
Abstract
During the last glacial–interglacial cycle, Arctic biotas experienced substantial climatic changes, yet the nature, extent and rate of their responses are not fully understood1,2,3,4,5,6,7,8. Here we report a large-scale environmental DNA metagenomic study of ancient plant and mammal communities, analysing 535 permafrost and lake sediment samples from across the Arctic spanning the past 50,000 years. Furthermore, we present 1,541 contemporary plant genome assemblies that were generated as reference sequences. Our study provides several insights into the long-term dynamics of the Arctic biota at the circumpolar and regional scales. Our key findings include: (1) a relatively homogeneous steppe–tundra flora dominated the Arctic during the Last Glacial Maximum, followed by regional divergence of vegetation during the Holocene epoch; (2) certain grazing animals consistently co-occurred in space and time; (3) humans appear to have been a minor factor in driving animal distributions; (4) higher effective precipitation, as well as an increase in the proportion of wetland plants, show negative effects on animal diversity; (5) the persistence of the steppe–tundra vegetation in northern Siberia enabled the late survival of several now-extinct megafauna species, including the woolly mammoth until 3.9 ± 0.2 thousand years ago (ka) and the woolly rhinoceros until 9.8 ± 0.2 ka; and (6) phylogenetic analysis of mammoth environmental DNA reveals a previously unsampled mitochondrial lineage. Our findings highlight the power of ancient environmental metagenomics analyses to advance understanding of population histories and long-term ecological dynamics.
Similar content being viewed by others

Main
Climate changes are amplified at high latitudes and have pronounced effects on Arctic ecosystems1. Their effects on Arctic plant and animal communities, as well as the human populations who are dependent on them, would have been especially pronounced during the extremely cold and arid Last Glacial Maximum (LGM) (26.5–19 ka)2 and later during the rapid warming that preceded the Holocene. However, precisely what those effects were, and how they played out across the Arctic, are not fully understood. These dynamics were further complicated by differences in the timing and extent of glaciation in different regions across this vast and topographically complex landscape. Previous studies based on pollen and plant macrofossils have documented substantial spatiotemporal variations in Arctic vegetation over the past 50,000 years (50 kyr)1,3, yet it continues to be debated how climatic changes during this period affected plant communities in different regions of the Arctic, and how changes in climate and vegetation may have affected large mammals (that is, megafauna)4,5,6. Skeletal remains show that several megafaunal species, including woolly mammoth (Mammuthus primigenius), woolly rhinoceros (Coelodonta antiquitatis), steppe bison (Bison priscus) and horse (Equus spp.), were abundant in the Arctic during the Pleistocene epoch, but are thought to have become regionally or globally extinct by the onset of the Holocene4,5. However, the precise timing of megafaunal extinctions, and whether and to what extent some of these taxa survived into the Holocene, is uncertain. Similarly, the contribution of various abiotic and biotic drivers to the extinction process of different taxa remains an open question7,8.
To address these knowledge gaps, we performed a metagenomics analysis of ancient environmental DNA (eDNA) of plants and animals recovered from sediments from sites distributed across much of the Arctic covering the past 50 kyr. Relative to other palaeoecological proxies (such as pollen and macrofossils), ancient eDNA offers distinct advantages—including greater taxonomic resolution across the full tree of life9 and higher spatial and temporal precision than pollen—as eDNA mainly derives from the local community10. We used metagenomic analysis rather than the widely used metabarcoding approach because it enables the sequencing of DNA fragments from entire genomes without taxon-specific amplifications, therefore improving the specificity and sensitivity of taxonomic identification, as well as facilitating the authentication of endogenous ancient DNA from modern contaminants9. However, metagenomic analysis requires genome-scale reference data, which are limited for most regions of the world, including the Arctic. Thus, a key component of our study is the generation of a substantial corpus of plant reference sequences.
Metagenomic dataset and database
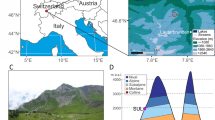
We generated the eDNA metagenomic dataset from 535 sediment samples obtained at 74 circumpolar sites (Fig. 1). Samples come from lake sediments and stratigraphic exposures (unconsolidated permafrost). For the purpose of understanding regional variability, we grouped sites into four regions: North Atlantic; northwest and central Siberia; northeast Siberia; and North America (Fig. 1). Sample ages span the past 50 kyr, albeit in varying numbers, from all regions with the notable exception of the North Atlantic, which was largely covered by ice sheets that often erased pre-LGM deposits2,11.

From the 535 samples, we generated 10.2 billion sequencing reads that passed the filtering criteria and were used for analysis (Methods). We created a comprehensive reference database for taxonomic identification by merging the NCBI-nt and NCBI-RefSeq databases, and supplemented the limited genomic-scale public reference data for Arctic species with 12 Arctic animals and an extensive sequencing effort of 1,541 modern Holarctic plant genome skims (PhyloNorway; Methods). These new sequences comprise 311.3 million whole-genome contigs and provide a broader and more reliable plant reference database than previously available. The merged reference database contains a total of 380.4 million entries and covers about 1.47 million organisms. We developed a k-mer-based method to evaluate the availability and coverage of our combined reference database for different taxa (Methods) and found that it covers a wide range of both Arctic and non-Arctic species (Supplementary Information 9.2.3). Accordingly, the addition of our new reference genomes did not cause bias towards Arctic taxa, providing confidence in our identifications. We used robust approaches to identify taxa from individual reads and collated the resulting taxonomic composition at the generic or familial level (Methods). We applied several methods to authenticate the plant and animal taxonomic profiles; the identifications were reliably classified despite the short DNA sequences that were preserved in these samples (Methods).
Moreover, 131 samples in this dataset were processed for metabarcoding, targeting the short DNA barcodes of plants12, enabling a comparison between the two approaches (Methods). The results showed that the metagenomic analysis captured greater floristic and faunal diversity and achieved better taxonomic resolution (Supplementary Information 11.2). We also found that only about 1.26% of the plant DNA reads are of ribosomal and chloroplast origin (Supplementary Information 9.2.5), suggesting that the metabarcoding approach—which relies on organelle DNA—makes use of only a small fraction of preserved DNA. However, we acknowledge that these comparisons are sample- and method-specific; more studies are needed before broader conclusions about the relative merits of the two approaches can be reached.
Circum-Arctic vegetation dynamics
We combined plant assemblages that were reconstructed from all of the samples to describe the temporal changes in floristic composition, diversity and community structure across the Arctic (Fig. 2a and Extended Data Fig. 1). Our results show substantial and repeated responses of Arctic vegetation to changing climates over the past 50 kyr.

a, Pan-Arctic climate changes and vegetation variations. LGM (26.5−19 ka) and Younger Dryas (YD) (12.9−11.7 ka) are indicated by grey bars. The six time intervals are indicated by light blue bars (Supplementary Information 2). The error bands denote s.e. From top to bottom (see Methods for detailed calculations): the Greenlandic ice-core δ18O ratio and snow accumulation rate; the plant Shannon diversity and the Greenlandic ice-core calcium concentration; the average modelled annual temperature and precipitation for all eDNA sampling sites; the proportion of plant growth forms; the proportion of the herbaceous plant growth forms; and the vegetation turnover rates. b, The number of observed genera in different regions. c, Regional vegetation turnovers. d, Regional vegetation morphological compositions. The sample sizes for each region and time interval are provided in Supplementary Information 2. Calculations are supplied in the Methods.
The overall floristic diversity increased steadily from 50 ka and reached its highest levels at the onset of the LGM (about 26.5 ka), when the climate reached its coldest and driest point at many locations2,11 (Fig. 2a). Vegetation turnover was high before about 38 ka, and the identified shrubs, forbs and grasses suggest a shifting mosaic of steppe–tundra vegetation. Herbaceous plants were the dominant plant group until about 19 ka, with forbs more abundant than graminoids (Fig. 2a), but not as dominant as suggested by a previous metabarcoding study12. Trees and aquatic plants were limited in distribution to lower-latitude sites—consistent with overall dry and cold climate conditions during this period. The scarcity of cold-tolerant trees such as Pinus and Picea, and absence of Larix, reflect low precipitation and strong winds (Fig. 2a and Extended Data Fig. 1a).
The transition into the LGM featured declining temperature and precipitation (Fig. 2a). Across the Arctic, trees remained absent, and there was a sharp decrease in floristic diversity, mainly caused by the decline in herbaceous taxa. Overall, vegetation turnover was consistently high during this decline in diversity, suggesting that cold and dry extremes caused the loss of taxa from all plant communities, although the taxa that were dominant in the pre-LGM period remained (Extended Data Fig. 1a). LGM vegetation dissimilarity was the lowest of all time periods (Extended Data Fig. 1b, c), indicating considerable homogeneity across much of the unglaciated Arctic.
After the LGM, warming towards the Bølling–Allerød interstadial (approximately 14.6–12.9 ka)13 led to vegetation divergence among sites (Extended Data Fig. 1b, c). There was a substantial increase in the abundance of woody plants (such as Salix and Betula), whereas the herbaceous diversity continued to decline, causing the overall diversity to reach its lowest point at the beginning of the cold Younger Dryas stadial (approximately 12.9–11.7 ka)14 (Fig. 2a). The abundance of woody taxa and vegetation turnover rate reached the highest point during the Younger Dryas; the latter is consistent with the intensive climate changes that mark the transition from the Pleistocene to the Holocene.
Shortly after the Younger Dryas, summer insolation peaked and atmospheric CO2 reached Holocene levels15. Previously abundant plant taxa such as Artemisia and Poa rapidly declined or vanished locally. Other plant taxa, particularly boreal trees and prostrate shrubs (such as Vaccinium), appeared and later became abundant (Extended Data Fig. 1a), suggesting that there was a shift from open, cold-adapted tundra–steppe to a mosaic of herbaceous and woody plant communities. The floristic diversity of this more mesophilic vegetation increased during the Early Holocene as climate continued to warm and effective precipitation increased, but then declined during the middle Holocene (Fig. 2a).
Owing to dating uncertainties and limits on the temporal resolution of palaeoclimatic simulations, our results captured only broader changes in vegetation dynamics under climate change. During much of the past 50 kyr, overall plant diversity decreased when the proportion of trees and shrubs increased, as they outcompete herbaceous taxa through shading16. By contrast, when climate became more suitable for herbaceous taxa, diverse taxa expanded to share the landscape, and the overall diversity therefore increased.
Regional vegetation dynamics
Underlying the generalized pattern of Holarctic vegetation changes are significant geographical differences. Early in postglacial times, the North Atlantic experienced the sharpest rises in taxonomic richness (Fig. 2b), along with the steepest temperature increase (Extended Data Fig. 2b). The increase in postglacial richness was probably driven by species dispersals coupled with habitat diversification17, that is, gynomorphically dynamic substrates that were exposed by glacial retreat and shaped by meltwater. The resultant vegetation initially had low diversity but was rich in aquatic taxa (Fig. 2b, d). The abundance of aquatic taxa relates in part to the prevalence of samples from lakes in the North Atlantic (Supplementary Information 10), but nonetheless highlights the ability of aquatic plants to disperse rapidly into newly deglaciated terrain containing abundant streams and lake basins18. As the postglacial climate continued to warm, the overall proportion of aquatic taxa declined as trees and shrubs (for example, Betula, Salix and Vaccinium) became abundant in this region (Fig. 2d and Extended Data Fig. 3).
Northeast Siberia and North America experienced less radical postglacial changes in vegetation type (Fig. 2c, d). During the Late Glacial, trees and shrubs became more widely distributed, and floristic diversity started to decline—a trend that was especially pronounced in North America (Fig. 2b, d). By about 12 ka, rising sea levels had flooded the Bering Strait, and the vegetation on each side started to diverge (Extended Data Fig. 2a). In northeast Siberia, greater effective moisture within the Holocene led to the expansion of aquatic plants (such as Hippuris and Menyanthes). The previously dominant steppe taxa (for example, Poa and Artemisia) declined, although sedges, of which many species are hygrophilous, continued to be abundant (Extended Data Fig. 3). The vegetation of this region became a mosaic of steppe and tundra elements. In North America, trees such as Populus and Picea became more widespread during the Early Holocene and previously widespread steppe species declined (Fig. 2d and Extended Data Fig. 3). A broad, southern swath of eastern Beringia became boreal forest.
In contrast to the changes observed in these regions, vegetation in northwest and central Siberia remained relatively unchanged through the Pleistocene–Holocene transition (Fig. 2c, d). However, some cold- and/or dry-adapted taxa (such as Artemisia and Poa) were replaced by forbs that were better adapted to warmer climates, and Salix was partially replaced by Betula and Alnus (Extended Data Fig. 3). The vegetation in this region persisted as a steppe–tundra mosaic through much of the Holocene, probably due to central Siberia’s extreme climatic continentality caused by the Siberian anticyclone19, which created largely ice-free conditions during the LGM and fostered dry hydrogeological conditions in postglacial times that mitigated the effects of rising global temperatures on vegetation11.
Overall, these results show that postglacial plant communities regionally diverged in response to warming temperatures, increasing moisture, retreating ice sheets and marine transgressions. Although regions that were once overridden by continental ice sheets experienced extreme vegetation changes, the vegetation in unglaciated interior regions remained rather stable. This maritime–continental contrast highlights the importance of moisture in driving ecosystem changes in the Arctic7,20. We next incorporate these insights into vegetation dynamics, together with other potential drivers, into a model to identify the factors influencing animal distributions.
Animal distribution drivers
We developed a model using reconstructed animal distributions and floristic compositions, modelled palaeoclimate variables and inferred human occurrences (Methods) to examine the relative effects of abiotic and biotic factors on Arctic mammal distributions over the past 50 kyr.
We found that certain herbivores tend to co-occur in time and space. For example, the eDNA presences of caribou, hare and vole are statistically strong co-indicators for the presence of horse and mammoth eDNA (Fig. 3). This suggests that co-existence was more common among Arctic herbivores than interspecies exclusion21. By contrast, the distribution of humans over time was almost entirely unrelated to the presence of most herbivores (apart from hares) (Fig. 3). Given that the model purposefully overestimated the presence of humans (Methods), their largely independent distributions from megafauna, their sparseness in the high Arctic before 4 ka (Supplementary Data 7) and the scarcity of kill sites in archaeological records, the notion of human overkill as the cause of Arctic megafaunal extinction is highly improbable6,8. Interestingly, the only predator–prey relationship of note in the model is the significant positive effect of caribou on the distribution of wolves (Fig. 3), probably reflecting that the wolf is well-adapted to hunt caribou.

The values indicate posterior parameter estimates of covariate effects for the models explaining the presence–absence of each animal’s eDNA. Only covariates included in the model with lowest Watanabe–Aikaike information criterion are shown (Methods). The dots represent the posterior means, and the whiskers represent the posterior 2.5% and 97.5% quantiles. Covariate effects of which the 2.5% and 97.5% quantiles are both negative (red), and effects of which the 2.5% and 97.5% quantiles are both positive (blue) are indicated.
To better gauge the explanatory power of environmental variables, we removed the effects of the presence of the eDNA of other animals (Extended Data Fig. 4a and Methods). The most consistent and widely prevalent patterns are the generally negative effects of plant NMDS1 and NMDS3—the first and third components of the non-metric multidimensional scaling (NMDS) of the vegetation compositions (Methods)—on the presence of animal eDNA. Plant NMDS1 reflects an aquatic-to-terrestrial plant gradient, and plant NMDS3 reflects a graminoids-to-woody plant gradient, particularly sedges within the graminoids, which include species that are prominent in present-day wetland communities (Extended Data Fig. 4b). These two negative covariates apply to the distribution of both small (vole and hare) and large (horse and mammoth) mammals, indicating that a wetter environment with a high proportion of hygrophilous plants (that is, moisture-loving plants) was a key factor restricting animal distributions. The distribution of mammoths tends to be positively affected by plant NMDS2, which mainly reflects the proportion of woody plants (particularly shrubs and subshrubs) as opposed to herbaceous plants, whereas the reverse is true for horses (Fig. 3). We also found that horses are more sensitive to vegetation composition compared with other herbivores (Supplementary Information 13.3). These findings support the hypothesis that horses were more restricted to a grassland environment and may also indicate a greater dietary flexibility in mammoths.
When each herbivore species is considered individually, the only climate variable that is consistently and positively associated with the presence of their eDNA is temperature seasonality (Fig. 3 and Extended Data Fig. 4a), consistent with expectations based on the continental climate associated with the Mammoth Steppe, a biome that is associated with extremely cold and dry conditions that supported abundant large mammal grazers19. The importance of climatic variables becomes more evident when herbivores are considered as a group. Precipitation—in greater amounts and seasonality—is a principal negative factor in the distribution of Arctic herbivores (Fig. 3), presumably because increased snow cover during winter limited the food access of grazers, and a wetter substrate is more difficult for them to exploit, in contrast to the firm and dry ground of the steppe–tundra7,19.
Late-surviving megafauna
The timing of Arctic megafaunal extinction is a matter of debate, not least because last appearance dates (LADs) are repeatedly revised as younger fossils are reported5,6, and also because discovering the remains of the last surviving individuals of a species is extremely unlikely22. As a result, LADs systematically underestimate when a species disappeared, raising the possibility that populations persisted longer than is now evident4,23. The extinction timing can be better gauged with eDNA; an animal leaves behind only a single skeleton, which is much less likely to be preserved, recovered and dated, when compared with the amount of DNA it continuously spread into the environment while it was alive.
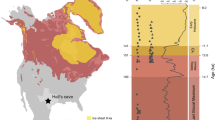
Our data indicate that mammoths survived into the Early Holocene in present-day continental northeast Siberia until 7.3 ± 0.2 ka (seven samples younger than 10 ka) and North America until 8.6 ± 0.3 ka. Notably, we recovered mammoth DNA from a series of samples from the Taimyr Peninsula that indicate the presence of mammoths in north central Siberia as late as 3.9 ± 0.2 ka (site LUR10) (Fig. 4 and Supplementary Information 3.3). The survival of mammoths into the Holocene in these regions is probably attributable to the persistence of the steppe–tundra vegetation of dry- and cold-adapted herbaceous plants that was present during the Pleistocene (Fig. 2d). This vegetation would have provided a suitable habitat for mammoths and possibly other dryland grazers such as horses (Extended Data Fig. 5), which are known to have survived in the region until at least 5 ka (ref. 24). Together, these eDNA results indicate that mammoths survived much longer than previously thought—which, on the basis of skeletal remains, was around 10.7 ka on continental Eurasia25 and around 13.8 ka in Alaska8. Given that humans occupied northern Eurasia sporadically from at least 40 ka and continuously after 16 ka (refs. 26,27), the late-surviving Taimyr mammoths potentially encountered and co-existed with humans over at least a 20-kyr interval, therefore giving no support to the human overkill (blitzkrieg) model that postulates the mammoth extinction occurred within centuries after the first human contact6.

A total of 78 mammoth mitochondrial genomes and 159 eDNA-identified mammoths (79 among them were assigned to mitochondrial haplotypes) are shown. Records of dated mammoth fossils62 are also plotted. All samples older than 26.5 ka were combined into the pre-LGM interval.
We also detected woolly rhinoceros DNA as late as 9.8 ± 0.2 ka in northeast Kolyma, horse DNA in Alaska and the Yukon as late as 7.9 ± 0.2 ka, and bison as late as 6.4 ± 0.6 ka in high-latitude localities of northeast Siberia (Extended Data Fig. 5). All of these instances represent substantially later LADs than fossil-based dates (that is, for woolly rhinoceros in Eurasia, about 14 ka (ref. 28); and for horses and steppe bison in Alaska, 12.5 ka (refs. 5,8)). Collectively, these findings highlight the value of eDNA in improving megafauna extinction chronologies.
Population diversity of megafauna
Megafaunal eDNA from across the Arctic also enables us to resolve population-level patterns, which is crucial for uncovering species-specific demographic and evolutionary responses to past climatic and environmental changes. We applied a method for phylogenetically assigning the identified eDNA to mitochondrial haplogroups of mammoth and horse, the two most abundant species detected in our dataset (Methods).
A mammoth phylogeny composed of four previously described major mitochondrial clades (clade 1, including 1C and 1DE, and clades 2 and 3)29 was reconstructed from 78 mammoth mitochondrial genomes. The recovered mammoth eDNA was then assigned to a best-fit node on the tree based on single-nucleotide polymorphism (SNP) support/conflict, enabling clade assignment for 79 eDNA samples (Extended Data Fig. 6).
The mammoth haplogroups that we identified are consistent with those that were previously identified from fossil remains and have comparable biogeographical and biostratigraphic distributions (Fig. 4). Overall, clade 3 was present mainly in Europe and northwest Siberia, whereas clade 2 occurred mostly in central and northeast Siberia. Clade 1 was widely scattered across North America and the Asian Arctic, with 1DE occurring throughout Siberia and 1C in North America. Temporally, clades 2 and 3 were the older lineages, and disappeared between 40 ka and 30 ka. Only clade 1 survived past the LGM, with the last 1C individual dating to 10.35 ka. Like the late-surviving mammoths on Wrangel Island30, the late-surviving mammoths on mainland Siberia were also members of 1DE, the only clade detected to date that postdates the Early Holocene (that is, after 8.2 ka). However, despite belonging to the same clade, none of the mainland late-surviving populations is placed in the Wrangel Island haplogroup (Extended Data Fig. 6). Furthermore, we note that two mammoth eDNA samples (cr5_11 and tm4_13) attach to the existing tree at the shared root of clades 2 and 3 (Extended Data Fig. 6), with cr5_11 containing many sequence variants not found in previously sequenced samples (Supplementary Information 14.1.2), suggesting that they represent a separate and previously unrecorded mitochondrial lineage. The distinctive mitochondrial genome haplogroups, together with the shrinking and increasingly isolated occurrences of mammoths (Fig. 4), hint that Siberian mainland mammoths experienced a similar fate to those on Wrangel and St Paul Islands. However, whether the precise causes of their disappearance were the same4,30, and whether the mainland mammoth also accumulated detrimental mutations consistent with genetic decline31, will require further data to resolve.
The reliability of our method was further corroborated on the horse phylogeny (Supplementary Information 14.2). Successful assignment of ancient eDNA data to mitochondrial haplogroups, even when the DNA is highly degraded, highlights the potential for applying eDNA analysis to uncover population histories in regions in which fossils are rare or absent.
Concluding remarks
Controversy has persisted for decades over the nature of the Mammoth Steppe, a distinctive, now-vanished biome dominated by large mammal grazers1,19,32. Some studies, emphasizing the abundance of grazers (and the absence of large browsers), suggest that broad swaths of the unglaciated Late Pleistocene Arctic were covered by an extensive steppe dominated by low-sward herbaceous plants that were well-suited for megafaunal grazers19,32. Others, on the basis of pollen and plant macrofossil records, suggest that Arctic vegetation during this period was regionally diverse and included both tundra and steppe taxa3,33. Our results suggest the nature of the Mammoth Steppe lies in between these two seemingly conflicting interpretations. Consistent with the view of the Mammoth Steppe as a biome of intercontinental extent, our data show that various regions of the Arctic supported a more homogenous vegetation cover before and during the LGM (Extended Data Fig. 1b, c). We also found evidence of an elevated and episodic turnover of plant taxa during the Late Pleistocene compared with during the Holocene (Fig. 2a), consistent with inferences about changeable vegetation types during the glacial age based on the network of palaeobotanical (and fossil insect) sites presently available3,12. Jointly, our results suggest that the Mammoth Steppe was a regionally complex cryo-arid steppe, composed of forbs, graminoids and willow shrubs.
Our findings relating to the late survival of megafauna have important implications for the debate over the causes of Late Quaternary extinctions. Megafaunal survival into the Holocene indicates that, at least in certain parts of the Arctic and Subarctic, humans coexisted with these species for tens of thousands of years, which implies that human hunting was not an important factor in their extinction6,25. Instead, our results suggest that their extinction came when the last pockets of the steppe–tundra vegetation finally disappeared, when the Arctic-wide paludification was brought on by warmer and wetter climates7,20.
What we have mined from this substantial dataset does not exploit its full potential. For example, we detected DNA of Camelidae (most probably the Arctic camel34) and Panthera (possibly the steppe lion). However, due to a lack of reference genomes for these species, we could not confirm these identifications. This constraint also applies to other species because our reference database—large as it is—is far from complete, despite our extensive sequencing efforts. With more species sequenced and new bioinformatics methods developed, this dataset can be reanalysed to explore more questions of Arctic biotic history.
Our study demonstrates how metagenomic analysis of eDNA extracted from ancient sediments can provide diverse insights, from detailed records of past flora and fauna to reconstructions of population histories and biotic interactions, to a greatly expanded spatiotemporal network of palaeoecological records. These advances are important in the context of continuous efforts to elucidate the past 50 kyr of Arctic biotic dynamics, especially given that the coevolution of plant and animal species, and their responses to the past climatic changes across this vast region, have previously been challenging to address at this resolution and at this scale using classical palaeobotanical and palaeontological data.
Methods
Sampling, chronology and eDNA taphonomy
Sampling and subsampling methods are described in Supplementary Information 1. Sample ages were determined through conventional or accelerator mass spectrometer radiocarbon (14C) as well as optically stimulated luminescence. In total, 631 radiocarbon ages and 81 optically stimulated luminescence dates were used. For sedimentary sections with multiple contiguous dates without stratigraphic inversions, age–depth models were built to calculate sedimentation rates and estimate the ages of undated samples within these sections. All radiocarbon ages are in calibrated years before present, calibrated using IntCal20 (ref. 35). Chronological information is provided in Supplementary Information 2 and Supplementary Data 1 and 2.
To determine whether DNA was in situ, control samples were obtained from modern surfaces, from water in adjacent rivers and lakes, and from stratigraphic layers bracketing the samples. Consistent with previous eDNA studies in the Arctic12,23,36, we found no evidence of DNA leaching or redeposition in either terrestrial or lake sediment samples (Supplementary Information 5).
DNA extraction and sequencing
We tested the performance of different operations included in the widely used ancient eDNA extraction protocols36,37,38 and a variety of purification methods on different sediment sample types. On the basis of these tests, we developed two new eDNA-extraction protocols that were optimized for isolating and purifying eDNA from our sediment samples (Supplementary Information 6.1 and 6.2). The InhibitEx-based protocol was then applied for extracting DNA from all samples. DNA extracts were thereafter converted into sequencing libraries according to the standard protocol39, and sequenced using Illumina platforms after quality controls (Supplementary Information 6.3). All DNA extractions and pre-index analyses were performed in the dedicated ancient DNA laboratories at the Centre for GeoGenetics, University of Copenhagen, according to established ancient DNA protocols40.
PhyloNorway plant genome database construction
The PhyloNorway plant genome database was constructed by sequencing 1,541 Arctic and boreal plant specimens collected from herbaria. DNA was extracted from the selected specimens using a modified Macherey–Nagel Nucleospin 96 Plant II protocol. Two different library preparation protocols were applied depending on DNA yields. All of the libraries were then sequenced. Nuclear ribosomal DNA and chloroplast genome from each plant were assembled to evaluate the data quality. Whole-genome contigs for each plant were assembled and annotated as the final reference database. A list of plant species, herbarium information, DNA extraction, sequencing and database statistics are supplied in Supplementary Data 3. Data for three standard barcodes skimmed from this database were also used in ref. 41. Details are provided in Supplementary Information 7.
Taxonomic identification, authentication and quantification
We performed taxonomic classification by mapping reads against a comprehensive genomic database that was annotated with taxonomic information according to the principle of the Holi pipeline36. Details of the composition of the reference database are provided in Supplementary Information 9.2.1.
All reads were first quality-controlled, and each read was then offered an equal chance to be aligned against all entries in the database after duplicate removal (Supplementary Information 9.1 and 9.2). No limitation to specific taxonomic group, geography or environment was applied for the alignment. The lowest common ancestor of all of the hits with 100% similarity was assigned to each read that had been aligned to multiple taxa. The taxonomic coverage of different database compositions and their effects on taxa identification were evaluated using a k-mer-based method (Supplementary Information 9.2.2). We found that using a proper reference database is important for eDNA metagenomics-based taxa identification, particularly for ancient datasets in which the DNA is highly fragmented. Even reference genome availability across taxa can improve the sensitivity and specificity of the identification by increasing the identified reads and correcting the misidentifications (Supplementary Information 9.2.5). Taxa that were detected in the laboratory controls were combined into a list, and all of the listed taxa were subtracted from samples (Supplementary Information 9.3). The resulting plant and animal taxonomic profiles were thereafter parsed for additional authentication using a series of conservative thresholds (Supplementary Information 9.4 and 9.6), on the basis of an Arctic flora and faunal checklist (Supplementary Information 8). Plant taxa that passed these filters all have Arctic or boreal distributions (Supplementary Information 9.4). All eDNA reads aligned to an animal were further confirmed as exclusive alignments, by requiring perfect alignment to that animal, and no alignment to any other organisms when allowing for 1 or 2 mismatches (Supplementary Information 9.6.3). The two extinct animals— mammoth and woolly rhinoceros— were also confirmed by the DNA-damage patterns (Supplementary Information 9.6.2).
Relative abundances for plants were estimated on the basis of the number of the assigned reads, by excluding the effects of DNA degradation in different samples, and eliminating the effects of the sequencing depth among different samples and the efficiency of the taxa-identification pipeline among different taxa (Supplementary Information 9.5).
Vegetation diversity and dissimilarity
The Shannon diversity index was calculated according to the method in ref. 42. Plant morphological forms were assigned at the genus level on the basis of the plant trait database of eFloras (http://www.efloras.org). Beta-diversity (dissimilarity) between every two plant assemblages was calculated according to the method in ref. 43. For the pan-Arctic vegetation turnover (Fig. 2a), plant genera identified in all samples in each 2,000-year interval were combined as an assemblage; beta-diversity between each two consecutive intervals was calculated. Regional vegetation turnover (Fig. 2c) was calculated at 5,000-year intervals. NMDS (k = 3; Extended Data Fig. 1c) was performed using the R package vegan44, allowing 100,000 iterations of random starting to find the best convergent solution. Correlations between the abundance of each plant genus (or proportion of each morphological form) and the values of each of the three NMDS components (Extended Data Fig. 4b) were assessed using the Pearson product–moment correlation and t-test (P < 0.05).
Comparison of eDNA shotgun metagenomics and metabarcoding
We applied two modules for comparing the metabarcoding and shotgun metagenomics in taxa identifications. (1) We conducted the two sequencing techniques in parallel on 14 DNA extracts to directly compare the retrieved taxonomic profiles. (2) We compared the floristic profiles reconstructed by this study and a previous metabarcoding study12 on 131 overlapping samples of the two datasets. The results show that metagenomics performed better on our samples in both captured floristic and faunal diversity. Details are provided in Supplementary Information 11.
Palaeoclimate panels and human distribution niche modelling
For the ice-core data from Greenland (Fig. 2a), we rescaled the available δ18O ratios (20-year slices) retrieved from NGRIP1 (ref. 45), NGRIP2 (ref. 46) and GISP2 (ref. 47) to the range of the corresponding ratio of GRIP48, for which there are valid values for all age slices, using the rescale function in the R package scales. The mean of the available ratios for each time slice from the four datasets was calculated and used. Calcium concentrations were calculated from refs. 49,50 using the same method as for δ18O. Snow-accumulation rates were based on GISP2 (ref. 51).
We also modelled monthly palaeoclimate anomalies at 1,000-year time steps using an emulator52 and downscaled them onto a modern baseline climatology (CHELSA)53 at a spatial resolution of 1°. From these data, we calculated four environmental variables—annual mean temperature, temperature seasonality, annual precipitation and precipitation seasonality—that were used to represent the climate for each of our eDNA sites. Details are provided in Supplementary Information 12.1.
We developed distribution models to map environmentally suitable conditions for Palaeolithic human occurrence in steps of 1,000 years from 5 ka to 31 ka and steps of 2,000 years from 32 ka to 47 ka. First, geo-references for human remains in the Arctic were collected and dates from 14C calibrations inferred from two databases CARD2.0 (ref. 54) and the Palaeolithic of Europe55. These data were filtered for quality, resulting in a final set of 6,497 occurrences. From 32 ka to 47 ka, we calculated 2,000-year averages of the four environmental variables. We then generated five-algorithm ensemble models at each time step to characterize the climatic niche of Palaeolithic humans. We validated all of the models by assessing the area under the receiver operating characteristic curve (AUC) and true skill statistic; we also used model AUCs to generate weighted ensemble models at each time step. Finally, we projected the ensemble models into geographic space to map climatic suitability for humans, expressed as the potential presence or absence at each time step at each of the eDNA sites. Details are provided in Supplementary Information 12.2.
Spatiotemporal models for animal eDNA
We combined our animal eDNA data with the modelled climate variables, projected human occurrence and the NMDS ordinations of vegetation to examine the relative impacts of climate, human activity and vegetation on the geographical distributions of a selected group of Arctic mammals. We developed a method to spatiotemporally model animal eDNA presence, using these three sets of variables, while accounting for auto-correlation in time and space. The method uses a hierarchical Bayesian model that includes a spatiotemporal Gaussian random field, and was implemented in R-INLA56,57. We used the Watanabe–Akaike information criterion to assess the model fit using different sets of covariates. Detailed methods are provided in Supplementary Information 13.
Mammoth and horse mitochondrial haplotyping
We placed eDNA mitochondrial reads for mammoth and horse into their respective mitochondrial reference phylogenies using recently developed software58. We used existing variation to assign informative markers onto branches of a mitochondrial phylogeny, then determined the number of supporting and conflicting single-nucleotide polymorphisms for each eDNA sample on each branch of the tree to place the sample onto the most likely branch. Detailed methods are provided in Supplementary Information 14.
Statistics and data visualization
Changing trends are illustrated against time (Fig. 2a–c and Extended Data Fig. 2b, c) or distance (Extended Data Fig. 1b) via the Loess Smooth (span = 4) function in the R package ggplot2 (ref. 59), with original data points or confidence intervals (s.e.) shown when other curves are not obstructed. The heat maps showing the mean of a genus’ proportions across all samples within an age interval were generated using the R package ComplexHeatmap60. The mammoth phylogenetic tree was illustrated using ggtree, which is included in the R package ggplot2. The base map source for Fig. 1 and Extended Data Fig. 5 was Arctic SDI and, for Fig. 4, was the R package maptools.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Adapter-removed plant or animal eDNA data were deposited at EMBL-ENA under project accession ERP127790. The raw data of PhyloNorway plant genome database are available at EMBL-ENA under project accession PRJEB43865. Assembled plant genome contigs of the PhyloNorway database are available at DataverseNO61. NCBI databases are available at the NCBI ftp server (https://ftp.ncbi.nlm.nih.gov). The Canadian Archaeological Radiocarbon Database (CARD2.0) is available online (https://www.canadianarchaeology.ca). The Radiocarbon Palaeolithic Europe Database is available online (https://ees.kuleuven.be/geography/projects/14c-palaeolithic). All other data are provided in the Supplementary Information and Supplementary Data 1–9.
Code availability
Scripts are archived at GitHub (https://github.com/wyc661217/Arctic_eDNA_2021).
Change history
26 September 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41586-022-05359-9
16 March 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41586-022-04628-x
References
Binney, H. et al. Vegetation of Eurasia from the last glacial maximum to present: key biogeographic patterns. Quat. Sci. Rev. 157, 80–97 (2017).
Clark, P. U. et al. The Last Glacial Maximum. Science 325, 710–714 (2009).
Bigelow, N. H. Climate change and Arctic ecosystems: 1. Vegetation changes north of 55°N between the last glacial maximum, mid-Holocene, and present. J. Geophys. Res. 108, https://doi.org/10.1029/2002jd002558 (2003).
Graham, R. W. et al. Timing and causes of mid-Holocene mammoth extinction on St. Paul Island, Alaska. Proc. Natl Acad. Sci. USA 113, 9310–9314 (2016).
Stuart, A. J. Late Quaternary megafaunal extinctions on the continents: a short review. Geol. J. 50, 338–363 (2015).
Koch, P. L. & Barnosky, A. D. Late Quaternary extinctions: state of the debate. Ann. Rev. Ecol. Evol. Syst. 37, 215–250 (2006).
Rabanus-Wallace, M. T. et al. Megafaunal isotopes reveal role of increased moisture on rangeland during late Pleistocene extinctions. Nat. Ecol. Evol. 1, 0125 (2017).
Mann, D. H., Groves, P., Kunz, M. L., Reanier, R. E. & Gaglioti, B. V. Ice-age megafauna in Arctic Alaska: extinction, invasion, survival. Quat. Sci. Rev. 70, 91–108 (2013).
Capo, E. et al. Lake sedimentary DNA research on past terrestrial and aquatic biodiversity: overview and recommendations. Quaternary 4, https://doi.org/10.3390/quat4010006 (2021).
Edwards, M. E. et al. Metabarcoding of modern soil DNA gives a highly local vegetation signal in Svalbard tundra. Holocene 28, 2006–2016 (2018).
Hughes, P. D., Gibbard, P. L. & Ehlers, J. Timing of glaciation during the last glacial cycle: evaluating the concept of a global ‘Last Glacial Maximum’ (LGM). Earth Sci. Rev. 125, 171–198 (2013).
Willerslev, E. et al. Fifty thousand years of Arctic vegetation and megafaunal diet. Nature 506, 47–51 (2014).
Rasmussen, S. O. et al. A new Greenland ice core chronology for the last glacial termination. J. Geophys. Res. 111, https://doi.org/10.1029/2005jd006079 (2006).
Mangerud, J. The discovery of the Younger Dryas, and comments on the current meaning and usage of the term. Boreas 50, 1–5 (2020).
Bauska, T. K. et al. Carbon isotopes characterize rapid changes in atmospheric carbon dioxide during the last deglaciation. Proc. Natl Acad. Sci. USA 113, 3465–3470 (2016).
Wesser, S. D. & Armbruster, W. S. Species distribution controls across a forest‐steppe transition: a causal model and experimental test. Ecol. Monogr. 61, 323–342 (1991).
Rijal, D. P. et al. Sedimentary ancient DNA shows terrestrial plant richness continuously increased over the Holocene in northern Fennoscandia. Sci. Adv. 7, eabf9557 (2021).
Birks, H. H. Aquatic macrophyte vegetation development in Kråkenes Lake, western Norway, during the late-glacial and early-Holocene. J. Paleolimnol. 23, 7–19 (2000).
Guthrie, R. D. Origin and causes of the mammoth steppe: a story of cloud cover, woolly mammal tooth pits, buckles, and inside-out Beringia. Quat. Sci. Rev. 20, 549–574 (2001).
Mann, D. H., Peteet, D. M., Reanier, R. E. & Kunz, M. L. Responses of an Arctic landscape to Lateglacial and early Holocene climatic changes: the importance of moisture. Quat. Sci. Rev. 21, 997–1021 (2002).
Ritchie, M. in Competition and Coexistence (eds Sommer, U. & Worm, B.) 109–131 (Springer, 2002).
Signor, P. W., Lipps, J. H., Silver, L. & Schultz, P. in Geological Implications of Impacts of Large Asteroids and Comets on the Earth vol. 190 (eds Silver, L. T. & Schultz, P. H.) 291–296 (1982).
Haile, J. et al. Ancient DNA reveals late survival of mammoth and horse in interior Alaska. Proc. Natl Acad. Sci. USA 106, 22352–22357 (2009).
Librado, P. et al. Tracking the origins of Yakutian horses and the genetic basis for their fast adaptation to subarctic environments. Proc. Natl Acad. Sci. USA 112, E6889–E6897 (2015).
Nikolskiy, P. A., Sulerzhitsky, L. D. & Pitulko, V. V. Last straw versus Blitzkrieg overkill: climate-driven changes in the Arctic Siberian mammoth population and the Late Pleistocene extinction problem. Quat. Sci. Rev. 30, 2309–2328 (2011).
Pavlov, P., Svendsen, J. I. & Indrelid, S. Human presence in the European Arctic nearly 40,000 years ago. Nature 413, 64–67 (2001).
Kuzmin, Y. V. & Keates, S. G. Siberia and neighboring regions in the Last Glacial Maximum: did people occupy northern Eurasia at that time? Archaeol. Anthropol. Sci. 10, 111–124 (2016).
Stuart, A. J. & Lister, A. M. Extinction chronology of the woolly rhinoceros Coelodonta antiquitatis in the context of late Quaternary megafaunal extinctions in northern Eurasia. Quat. Sci. Rev. 51, 1–17 (2012).
Chang, D. et al. The evolutionary and phylogeographic history of woolly mammoths: a comprehensive mitogenomic analysis. Sci. Rep. 7, 44585 (2017).
Vartanyan, S. L., Arslanov, K. A., Karhu, J. A., Possnert, G. & Sulerzhitsky, L. D. Collection of radiocarbon dates on the mammoths (Mammuthus primigenius) and other genera of Wrangel Island, northeast Siberia, Russia. Quat. Res. 70, 51–59 (2017).
Rogers, R. L. & Slatkin, M. Excess of genomic defects in a woolly mammoth on Wrangel island. PLoS Genet. 13, e1006601 (2017).
Zimov, S. A., Zimov, N. S., Tikhonov, A. N. & Chapin, F. S. Mammoth steppe: a high-productivity phenomenon. Quat. Sci. Rev. 57, 26–45 (2012).
Yurtsev, B. A. The Pleistocene “Tundra-Steppe” and the productivity paradox: the landscape approach. Quat. Sci. Rev. 20, 165–174 (2001).
Rybczynski, N. et al. Mid-Pliocene warm-period deposits in the High Arctic yield insight into camel evolution. Nat. Commun. 4, 1550 (2013).
Reimer, P. J. et al. The IntCal20 Northern Hemisphere radiocarbon age calibration curve (0–55 cal kBP). Radiocarbon 62, 725–757 (2020).
Pedersen, M. W. et al. Postglacial viability and colonization in North America’s ice-free corridor. Nature 537, 45–49 (2016).
Slon, V. et al. Neandertal and Denisovan DNA from Pleistocene sediments. Science 356, 605–608 (2017).
Lorenz, M. G. & Wackernagel, W. Adsorption of DNA to sand and variable degradation rates of adsorbed DNA. Appl. Environ. Microb. 53, 2948–2952 (1987).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, pdb.prot5448 (2010).
Willerslev, E., Hansen, A. J. & Poinar, H. N. Isolation of nucleic acids and cultures from fossil ice and permafrost. Trends Ecol. Evol. 19, 141–147 (2004).
Alsos, I. G. et al. The treasure vault can be opened: large-scale genome skimming works well using herbarium and silica gel dried material. Plants 9, https://doi.org/10.3390/plants9040432 (2020).
Hill, M. O. Diversity and evenness: a unifying notation and its consequences. Ecology 54, 427–432 (1973).
Koleff, P., Gaston, K. J. & Lennon, J. J. Measuring beta diversity for presence-absence data. J. Anim. Ecol. 72, 367–382 (2003).
Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930 (2003).
Grootes, P. M. & Stuiver, M. Oxygen 18/16 variability in Greenland snow and ice with 10−3- to 105-year time resolution. J. Geophys. Res. Oceans 102, 26455–26470 (1997).
Andersen, K. K. et al. High-resolution record of Northern Hemisphere climate extending into the last interglacial period. Nature 431, 147–151 (2004).
Stuiver, M. & Grootes, P. M. GISP2 oxygen isotope ratios. Quat. Res. 53, 277–284 (2017).
Johnsen, S. J. et al. The δ18O record along the Greenland Ice Core Project deep ice core and the problem of possible Eemian climatic instability. J. Geophys. Res. Oceans 102, 26397–26410 (1997).
Fuhrer, K., Neftel, A., Anklin, M. & Maggi, V. Continuous measurements of hydrogen peroxide, formaldehyde, calcium and ammonium concentrations along the new grip ice core from summit, Central Greenland. Atmos. Environ. A 27, 1873–1880 (1993).
Mayewski, P. A. et al. Major features and forcing of high-latitude northern hemisphere atmospheric circulation using a 110,000-year-long glaciochemical series. J. Geophys. Res. Oceans 102, 26345–26366 (1997).
Alley, R. B. et al. Abrupt increase in Greenland snow accumulation at the end of the Younger Dryas event. Nature 362, 527–529 (1993).
Holden, P. B. et al. PALEO-PGEM v1.0: a statistical emulator of Pliocene–Pleistocene climate. Geosci. Model Dev. 12, 5137–5155 (2019).
Karger, D. N. et al. Climatologies at high resolution for the earth’s land surface areas. Sci. Data 4, 170122 (2017).
Martindale, A. et al. Canadian Archaeological Radiocarbon Database (CARD 2.1) (Laboratory of Archaeology at the University of British Columbia, and the Canadian Museum of History, accessed 6 February 2020).
Vermeersch, P. M. Radiocarbon Palaeolithic Europe database: a regularly updated dataset of the radiometric data regarding the Palaeolithic of Europe, Siberia included. Data Brief 31, 105793 (2020).
Rue, H., Martino, S. & Chopin, N. Approximate Bayesian inference for latent Gaussian models by using integrated nested Laplace approximations. J. R. Stat. Soc. B 71, 319–392 (2009).
Lindgren, F. & Rue, H. Bayesian spatial modelling with R-INLA. J. Stat. Softw. 63, 1–25 (2015).
Martiniano, R., De Sanctis, B., Hallast, P. & Durbin, R. Placing ancient DNA sequences into reference phylogenies. Preprint at https://doi.org/10.1101/2020.12.19.423614 (2020).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Wang, Y. et al. Supporting data for: Late Quaternary dynamics of Arctic Biota revealed by ancient environmental metagenomics. https://doi.org/10.18710/3CVQAG, DataverseNO, V1 (2021).
Theodoridis, S. et al. Climate and genetic diversity change in mammals during the Late Quaternary. Preprint at https://doi.org/10.1101/2021.03.05.433883 (2021).
Acknowledgements
We thank D. H. Mann for his detailed and constructive comments; and T. Ager, J. Austin, T. B. Brand, A. Cooper, S. Funder, M. T. P. Gilbert, T. Jørgensen, N. J. Korsgaard, S. Liu, M. Meldgaard, P. V. S. Olsen, M. L. Siggaard-Andersen, J. Stenderup, S. A. Woodroffe and staff at the GeoGenetics Sequencing Core and National Park Service-Western Arctic National Parklands for help and support. E.W. and D.J.M. thank the staff at St. John’s College, Cambridge, for providing a stimulating environment for scientific discussion of the project. E.W. thanks Illumina for collaboration. The Lundbeck Foundation GeoGenetics Centre is supported by the Carlsberg Foundation (CF18-0024), the Lundbeck Foundation (R302-2018-2155), the Novo Nordisk Foundation (NNF18SA0035006), the Wellcome Trust (UNS69906) and GRF EXC CRS Chair (44113220)—Cluster of Excellence. The PhyloNorway plant genome database is part of the Norwegian Barcode of Life Network (https://www.norbol.org) funded by the Research Council of Norway (226134/F50), the Norwegian Biodiversity Information Centre (14-14, 70184209) and The Arctic University Museum of Norway. Metabarcoding sequencing was funded by the Central Public-Interest Scientific Institution Basal Research Fund, CAFS (2017B001 and 2020A001). B.D.S. is supported by the Wellcome Trust programme in Mathematical Genomics and Medicine (WT220023); F.R. by a Villum Fonden Young Investigator award (no. 00025300); D.J.M. by the Quest Archaeological Research Fund; P.M. by the Swedish Research Council (VR); R.D. by the Wellcome Trust (WT207492); and A.R. by a Marie Skłodowska-Curie Actions Individual Fellowship (MSCA-IF, 703542) and the Research Council of Norway (KLIMAFORSK, 294929). L.O. has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (no. 681605); I.G.A. and Y.L. from the ERC under the European Union’s Horizon 2020 research and innovation programme (no. 819192). J.I.S. and J.M. are supported by the Research Council of Norway. P.B.H. and N.R.E. acknowledge NERC funding (grant NE/P015093/1). D.W.B. was supported by a Marie Skłodowska-Curie Actions Incoming International Fellowship (MCIIF-40974). T.S.K. is funded by a Carlsberg Foundation Young Researcher Fellowship (CF19-0712).
Author information
Authors and Affiliations
Contributions
B.D.S., F.R., A.P. and E.C. contributed equally to this work. E.W. and Y.W. initiated and led the study. E.W., Y.W., M.W.P. and K.H.K. designed the study. P.M., K.H.K., E.W., I.G.A., Y.W., J.I.S., D.G.F., J.M., A.R., M.E.E., J.H., D.W.B., A.A.B., J.E., K.K.K., J.T.R., A.T., A.A.C. and G.B.F. provided samples. Y.W., N.K.L. and A.R. revised the age of samples. Y.W. performed the DNA laboratory work. Y.W., B.D.S., A.F.-G., R.D., D.M. and T.S.K. did the bioinformatics and statistical analyses. I.G.A., Y.W. and A.R. evaluated the plant taxonomic profiles. For the PhyloNorway plant genome database: I.G.A. and E.C. designed the study and led the work; M.K.F.M. and A.A. did the laboratory work; P.W. led the sequencing; E.C., Y.W., Y.L. and F.D. did the bioinformatics analyses. H.L.O., D.N.B., Y.W., C.R. and D.J.M. modelled the human distribution. P.B.H. and N.R.E. modelled the climate. F.R. developed the animal spatiotemporal model. Y.W., M.W.P., D.J.M., I.G.A., E.W. and A.P. drafted the manuscript which all of the co-authors commented on.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Patricia Fall, Brian Huntley, Paul Valdes and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1
Circum-Arctic plant abundance variations and vegetation similarity clustering. a, Pan-Arctic plant abundance heatmap. b, Spatial vegetation dissimilarities. Pairwise spatial beta-diversities (dissimilarities between every two plant communities) against the geographical distances between the two communities. c, Non-metric Multidimensional Scaling (NMDS, k=3) on vegetation communities.
Extended Data Fig. 2
Regional vegetation differences and climate changes. a, Vegetation similarities between each two regions. All identified plant genera across sites in a region during a time interval were merged as a plant assemblage. Spatial beta-diversity between every two assemblages were calculated and illustrated. NAt, North Atlantic; WcS, Northwest and central Siberia; ES, Northeast Siberia; Nam, North America. b and c, Modelled annual temperature and precipitation in different regions. Means of the modelled annual temperature and precipitation values (Methods) at all eDNA sampling sites within a region at each 1,000-year time step were calculated. The changing trends are illustrated.
Extended Data Fig. 3
Regional plant abundance heatmaps. Heatmaps show the relative abundances of the 40 abundant plant genera in each region.
Extended Data Fig. 4 Environmental explanatory factors for animal distribution, and plant NMDS components.
a, Posterior parameter estimates of covariate effects for the models explaining the presence/absence of each animal’s eDNA using climate, human presence and plant NMDS as explanatory variables. The dots represent the posterior means, and the whiskers represent the posterior 2.5% and 97.5% quantiles. The colour red denotes covariate effects whose 2.5% and 97.5% quantiles are both negative, while the colour blue denotes covariate effects 2.5% and 97.5% quantiles are both positive. b, The plant genera and morphological forms correlated to the 3 components of plant NMDS. Plant genera (morphological forms) are ranked by the p-value of t-test, and only the top 20 Pearson correlations are shown. The colour red denotes negative correlations while the colour blue denotes positive correlations.
Extended Data Fig. 5 Distribution chronologies for woolly rhinoceros, bison, horse, caribou, hare, wolf, and vole.
We combined our DNA results and the fossil records62 (available for woolly rhinoceros, bison, and caribou). Samples older than 26.5 ka were combined into Pre-LGM; samples younger than 4.2 ka were combined into the Late Holocene.
Extended Data Fig. 6 Mammoth mitochondrial phylogenetic tree.
For placed eDNA samples the number of supporting single-nucleotide polymorphisms is given in braces (Methods). IDs for the Wrangel Island population are underlined.
Supplementary information
Supplementary Information
See SI guide for full description of contents.
Supplementary Data 1 Site description
An Excel table covering the site metadata for all 74 eDNA sampling sites.
Supplementary Data 2 Sample metadata & age-dpeth models.xlsx
An Excel table covering the sample metadata for all 535 eDNA samples, and the age–depth models.
Supplementary Data 3 PhyloNorway metadata
An Excel table covering the metadata for all 1,541 PhyloNorway herbarium specimens.
Supplementary Data 4 Arctic flora and fauna checklist
An Excel table covering the assembled Arctic plant and animal checklist.
Supplementary Data 5 Plant abundance
An Excel table covering the reconstructed plant genera abundance.
Supplementary Data 6 Animal distribution & DNA damage
An Excel table covering the animal existence/absence matrix and the animal ancient DNA damage rate.
Supplementary Data 7 Human Presence/Absence
A PDF file showing the modelled environmentally suitable conditions for Palaeolithic human occurrence.
Supplementary Data 8 Human Presence/Absence for eDNA sites
An Excel table covering the modelled human existence/absence matrix for eDNA sampling sites.
Supplementary Data 9 Horse mitochondrial genomes
An Excel table covering the metadata for the horse mitochondrial genomes used for constructing the horse phylogeny.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Pedersen, M.W., Alsos, I.G. et al. Late Quaternary dynamics of Arctic biota from ancient environmental genomics. Nature 600, 86–92 (2021). https://doi.org/10.1038/s41586-021-04016-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-04016-x
This article is cited by
-
Genomics for monitoring and understanding species responses to global climate change
Nature Reviews Genetics (2024)
-
Archaeology meets environmental genomics: implementing sedaDNA in the study of the human past
Archaeological and Anthropological Sciences (2024)
-
Reframing Prehistoric Human-Proboscidean Interactions: on the Use and Implications of Ethnohistoric Records for Understanding the Productivity of Hunting Megaherbivores
Journal of Archaeological Method and Theory (2024)
-
Paleoecology of extinct species
BMC Ecology and Evolution (2023)
-
Plants under pressure: the impact of environmental change on plant ecology and evolution
BMC Ecology and Evolution (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
