red numbers indicate conflicting (−) SNPs in the ancient Anatidae environmental mitochondrial genome overlapping the reference SNPs assigned to the respective edge. There is a clear placement for the ancient Anatidae environmental mitochondrial genome on the edge marked +3, basal to the Branta genus.
Extended Data Fig. 6 Phylogenetic placement results of Cricetidae mitochondrial reads, using transversion SNPs only.
Reads have been merged from all layers and sites. The green numbers on each edge represent the number of supporting (+) SNPs, whereas the red numbers in the red circles indicate conflicting (−) SNPs in the ancient Cricetidae environmental mitochondrial genome overlapping the reference SNPs assigned to the respective edge. There is a placement for the ancient Cricetidae environmental mitochondrial genome on the edge marked +1, basal to the Arvicolinae subfamily.
Extended Data Fig. 7 Phylogenetic placement results for our Populus chloroplast reads, using both transition and transversion SNPs, and using reads merged from all layers and sites.
The numbers on each edge represent the number of supporting (+) and conflicting (−) SNPs in the ancient Populus environmental genome overlapping the reference SNPs assigned to that edge. The ancient Populus environmental genome clearly contains a mixture of different species. The most likely placement is on the edge above Populus trichocarpa (NC 009143.1) and Populus balsamifera (NC 024735.1), with +71/−15 supporting and conflicting SNPs. However, we find some support for both branches directly leading to these species as well. Populus balsamifera and P. trichocarpa are considered sister species. They are both distributed in North America, as far North as Alaska, are known to hybridise both among themselves and other Populus species and are morphologically very similar93,99,100. Previous analyses found a very recent nuclear genome divergence time of only 75000 years ago for Populus trichocarpa and P. balsamifera100, but a deep chloroplast genome divergence time of at least 6-7 Ma99, which is an uncommon pattern in plants. Our ancient Populus sample could contain individuals either ancestral to, or hybridized from, the modern Populus trichocarpa and P. balsamifera species.
Extended Data Fig. 8 Phylogenetic placement results for our Salix chloroplast reads, using both transition and transversion SNPs, and using reads merged from all layers and sites.
The numbers on each edge represent the number of supporting (+) and conflicting (−) SNPs in the ancient Salix environmental genome overlapping the reference SNPs assigned to that edge. The ancient Salix environmental genome falls basal to a main Salix clade. Our ancient Salix sample is phylogenetically placed, with 356 supporting SNPs and 22 conflicting SNPs, on a basal branch leading to the main clade. Although the Salix chloroplast phylogeny is not considered fully resolved93, the difficulties in resolution lie underneath our placement branch, and this along with the high number of SNPs on the placement branch allow us to be confident in the placement position. Our chloroplast phylogeny agrees roughly with101, which estimated a divergence date between these two main Salix clades at 16.9 Ma, and a root age of the first clade, to which our ancient sample is basal to, of 8.1 Ma. It is reasonable, then, to conclude that our ancient Salix sample is at least 8.1 Ma diverged from modern Salix species, and probably represents an extinct species, or extant species without a reference genome sequenced, or a pool thereof.
Extended Data Fig. 9 Phylogenetic placement results for our Betula chloroplast reads, using both transition and transversion SNPs, and using reads merged from all layers and sites.
Our ancient Betula sample was placed basal to a main Betula clade, based on 29 supporting (green) and 13 conflicting (red) SNPs on its placement branch, and with very low numbers of supporting SNPs elsewhere in the tree other than those leading to this branch. This placement agrees with the BEAST molecular dating analysis (see Molecular Dating Methods).
Extended Data Fig 10 Molecular age distribution.
Results of running the ancient Betula chloroplast molecular dating analysis BEAST v1.10.4 (ref. 47) with different priors and nucleotide substitution models. Using only coding regions, and therefore fewer total sites, gives a larger confidence interval as expected. Results reported in the text are for the red curve, with a flat prior and a GTR+Γ4 substitution model.
Supplementary information
Supplementary Information
This file contains background as well as additional information and discussion on the methods and intermediate results related to the main findings of Palaeomagnetism, previous age control, cosmogenic dating, minerology, micro- and macrofossil analysis, and eDNA presented in the main text. It also lists the PhyloNorway consortium members.
Supplementary Data 1
This file contains tables with the DNA sequence metadata and sample-geological unit information, the comparisons between DNA, macrofossils, phylogenetic placements, additional references added for the genomic databases, the digital droplet PCR results, and the tables used for plotting the taxonomic profiles (both raw counts and the percentages).
Supplementary Data 2
Supplementary Data 2 contains all X-ray diffraction results, mineral experiments treatments and adsorption, as well as the thermal age calculation.
Supplementary Data 3
Supplementary Data 3 is the .xml file with the Betula molecular age estimate.
Supplementary Data 4
The individual X-ray diffraction profiles obtained.
Supplementary Data 5
Supplementary Data 5 are the calculated intensity and their differences as .tiff files.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kjær, K.H., Winther Pedersen, M., De Sanctis, B. et al. A 2-million-year-old ecosystem in Greenland uncovered by environmental DNA. Nature 612, 283–291 (2022). https://doi.org/10.1038/s41586-022-05453-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05453-y
This article is cited by
-
Genomics for monitoring and understanding species responses to global climate change
Nature Reviews Genetics (2024)
-
Genetic gains underpinning a little-known strawberry Green Revolution
Nature Communications (2024)
-
Archaeology meets environmental genomics: implementing sedaDNA in the study of the human past
Archaeological and Anthropological Sciences (2024)
-
Cladocerans and diatoms from an Early Pleistocene interglacial deposit at Pingorsuit, North-West Greenland
Journal of Paleolimnology (2024)
-
Paleoecology of extinct species
BMC Ecology and Evolution (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
