
Abstract
Enzymatic stereoselectivity has typically been unrivalled by most chemical catalysts, especially in the conversion of small substrates. According to the ‘lock-and-key theory’1,2, enzymes have confined active sites to accommodate their specific reacting substrates, a feature that is typically absent from chemical catalysts. An interesting case in this context is the formation of cyanohydrins from ketones and HCN, as this reaction can be catalysed by various classes of catalysts, including biological, inorganic and organic ones3,4,5,6,7. We now report the development of broadly applicable confined organocatalysts for the highly enantioselective cyanosilylation of aromatic and aliphatic ketones, including the challenging 2-butanone. The selectivity (98:2 enantiomeric ratio (e.r.)) obtained towards its pharmaceutically relevant product is unmatched by any other catalyst class, including engineered biocatalysts. Our results indicate that confined chemical catalysts can be designed that are as selective as enzymes in converting small, unbiased substrates, while still providing a broad scope.
Similar content being viewed by others


Main
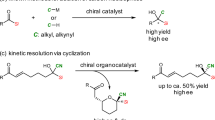
The amount of discrimination that enzymes show when operating on their substrates is unique. Frequently, only a single substrate is even accepted. Regioselectivities, diastereoselectivities and enantioselectivities typically achieved by enzymes have served as an inspiration for chemists in their aim to create perfect catalysts. This quest is particularly formidable when it comes to small substrates, which are notoriously difficult to handle in enantioselective processes. An example of this category concerns the catalytic enantiofacial differentiation of 2-butanone. Given the similar steric bulk of the ketone substituents, for example, as expressed with their corresponding A values (Me 1.74, Et 1.75)8,9, such a stereoselective process represents a daunting task (Fig. 1a). Although enzymes and an iridium complex have been described that reduce 2-butanone to the corresponding alcohol with high enantioselectivity10,11, nucleophilic additions of carbon nucleophiles generally give poor results12,13,14. This also applies for the synthesis of the cyanohydrin that is produced in the addition of HCN to 2-butanone. Notably, the hydrolysis product of this particular cyanohydrin, 2-hydroxy-2-methyl-butyric acid, is a privileged pharmacophore contained in three marketed drugs, beclobrate, clinofibrate and paramethadione15,16,17. Furthermore, the (S)-enantiomer is used in the preparation of a COX-2 inhibitor and PPAR agonist, whereas the (R)-enantiomer builds the skeletons or side chain of several biologically active natural products18,19,20,21,22,23. To the best of our knowledge, until now, only hydroxynitrile lyases (HNL) delivered satisfactory results in the asymmetric hydrocyanation of 2-butanone, with a maximum enantioselectivity of 93.5:6.5 (87% enantiomeric excess), when the engineered enzyme LuHNL was used24. Previously developed chiral thiourea organocatalysts and a chiral salen titanium complex gave only poor amounts of enantiocontrol with this important substrate25,26 (Fig. 1b). We were inspired by recent studies on using strong and confined acid catalysts to control difficult substrates and reactions. For example, we have shown that the acetaldehyde enolsilane only reacts once with another aldehyde, with high enantioselectivity, in the presence of one such confined imidodiphosphorimidate (IDPi) catalyst27. Further encouragement came from our studies on Diels–Alder and hydroarylation reactions, in which we noticed that IDPi catalysts can differentiate between ethyl and methyl groups, although with moderate enantioselectivities28,29. With these results, demonstrating the control that confined catalysts can show, and the underlying catalysis principle of silylium ion asymmetric counteranion directed catalysis (Si-ACDC)30,31, we reasoned that a tailored, strong and perhaps even more confined silylium-IDPi organocatalyst could accomplish the targeted highly enantioselective cyanosilylation of 2-butanone (Fig. 1c). We suggested that a critical silyl oxocarbenium cation may ion pair with its confined IDPi counteranion in such a way that only one of the two enantiofaces would be exposed towards attack by the cyanide sp-nucleophile.

a, Enantiofacial differentiation in 2-butanone. b, Previously reported asymmetric cyanations of 2-butanone with an enzyme, an organocatalyst and a transition-metal catalyst. c, This work: an IDPi-catalysed, highly enantioselective silylcyanation of 2-butanone and a broad scope of other ketones.
Indeed, after extensive catalyst evaluation (see Supplementary Information Tables S1–S6), IDPi 2 was found to be a particularly promising motif and afforded product 6 in quantitative yield with an e.r. of 98:2, which constitutes the highest enantiofacial selectivity ever obtained with 2-butanone. Additionally, IDPis 3–5 also emerged as privileged catalysts. Using catalysts 2–5, the scope of ketones was explored (Fig. 2). Aliphatic ketones bearing a methyl group and a longer n-alkyl group were tested at the outset. Our IDPi catalysts were found to be competent, affording the corresponding cyanohydrin silyl ethers in 90–97% yield, with e.r. values ranging from 93:7 to 98:2, independent of the length of the alkyl chain (products 6–12). Chlorinated ketones were also found to be suitable substrates, furnishing the corresponding silylcyanohydrins 13 and 14 in 94% and 95% yield with 95:5 e.r., respectively. A substrate bearing an alkenyl substituent was also well tolerated, providing product 15 with an e.r. of 92.5:7.5 in 92% yield. Moreover, ketone bearing a protected hydroxy substituent readily gave the desired product 16 in 95% isolated yield with an e.r. of 87:13. In addition, a cyclic ketone could also be successfully used and product 17 was obtained in high yield and with high enantioselectivity when 3,3-dimethyl cyclohexanone was subjected to the reaction conditions. Remarkably, 1,4-addition product 18 was observed as the main product with an e.r. of 91:9 in 45% yield when we reacted 2-cyclohexenone. By contrast, 1,2-adducts were exclusively obtained in moderate e.r. when conjugated acyclic ketones were subjected to the reaction conditions (see Supplementary Information Fig. S6). Aryl-substituted aliphatic ketones reacted similarly and neither electronic effects nor the substitution pattern on the aromatic group notably influenced the enantioselectivity (91:9–99:1 e.r.) (products 19–29).

Performed with TMSCN 1 (2.0 equiv.) and IDPi (0.5 mol%) in toluene (0.2 M) at rt for 0.5 h, then different ketones (0.2 mmol) were added at the indicated temperature for 24 h. Isolated yields after chromatographic purification. The e.r. was determined by high-performance liquid chromatography or gas chromatography analysis. aWith catalyst 2 at −100 °C. bWith catalyst 3 at −100 °C. cWith catalyst 4 at −80 °C. dWith catalyst 2 at −80 °C. eWith catalyst 5 (5.0 mol%) in Et2O at −100 °C for 5 days. fWith catalyst 5 (2.5 mol%) in Et2O at −100 °C for 5 days. gWith catalyst 2 at 25 °C for 12 h. Applications and functionalizations. Performed with TMSCN 1 (2.0 equiv.) and IDPi 4 (0.5 mol%) in Et2O (0.4 M) at rt for 3.0 h, followed by the addition of 4-phenylbutan-2-one (1.0 g, 6.7 mmol) at −60 °C for 24 h. (1) LiAlH4 (2.0 equiv.), Et2O, rt, 1 h. (2) TFA, DCM, rt, 6 h. (3) DIBAL-H (1.5 equiv.), Et2O, rt, 12 h. (4) One-pot synthesis: LiAlH4 (2.0 equiv.), Et2O, rt, 1 h, then carbonyldiimidazole (1.0 equiv.), THF, rt, 12 h. Synthesis of the key intermediate 50 for COX-2 inhibitor: product 6 (95:5 e.r.) made from gram-scale synthesis was treated with HCl (12.0 M), 80 °C, 3 h.
Subsequently, we explored the reactions of aromatic ketones with both electron-donating (Me, OMe) and electron-withdrawing (F, Cl, Br, CF3) groups in different positions on the phenyl ring, delivering the desired products 30–41 with moderate to high yields and high enantioselectivities (up to 98:2 e.r.), although higher catalyst loading was required. Cyanosilylations of ketones bearing furyl and thienyl groups, which are moderately basic heterocyclic substituents, also proceeded smoothly, delivering the corresponding products 42–44 in moderate to good yields and with high enantioselectivities. To our delight, steroid derivative 45 could also be obtained in 92% yield with a remarkable diastereoselectivity (>95:5 d.r.). It is noteworthy that a shorter reaction time could be observed when the reaction was performed under anhydrous conditions, on pre-drying at room temperature (rt) (see Supplementary Information Figs. S1–S5).
To demonstrate the synthetic utility of our reaction, we performed the cyanosilylation of 4-phenylbutan-2-one on a gram scale, furnishing product 23 in 95% yield with 95:5 e.r. Enantioenriched tertiary cyanohydrins are synthetically important building blocks, which can be easily transformed into natural products and biologically or pharmaceutically active compounds32. We investigated the synthetic potential of cyanohydrin precursor 23. The corresponding amino alcohol 46, free cyanohydrin 47, aldehyde 48 and oxazoline 49 could be prepared in good to high yields without erosion of enantiopurity under simple, mild and concise conditions. Moreover, silylcyanohydrin 6, which we also made on a gram scale, can be easily and efficiently converted into 2-hydroxy-2-methylbutyric acid 50, which is an important intermediate in the synthesis of COX-2 inhibitor 51, an effective anti-inflammatory drug22, as well as—potentially—various other enantiopure pharmaceuticals, applied as racemates at present33,34.
Enol silanes were exclusively obtained under disulfonimide (DSI) catalysis, in accordance with our previously established silicon–hydrogen exchange reaction35. For example, enolsilanes were formed in 75% yield and a ratio of 15:1:1 ((Z)-53:(E)-53:54) from 4-phenylbutan-2-one (Fig. 3a, eq. 1). Instead, the cyanosilylation of ketone 52 enabled by IDPi 4 furnished the desired silyl cyanohydrin product 23 in 92% yield with 92:8 e.r. (Fig. 3a, eq. 2). Unexpectedly, we found that, even under these conditions, using IDPi 4 as the catalyst, the corresponding enol silanes (Z)-53 and 54 could clearly be detected during the reaction by 1H NMR spectroscopy (Fig. 3b). A further control experiment in which an enol silane mixture was directly reacted with HCN and 0.5 mol% of catalyst 4 was performed next in toluene-d8 at −80 °C for 24 h, affording silylcyanohydrin 23 in 70% yield with 96:4 e.r. (Fig. 3a, eq. 3). Towards a deeper understanding of the reaction mechanism, we monitored the reaction progress by 1H NMR spectroscopy. As shown in Fig. 3c, enol silane 54 readily reacted with HCN and was found to be fully consumed within 10 min. The reaction of the (Z)-enol silane reached completion within 20 h, whereas the corresponding (E)-enol silane, which is not actually generated during the IDPi-catalysed silyl cyanation, hardly reacted with HCN under the standard conditions, leading to an incomplete consumption of the starting material. We also analysed the reaction by variable time normalization analysis with kinetic data obtained from 1H NMR (Fig. 3d). When following the procedures described by Burés36, we found that the overall reaction is first order in catalyst37.

a, Reactivity comparison between DSI-I and confined IDPi 4 catalyst (eq. 1 and eq. 2); reaction of enol silane mixture and HCN catalysed by IDPi 4 (eq. 3). b, Enol silane formation captured by 1H NMR in the reaction of 4-phenylbutan-2-one 52 and TMSCN 1 catalysed by IDPi 4. c, Reaction profile of different enol silanes with HCN. d, Reaction order determination of the reaction components by variable time normalization analysis following the method of Burés: concentration plots obtained from 1H NMR showing the formation of silylcyanohydrin 23 with a timescale normalized to a first-order dependence on catalyst concentration.
Density functional theory (DFT) calculations38 were performed to study the mechanism of the deprotosilylation and cyanosilylation of 2-butanone with TMSCN catalysed by IDPi 2. As shown in Fig. 4a, the IDPi catalyst first undergoes a reversible in situ silylation with TMSCN to generate the active catalyst INT1 ([X-TMS][HNC], X = IDPi−), which has also been detected by NMR studies (see Supplementary Information Figs. S17–S20). Notably, this process affords the hydrogen isocyanide (HNC) instead of HCN (ΔG≠TS0 = 11.1 versus ΔG≠TS0′ = 38.4 kcal mol−1 (ref. 39) (Fig. 4a and Fig. S22a). The ketone substrate was then activated by the silylated catalyst (ΔG≠TS1 = 12.8, Fig. S22b) to form an oxocarbenium intermediate INT2, in which a cationic species, [TMS-2-butanone]+, ion pairs with the counteranion [X-HNC]−. Under the catalysis of IDPi, ΗΝC readily interconverts with HCN (ΔG≠HNC→HCN = 2.4 and ΔG≠HCN→HNC = 16.3 kcal mol−1, Fig. 4d and Fig. S22c), which enables the interconversions of INT1 and INT2 with their HCN-containing counterpart intermediates, that is INT1′ and INT2′, respectively. Although the less stable tautomer HNC leads to higher energies of INT1 and INT2, it is much more reactive than HCN towards the nucleophilic attack to the oxocarbenium intermediate. The calculated activation free energy for the transition state (TS2-S) of cyanation with HNC is 17.0 kcal mol−1, which is much lower than that with HCN (30.0 kcal mol−1 for TS2′-S) and is able to be overcome under the reaction condition. Once the activation barrier is surmounted, the silylated cyanohydrin products could be generated with the concomitant release of energy (ΔΔG(6-INT2) = −12.7 kcal mol−1). In an alternative pathway, the enol silane product (Z)-53 is readily formed by means of a facile deprotonation process (TS3), in line with NMR detection of enol silanes (Fig. 3a, eq. 2 and Fig. 3b). More importantly, the deprotonation step is endogonic and reversible, thus enol silane can be readily reprotonated to afford oxocarbenium intermediate INT2′ for further conversions—for example, cyanation—which is in agreement with the experiments (Fig. 3a, eq. 2 and eq. 3). Therefore, the computational results support that the formation of enol silane is fast and reversible, and the enol silane could be reprotonated and converted to the thermodynamically favoured silylcyanohydrin product 6.

a, Possible reaction pathways of the deprotosilylation and cyanosilylation of 2-butanone with TMSCN 1 catalysed by IDPi 2. Calculated relative free energies are indicated in parentheses and in kcal mol−1. b, Steric map of IDPi on the basis of the DFT-optimized structure of TS2-S. The steric map is viewed down the z axis; the orientation of IDPi is indicated in the left panel. The red and blue zones indicate the more-hindered and less-hindered zones in the catalytic pocket, respectively. %VBur, percentage of buried volume. c, Optimized transition state structures TS2-S and TS2-R. Their activation free energies are in kcal mol−1. d, Proposed reaction mechanism.
Additionally, we computationally investigated the origin of enantioselectivity. The results indicate that TS2-R is 3.0 kcal mol−1 higher than TS2-S, which matches the experimentally observed enantioselectivity. As depicted in Fig. 4b, the 3D structure and steric map of (S, S)-IDPi 2 shows that the N–P–N–P–N bonding, the sterically demanding Ar substituents and Tf groups constitute a confined and deep chiral pocket (the percentage of buried volume is as high as 73.4). In the lowest-energy transition states (TSs) that lead to R and S products, the oxocarbenium ion intermediate accommodates within the chiral pocket in two different orientations. In TS2-S, the small methyl group is placed in the south-western quadrant and the TMS group occupies the less-hindered central region, orienting outward along the z axis (Fig. 4c, left), whereas TS2-R orients the bulky TMS group in the crowded south-western quadrant (Fig. 4c, right), which distorts the TMS to destroy its coplanar arrangement with the carbonyl group (α(Me-C-O-Si) = −40°). As a result, the overlapping between the p orbitals of oxygen and the carbonyl carbon is diminished, resulting in a higher energy of 1.6 kcal mol−1 compared with that in TS2-S (see Supplementary Information Fig. S24). Therefore, we speculate that the enantiocontrol of cyanation was enabled by the highly confined structure and steric bias of IDPi. The strong dependence of reaction outcome on the percentage of buried volume of IDPi catalysts is also supported by further DFT studies (see Supplementary Information Figs. S23, S25–S28).
On the basis of the accumulated experimental, spectroscopic and computational data, we can now propose a mechanism for the cyanosilylation of ketones enabled by IDPi catalysts. Accordingly, the initial silylation of the catalyst with TMSCN generates the silylated catalyst, which—in turn—activates the ketone to furnish a silyloxy carbenium intermediate, which can be readily deprotonated by the chiral anion of the catalyst to provide the corresponding enol silanes under the reaction conditions (Fig. 4d). We suggest this process to be merely an off-cycle phenomenon, as the enol silane can be readily reprotonated by the acidic IDPi species, affording the oxocarbenium ion intermediate, which is then captured by HNC to provide the final product, simultaneously regenerating the IDPi catalyst. The entire process features a kinetically favoured enol silane formation from the Si–H exchange reaction and the thermodynamically favoured silyl cyanohydrin formation from reaction of the silyloxocarbenium ion with HNC.
In conclusion, we show that properly designed chiral and confined acids can catalyse an asymmetric cyanosilylation reaction of both aromatic and aliphatic ketones, including the highly challenging 2-butanone. Our work can serve as an encouragement for chemists to create catalysts that rival the remarkable and sometimes extreme selectivities observed with enzymes. We also anticipate that our method could be of use in the synthesis of natural products and pharmaceuticals.
Methods
Reaction procedure for the gram-scale catalytic asymmetric cyanosilylation of 4-phenylbutan-2-one with TMSCN 1 in the presence of 0.5 mol% IDPi 4 catalyst. TMSCN 1 (1.7 ml, 13.4 mmol, 2.0 equiv.) was placed in a 25-ml flame-dried Schlenk flask equipped with a Teflon-coated magnetic stirring bar. A solution of IDPi 4 (52.6 mg, 0.034 mmol, 0.005 equiv.) in diethyl ether (0.4 M, 16.8 ml) was added at rt. After stirring for 3.0 h at rt, the reaction mixture was cooled to −60 °C for 0.5 h, and 4-phenylbutan-2-one (1.0 g, 6.7 mmol, 1.0 equiv.) was slowly added. The resultant mixture was stirred at −60 °C for 24 h until the ketone was fully consumed (monitored by thin-layer chromatography). On completion, the reaction mixture was treated with three drops of triethylamine. Organic volatiles were evaporated in vacuo and the crude mixture was purified by column chromatography with neutral aluminium oxide (activated by 20 wt% water and washed by triethylamine) to afford the desired cyanohydrin silyl ether 23 (1.6 g, 95% yield, 95:5 e.r.).
Data availability
The experimental procedures and analytical data supporting the findings of this study are available in the manuscript and its Supplementary Information file. Raw and unprocessed NMR data are available from the corresponding author on reasonable request.
References
Bender, M. L., Van Etten, R. L., Clowes, G. A. & Sebastian, J. F. A pictorial description of the “lock and key” theory. J. Am. Chem. Soc. 88, 2318–2319 (1966).
Koshland Jr, D. E. The key–lock theory and the induced fit theory. Angew. Chem. Int. Ed. 33, 2375–2378 (1995).
Gregory, R. J. Cyanohydrins in nature and the laboratory: biology, preparations, and synthetic applications. Chem. Rev. 99, 3649–3682 (1999).
Brunel, J. M. & Holmes, I. P. Chemically catalyzed asymmetric cyanohydrin syntheses. Angew. Chem. Int. Ed. 43, 2752–2778 (2004).
North, M., Usanov, D. L. & Young, C. Lewis acid catalyzed asymmetric cyanohydrin synthesis. Chem. Rev. 108, 5146–5226 (2008).
Kurono, N. & Ohkuma, T. Catalytic asymmetric cyanation reactions. ACS Catal. 6, 989–1023 (2016).
Bracco, P., Busch, H., von Langermann, J. & Hanefeld, U. Enantioselective synthesis of cyanohydrins catalysed by hydroxynitrile lyases–a review. Org. Biomol. Chem. 14, 6375–6389 (2016).
Hirsch, J. A. Topics in Stereochemistry 1st edn (Wiley, 1967).
Eliel, E. L. & Wilen, S. H. Stereochemistry of Organic Compounds (Wiley, 1994).
Velonia, K., Tsigos, I., Bouriotis, V. & Smonou, I. Stereospecificity of hydrogen transfer by the NAD+-linked alcohol dehydrogenase from the Antarctic psychrophile Moraxella sp. TAE123. Bioorg. Med. Chem. Lett. 9, 65–68 (1999).
Zhang, F.-H., Zhang, F.-J., Li, M.-L., Xie, J.-H. & Zhou, Q.-L. Enantioselective hydrogenation of dialkyl ketones. Nat. Catal. 3, 621–627 (2020).
Denmark, S. E., Fan, Y. & Eastgate, M. D. Lewis base catalyzed, enantioselective aldol addition of methyl trichlorosilyl ketene acetal to ketones. J. Org. Chem. 70, 5235–5248 (2005).
Forrat, V. J., Prieto, O., Ramón, D. J. & Yus, M. trans-1-Sulfonylamino-2-isoborneolsulfonylaminocyclohexane derivatives: excellent chiral ligands for the catalytic enantioselective addition of organozinc reagents to ketones. Chem. Eur. J. 12, 4431–4445 (2006).
Harper, K. C. & Sigman, M. S. Predicting and optimizing asymmetric catalyst performance using the principles of experimental design and steric parameters. Proc. Natl Acad. Sci. 108, 2179–2183 (2011).
Wanner, C., Wieland, H., Schollmeyer, P. & Hörl, W. Beclobrate: pharmacodynamic properties and therapeutic use in hyperlipidemia. Eur. J. Clin. Pharmacol. 40, S85–S89 (1991).
Avellone, G., Di Garbo, V. & Strano, A. Therapy of hyperlipidemia. Clinical experience with clinofibrate. Clin. Ter. 129, 25–29 (1989).
Avolio, J., Myers, C., Rothchild, R. & Valentin, I. 1H NMR spectral simplification with achiral and chiral lanthanide shift reagents. Paramethadione, 5-ethyl-3,5-dimethyl-2,4-oxazolidinedione. Spectrosc. Lett. 23, 459–479 (1990).
Kato, N., Shibayama, S., Munakata, K. & Katayama, C. Structure of the diterpene clerodendrin A. J. Chem. Soc. D 24, 1632–1633 (1971).
Kawada, K., Kim, M. & Watt, D. S. Synthesis of quassinoids. A review. Org. Prep. Proced. Int. 21, 521–618 (1989).
Merritt, A. & Ley, S. Clerodane diterpenoids. Nat. Prod. Rep. 9, 243–287 (1992).
Kawai, K., Amano, T., Nishida, R., Kuwahara, Y. & Fukami, H. Clerodendrins from Clerodendron trichotomum and their feeding stimulant activity for the turnip sawfly. Phytochemistry 49, 1975–1980 (1998).
Tan, L. et al. Practical enantioselective synthesis of a COX-2 specific inhibitor. Tetrahedron 58, 7403–7410 (2002).
Pochetti, G. et al. Structural insight into peroxisome proliferator-activated receptor γ binding of two ureidofibrate-like enantiomers by molecular dynamics, cofactor interaction analysis, and site-directed mutagenesis. J. Med. Chem. 53, 4354–4366 (2010).
Cabirol, F. L. et al. Linum usitatissimum hydroxynitrile lyase cross-linked enzyme aggregates: a recyclable enantioselective catalyst. Adv. Synth. Catal. 350, 2329–2338 (2008).
Zuend, S. J. & Jacobsen, E. N. Cooperative catalysis by tertiary amino-thioureas: mechanism and basis for enantioselectivity of ketone cyanosilylation. J. Am. Chem. Soc. 129, 15872–15883 (2007).
Lv, C. W., Cheng, Q. G., Wang, S. F. & Sun, W. Asymmetric cyanosilylation of ketones catalyzed by monometallic bifunctional salen N-oxide catalyst. J. Mol. Catal. 25, 295–300 (2011).
Schreyer, L. et al. Confined acids catalyze asymmetric single aldolizations of acetaldehyde enolates. Science 362, 216–219 (2018).
Ghosh, S. et al. Strong and confined acids control five stereogenic centers in catalytic asymmetric Diels–Alder reactions of cyclohexadienones with cyclopentadiene. Angew. Chem. Int. Ed. 59, 12347–12351 (2020).
Zhang, P., Tsuji, N., Ouyang, J. & List, B. Strong and confined acids catalyze asymmetric intramolecular hydroarylations of unactivated olefins with indoles. J. Am. Chem. Soc. 143, 675–680 (2021).
Schreyer, L., Properzi, R. & List, B. IDPi catalysis. Angew. Chem. Int. Ed. 58, 12761–12777 (2019).
Zhang, Z. et al. Asymmetric counteranion-directed Lewis acid organocatalysis for the scalable cyanosilylation of aldehydes. Nat. Commun. 7, 12478 (2016).
Wu, W.-B., Yu, J.-S. & Zhou, J. Catalytic enantioselective cyanation: recent advances and perspectives. ACS Catal. 10, 7668–7690 (2020).
Aicher, T. D. et al. Secondary amides of (R)-3,3,3-trifluoro-2-hydroxy-2-methylpropionic acid as inhibitors of pyruvate dehydrogenase kinase. J. Med. Chem. 43, 236–249 (2000).
Sayegh, C. E. et al. Bicyclic heteroaryl compounds useful as inhibitors of the PAR-2 signaling pathway. US Patent Application US15/559,761 (2018).
Zhou, H. et al. The silicon–hydrogen exchange reaction: a catalytic σ-bond metathesis approach to the enantioselective synthesis of enol silanes. J. Am. Chem. Soc. 142, 13695–13700 (2020).
Burés, J. A simple graphical method to determine the order in catalyst. Angew. Chem. Int. Ed. 55, 2028–2031 (2016).
Zhang, Z., Klussmann, M. & List, B. Kinetic study of disulfonimide-catalyzed cyanosilylation of aldehydes by using a method of progress rates. Synlett 31, 1593–1597 (2020).
Frisch, M. J. et al. Gaussian 09 (Gaussian, Inc., 2009).
McCrosky, C., Bergstrom, F. & Waitkins, G. On the structure of hydrogen cyanide. J. Am. Chem. Soc. 64, 722–724 (1942).
Acknowledgements
Generous support from the Max Planck Society, the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Leibniz Award to B.L. and under Germany’s Excellence Strategy – EXC 2033 – 390677874 – RESOLV, and the European Research Council (ERC, European Union’s Horizon 2020 research and innovation program ‘C–H Acids for Organic Synthesis, CHAOS’ Advanced Grant Agreement No. 694228) and National Natural Science Foundation of China (22173077) is gratefully acknowledged. The authors thank B. Mitschke for his help during the preparation of this manuscript and several members of the group for crowd reviewing. We thank R. Properzi and T. Amatov for thoughtful discussions. We thank J. T. Han and T. Ito for their help. We also thank the technicians of our group and the members of our NMR, mass spectrometry, crystallography and chromatography groups for their excellent service.
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
H.Z. developed the reaction, investigated the substrate scope and studied the reaction mechanism. Y.L. determined the absolute configuration of product 6. H.Y.B. first observed the reactivity and performed the initial screening of the cyanosilylation of 4-phenylbutan-2-one. M.L. conducted the kinetic studies using NMR spectroscopy. C.K.D. designed and developed the IDPi catalysts 3 and 5. Y.Z. and G.-J.C. performed the computational studies. B.L. designed and oversaw the project. H.Z. and B.L. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
B.L. is one of the inventors on patent WO2017037141 (A1) filed by the Max-Planck-Institut für Kohlenforschung covering the IDPi catalyst class and its applications in asymmetric synthesis.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
This file contains Supplementary Sections 1–13 — see contents page for details.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, H., Zhou, Y., Bae, H.Y. et al. Organocatalytic stereoselective cyanosilylation of small ketones. Nature 605, 84–89 (2022). https://doi.org/10.1038/s41586-022-04531-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04531-5
This article is cited by
-
Catalysis of an SN2 pathway by geometric preorganization
Nature (2024)
-
Asymmetric hydrogenation of ketimines with minimally different alkyl groups
Nature (2024)
-
Photovoltaic nanocells for high-performance large-scale-integrated organic phototransistors
Nature Nanotechnology (2024)
-
Dynamic kinetic asymmetric allylation, propargylation and crotylation of ketones using copper catalysis
Nature Synthesis (2024)
-
Trialkoxysilane-Induced Iridium-Catalyzed para-Selective C–H Bond Borylation of Arenes
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
