
Abstract
The extent to which Omicron infection1,2,3,4,5,6,7,8,9, with or without previous vaccination, elicits protection against the previously dominant Delta (B.1.617.2) variant is unclear. Here we measured the neutralization capacity against variants of severe acute respiratory syndrome coronavirus 2 in 39 individuals in South Africa infected with the Omicron sublineage BA.1 starting at a median of 6 (interquartile range 3–9) days post symptom onset and continuing until last follow-up sample available, a median of 23 (interquartile range 19–27) days post symptoms to allow BA.1-elicited neutralizing immunity time to develop. Fifteen participants were vaccinated with Pfizer's BNT162b2 or Johnson & Johnson's Ad26.CoV2.S and had BA.1 breakthrough infections, and 24 were unvaccinated. BA.1 neutralization increased from a geometric mean 50% focus reduction neutralization test titre of 42 at enrolment to 575 at the last follow-up time point (13.6-fold) in vaccinated participants and from 46 to 272 (6.0-fold) in unvaccinated participants. Delta virus neutralization also increased, from 192 to 1,091 (5.7-fold) in vaccinated participants and from 28 to 91 (3.0-fold) in unvaccinated participants. At the last time point, unvaccinated individuals infected with BA.1 had low absolute levels of neutralization for the non-BA.1 viruses and 2.2-fold lower BA.1 neutralization, 12.0-fold lower Delta neutralization, 9.6-fold lower Beta variant neutralization, 17.9-fold lower ancestral virus neutralization and 4.8-fold lower Omicron sublineage BA.2 neutralization relative to vaccinated individuals infected with BA.1. These results indicate that hybrid immunity formed by vaccination and Omicron BA.1 infection should be protective against Delta and other variants. By contrast, infection with Omicron BA.1 alone offers limited cross-protection despite moderate enhancement.
Similar content being viewed by others

Main
The Omicron variant of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), first identified in November 2021 in South Africa and Botswana10, has been shown by us1 and others2,3,4,5,6,7,8,9 to have extensive but incomplete escape from neutralizing immunity elicited by vaccines and previous infection, with boosted individuals showing better neutralization. In South Africa, Omicron infections led to a lower incidence of severe disease relative to other variants11,12, although this can be at least partly explained by pre-existing immunity13. The first Omicron sublineage to appear was BA.1, which was supplanted by the BA.2 sublineage in many countries14.
How Omicron BA.1 infection will interact with vaccination to protect against the previously dominant Delta variant, emerging variants such as BA.2 and other variants is still unclear. We isolated live Omicron BA.1, Omicron BA.2, ancestral, Beta and Delta viruses and neutralized viruses with plasma from participants enrolled and longitudinally sampled during the Omicron BA.1 infection wave in South Africa, with all participants having a confirmed diagnosis of SARS-CoV-2 by quantitative PCR. To quantify neutralization, we used a live virus neutralization assay and calculated the focus reduction neutralization test (FRNT50) titre, the inverse of the plasma dilution required for 50% neutralization, as measured by the reduction in the number of infection foci. We enrolled 41 participants who reported symptoms from late November 2021 to January 2022. We successfully sequenced the infecting virus in 26 participants, and all sequences corresponded to Omicron BA.1 (Extended Data Table 1). Two participants had advanced human immunodeficiency virus (HIV) disease on the basis of a low CD4 count (<200 cells μl−1 throughout the study) and unsuppressed HIV infection, and we excluded these participants because of our previous data showing an atypical response to SARS-CoV-2 in advanced HIV disease15. Extended Data Table 2 summarizes the characteristics of the remaining 39 participants.
Of the 39 participants, 27 were admitted to hospital because of coronavirus disease 2019 symptoms. Seven required supplemental oxygen and one died. Fifteen participants were vaccinated and had a breakthrough Omicron BA.1 infection. The median time post vaccination was 139 days (interquartile range (IQR) 120–178), a time interval that would predict considerable waning of the vaccine-elicited immune response16, which may have contributed to the breakthrough infections. Eight participants were vaccinated with two doses of Pfizer's BNT162b2 and seven were vaccinated with Johnson & Johnson's Ad26.CoV2.S (six with a single dose and one with two doses; Extended Data Table 1). The length of hospital stay was shorter in the vaccinated (3.5 days) relative to unvaccinated (8 days; Extended Data Table 2) participants. Three participants self-reported having a previous SARS-CoV-2 infection (Extended Data Table 1).
Participants were sampled at enrolment at a median of 6 days (IQR 3–9 days) after symptom onset, and again at weekly follow-up visits that were attended as practicable because of the Christmas holidays in South Africa. The last follow-up visit was a median of 23 days (IQR 19–27 days) post-symptom onset (Extended Data Table 1). Examining neutralization at all available time points per study participant showed that neutralization of the Omicron BA.1 variant increased substantially in most participants from enrolment to the time of the last follow-up (Extended Data Fig. 1), consistent with developing a neutralizing antibody response to Omicron BA.1 infection. We therefore analysed neutralization at enrolment (baseline for the study) and the last follow-up visit to quantify the increase in neutralization capacity after Omicron infection.
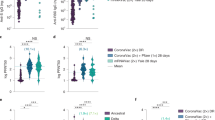
We observed that Omicron BA.1 neutralization increased in vaccinated individuals from a low geometric mean titre (GMT) FRNT50 of 42 at the enrolment visit to 575 at the last follow-up visit about 2 to 3 weeks later, a 13.6-fold change (95% CI confidence interval (CI) 3.7–50.2; Fig. 1a). The samples from unvaccinated participants at the study baseline neutralized Omicron BA.1 at a similar starting level of 46 and reached a final level of 272 at the last follow-up, a 6.0-fold increase (95% CI 2.2–16.1; Fig. 1b). Neutralization of the Delta virus also increased during this period. At enrolment, neutralization capacity against the Delta virus was 192 and reached a final level of 1,091 at the last follow-up visit in vaccinated participants, a 5.7-fold increase (95% CI 1.7–18.4; Fig. 1c). Unvaccinated participants had lower Delta neutralization at baseline with Delta virus FRNT50 = 28, and reached FRNT50 = 91, a 3.2-fold increase (95% CI 1.3–8.1; Fig. 1d).

a,b, Neutralization of the Omicron BA.1 virus over time for n = 15 vaccinated (a) and n = 24 unvaccinated (b) participants infected with Omicron BA.1. c,d, Neutralization of the Delta virus over time for the same vaccinated (n = 15; c) and unvaccinated (n = 24; d) participants as in a,b. For each participant, the sample collected at the initial enrolment visit (median 6 days post symptom onset) was compared with that collected at the last follow-up visit (median 23 days post symptom onset). The neutralization capacity per participant was determined in two independent experiments, and the numbers and horizontal bars are GMTs over all participants per group of the reciprocal plasma dilution (FRNT50) resulting in 50% neutralization. The fold change was calculated by dividing the GMT from the follow-up by the GMT from the enrolment visit. The dashed line is the most concentrated plasma tested. The P values were determined by a left-sided Wilcoxon rank sum test measuring the significance of the increase; **P = 0.01–0.001; NS, not significant. The exact P values are 0.0012 (a), 0.0081 (b), 0.0021 (c) and 0.11 (d).
We next compared Omicron BA.1 to Omicron BA.2, Delta, Beta (ref. 17) and ancestral virus neutralization at the last available follow-up visit in three sets of paired experiments, each comparing Omicron BA.2, Delta or ancestral and Beta virus neutralization to Omicron BA.1 neutralization. The range of Omicron BA.1 neutralization shown in Fig. 2a for different experiments (FRNT50 = 516 to 646 for vaccinated samples and 266 to 271 for unvaccinated samples) is the result of experimental variation. BA.2 neutralization was moderately and not significantly lower relative to BA.1 neutralization in both vaccinated and unvaccinated participants. Testing only participants with sequence-confirmed Omicron BA.1 infection gave a similar result (Extended Data Fig. 2). The trend for the other variants and the ancestral virus was that neutralization was higher relative to Omicron BA.1 in vaccinated participants but lower relative to Omicron BA.1 in unvaccinated participants, although the differences were mostly not significant (Fig. 2a). As a result of the relatively moderate fold change, higher participant numbers would probably be required to make the trends statistically significant.

a, Neutralization of Omicron BA.2, Beta, Delta and ancestral (with the D614G substitution) viruses compared to the Omicron BA.1 virus at the last available follow-up time point in n = 15 vaccinated or n = 24 unvaccinated participants infected with Omicron BA.1. The neutralization capacity per participant was determined in two independent experiments, and the numbers and horizontal bars are GMT FRNT50. The fold change was calculated by dividing the larger by the smaller GMT. The dashed line is the most concentrated plasma tested. The P values were determined by a two-sided Wilcoxon rank sum test; *P = 0.05–0.01; NS, not significant. The exact P values (vaccinated/unvaccinated) are: 0.22/0.087 for BA.2, 0.36/0.071 for Beta, 0.15/0.25 for Delta and 0.014/0.20 for ancestral. b, Comparison of the neutralization capacity against the Omicron BA.1, Omicron BA.2, Beta, Delta and ancestral (D614G) viruses in vaccinated (n = 15) versus unvaccinated (n = 24) participants infected with Omicron BA.1. The neutralization capacity per participant was determined in two independent experiments for all strains except for Omicron BA.1, for which six experiments were available and were used in the calculation. The points represent GMT FRNT50 per group and the error bars are GMT 95% CIs. The P values were determined by a two-sided Wilcoxon rank sum test; *P = 0.05–0.01; **P = 0.01–0.001; ***P = 0.001–0.0001. The exact P values are 0.025 (BA.1), 0.0026 (BA.2), 4.1 × 10−4 (Beta), 0.0012 (Delta) and 3.3 × 10−4 (ancestral).
The comparison of the other variants to Omicron BA.1 within the vaccinated or unvaccinated group does not indicate the differences in neutralization capacity elicited by Omicron BA.1 between the vaccinated and unvaccinated participants. We therefore compared neutralization of each variant between the vaccinated and unvaccinated groups at the last time point directly (Fig. 2b). The smallest difference between vaccinated and unvaccinated participants was in neutralization of Omicron BA.1, the infecting variant, with the vaccinated participants showing 2.2-fold higher neutralization. For the other variants, neutralization was higher in vaccinated participants by a factor of 4.8-fold for Omicron BA.2, 9.6-fold for Beta, 12.0-fold for Delta and 17.9-fold for ancestral (Fig. 2b). All differences were significant, and the 95% CIs for the GMT FRNT50 of vaccinated and unvaccinated participants did not overlap for BA.2, Beta, Delta or ancestral virus neutralization (Fig. 2b). For the unvaccinated participants, the absolute neutralization capacity against the BA.2, Beta, Delta and ancestral viruses was low18, with GMT FRNT50 being about or below FRNT50 = 100 (Fig. 2b).
We also tested neutralization of Omicron BA.1 by Delta-variant-elicited immunity. We collected 18 plasma samples from 14 participants (including pre-vaccination and post-vaccination samples from 4 participants) previously infected in the Delta variant wave in South Africa, 8 of whom were vaccinated either before or after infection (Extended Data Table 3). Confirming previously reported results19, we observed similar escape of Omicron BA.1 from Delta-elicited immunity across all samples tested, manifested as a 22.5-fold decrease (95% CI 14.4–35.0) in Omicron BA.1 neutralization compared to Delta virus neutralization (Fig. 3).

Neutralization of Delta compared to the Omicron BA.1 virus by Delta-infection-elicited plasma immunity in vaccinated and unvaccinated participants. A total of 18 samples were tested from n = 14 participants infected during the Delta infection wave in South Africa. The neutralization capacity per participant was determined in two independent experiments, and the numbers and horizontal bars are GMT FRNT50. The fold change was calculated by dividing the larger by the smaller GMT. The dashed line is the most concentrated plasma tested. ****P = 3.2 × 10−7 as determined by a two-sided Wilcoxon rank sum test.
The large fold drop in Delta-infection-elicited neutralization capacity against Omicron BA.1 contrasts with the moderate and nonsignificant fold drops, or even fold increases, in neutralization of other variants relative to Omicron BA.1 in individuals infected with Omicron BA.1. However, in unvaccinated individuals, even though fold drops in neutralization were moderate and nonsignificant, the absolute levels of neutralization of the other variants, and of Omicron BA.1 itself, were low and on a similar scale to the cross-neutralization capacity against Omicron in Delta-infection-elicited immunity. This is consistent with other recently reported results20, and possibly indicates that Omicron is poorly immunogenic. In agreement with recent reports21,22, our observations show moderately and nonsignificantly lower neutralization of BA.2 by BA.1-elicited immunity. The results explain epidemiological observations showing that Omicron BA.2 reinfection is relatively rare soon after Omicron BA.1 infection23,24.
Our results may be supportive of a scenario in which hybrid immunity formed by Omicron infection combined with vaccination protects as well or better against reinfection with variants such as Delta relative to reinfection with Omicron itself. By contrast, unvaccinated participants infected with Omicron BA.1 only, have low neutralization capacity against the Omicron BA.2, Beta, Delta and ancestral viruses.
Limitations of this study include heterogeneity in participant immune history, including two vaccination types and one boost. On the basis of the high seroprevalence observed in South Africa25,26, some participants may also have had unreported previous infection. However, including two vaccine types did not mask the differences between vaccinated and unvaccinated participants, and the low levels of neutralization in unvaccinated participants against the ancestral, Beta and Delta viruses (the dominant strains in the preceding South African infection waves) support the notion that these participants were either not previously infected, or that immunity has waned completely. Participants were also mostly hospitalized, which may not be typical of Omicron infection13,27. Increased disease severity has been shown to lead to higher anti-SARS-CoV-2 antibody titres28. This should help in the detection of the neutralization response, but whether it would affect the trend we observed is unclear. Omicron infection is unlikely benign to the extent that hospitalization is an outlier outcome: in the USA, the number of individuals with coronavirus disease 2019 who died in the Omicron wave was similar to the number who died in the Delta wave27. Neutralizing immunity may have increased further in some participants had we sampled later: the neutralizing capacity did not plateau at the last time point in 8 of the 24 (33%) unvaccinated participants (participants 10, 14, 21, 27, 30, 31, 34 and 38; Extended Data Fig. 1) and 6 of the 15 (40%) vaccinated participants (participants 4, 6, 15, 16, 25 and 26). Therefore, the temporal dynamics give no clear indication that the immunity in the unvaccinated participants was delayed and would have reached similar levels to that of vaccinated participants if sampled later. However, the consequences of waning immunity several months post Omicron infection should be investigated.
The gap in immunity between unvaccinated individuals infected with Omicron BA.1 and vaccinated individuals with BA.1 breakthrough infection is concerning. Especially as immunity wanes, unvaccinated individuals post Omicron infection are likely to have poor cross-protection against existing and possibly emerging SARS-CoV-2 variants, despite acquiring some neutralizing immunity to the infecting Omicron BA.1 sub-lineage variant. The implication may be that Omicron BA.1 infection alone is not sufficient for protection and vaccination should be administered even in areas with a high prevalence of Omicron infection to protect against other variants.
Methods
Informed consent and ethics
Blood samples and the Delta isolate were obtained after written informed consent from adults with PCR-confirmed SARS-CoV-2 infection who were enrolled in a prospective cohort study at the Africa Health Research Institute approved by the Biomedical Research Ethics Committee at the University of KwaZulu-Natal (reference BREC/00001275/2020). Omicron BA.1 was isolated from a residual swab sample for SARS-CoV-2 where isolation from the sample was approved by the University of the Witwatersrand Human Research Ethics Committee (HREC; reference M210752). The sample to isolate Omicron BA.2 was collected after written informed consent as part of the study “COVID-19 transmission and natural history in KwaZulu-Natal, South Africa: Epidemiological Investigation to Guide Prevention and Clinical Care” of the Centre for the AIDS Programme of Research in South Africa (CAPRISA) and isolation from the sample approved by the Biomedical Research Ethics Committee at the University of KwaZulu-Natal (reference BREC/00001195/2020, BREC/00003106/2021).
Reagent availability
Virus isolates and the cell line are available from the corresponding author. A Biosafety Level 3 facility is required for laboratories receiving live SARS-CoV-2.
Whole-genome sequencing, genome assembly and phylogenetic analysis
RNA was extracted on an automated Chemagic 360 instrument, using the CMG-1049 kit (Perkin Elmer). The RNA was stored at −80 °C before use. Libraries for whole-genome sequencing were prepared using either the Oxford Nanopore Midnight protocol with Rapid Barcoding or the Illumina COVIDseq Assay. For the Illumina COVIDseq assay, the libraries were prepared according to the manufacturer’s protocol. Briefly, amplicons were tagmented, followed by indexing using the Nextera UD Indexes Set A. Sequencing libraries were pooled, normalized to 4 nM and denatured with 0.2 N sodium acetate. An 8 pM sample library was spiked with 1% PhiX (PhiX Control v3 adaptor-ligated library used as a control). We sequenced libraries on a 500-cycle v2 MiSeq Reagent Kit on the Illumina MiSeq instrument (Illumina). On the Illumina NextSeq 550 instrument, sequencing was performed using the Illumina COVIDSeq protocol (Illumina), an amplicon-based next-generation sequencing approach. The first-strand synthesis was carried using random hexamer primers from Illumina, and the synthesized cDNA underwent two separate multiplex PCR reactions. The pooled PCR-amplified products were processed for tagmentation and adaptor ligation using IDT for Illumina Nextera UD Indexes. Further enrichment and cleanup was performed as per protocols provided by the manufacturer (Illumina). Pooled samples were quantified using a Qubit 3.0 or 4.0 fluorometer (Invitrogen) through the Qubit dsDNA High Sensitivity Assay according to the manufacturer’s instructions. The fragment sizes were analysed using TapeStation 4200 (Invitrogen). The pooled libraries were further normalized to 4 nM concentration and 25 μl of each normalized pool containing unique index adaptor sets was combined in a new tube. The final library pool was denatured and neutralized with 0.2 N sodium hydroxide and 200 mM Tris-HCl (pH 7), respectively. A 1.5 pM sample library was spiked with 2% PhiX. Libraries were loaded onto a 300-cycle NextSeq 500/550 HighOutput Kit v2 and run on the Illumina NextSeq 550 instrument (Illumina). For Oxford Nanopore sequencing, the Midnight primer kit was used: cDNA synthesis was performed on the extracted RNA using LunaScript RT mastermix (New England BioLabs) followed by gene-specific multiplex PCR using the Midnight Primer pools that produce 1,200-base-pair amplicons that overlap to cover the 30-kb SARS-CoV-2 genome. Amplicons from each pool were pooled and used neat for barcoding with the Oxford Nanopore Rapid Barcoding kit as per the manufacturer’s protocol. Barcoded samples were pooled and bead purified. After the bead cleanup, the library was loaded on a prepared R9.4.1 flow cell. A GridION X5 or MinION sequencing run was initiated using MinKNOW software with the base-call setting switched off. We assembled paired-end and nanopore.fastq reads using Genome Detective 1.132 (https://www.genomedetective.com), which was updated for the accurate assembly and variant calling of tiled primer amplicon Illumina or Oxford Nanopore reads, and the Coronavirus Typing Tool. For Illumina assembly, the GATK HaploTypeCaller –min-pruning 0 argument was added to increase mutation calling sensitivity near sequencing gaps. For Nanopore, low-coverage regions with poor alignment quality (<85% variant homogeneity) near sequencing/amplicon ends were masked to be robust against primer drop-out experienced in the spike gene, and the sensitivity for detecting short inserts using a region-local global alignment of reads was increased. In addition, we also used the wf_artic (ARTIC SARS-CoV-2) pipeline as built using the nextflow workflow framework. In some instances, mutations were confirmed visually with .bam files using Geneious software V2020.1.2 (Biomatters). The reference genome used throughout the assembly process was NC_045512.2 (numbering equivalent to MN908947.3). For lineage classification, we used the widespread dynamic lineage classification method from the Phylogenetic Assignment of Named Global Outbreak Lineages software suite (https://github.com/hCoV-2019/pangolin).
Cells
Vero E6 cells (originally ATCC CRL-1586, obtained from Cellonex in South Africa) were propagated in complete growth medium consisting of Dulbecco's modified Eagle medium with 10% fetal bovine serum (Hyclone) containing 10 mM HEPES, 1 mM sodium pyruvate, 2 mM l-glutamine and 0.1 mM nonessential amino acids (Sigma-Aldrich). Vero E6 cells were passaged every 3–4 days. The H1299-E3 cell line (H1299 originally from ATCC as CRL-5803) was propagated in growth medium consisting of complete Roswell Park Memorial Institute 1640 medium with 10% fetal bovine serum containing 10 mM HEPES, 1 mM sodium pyruvate, 2 mM l-glutamine and 0.1 mM nonessential amino acids. Cells were passaged every second day. The H1299-E3 (H1299-ACE2, clone E3) cell line was derived from H1299 as described in our previous work1,17. Cell lines were not authenticated. Cell lines tested negative for mycoplasma contamination.
Virus expansion
All work with live virus was performed in Biosafety Level 3 containment using protocols for SARS-CoV-2 approved by the Africa Health Research Institute Biosafety Committee. ACE2-expressing H1299-E3 cells were seeded at 4.5 × 105 cells in a 6-well plate well and incubated for 18–20 h. After one Dulbecco's phosphate-buffered saline (PBS) wash, the subconfluent cell monolayer was inoculated with 500 μl universal transport medium diluted 1:1 with growth medium filtered through a 0.45-μm filter. Cells were incubated for 1 h. Wells were then filled with 3 ml complete growth medium. After 4 days of infection (completion of passage 1 (P1)), cells were trypsinized, centrifuged at 300 RCF for 3 min and resuspended in 4 ml growth medium. All cells from the P1 infection were added to Vero E6 cells that had been seeded at 2 × 105 cells ml−1, 20 ml total, 18–20 h earlier in a T75 flask for cell-to-cell infection. The coculture of ACE2-expressing H1299-E3 and Vero E6 cells was incubated for 1 h, and the flask was filled with 20 ml of complete growth medium and incubated for 4 days. The viral supernatant from this culture (passage 2 (P2) stock) was used for experiments.
Live virus neutralization assay
H1299-E3 cells were plated in a 96-well plate (Corning) at 30,000 cells per well 1 day pre-infection. Plasma was separated from EDTA-anticoagulated blood by centrifugation at 500 RCF for 10 min and stored at −80 °C. Aliquots of plasma samples were heat-inactivated at 56 °C for 30 min and clarified by centrifugation at 10,000 RCF for 5 min. Virus stocks were used at approximately 50–100 focus-forming units per microwell and added to diluted plasma. Antibody–virus mixtures were incubated for 1 h at 37 °C, 5% CO2. Cells were infected with 100 μl of the virus–antibody mixtures for 1 h, and then 100 μl of a 1× Roswell Park Memorial Institute 1640 (Sigma-Aldrich, R6504), 1.5% carboxymethylcellulose (Sigma-Aldrich, C4888) overlay was added without removing the inoculum. Cells were fixed 18 h after infection using 4% paraformaldehyde (Sigma-Aldrich) for 20 min. Foci were stained with a rabbit anti-spike monoclonal antibody (BS-R2B12, GenScript A02058) at 0.5 μg ml−1 in a permeabilization buffer containing 0.1% saponin (Sigma-Aldrich), 0.1% BSA (Sigma-Aldrich) and 0.05% Tween-20 (Sigma-Aldrich) in PBS. Plates were incubated with primary antibody overnight at 4 °C, and then washed with wash buffer containing 0.05% Tween-20 in PBS. Secondary goat anti-rabbit antibody conjugated to horseradish peroxidase (Abcam ab205718) was added at 1 μg ml−1 and incubated for 2 h at room temperature with shaking. TrueBlue peroxidase substrate (SeraCare 5510-0030) was then added at 50 μl per well and incubated for 20 min at room temperature. Plates were imaged in an ImmunoSpot Ultra-V S6-02-6140 Analyzer ELISPOT instrument with BioSpot Professional built-in image analysis (Cellular Technology Limited).
Statistics and fitting
Statistical methods were not used to predetermine sample size, and blinding and randomization were not used. All statistics and fitting were performed using custom code in MATLAB v.2019b. Neutralization data were fitted to:
\({\rm{Tx}}=1/1+(D/{{\rm{ID}}}_{50}).\)
Here Tx is the number of foci normalized to the number of foci in the absence of plasma on the same plate at dilution D, and ID50 is the plasma dilution giving 50% neutralization. FRNT50 = 1/ID50. Values of FRNT50 < 1 are set to 1 (undiluted), the lowest measurable value. We note that the most concentrated plasma dilution was 1:25 and therefore FRNT50 < 25 data were extrapolated. To calculate CIs, FRNT50 or fold change in FRNT50 per participant was log transformed and the arithmetic mean plus 2 s.d. and the arithmetic mean minus 2 s.d. were calculated for the log-transformed values. These were exponentiated to obtain the upper and lower 95% CIs on the geometric mean FRNT50 or the fold change in FRNT50 geometric means.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Sequences of outgrown Omicron sublineages have been deposited in GISAID (https://www.gisaid.org/) with accessions EPI_ISL_7886688 (Omicron BA.1), EPI_ISL_9082893 (Omicron BA.2) and EPI_ISL_602626.1 (ancestral/D614G). Delta (EPI_ISL_3118687) and Beta (EPI_ISL_678615) isolates have been described previously15. Raw images of the data are available upon reasonable request. Source data are provided with this paper.
References
Cele, S. et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature https://doi.org/10.1038/s41586-021-04387-1 (2021).
Andrews, N. et al. Covid-19 vaccine effectiveness against the omicron (B.1.1.529) variant. N. Engl. J. Med. 386, 1532–1546 (2022).
Cao, Y. et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 602, 657–663 (2022).
Dejnirattisai, W. et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 185, 467–484 (2022).
Dejnirattisai, W. et al. Reduced neutralisation of SARS-CoV-2 omicron B.1.1.529 variant by post-immunisation serum. Lancet 399, 234–236 (2022).
Garcia-Beltran, W. F. et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 185, 457–466 (2022).
Lu, L. et al. Neutralization of SARS-CoV-2 Omicron variant by sera from BNT162b2 or Coronavac vaccine recipients. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciab1041 (2021).
Planas, D. et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 602, 671–675 (2022).
VanBlargan, L. A. et al. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 28, 490–495 (2022).
Viana, R. et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature https://doi.org/10.1038/d41586-021-03832-5 (2021).
Davies, M.-A. et al. Outcomes of laboratory-confirmed SARS-CoV-2 infection in the Omicron-driven fourth wave compared with previous waves in the Western Cape Province, South Africa. Trop. Med. Int. Health https://doi.org/10.1111/tmi.13752 (2022).
Wolter, N. et al. Early assessment of the clinical severity of the SARS-CoV-2 omicron variant in South Africa: a data linkage study. Lancet 399, 437–446 (2022).
Sigal, A. Milder disease with Omicron: is it the virus or the pre-existing immunity? Nat. Rev. Immunol. 22, 69–71 (2022).
Lyngse, F. P. et al. Transmission of SARS-CoV-2 Omicron VOC subvariants BA.1 and BA.2: evidence from Danish households. Preprint at medRxiv https://doi.org/10.1101/2022.01.28.22270044 (2022).
Cele, S. et al. SARS-CoV-2 prolonged infection during advanced HIV disease evolves extensive immune escape. Cell Host Microbe 30, 154–162 (2022).
Goldberg, Y. et al. Waning immunity after the BNT162b2 vaccine in Israel. N. Engl. J. Med. 385, e85 (2021).
Cele, S. et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature 593, 142–146 (2021).
Khoury, D. S. et al. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 27, 1205–1211 (2021).
Rössler, A., Riepler, L., Bante, D., von Laer, D. & Kimpel, J. SARS-CoV-2 omicron variant neutralization in serum from vaccinated and convalescent persons. N. Engl. J. Med. 386, 698–700 (2022).
Servellita, V. et al. Neutralizing immunity in vaccine breakthrough infections from the SARS-CoV-2 Omicron and Delta variants. Cell 185, 1539–1548 (2022).
Yu, J. et al. Neutralization of the SARS-CoV-2 Omicron BA.1 and BA.2 Variants. N. Engl. J. Med. 386, 1579–1580 (2022).
Richardson, S. I. et al. SARS-CoV-2 Omicron triggers cross-reactive neutralization and Fc effector functions in previously vaccinated, but not unvaccinated, individuals. Cell Host Microbe https://doi.org/10.1016/j.chom.2022.03.029 (2022).
Chemaitelly, H. et al. Protection of Omicron sub-lineage infection against reinfection with another Omicron sub-lineage. Preprint at medRxiv https://doi.org/10.1101/2022.02.24.22271440 (2022).
Stegger, M. et al. Occurrence and significance of Omicron BA.1 infection followed by BA.2 reinfection. Preprint at medRxiv https://doi.org/10.1101/2022.02.19.22271112 (2022).
Khan, K. et al. Immunogenicity of SARS-CoV-2 infection and Ad26.CoV2.S vaccination in people living with HIV. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciab1008 (2021).
Madhi, S. A. et al. Population immunity and Covid-19 severity with Omicron Variant in South Africa. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2119658 (2022).
Sigal, A., Milo, R. & Jassat, W. Estimating disease severity of Omicron and Delta SARS-CoV-2 infections. Nat. Rev. Immunol. 22, 267–269 (2022).
Smith, M. et al. Age, disease severity and ethnicity influence humoral responses in a multi-ethnic COVID-19 cohort. Viruses 13, 786 (2021).
Acknowledgements
This study was supported by the Bill and Melinda Gates award INV-018944 (A.Sigal), the National Institutes of Health award R01 AI138546 (A.Sigal), South African Medical Research Council awards including 6084COAP2020 (A.Sigal, T.d.O. and P.L.M.), the UK Foreign, Commonwealth and Development Office and the Wellcome Trust (grant number 221003/Z/20/Z; P.L.M.). P.L.M. is also supported by the South African Research Chairs Initiative of the Department of Science and Innovation and the NRF (grant number 98341). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
A.Sigal., K.K. and F.K. conceived the study and designed the study and experiments. A.v.G., Q.A.K., S.S.A.K., G.L., A.Sivro and N.S. identified and provided virus samples. M.-Y.S.M., F.K., B.I.G., M.B., K.K., T.N., M.M., N. Mthabela, Z.M., N.N., Y.M., N. Mbatha, N. Manickchund, N. Magula, Z.J., K.R. and Y.G. set up and managed the cohort and cohort data. K.K., Z.J., K.R., S.C., H.T., J.E.S., Y.G., J.G., Y. Ramphal, A.B.M.K., D.A. and J.N.B. performed experiments and sequence analysis with input from A.Sigal, T.d.O., R.J.L. and J.N.B., A.Sigal, K.K., F.K., R.M. and Y. Rosenberg interpreted data with input from M.-Y.S.M., G.G., S.S.A.K., W.H., T.d.O., N. Magula, R.J.L. and P.L.M., A.Sigal, K.K., G.L., F.K. and M.B. prepared the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Longitudinal Omicron/BA.1 and Delta neutralization capacity in Omicron/BA.1 infected participants.
Neutralization of Omicron (blue) and Delta (red) at all study visits. Participant number is as in Extended Data Table 1. Top three rows are participants vaccinated with Pfizer BNT162b2 (n = 8) or Johnson and Johnson Ad26.CoV2.S (n = 7) and bottom five rows are unvaccinated participants (n = 24). X-axis is the time post-symptom onset when sample was collected, and y-axis is neutralization as FRNT50. Dashed line is the most concentrated plasma tested (LOQ, limit of quantification below which FRNT50 values are extrapolated). All participants recovered except participant 29, who died.
Extended Data Fig. 2 Fold-drop in BA.2 versus BA.1 neutralization in all and sequence confirmed samples.
Neutralization of Omicron BA.2 compared to BA.1 in participants described in Extended Data Table 1, excluding participant 14 for technical reasons and participants 40 and 41 because of advanced HIV disease. Left panel shows neutralization capacity in all n = 38 participants and right panel shows neutralization capacity for n = 25 participants where infection was successfully sequenced and determined to be BA.1. Dashed line is the most concentrated plasma tested. p-values were 0.077 for all and 0.15 for BA.1 sequence confirmed participants as determined by a two-sided Wilcoxon rank sum test. ns, not significant.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, K., Karim, F., Cele, S. et al. Omicron infection enhances Delta antibody immunity in vaccinated persons. Nature 607, 356–359 (2022). https://doi.org/10.1038/s41586-022-04830-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04830-x
This article is cited by
-
Modelling optimal lockdowns with waning immunity
Economic Theory (2024)
-
Clearance of persistent SARS-CoV-2 associates with increased neutralizing antibodies in advanced HIV disease post-ART initiation
Nature Communications (2024)
-
Monovalent vaccination with inactivated SARS-CoV-2 BA.5 protects hamsters against Omicron but not non-Omicron variants
npj Vaccines (2023)
-
Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations
Nature Methods (2023)
-
Prior vaccination promotes early activation of memory T cells and enhances immune responses during SARS-CoV-2 breakthrough infection
Nature Immunology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
