
Abstract
Despite the existence of good catalogues of cancer genes1,2, identifying the specific mutations of those genes that drive tumorigenesis across tumour types is still a largely unsolved problem. As a result, most mutations identified in cancer genes across tumours are of unknown significance to tumorigenesis3. We propose that the mutations observed in thousands of tumours—natural experiments testing their oncogenic potential replicated across individuals and tissues—can be exploited to solve this problem. From these mutations, features that describe the mechanism of tumorigenesis of each cancer gene and tissue may be computed and used to build machine learning models that encapsulate these mechanisms. Here we demonstrate the feasibility of this solution by building and validating 185 gene–tissue-specific machine learning models that outperform experimental saturation mutagenesis in the identification of driver and passenger mutations. The models and their assessment of each mutation are designed to be interpretable, thus avoiding a black-box prediction device. Using these models, we outline the blueprints of potential driver mutations in cancer genes, and demonstrate the role of mutation probability in shaping the landscape of observed driver mutations. These blueprints will support the interpretation of newly sequenced tumours in patients and the study of the mechanisms of tumorigenesis of cancer genes across tissues.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
Cancer genes and their observed mutations across ~28,000 tumours of 66 cancer types (the starting point to train boostDM models) are available through IntOGen (https://intogen.org/). Mutational features computed across cancer genes, and positive and negative sets needed to train boostDM models, are available in a Zenodo repository (https://doi.org/10.5281/zenodo.4813082). Data source files point towards this repository. Blueprints may be browsed and downloaded at https://www.intogen.org/boostdm/ (including all protein sequence affecting mutations), and newly sequenced tumours may be analysed using boostDM models via the Cancer Genome Interpreter (https://www.cancergenomeinterpreter.org/). Source data are provided with this paper.
Code availability
All code needed to train and test boostDM models, as well as python scripts and jupyter notebooks necessary to reproduce all figures of the paper in conjunction with the data, are available in the aforementioned Zenodo repository (https://doi.org/10.5281/zenodo.4813082).
References
Sondka, Z. et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705 (2018).
Martínez-Jiménez, F. et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 20, 555–572 (2020).
Tamborero, D. et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 10, 25 (2018).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Gonzalez-Perez, A., Sabarinathan, R. & Lopez-Bigas, N. Local determinants of the mutational landscape of the human genome. Cell 177, 101–114 (2019).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Polak, P. et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 518, 360–364 (2015).
Supek, F. & Lehner, B. Scales and mechanisms of somatic mutation rate variation across the human genome. DNA Repair 81, 102647 (2019).
Martincorena, I. et al. Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041.e21 (2017).
Sabarinathan, R. et al. The whole-genome panorama of cancer drivers. Preprint at https://doi.org/10.1101/190330 (2017).
Kato, S. et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl Acad. Sci. USA 100, 8424–8429 (2003).
Giacomelli, A. O. et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 50, 1381–1387 (2018).
Berger, A. H. et al. High-throughput phenotyping of lung cancer somatic mutations. Cancer Cell 30, 214–228 (2016).
Kim, E. et al. Systematic functional interrogation of rare cancer variants identifies oncogenic alleles. Cancer Discov. 6, 714–726 (2016).
Mighell, T. L., Evans-Dutson, S. & O’Roak, B. J. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am. J. Hum. Genet. 102, 943–955 (2018).
Boettcher, S. et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604 (2019).
Chen, H. et al. Comprehensive assessment of computational algorithms in predicting cancer driver mutations. Genome Biol. 21, 43 (2020).
Tokheim, C. & Karchin, R. CHASMplus reveals the scope of somatic missense mutations driving human cancers. Cell Syst. 9, 9–23.e8 (2019).
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M. & Ng, P. C. SIFT missense predictions for genomes. Nat. Protocols 11, 1–9 (2016).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 76, 7.20.1–7.20.41 (2013).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Bandaru, P. et al. Deconstruction of the Ras switching cycle through saturation mutagenesis. eLife 6, e27810 (2017).
Shihab, H. A. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65 (2013).
Carter, H., Douville, C., Stenson, P. D., Cooper, D. N. & Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14 (Suppl. 3), S3 (2013).
Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39, e118 (2011).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Chakravarty, D. et al. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. https://doi.org/10.1200/PO.17.00011 (2017).
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R. & Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 (2010).
Hornbeck, P. V. et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015).
McLaren, W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010).
Lundberg, S. M. & Lee, S.-I. A unified approach to interpreting model predictions. In Advances in Neural Information Processing Systems 30 (eds. Guyon, I. et al.) 4765–4774 (Curran, 2017).
Ng, P. C. & Henikoff, S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Liu, X., Li, C., Mou, C., Dong, Y. & Tu, Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 12, 103 (2020).
Chen, T. & Guestrin, C. XGBoost: a scalable tree boosting system. In Proc. 22nd ACM SIGKDD Intl Conf. on Knowledge Discovery and Data Mining 785–794 (Association for Computing Machinery, 2016).
Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017).
Rosenthal, R., McGranahan, N., Herrero, J., Taylor, B. S. & Swanton, C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31 (2016).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Pich, O. et al. Somatic and germline mutation periodicity follow the orientation of the DNA minor groove around nucleosomes. Cell 175, 1074–1087.e18 (2018).
Hunter, J. D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007).
Oliphant, T. E. Guide to NumPy (Open Source Book, 2006).
McKinney, W. Python for Data Analysis: Data Wrangling with Pandas, NumPy, and IPython (O’Reilly Media, 2017).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Acknowledgements
N.L.-B. acknowledges funding from the European Research Council (consolidator grant 682398) and ERDF/Spanish Ministry of Science, Innovation and Universities – Spanish State Research Agency/DamReMap Project (RTI2018-094095-B-I00) and Asociación Española Contra el Cáncer (AECC) (GC16173697BIGA). IRB Barcelona is a recipient of a Severo Ochoa Centre of Excellence Award from the Spanish Ministry of Economy and Competitiveness (MINECO; Government of Spain) and is supported by CERCA (Generalitat de Catalunya). O.P. is the recipient of a BIST PhD fellowship supported by the Secretariat for Universities and Research of the Ministry of Business and Knowledge of the Government of Catalonia, and the Barcelona Institute of Science and Technology (BIST). This publication and the underlying research were partly facilitated by the Cancer Genome Atlas project, the TARGET project and the Hartwig Medical Foundation and the Center for Personalized Cancer Treatment (CPCT), which have generated, analysed and made available data for this purpose. The schematic DNA double helices in Extended Data Fig. 9a, e were created using Biorender (N.L.-B. premium membership).
Author information
Authors and Affiliations
Contributions
A.G.-P. and N.L.-B. conceptualized the project. F.M. conceived, implemented and tested boostDM. F.M.-J. participated in the implementation and testing of boostDM code. All authors participated in the conceptualization of boostDM. F.M. and O.P. benchmarked boostDM. F.M. computed the mutation probability bias. O.P. performed the analyses of mutational signatures. F.M.-J. performed the analysis of variants of unknown significance. F.M. and F.M.-J. designed the visualization of blueprints and radial plots to interpret the results of boostDM. F.M., F.M.-J. and O.P. prepared the figures. All authors participated in the design of the analyses and in the interpretation of the results. A.G.-P. and N.L.-B. drafted the manuscript. F.M., F.M.-J. and O.P. edited the manuscript. A.G.-P. and N.L.-B. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Moritz Gerstung and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Specific models encapsulate the features of driver mutations.
a, Flow chart describing the construction of specific models from all possible cancer gene–tumour type combinations in the compendium of mutational cancer genes. b, Discovery index of ten cancer gene–tumour type combinations (trend of the number of unique mutations as a function of the size of bootstrapped subsets of samples). c, As in b for TP53 across ten tumour types. d, Distribution of discovery index resulting from 100 iterations in each example (see Methods). e, As in d for TP53 across 10 tumour types. f, Schematic depiction of the calculation of the F50 value used to measure the performance of the models. Fifty subsets of observed and synthetic mutations are randomly divided into training and test sets, and used in cross validations, from which a family of values of F50 are derived (Fig. 1c and e). g, Fraction of mutations observed in 50 cancer gene–tissue combinations that are classified as drivers by the corresponding boostDM models. These include all mutations observed in the gene (including synonymous variants), not only those employed in the training. h, Performance (median and IQR F50 of base classifiers) of 282 gene–tissue models as a function of the number of observed mutations. Boxplots: centre line, median; box limits, first and third quartiles; whiskers, lowest/highest data points at first quartile minus/plus 1.5× IQR.
Extended Data Fig. 2 The complexity of trained models.
a, Schematics depicting the calculation of the complexity index of models from the assessed compressibility of SHAP values for the 18 mutational features of a given model. Specifically, the smaller the number of principal components explaining a larger fraction of variance, the smaller the complexity index. b, Effect of reducing the size of observed mutations available for training on the performance and complexity index of three models. The circle and bars represent the median and IQR of F50 values computed from the cross-validation. As the size of the set of mutations available to train the models is reduced, the performance and complexity of built models decrease, demonstrating that as more cancer genomes are sequenced, more good-quality specific boostDM models will be within reach. c, Comparison of the performance of the cross-validation of TP53 high-confidence models (median F50 and IQR of values obtained from the base classifiers) with models trained and tested on randomly drawn 90% of the samples of each tumour type, leaving the remaining 10% held out as external datasets for validation. d, Distribution of median F50 of cross-validation and re-trained models (as in c) for 155 cancer gene–tumour type combinations for which re-training with 90% of the original samples is possible, respecting the conditions set out in Extended Data Fig. 1a. Boxplots: centre line, median; box limits, first and third quartiles; whiskers, lowest/highest data points at first quartile minus/plus 1.5× IQR.
Extended Data Fig. 3 Performance of boostDM models on observed cancer mutations.
a, Comparison of performance of boostDM TP53 models and saturation mutagenesis experiments (left) or computational tools (right) using PRCs. The PRCs represent median values of the base classifiers. b, Comparison of the performance of absolute classification (F50) of TP53 boostDM models and two saturation mutagenesis experiments11,12 (Supplementary Note). F50 values represent the median of the base classifiers. The F50 values for all base classifiers of the colorectal model are compared to both experimental saturation mutagenesis assays (middle). BoostDM score and classification of six TP53 driver mutations (experimentally validated in Boettcher et al.16) by 33 TP53-specific models. Points represent the scores from models and boxplots their distribution (bottom). c, PRCs (as in a) resulting from scoring KRAS, HRAS and NRAS mutations using boostDM models, a saturation mutagenesis experiment, and computational tools. d, Distribution of auPRC of KRAS, HRAS and NRAS boostDM models, a saturation mutagenesis experiment and bioinformatics tools across seven tumour types. e, As in a, b for PTEN. f, Comparison of performance (PRCs) of boostDM models and computational tools on mutations in several cancer genes. g, Comparison of the performance of absolute classification (F50 values) of boostDM models and the CHASMplus tool (using two different CHASMplus q values as threshold). h, Comparison of the performance of absolute classification (F50 values) of boostDM models and CHASMplus (q-value = 0). i, As in h for CADD score (CADD > 10). Boxplots: centre line, median; box limits, first and third quartiles; whiskers, lowest/highest data points at first quartile minus/plus 1.5× IQR.
Extended Data Fig. 4 Performance of boostDM models on other sets of variants.
a, Left, boostDM scores of rare experimentally validated oncogenic and benign variants13,14 and absolute performance of boostDM models (precision, recall and F50). Right, performance of specific models. In parentheses, sizes of positive and negative sets of mutations. b, Performance of boostDM models (precision, recall and F50) in the discrimination between pathogenic somatic and benign germline variants (ClinVar). c, Distribution of predicted drivers across polymorphisms with different allele frequencies across the population. The bars represent the effect size of a logistic regression of the categorical frequency of the polymorphisms on their boostDM classification. Bars with positive effect size represent genes (or pooled cancer genes, in red, with regression P value indicated) across which very rare polymorphisms have an increased likelihood of being classified as drivers by boostDM models. The P value of the logistic regression corresponding to polymorphisms across all cancer genes is shown. Boxplots: centre line, median; box limits, first and third quartiles; whiskers, lowest/highest data points at first quartile minus/plus 1.5× IQR.
Extended Data Fig. 5 Features of driver mutations.
a, Radial plots representing the contributions of different features (via SHAP values) to the classification of five selected driver mutations. b, Two-dimensional representation (via t-distributed stochastic neighbour embedding; t-SNE) of the combination of features relevant to the classification of all driver mutations across genes and tumour types. Mutations are coloured according to the mode of action (left) of the genes they affect, clusters (centre) resulting from the similarity of their SHAP values (Fig. 2d) and the complexity index (right) of the model used to classify them
Extended Data Fig. 6 Blueprints of driver mutations of cancer genes.
a–d, Blueprints of potential driver mutations in EGFR across lung adenocarcinomas (a), in KRAS across pancreatic adenocarcinomas (b), in RB1 across bladder tumours (c), and in PIK3CA across breast adenocarcinomas (d). Blueprints of all cancer genes with models are available at https://intogen.org/boostdm. e, Comparison of the fraction of potential driver mutations (from all possible) in tumour suppressors and oncogenes. The former exhibit significantly larger fractions of potential driver mutations than the latter. f, Distribution of potential driver mutations along the sequence of TP53 across 33 tumour types. The dendrogram represents the similarity in distribution of potential driver mutations between malignancies. Boxplots: centre line, median; box limits, first and third quartiles; whiskers, lowest/highest data points at first quartile minus/plus 1.5× IQR.
Extended Data Fig. 7 Mutational processes.
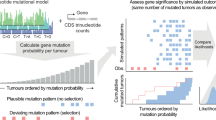
a, Left, distribution of mutation probability bias of observed versus unobserved mutations across all non-cancer genes. Middle, distribution of mutation probability bias of observed versus unobserved mutations (irrespective of their classification by boostDM models) across all cancer genes. Right, comparison of the distributions of mutation probability bias of all observed versus unobserved mutations in cancer genes (185) and non-cancer genes (3,035) and the distributions of mutation probability bias of potential driver observed versus unobserved mutations and all observed and unobserved mutations (irrespective of their classification by boostDM models) in cancer genes. Potential driver mutations show the lowest mutation probability bias of the three, which is evidence of the activity of selection. b, Selected driver mutations and the most likely mutational signature contributing them (colour) across tissues. The diameter of the circles is proportional to their frequency. c, Selected driver mutations across bladder adenocarcinomas are likely to be contributed by different mutational processes. The diameter of each circle represents the likelihood that the mutation is contributed by the corresponding (colour-coded) signature. d, Frequency (top) of different mutations affecting a section of the sequence of PIK3CA (bottom) in breast adenocarcinomas. Middle, the probability that each mutation is contributed by different signatures that are active in breast tumours. Boxplots: centre line, median; box limits, first and third quartiles; whiskers, lowest/highest data points at first quartile minus/plus 1.5× IQR.
Extended Data Fig. 8 General models.
a, Schematic representation of the process followed to build and select gene-specific models or gene–other tumour type models. If a high-confidence model is available for a cancer gene in a specific tumour type (that is, through completion of the process represented in Extended Data Fig. 1a), no general gene model is built to represent this gene–tumour type combination (FBXW7 in uterine carcinosarcomas). We call these models gene–tumour type-specific. Conversely, if no specific model is available (FBXW7 in oesophageal adenocarcinomas), a gene–general model is trained on mutations pooled from tumour types at the leaves of the oncotree (Supplementary Note). For example, a model of FBXW7 across solid tumours may be trained on the features computed for the gene in oesophageal adenocarcinomas. If several gene-specific models can be built for the gene–tissue combination (for example, FBXW7 across solid tumours and FBXW7 across all cancer), the model built on a general tumour type that is closest to the tumour type in question and with F50 greater than 0.8 will be chosen. In total, 48 gene-specific models represent 72 gene–tumour type cases not covered by gene–tumour type-specific models. Finally, if neither a gene–tumour type-specific model nor a gene-specific model is available, a model trained on a different tumour type may be used (gene–other tumour type model). b, The types of model selected to classify the mutations observed in 20 cancer genes across 32 tumour types.
Extended Data Fig. 9 Interpretation of variants using boostDM models.
a, BoostDM models can be used to interpret the mutations observed in a newly sequenced tumour genome. Classifications and their explanation (represented via radial plots) may be useful for an expert with other ancillary information. b, Number of models of each type (gene–tumour type-specific, gene-specific or gene–other tumour type) selected across tumour types in a pan-cancer cohort of about 28,000 tumours to classify mutations observed in cancer genes (numbers below the plot). At least one model is available to classify mutations across 2,080 cancer gene–tissue combinations. c, Fraction of all mutations in cancer genes (variants of unknown significance, or VUS) that are covered by either type of model across tumour types. Fractions for the entire cohort appear in the right-hand barplot. Specifically, 14,757 (26%) are classified by specific boostDM models, while a further 2,588 (4%) may be classified by models trained by pooling mutations and features of several related tumour types; 20% more may be classified by a model trained on a different tumour type. In the entire cohort, 28,080 VUSs (about 50%) are covered for interpretation by at least one boostDM model. These are compared to the fraction of VUSs covered by interpretation using two curated datasets of known oncogenic mutations (ClinVar and OncoKB). d, Comparison of the number of driver mutations identified by boostDM per sample across the cohort with the number of excess mutations (over the expectation provided under the hypothesis of neutrality) identified by a dNdS approach. The numbers identified are of the same order of magnitude as the numbers of driver mutations predicted by dNdScv. Points, median; bars, extending between 10th and 90th percentiles of the distribution. e, The interpretation of newly sequenced tumour genomes is implemented within the Cancer Genome Interpreter platform. VUSs not covered by boostDM models are interpreted following a simple rule-based approach (OncodriveMut).
Supplementary information
Supplementary Information
This file contains Supplementary Notes, Supplementary Figures 1-4 and Supplementary References – see Contents page for details.
Supplementary Tables
This file contains Supplementary Tables 1-2.
Source data
Rights and permissions
About this article

Cite this article
Muiños, F., Martínez-Jiménez, F., Pich, O. et al. In silico saturation mutagenesis of cancer genes. Nature 596, 428–432 (2021). https://doi.org/10.1038/s41586-021-03771-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03771-1
This article is cited by
-
New pan-ALK inhibitor-resistant EML4::ALK mutations detected by liquid biopsy in lung cancer patients
npj Precision Oncology (2024)
-
A novel bioinformatic approach reveals cooperation between Cancer/Testis genes in basal-like breast tumors
Oncogene (2024)
-
A novel patient-derived meningioma spheroid model as a tool to study and treat epithelial-to-mesenchymal transition (EMT) in meningiomas
Acta Neuropathologica Communications (2023)
-
Neurodevelopmental disorders and cancer networks share pathways, but differ in mechanisms, signaling strength, and outcome
npj Genomic Medicine (2023)
-
Actionability classification of variants of unknown significance correlates with functional effect
npj Precision Oncology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
