
Abstract
The spike protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is critical for virus infection through the engagement of the human ACE2 protein1 and is a major antibody target. Here we show that chronic infection with SARS-CoV-2 leads to viral evolution and reduced sensitivity to neutralizing antibodies in an immunosuppressed individual treated with convalescent plasma, by generating whole-genome ultra-deep sequences for 23 time points that span 101 days and using in vitro techniques to characterize the mutations revealed by sequencing. There was little change in the overall structure of the viral population after two courses of remdesivir during the first 57 days. However, after convalescent plasma therapy, we observed large, dynamic shifts in the viral population, with the emergence of a dominant viral strain that contained a substitution (D796H) in the S2 subunit and a deletion (ΔH69/ΔV70) in the S1 N-terminal domain of the spike protein. As passively transferred serum antibodies diminished, viruses with the escape genotype were reduced in frequency, before returning during a final, unsuccessful course of convalescent plasma treatment. In vitro, the spike double mutant bearing both ΔH69/ΔV70 and D796H conferred modestly decreased sensitivity to convalescent plasma, while maintaining infectivity levels that were similar to the wild-type virus.The spike substitution mutant D796H appeared to be the main contributor to the decreased susceptibility to neutralizing antibodies, but this mutation resulted in an infectivity defect. The spike deletion mutant ΔH69/ΔV70 had a twofold higher level of infectivity than wild-type SARS-CoV-2, possibly compensating for the reduced infectivity of the D796H mutation. These data reveal strong selection on SARS-CoV-2 during convalescent plasma therapy, which is associated with the emergence of viral variants that show evidence of reduced susceptibility to neutralizing antibodies in immunosuppressed individuals.
Similar content being viewed by others

Main
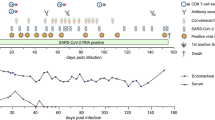
A septuagenarian male was admitted to a tertiary hospital in the summer of 2020 and had tested positive for SARS-CoV-2 using reverse-transcription quantitative PCR (RT–qPCR) 35 days previously in a nasopharyngeal swab (day 1) at a local hospital (Extended Data Figs. 1, 2). His past medical history included marginal B cell lymphoma diagnosed in 2012, with previous chemotherapy including vincristine, prednisolone, cyclophosphamide and anti-CD20 B cell depletion with rituximab. It is likely that both chemotherapy and underlying lymphoma contributed to combined immunodeficiency of B and T cells (Extended Data Figs. 2, 3 and Supplementary Table 1). Computed tomography of the chest showed widespread abnormalities consistent with pneumonia associated with coronavirus disease 2019 (COVID-19) (Supplementary Fig. 1). Treatment included two 10-day courses of remdesivir with a 5-day gap in between (Extended Data Fig. 1). Two units of convalescent plasma were administered on days 63 and 65 (Extended Data Fig. 3). After clinical deterioration, remdesivir and a unit of convalescent plasma were administered on day 95, but the individual died on day 102 (Supplementary Note).
Comparative analysis of sequential virus samples
Most samples were respiratory samples from the nose and throat or endotracheal aspirates during the period of intubation (Supplementary Table 3). Cycle threshold (Ct) values ranged from 16 to 34 and all 23 respiratory samples were successfully sequenced by a standard single-molecule sequencing approach as per the ARTIC protocol implemented by The COVID-19 Genomics UK (COG-UK; https://www.cogconsortium.uk/) Consortium; of these samples, 20 additionally underwent short-read deep sequencing using the Illumina platform (Supplementary Table 4). There was general agreement between the two methods (Extended Data Fig. 4). However, owing to the higher reliability of Illumina for low frequency variants, this was used for formal analysis2,3. Additionally, single-genome amplification and sequencing of spike using extracted RNA from respiratory samples was used as an independent method to detect the observed mutations (Extended Data Fig. 4). Finally, we detected no evidence of recombination, based on two independent methods (data not shown).
Maximum-likelihood analysis of patient-derived whole-genome consensus sequences demonstrated clustering with other local sequences from the same region (Fig. 1). The infecting strain was assigned to lineage 20B bearing the spike(D614G) variant. Environmental sampling showed evidence of virus on surfaces such as the telephone and call bell. Sequencing of these surface viruses showed clustering with those derived from the respiratory tract (Extended Data Fig. 2). All samples were consistent with having arisen from a single underlying viral population. In our phylogenetic analysis, we included sequential sequences from three other local patients identified with persistent viral RNA shedding over a period of 4 weeks or more as well as two recently reported long-term immunosuppressed individuals with SARS-CoV-24,5 (Extended Data Fig. 2 and Supplementary Table 2). Whereas the sequences from the three local patients as well as one of the previous studies5 showed little divergence with no amino acid changes in the spike protein over time, the case patient (hereafter, patient X1) showed considerable diversification. The other, previously reported patient4 showed a similar degree of diversification as patient X1. Further investigation of the sequence data suggested the existence of an underlying structure to the viral population in patient X1, with samples collected at days 93 and 95 being rooted within, but divergent from, the original population (Extended Data Figs. 5, 6). The relationship of the divergent samples to those at earlier time points argues against superinfection.

The circularized maximum-likelihood phylogenetic tree is rooted using the Wuhan-Hu-1 reference sequence, showing a subset of 250 local SARS-CoV-2 genomes from GISAID. This diagram highlights the considerable diversity of patient X1 (green) compared with three other local patients with prolonged shedding (patients C1–C3 are shown as red, blue and purple sequences, respectively). All ‘United Kingdom/English’ SARS-CoV-2 genomes were downloaded from the GISAID database and a random subset of 250 sequences were selected as background (shown in black).
SARS-CoV-2 viral diversity
All samples tested positive for SARS-CoV-2 by RT–qPCR and there was no sustained change in Ct values throughout the 101 days after the first two courses of remdesivir (days 41 and 54) or the first two units of convalescent plasma with polyclonal antibodies (days 63 and 65) (Extended Data Fig. 3). Notably, we were not able to culture virus from stored swab samples. Consensus sequences from short-read deep-sequencing Illumina data revealed dynamic population changes after day 65, as shown in a highlighter plot (Extended Data Fig. 6). In addition, we were also able to follow the dynamics of virus populations down to low frequencies during the entire period (Fig. 2 and Supplementary Table 4). After treatment with remdesivir on day 41, the low frequency variant analysis showed transient amino acid changes in populations at below 50% abundance in open reading frame (ORF) 1b, 3a and spike, with a C27509T (causing a T39I substitution) mutation in orf7a reaching 79% on day 45 (Fig. 2, and Supplementary Information). We found that an I513T substitution (encoded by T2343C) in NSP2 and a V157L substitution (encoded by G13936T) in RdRp had emerged from undetectable at day 54 to almost 100% frequency on day 66 (Fig. 2), with the mutation in the polymerase being the more plausible candidate for driving this sweep. Notably, spike(N501Y)—which can increase the affinity for the ACE2 receptor6 and which is present in the UK B1.1.7 lineage7—was observed on day 55 at a frequency of 33%, but was eliminated by the sweep of the NSP2 and RdRp variant.

Data are based on Illumina short-read ultra-deep sequencing values at 1,000× coverage. Variants shown reached a frequency of at least 10% in at least two samples. CP, convalescent plasma; RDV, remdesivir. Amino acid substitutions described in the text are indicated by labels using the same colouring as the position in the genome (associated proteins are included for each name). Labelled variants are represented by dashed lines. a, Variants detected in patient X1 on days 1–82 after the first positive RT–qPCR test for SARS-CoV-2. Spike(D796H*) (light blue) has the same frequency as NSP3(K902N) (orange) and is therefore hidden beneath the orange line. b, Variants detected in patient X1 on days 82–101.
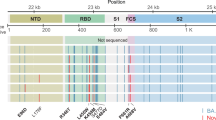
In contrast to the early period of infection, between days 66 and 82, after the first two administrations of convalescent sera, a shift in the virus population was observed, with a variant bearing a D796H substitution in S2 and a ΔH69/ΔV70 deletion in the S1 N-terminal domain (NTD) of spike becoming the dominant population at day 82. This was identified in a nose and throat swab sample with high viral load as indicated by a Ct value of 23 (Fig. 3a). The deletion was detected transiently at baseline on the basis of to short-read deep sequencing. The ΔH69/ΔV70 deletion was due to an out-of-frame six-nucleotide deletion, which results in the sequence of codon 68 changing from ATA to ATC.

a, At baseline, all six spike variants (Illumina sequencing) except for spike(ΔH69/ΔV70) were absent (less than 1% and fewer than 20 reads). Approximately two weeks after receiving two units of convalescent plasma, viral populations carrying spike(ΔH69/ΔV70) and spike(D796H) increased to frequencies of more than 80% but decreased significantly 4 days later. This population was replaced by a population bearing spike(Y200H) and spike(T240I), detected in two samples over a period of 6 days. These viral populations were then replaced by virus carrying spike(W64G) and spike(P330S), which both dominated at day 93. Following a third course of remdesivir and an additional unit of convalescent plasma, the spike(ΔH69/ΔV70) and spike(D796H) virus population re-emerged to become the dominant viral strain reaching variant frequencies of more than 75%. Pairs of mutations arose and disappeared simultaneously, indicating linkage on the same viral haplotype. Ct values from respiratory samples are indicated on the right y axis (black dashed line and triangles). In cases in which there were duplicate readings on the same day, to remain consistent, samples from nose and throat swabs were plotted. b, Maximum-likelihood phylogenetic tree of patient X1 with the day of sampling indicated. Spike mutations defining each of the clades are shown ancestrally on the branches on which they arose. On dates for which multiple samples were collected, these are indicated as endotracheal aspirate (ETA) and nose and throat swabs (N+T).
On days 86 and 89, viruses obtained from upper respiratory tract samples were characterized by the spike(Y200H, T240I) double mutant, with the mutation–deletion pair (spike(D796H, ΔH69/ΔV70)) observed on day 82 having decreased to frequencies of 10% or less (Figs. 2, 3). Spike(Y200H, T240I) were accompanied at high frequency by three other nonsynonymous variants with similar allele frequencies, encoding I513T in NSP2, V157L in RdRp and N177S in NSP15 (Fig. 2a). Both of these were also previously observed at a frequency of more than 98% in the sample on day 66 (Fig. 2a), suggesting that this new lineage emerged out of a population that existed previously.
Sequencing of a nose and throat swab sample on day 93 identified viruses characterized by spike(P330S) at the edge of the receptor-binding domain (RBD) and spike(W64G) in the S1 NTD at close to 100% abundance, whereas spike(D796H, ΔH69/ΔV70) showed an abundance of less than 1% and the variants spike(Y200H) and spike(T240I) had frequencies of less than 2%. Viruses with the spike(P330S) variant were detected in two independent samples from different sampling sites, arguing against the possibility of contamination. The divergence of these samples from the remainder of the population (Figs. 2, 3b and Extended Data Figs. 5, 6) suggests the possibility that they represent a compartmentalized subpopulation.
Patterns in the variant frequencies suggest competition between virus populations carrying different mutations; viruses with the mutation–deletion pair spike(D796H, ΔH69/ΔV70) rose to high frequency during convalescent plasma therapy, but were then outcompeted by another population in the absence of therapy. Specifically, these data are consistent with a lineage of viruses with the NSP2(I513T) and RdRp(V157L) variant, which was dominant on day 66 but was outcompeted during therapy by the mutation–deletion variant. With the lapse in therapy, the original strain—which had acquired NSP15(N1773S) and the spike(Y200H, T240I)—regained dominance, followed by the emergence of a separate population with the spike(W64G, P330S) variant.
In a final attempt to reduce the viral load, a third course of remdesivir (day 93) and a third dose of convalescent plasma (day 95) were administered. We observed the re-emergence of the spike(D796H, ΔH69/ΔV70) viral population (Figs. 2, 3). The inferred linkage of spike(D796H) and spike(ΔH69/ΔV70) was maintained as evidenced by the highly similar frequencies of the two variants, suggesting that the third unit of convalescent plasma led to the re-emergence of this population under renewed positive selection. In further support of our proposed idea of competition, frequencies of these two variants appeared to mirror changes in the NSP2(I513T) variant (Fig. 2), suggesting that these variants are markers of opposing clades in the viral population. Ct values remained low throughout this period with hyperinflammation, which eventually led to multi-organ failure and death on day 102. The repeated increase in the frequency of the viral population with convalescent plasma therapy strongly supports the hypothesis that the combination of the deletion and mutation in the spike protein conferred a selective advantage.
Spike mutations impair neutralizing antibody potency
Using lentiviral pseudotyping we generated wild-type spike, spike(D796H, ΔH69/ΔV70) and single mutant spike proteins in enveloped virions to measure the neutralization activity of convalescent plasma against these viruses (Fig. 4). This system has been shown to give generally similar results to replication-competent viruses8,9. Spike protein from each mutant was detected in pelleted virions (Fig. 4a). We also used an HIV-1(p24) antibody to monitor the levels of lentiviral particle production (Fig. 4a and Supplementary Fig. 2). We then measured the infectivity of the pseudoviruses, correcting for virus input using measurements of the reverse transcriptase activity, and found that spike(ΔH69/ΔV70) appeared to have twofold higher infectivity over a single round of infection compared to wild-type spike (Fig. 4b and Extended Data Fig. 7). By contrast, the spike(D796H) single mutant had significantly lower infectivity compared to the wild-type spike protein and the double mutant (spike(D796H, ΔH69/ΔV70)) had a similar infectivity to wild-type spike (Fig. 4b and Extended Data Fig. 7).

a, Western blot of virus pellets after centrifugation of supernatants from cells transfected with lentiviral pseudotyping plasmids that included the spike protein. Blots are representative of two independent transfections. b, Single-round infectivity of luciferase-expressing lentiviruses pseudotyped with the spike protein (wild-type (WT) or mutant) of SARS-CoV-2 in HEK293T cells co-transfected with ACE2 and TMPRSS2 plasmids. Infectivity is corrected for reverse transcriptase activity in the virus supernatant as measured by qPCR. Data points represent technical replicates (n = 3). Data are mean ± s.e.m. of two independent experiments. RLU, relative light units; nU, nanounit. c–e, Neutralization potency of convalescent plasma units (CP1–CP3) against pseudotyped viruses bearing spike(D796H), spike(ΔH69/ΔV70) and spike(D796H, ΔH69/ΔV70). f, g, Neutralizing potency of the serum of patient X1 against pseudotyped virus bearing spike(D796H), spike(ΔH69/ΔV70) and spike(D796H, ΔH69/ΔV70). Patient serum was taken on the indicated days. The serum dilution required to inhibit 50% of virus infection (ID50) is shown, expressed as a fold change relative to the wild-type virus. Data points represent means of technical replicates (horizontal bars) obtained an independent experiments (n = 2–6).
We found that spike(D796H) alone and the spike(D796H, ΔH69/ΔV70) double mutant were less sensitive to neutralization by convalescent plasma samples (Fig. 4c–e and Extended Data Fig. 7). By contrast, the spike(ΔH69/ΔV70) single mutant did not reduce neutralization sensitivity. In addition, patient-derived serum from days 64 and 66 (one day before and after the infusion with the second convalescent plasma therapy) similarly showed lower potency against the spike(D796H, ΔH69/ΔV70) double mutant (Fig. 4f, g).
A panel of nineteen monoclonal antibodies isolated from three donors was previously identified to neutralize SARS-CoV-2. To establish whether the mutations that occurred in vivo (spike(D796H) and spike(ΔH69/ΔV70)) resulted in a global change in neutralization sensitivity, we tested neutralizing monoclonal antibodies targeting the seven major epitope clusters that have previously been described (excluding the non-neutralizing clusters II and V and the small (n ≤2) neutralizing clusters IV and X). The eight RBD-specific monoclonal antibodies (Extended Data Fig. 8) exhibited no major change in neutralization potency and the non-RBD-specific antibody COVA1-21 showed a 3–5-fold reduction in potency against spike(D796H, ΔH69/ΔV70) and spike(ΔH69/ΔV70), but not against spike(D796H) alone9 (Extended Data Fig. 8). We observed no differences in neutralization between single or double mutants and the wild-type spike protein, suggesting that the mechanism of escape was probably outside of these epitopes in the RBD. These data confirm the specificity of the findings from convalescent plasma and suggest that the observed mutations are related to antibodies that target regions outside of the RBD. Notably, spike(ΔH69/ΔV70)-containing viruses showed reduced neutralization sensitivity to the monoclonal antibody COVA1-21, targeting an as-yet undefined epitope outside of the RBD10.
To understand how spike(ΔH69/ΔV70) and spike(D796H) might confer antibody resistance, we assessed how these mutations might affect the spike structure (Extended Data Fig. 9). We based this analysis primarily on a structure that lacked stabilizing modifications (PDB 6XR8)11, but also referred to stabilized structures determined at different pH values12. ΔH69/ΔV70 is located in a disordered, glycosylated loop at the distal surface of the NTD, near the binding site of polyclonal antibodies derived from COV57 plasma13,14 (Extended Data Fig. 9). As this loop is flexible and highly accessible, ΔH69/ΔV70 could in principle affect antibody binding in this region. D796 is located near the base of the spike protein, in a surface loop that is structurally disordered in the prefusion conformation and becomes part of a large disordered region in the post-fusion S2 trimer11 (Extended Data Fig. 9). The loop containing residue 796 is proposed to be targeted by antibodies15, despite mutations at position 796 being relatively uncommon (Extended Data Fig. 9). In the RBD-down spike structures11,12, D796 forms contacts with residues in the neighbouring protomer, including the glycosylated residue N709 (Extended Data Fig. 9).
Discussion
Here we documented a repeated evolutionary response by SARS-CoV-2 in the presence of antibody therapy during the course of a persistent infection in an immunocompromised host. The observation of potential selection for specific variants coinciding with the presence of antibodies from convalescent plasma is supported by the experimental finding of the twofold reduced susceptibility of these viruses to convalescent plasma containing polyclonal antibodies. In this case, the emergence of the variant was not the primary reason for treatment failure.
We noted in our analysis signs of compartmentalized viral replication based on the sequences recovered in upper respiratory tract samples. Both population-genetic and small-animal studies have shown a lack of reassortment between influenza viruses within a single host during an infection, suggesting that acute respiratory viral infection may be characterized by spatially distinct viral populations16,17. In the analysis of data, it is important to distinguish genetic changes that occur in the primary viral population from apparent changes that arise from the stochastic observation of spatially distinct subpopulations in the host. Although the samples that we obtained on days 93 and 95 of infection are genetically distinct from the other samples, the remaining samples are consistent with arising from a consistent viral population. We note that detection of viral RNA in post-mortem tissue was observed previously not only in lung tissue, but also in the spleen, liver, and heart4. Mixing of virus from different compartments—for example, via blood or the movement of secretions from lower to upper respiratory tract—could lead to fluctuations in viral populations at particular sampling sites.
This is a report of a single case and therefore limited conclusions can be drawn about generalizability.
An important limitation is that the data were derived from sampling from the upper respiratory tract and not the lower tract, thus limiting the inferences that can be drawn regarding viral populations in this single patient.
In addition to documenting the emergence of SARS-CoV-2 spike(ΔH69/ΔV70) in vivo, we show that this variant modestly increases infectivity of the spike protein in a pseudotyping assay. The deletion was observed contemporaneously with the rare S2 variant spike(D796H) after two separate courses of convalescent plasma, with other viral populations emerging. D796H, but not ΔH69/ΔV70, conferred a reduction in susceptibility to polyclonal antibodies in the units of convalescent plasma administered, although we cannot speculate as to their individual effect on sera from other individuals. It is notable that the spike(D796H, ΔH69/ΔV70) double mutant decreased in prevalence in between convalescent plasma courses, suggesting that there were other selective forces at play in the intervening period, which are possibly driven by the inflammation observed in the individual. This includes the possibility that the haplotype comprising spike (D796H, ΔH69/ΔV70) may have carried mutations in other regions that were deleterious during that intervening period. Although ΔH69/ΔV70 is expanding at a high rate18, D796 mutations are also increasing. D796H has been documented in 0.02% of global sequences and D796Y appears in 0.05% of global sequences (Extended Data Fig. 9).
The effects of convalescent plasma on virus evolution found here are unlikely to apply in immunocompetent hosts in whom viral diversity is likely to be lower owing to better immune control. Our data highlight that infection control measures may need to be tailored to the needs of immunocompromised patients and also caution in the interpretation of guidelines from the US Centres of Disease Control that recommend 20 days as the upper limit of infection-prevention precautions in immunocompromised patients who are afebrile19. Owing to the difficulty of culturing clinical isolates, use of surrogate experiments are warranted20. However, in cases in which the detection of ongoing viral evolution is possible, this serves as a clear proxy for the existence of infectious virus. In our case, we detected environmental contamination while in a single-occupancy room and the patient was moved to a negative-pressure high air-change infectious-disease isolation room.
The clinical efficacy of convalescent plasma in patients with severe COVID-19 has not been demonstrated21, and its use in different stages of infection and disease remains experimental; as such, we suggest that it should be reserved for use within clinical trials, and with rigorous monitoring of clinical and virological parameters. The data from this single patient indicate caution should be used for convalescent plasma therapy in patients with immunosuppression of both T cell and B cell arms; in these patients, the administered antibodies have little support from cytotoxic T cells, thereby reducing the chances of clearance and theoretically raising the potential for the evolution of escape mutations in SARS-CoV-2. Although we await further data, in cases in which clinical trial enrolment is not possible, convalescent plasma administered for clinical need in immunosuppressed individuals should ideally only be considered as part of observational studies, undertaken preferably in single-occupancy rooms with enhanced infection-control precautions, including environmental sampling of SARS-CoV-2 and real-time sequencing. Understanding the viral dynamics and characterizing the viral evolution in response to different selection pressures in an immunocompromised individual are necessary not only for improved patient management but also for public health benefit.
Methods
Clinical-sample collection and next-generation sequencing
Serial samples were collected from the patient periodically from the lower respiratory tract (sputum or endotracheal aspirate), upper respiratory tract (throat and nasal swab) and from stool. Nucleic acid extraction was done from 500 μl of sample with a dilution of MS2 bacteriophage to act as an internal control, using the easyMAG platform (Biomerieux) according to the manufacturers’ instructions. All samples were tested for presence of SARS-CoV-2 with a validated one-step RT–qPCR assay developed in conjunction with the Public Health England Clinical Microbiology22. Amplification reactions were all performed on a Rotorgene PCR instrument. Samples with a Ct value of ≤36 were considered to be positive.
Sera from recovered patients in the COVIDx study23 were used for testing of neutralization activity by SARS-CoV-2 mutants.
SARS-CoV-2 serology by multiplex particle-based flow cytometry
Recombinant SARS-CoV-2 nucleocapsid, spike and RBD proteins were covalently coupled to distinct carboxylated bead sets (Luminex) to form a triplex and analysed as previously described24. Specific binding was reported as mean fluorescence intensities.
Whole-blood T cell and innate stimulation assay
Whole blood was diluted 1:5 in RPMI into 96-well F plates (Corning) and activated by single stimulation with phytohaemagglutinin (10 μg ml−1; Sigma-Aldrich), lipopolysaccharide (1 μg ml−1, List Biochemicals) or by costimulation with anti-CD3 (MEM57, Abcam, 200 ng ml−1, 1:1,000) and IL-2 (Immunotools, 1,430 U ml−1, 1:1,000) antibodies. Supernatants were taken after 24 h. Levels (pg ml−1) are shown for IFNγ, IL-17, IL-2, TNF, IL-6, IL-1b and IL-10. Cytokines were measured by multiplexed particle-based flow cytometry on a Luminex analyser (Bio-Plex, Bio-Rad) using an R&D Systems custom kit (R&D Systems).
For viral genomic sequencing, total RNA was extracted from samples as described previously25. Samples were sequenced using MinION flow cells v.9.4.1 (Oxford Nanopore Technologies) following the ARTICnetwork v.3 protocol26 and BAM files assembled using the ARTICnetwork assembly pipeline27. A representative set of 10 sequences was selected and also sequenced using the Illumina MiSeq platform. Amplicons were diluted to 2 ng μl−1 and 25 μl (50 ng) were used as input for each library preparation reaction. The library preparation used KAPA Hyper Prep kit (Roche) according to the manufacturer’s instructions. In brief, amplicons were end-repaired and had A-overhang added; these were then ligated with 15 mM of NEXTflex DNA Barcodes (Bio Scientific). Post-ligation products were cleaned using AMPure beads and eluted in 25 μl. Then, 20 μl was used for library amplification by 5 cycles of PCR. For the negative controls, 1 ng was used for ligation-based library preparation. All libraries were assayed using TapeStation (Agilent Technologies) to assess fragment size and quantified by qPCR. All libraries were then pooled in equimolar ratios accordingly. Libraries were loaded at 15 nM and spiked in 5% PhiX (Illumina) and sequenced on for MiSeq 500 cycle using a Miseq Nano v.2 with 2 × 250 bp paired-end sequencing. A minimum of ten reads was required for a variant call.
Bioinformatics processes
For long-read sequencing, genomes were assembled with reference-based assembly and a curated bioinformatics pipeline with 20× minimum coverage across the whole genome27. For short-read sequencing, FASTQ files were downloaded, poor-quality reads were identified and removed, and both Illumina and PHiX adapters were removed using TrimGalore v.0.6.628. Trimmed paired-end reads were mapped to the National Center for Biotechnology Information SARS-CoV-2 reference sequence MN908947.3 using MiniMap2-2.17 with arguments -ax and sr29. BAM files were then sorted and indexed with Samtools v.1.11 and PCR optical duplicates were removed using Picard (http://broadinstitute.github.io/picard). Consensus sequences of nucleic acids with a minimum whole-genome coverage of at least 20× were generated with BCFtools using a 0% majority threshold.
Variant calling
Variant frequencies were validated using custom code as part of the AnCovMulti package (https://github.com/PollockLaboratory/AnCovMulti). The main idea behind this validation was to identify and remove consistent potential amplification errors and mutability near the end of Illumina reads. Furthermore, stringent filtering was applied to remove biased amplification of early laboratory-induced mutations or very low copy variations.
Filtering consisted of requiring exact initiation at a primer within 2 bp of the start of a read, a minimum of 247 bp length read, fewer than four well-separated sites divergent from the reference sequence, a maximum insertion size of three nucleotides, a maximum deletion size of 11 bp, and resolution of conflicting signal from different primers.
Single-genome amplification and sequencing
Viral RNA extracts were reverse-transcribed from each sample to sufficiently capture the diversity of the viral population without introducing resampling bias. SuperScript IV (Thermo Fisher Scientific) and gene-specific primers were used for reverse transcription. Template RNA was degraded with RNase H (Thermo Fisher Scientific). All primers used were ‘in-house’ primers designed using the multiple sequence alignment of the patient’s consensus sequences obtained by next-generation sequencing. A partial gene sequence of spike (encoding amino acids 21–800) was amplified as one continuous length of DNA (the spike gene is approximately 1.8 kb) by nested PCR. Terminally diluted cDNA was amplified by PCR using Platinum Taq DNA Polymerase High Fidelity (Invitrogen) so that 30% of reactions were positive30. By Poisson statistics, sequences were deemed ≥80% likely to be derived from HIV-1 single genomes. We obtained between 20 and 60 single genomes at each sample time point to achieve 90% confidence of detecting variants present at ≥8% of the viral population in vivo31,32. Partial spike amplicons obtained from terminal-dilution PCR amplification were Sanger-sequenced to form a contiguous sequence using another set of eight in-house primers. Sanger sequencing was provided by Genewiz and manual sequence editing was performed using DNA Dynamo software (Blue Tractor Software).
Phylogenetic analysis
All available full-genome SARS-CoV-2 sequences were downloaded from the GISAID database (http://gisaid.org/)33 on 16 December 2020. Duplicate and low-quality sequences (>5% nucleocapsid regions) were removed, leaving a dataset of 212,297 sequences with a length of more than 29,000 bp. All sequences were sorted by name and only sequences sequenced with United Kingdom/England identifiers were retained. From this dataset, sequences were deduplicated and, in figures in which background sequences were required, randomly subsampled using seqtk (https://github.com/lh3/seqtk). All sequences were aligned to the SARS-CoV-2 reference strain MN908947.3, using MAFFT v.7.475 with automatic flavour selection34. Major SARS-CoV-2 clade memberships were assigned to all sequences using both the Nextclade server v.0.9 (https://clades.nextstrain.org/) and Phylogenetic Assignment Of Named Global Outbreak Lineages (PANGOLIN)35.
Maximum-likelihood phylogenetic trees were produced using the above curated dataset using IQ-TREE v.2.1.236. Evolutionary model selection for trees were inferred using ModelFinder37 and trees were estimated using the GTR + F + I model with 1,000 ultrafast bootstrap replicates38. All trees were visualized with Figtree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/), rooted on the SARS-CoV-2 reference sequence and nodes arranged in descending order. Nodes with bootstraps values of less than 50 were collapsed using an in-house script.
In-depth allele frequency variant calling
The SAMFIRE package v.1.0639 was used to call allele frequency trajectories from BAM file data. Reads were included in this analysis if they had a median PHRED score of at least 30, trimming the ends of reads to achieve this if necessary. Nucleotides were then filtered to have a PHRED score of at least 30; reads with fewer than 30 such reads were discarded. Distances between sequences, accounting for low-frequency variant information, were also obtained using SAMFIRE. The sequence distance metric, described in a previous paper40, combines allele frequencies across the whole genome. Where L is the length of the genome, we define q(t) as a 4 × L element vector describing the frequencies of each of the nucleotides A, C, G, and T at each locus in the viral genome sampled at time t. For any given locus i in the genome, we calculate the change in allele frequencies between the times t1 and t2 via a generalization of the Hamming distance
where the vertical lines indicate the absolute value of the difference. These statistics were then combined across the genome to generate the pairwise sequence distance metric
The Mathematica software package was used to conduct a regression analysis of pairwise sequence distances against time, leading to an estimate of a mean rate of within-host sequence evolution. In contrast to the phylogenetic analysis, this approach assumed that the samples collected on days 93 and 95 arose via stochastic emission from a spatially separated subpopulation within the host, leading to a lower inferred rate of viral evolution for the bulk of the viral population.
All variants were indecently validated using custom code as part of the AnCovMulti package (https://github.com/PollockLaboratory/AnCovMulti).
Western blot analysis
Cells were transfected with the indicated plasmid preparations and 48 h after transfection the culture supernatant was collected and passed through a 0.45-μm-pore-size filter to remove cellular debris. The filtrate was centrifuged at 15,000 rpm for 120 min to pellet virions. The pelleted virions were lysed in Laemmli reducing buffer (1 M Tris-HCl (pH 6.8), SDS, 100% glycerol, β-mercaptoethanol and bromophenol blue). Pelleted virions were subjected to electrophoresis on SDS 4–12% Bis-Tris protein gels (Thermo Fisher Scientific) under reducing conditions. This was followed by electroblotting onto polyvinylidene difluoride (PVDF) membranes. The SARS-CoV-2 spike proteins were visualized using a ChemiDoc MP imaging system (Biorad) using anti-spike S2 (Invitrogen; 1:1,000 dilution) and anti-p24 Gag (NIH AIDS Reagents; 1:1,000 dilution) antibodies.
Recombination detection
All sequences were tested for potential recombination, as this would affect evolutionary estimates. Potential recombination events were explored with nine algorithms (RDP, MaxChi, SisScan, GeneConv, Bootscan, PhylPro, Chimera, LARD and 3SEQ), implemented in RDP5 with default settings41. To corroborate any findings, ClonalFrameML v.1.1242 was also used to infer recombination breakpoints. Neither of the programs found evidence of recombination in our data.
Structural viewing
The PyMOL Molecular Graphics System v.2.4.0 (https://github.com/schrodinger/pymol-open-source/releases) was used to map the location of the four spike mutations of interested onto a previously published SARS-CoV-2 spike structure (PDB: 6ZGE)43.
Testing of convalescent plasma for antibody titres
The anti-SARS-CoV-2 ELISA (IgG) assay used to test convalescent plasma for antibody titres was Euroimmun Medizinische Labordiagnostika. This indirect ELISA-based assay uses a recombinant structural spike 1 (S1) protein of SARS-CoV-2 expressed in the human cell line HEK293 for the detection of SARS-CoV-2 IgG.
Generation of spike mutants
Amino acid substitutions were introduced into the D614G pCDNA_SARS-CoV-2_Spike plasmid as previously described44 using the QuikChange Lightening Site-Directed Mutagenesis kit, following the manufacturer’s instructions (Agilent Technologies).
Pseudotype virus preparation
Viral vectors were prepared by transfection of HEK293T cells using the Fugene HD transfection reagent (Promega). HEK293T cells were transfected with a mixture of 11 μl of Fugene HD, 1 μg of pCDNAΔ19Spike–HA, 1 μg of p8.91 HIV-1 gag–pol expression vector45,46 and 1.5 μg of pCSFLW (expressing the firefly luciferase reporter gene with the HIV-1 packaging signal). Viral supernatant was collected at 48 and 72 h after transfection, filtered through 0.45-μm filter and stored at −80 °C. The 50% tissue-culture infectious dose (TCID50) of SARS-CoV-2 pseudovirus was determined using the Steady-Glo Luciferase assay system (Promega).
Standardization of virus input by SYBR-Green-based product-enhanced PCR assay
The reverse transcriptase activity of virus preparations was determined by qPCR using a SYBR-Green-based product-enhanced PCR assay as previously described47. In brief, tenfold dilutions of virus supernatant were lysed in a 1:1 ratio in a 2× lysis solution (made up of 40% glycerol (v/v), 0.25% Triton X-100 (v/v), 100 mM KCl, RNase inhibitor 0.8 U ml−1, Tris HCL 100 mM, buffered to pH 7.4) for 10 min at room temperature.
Then, 12 μl of each sample lysate was added to 13 μl of a SYBR Green master mix (containing 0.5 μM of MS2 RNA forward and reverse primers, 3.5 pmol ml−1 of MS2 RNA and 0.125 U μl−1 of Ribolock RNase inhibitor and cycled in a QuantStudio. Relative amounts of reverse transcriptase activity were determined as the rate of transcription of bacteriophage MS2 RNA, with absolute reverse transcriptase activity calculated by comparing the relative amounts of reverse transcription to a reverse transcriptase standard of known activity.
Serum/plasma pseudotype neutralization assay
Spike pseudotype assays have been shown to have similar characteristics as neutralization testing using fully infectious wild-type SARS-CoV-28. Virus neutralization assays were performed on HEK293T cells transiently transfected with ACE2 and TMPRSS2 using SARS-CoV-2 spike pseudotyped virus expressing luciferase48. Pseudotyped virus was incubated with serial dilutions of heat-inactivated human serum samples or convalescent plasma in duplicate for 1 h at 37 °C. Virus- and cell-only controls were also included. Then, freshly trypsinized HEK293T ACE2- and TMPRSS2-expressing cells were added to each well. After 48 h incubation in a 5% CO2 environment at 37 °C, luminescence was measured using Steady-Glo Luciferase assay system (Promega).
Monoclonal antibody pseudotype neutralization assay
Virus neutralization assays were performed using HeLa cells stably expressing ACE2 and using SARS-CoV-2 spike pseudotyped virus expressing luciferase as previously described49. Pseudotyped virus was incubated with serial dilutions of purified monoclonal antibodies9 in duplicate for 1 h at 37 °C. Then, freshly trypsinized HeLa ACE2-expressing cells were added to each well. After 48 h incubation in a 5% CO2 environment at 37 °C, luminescence was measured using a Bright-Glo Luciferase assay system (Promega) and neutralization was calculated relative to virus-only controls. IC50 values were calculated in GraphPad Prism.
Ethics
The study was approved by the East of England–Cambridge Central Research Ethics Committee (17/EE/0025). Written informed consent was obtained from both the patient and his family. Additional control patients with COVID-19 were enrolled to the NIHR BioResource Centre Cambridge under ethics review board approval (17/EE/0025).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Long-read sequencing data that support the findings of this study have been deposited in the NCBI SRA database with accession codes SAMN16976824–SAMN16976846 under BioProject PRJNA682013. Short reads and data used to construct figures were deposited at https://github.com/Steven-Kemp/sequence_files. All data are also available from the corresponding author. Source data are provided with this paper.
Code availability
The SAMFIRE package v.1.06 was used for filtering and calling variants from the Illumina data. It is available at https://github.com/cjri/samfire/ for review. Additional code was used to validate the variant frequencies and can be found at https://github.com/PollockLaboratory/AnCovMulti.
Change history
21 July 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41586-022-05104-2
References
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Kim, K. W. et al. Respiratory viral co-infections among SARS-CoV-2 cases confirmed by virome capture sequencing. Preprint at https://doi.org/10.21203/rs.3.rs-105996/v1 (2020).
Bull, R. A. et al. Analytical validity of Nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 11, 6272 (2020).
Choi, B. et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N. Engl. J. Med. 383, 2291–2293 (2020).
Avanzato, V. A. et al. Case study: prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised cancer patient. Cell 183, 1901–1912 (2020).
Starr, T. N. et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 182, 1295–1310 (2020).
Rambaut, A. et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Preprint at https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (2020).
Schmidt, F. et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J. Exp. Med. 217, e20201181 (2020).
Brouwer, P. J. M. et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 369, 643–650 (2020).
Zussman, M. E., Bagby, M., Benson, D. W., Gupta, R. & Hirsch, R. Pulmonary vascular resistance in repaired congenital diaphragmatic hernia vs. age-matched controls. Pediatr. Res. 71, 697–700 (2012).
Cai, Y. et al. Distinct conformational states of SARS-CoV-2 spike protein. Science 369, 1586–1592 (2020).
Zhou, T. et al. Cryo-EM structures of SARS-CoV-2 spike without and with ACE2 reveal a pH-dependent switch to mediate endosomal positioning of receptor-binding domains. Cell Host Microbe 28, 867–879 (2020).
Robbiani, D. F. et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 584, 437–442 (2020).
Barnes, C. O. et al. Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell 182, 828–842 (2020).
Shrock, E. et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science 370, eabd4250 (2020).
Sobel Leonard, A. et al. The effective rate of influenza reassortment is limited during human infection. PLoS Pathog. 13, e1006203 (2017).
Richard, M., Herfst, S., Tao, H., Jacobs, N. T. & Lowen, A. C. Influenza A virus reassortment is limited by anatomical compartmentalization following coinfection via distinct routes. J. Virol. 92, e02063-17 (2018).
Kemp, S. A. et al. Recurrent emergence and transmission of a SARS-CoV-2 spike deletion H69/V70. Preprint at https://doi.org/10.1101/2020.12.14.422555 (2021).
CDC. Discontinuation of transmission-based precautions and disposition of patients with COVID-19 in healthcare settings (interim guidance). https://www.cdc.gov/coronavirus/2019-ncov/hcp/disposition-hospitalized-patients.html (2020).
Boshier, F. A. T. et al. Remdesivir induced viral RNA and subgenomic RNA suppression, and evolution of viral variants in SARS-CoV-2 infected patients. Preprint at https://doi.org/10.1101/2020.11.18.20230599 (2020).
Simonovich, V. A. et al. A randomized trial of convalescent plasma in COVID-19 severe pneumonia. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2031304 (2020).
Meredith, L. W. et al. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. Lancet Infect. Dis. 20, 1263–1271 (2020).
Collier, D. A. et al. Point of care nucleic acid testing for SARS-CoV-2 in hospitalized patients: a clinical validation trial and implementation study. Cell Rep. Med. 1, 100062 (2020).
Xiong, X. et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 27, 934–941 (2020).
Meredith, L. W. et al. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. Lancet Infect. Dis. 20, 1263–1271 (2020).
Quick, J. nCoV-2019 sequencing protocol. Protocols.io https://doi.org/10.17504/protocols.io.bbmuik6w (2020).
Loman, N., Rowe, W. & Rambaut, A. nCoV-2019 novel coronavirus bioinformatics protocol. Protocol version 1 https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (Artic Network, 2020).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Jordan, M. R. et al. Comparison of standard PCR/cloning to single genome sequencing for analysis of HIV-1 populations. J. Virol. Methods 168, 114–120 (2010).
Palmer, S. et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 43, 406–413 (2005).
Keele, B. F. et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl Acad. Sci. USA 105, 7552–7557 (2008).
Shu, Y. & McCauley, J. GISAID: global initiative on sharing all influenza data — from vision to reality. Euro Surveill. 22, 30494 (2017).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Rambaut, A. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 5, 1403–1407 (2020).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Minh, B. Q., Nguyen, M. A. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195 (2013).
Illingworth, C. J. SAMFIRE: multi-locus variant calling for time-resolved sequence data. Bioinformatics 32, 2208–2209 (2016).
Lumby, C. K., Zhao, L., Breuer, J. & Illingworth, C. J. A large effective population size for established within-host influenza virus infection. eLife 9, e56915 (2020).
Martin, D. P., Murrell, B., Golden, M., Khoosal, A. & Muhire, B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1, vev003 (2015).
Didelot, X. & Wilson, D. J. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11, e1004041 (2015).
Wrobel, A. G. et al. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 27, 763–767 (2020).
Gregson, J. et al. Human immunodeficiency virus-1 viral load is elevated in individuals with reverse-transcriptase mutation M184V/I during virological failure of first-line antiretroviral therapy and is associated with compensatory mutation L74I. J. Infect. Dis. 222, 1108–1116 (2020).
Naldini, L., Blömer, U., Gage, F. H., Trono, D. & Verma, I. M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl Acad. Sci. USA 93, 11382–11388 (1996).
Gupta, R. K. et al. Full-length HIV-1 Gag determines protease inhibitor susceptibility within in vitro assays. AIDS 24, 1651–1655 (2010).
Vermeire, J. et al. Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS ONE 7, e50859 (2012).
Mlcochova, P. et al. Combined point-of-care nucleic acid and antibody testing for SARS-CoV-2 following emergence of D614G spike variant. Cell Rep. Med. 1, 100099 (2020).
Seow, J. et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat. Microbiol. 5, 1598–1607 (2020).
Brouwer, P. J. M. et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 369, 643–650 (2020).
Acknowledgements
We are grateful to the patient and his family. We thank the staff at CUH and the NIHR Cambridge Clinical Research Facility; R. Kugathasan and W. Barclay for discussions; M. Curran, W. Hamilton and D. Sparkes, A. Floto and F. Gallagher; J. Voss for the gift of HeLa cells stably expressing ACE2; and J. Nathan for the RBD protein and L. James for the nucleocapsid protein. COG-UK is supported by funding from the Medical Research Council (MRC) part of UK Research & Innovation (UKRI), the National Institute of Health Research (NIHR) and Genome Research Limited, operating as the Wellcome Sanger Institute. R.K.G. is supported by a Wellcome Trust Senior Fellowship in Clinical Science (WT108082AIA). L.E.M. is supported by a Medical Research Council Career Development Award (MR/R008698/1). S.A.K. is supported by the Bill and Melinda Gates Foundation via PANGEA grant (OPP1175094). D.A.C. is supported by a Wellcome Trust Clinical PhD Research Fellowship. C.J.R.I. acknowledges MRC funding (MC_UU_00002/11). This research was supported by the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre, the Cambridge Clinical Trials Unit (CCTU) and by the UCL Coronavirus Response Fund and made possible through generous donations from UCL’s supporters, alumni, and friends (to L.E.M.). J.A.G.B. is supported by the Medical Research Council (MC_UP_1201/16). I.G.G. is a Wellcome Senior Fellow and supported by the Wellcome Trust (207498/Z/17/Z). D.D.P. is supported by NIH GM083127.
Author information
Authors and Affiliations
Consortia
Contributions
R.K.G., S.A.K., D.A.C., A.S., T.G. and E.G.-K. conceived the study. R.K.G., S.A.K., D.A.C., L.E.M., J.A.G.B., E.G.-K., N.T., A. Chandra, C.S., R.D., R.A.G., D.D.P. and Y.M. designed experiments. S.A.K., D.A.C., L.E.M., R.D., C.R.-S., A.J., I.A.T.M.F., K.S., T.G., C.J.R.I., J.R.B., J.P.S., M.J.v.G., L.G.-C., G.B.-M. and K.L. performed experiments. R.K.G., S.A.K., D.A.C., P.M., L.E.M., J.A.G.B., S.G., K.S., T.G., J.B., K.G.C.S., I.G.G., C.J.R.I., I.U.L., D.J.R., J.P.S., J.R.B., R.A.G., D.D.P., R.D., L.C.-G. and G.B.-M. interpreted data. R.K.G., D.A.C. and S.A.K wrote the first draft of the paper. All authors contributed to revising the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Richard Neher and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Clinical time line of events with longitudinal respiratory sample Ct values.
CPAP, continuous positive airway pressure; CT, cycle threshold; ITU, intensive therapy unit.
Extended Data Fig. 2 Blood parameters over time in patient X1.
a, White cell (WCC) and lymphocyte counts are expressed as 103 cells per mm3. CRP, C-reactive protein. b, Assessment of T cell and innate function. Cytokines were measured in whole blood after stimulation for 24 h either after T cell stimulation with phytohaemagglutinin (PHA) or anti-CD3/IL2 antibodies, or innate stimulation with lipopolysaccharide (LPS). Data from healthy control individuals are shown as grey circles (n = 15), data from patient X1 at days 71 and 98 are shown as blue and red circles, respectively. Cytokine levels are shown as pg ml−1. Data are mean ± s.d.
Extended Data Fig. 3 Serum antibody levels against SARS-CoV-2.
a, Anti-SARS-CoV-2 IgG antibodies in patient X1, before and after convalescent plasma treatment (days 66 and 95) compared with RNA+ patients with COVID-19 and pre-pandemic healthy control individuals. Red, grey and gold, IgG antibodies against the nucleocapsid (N) protein, trimeric spike (S) protein and RBD of SARS-CoV-2 were measured by multiplexed particle-based flow cytometry (Luminex) in RNA+ patients with COVID-19 (n = 20, red dots), pre-pandemic healthy control individuals (n = 20, grey dots) and convalescent donor plasma (orange dots). Patient sera over time are shown in blue: anti-SARS-CoV-2 IgG to nucleocapsid (blue squares), spike (blue circles) and RBD (blue triangles). Data are mean fluorescent intensity (MFI) ± s.d. The timing of convalescent plasma units (days after first positive test) is also shown. b, SARS-CoV-2 antibody titres in convalescent plasma. Measurement of SARS-CoV-2-specific IgG antibody titres in three units of convalescent plasma by Euroimmun assay. OD, optical density.
Extended Data Fig. 4 Cross-comparison of sequencing approaches.
a, Comparison between short-read (Illumina) and long-read single-molecule (Oxford Nanopore) sequencing methods for the six observed mutations in the spike protein. Concordance was generally good between the majority of time points; however, owing to large discrepancies in a number of time points, we suggest that due to the high base-calling error rate, the Nanopore method is not yet suitable for calling minority variants. As such, all figures in the main paper were produced using only the Illumina data. b, Single-genome sequencing data from respiratory samples at the indicated days. The number of single genomes obtained at each time point with the mutations of interest (identified by deep sequencing) are shown. *The denominator is 19, as the primer reads for two samples were poor quality at amino acid 796 on day 98. Amino acid variant and corresponding nucleotide position: spike(W64G), 21752; spike(Δ69), 21765–21770; spike(Y200H), 22160; spike(T240I), 22281; spike(P330S), 22550; spike(D795H), 23948.
Extended Data Fig. 5 Evidence of a within-host cladal structure.
a, Pairwise distances between samples measured using the all-locus distance metric plotted against pairwise distances in time (measured in days) between samples being collected. Internal distances between samples in the proposed main clade are shown in black, distances between samples in the main clade and samples collected on days 93 and 95 are shown in red, and internal distances between samples collected on days 93 and 95 are shown in green. b, Pairwise distances between samples in the larger clade (black) and between these samples and those collected on days 93 and 95 (red). The median values of the distributions of these values are significantly different according to a Mann–Whitney U-test. c, Pairwise distances between samples in the main clade, once the data collected on days 86, 89, 93 and 95 were removed (black) and between these samples and those collected on days 86 and 89 (red). The median values of the distributions of these values are not significantly different at the 5% level according to a Mann–Whitney U-test.
Extended Data Fig. 6 Sequence analyses and comparisons with other patients with SARS-CoV-2.
a, Magnified view of the maximum-likelihood phylogenetic tree showing the diversity of patient X1 and three other patients who showed long-term shedding of SARS-CoV-2 from the local area (red, blue and purple), compared to recently published sequences4,5 (orange and gold). Control patients with SARS-CoV-2 generally showed limited diversity temporally, although the sequences from one of the previous studies4 were highly divergent. Environmental samples (the call bell and mobile phone of the patient) are indicated. Tree branches have been collapsed where bootstrap support was <60. b, Highlighter plot showing the nucleotide changes at consensus level in sequential respiratory samples compared to the consensus sequence at first diagnosis of COVID-19. Each row indicates the time point the sample was collected (number of days after the first positive RT–qPCR result for SARS-CoV-2). Black dashed lines indicate the RNA-dependent RNA polymerase (RdRp) and spike regions of the genome. There were few nucleotide substitutions between days 1 and 54, despite the patient receiving two courses of remdesivir. The first major changes in the spike genome occurred on day 82, after convalescent plasma was administered on days 63 and 65. The amino acid deletion in S1 (ΔH69/ΔV70) is indicated by the black lines. Sample sites included endotracheal aspirate (ETA) and nose and throat swabs (N+T).
Extended Data Fig. 7 In vitro infectivity and neutralization sensitivity of spike-pseudotyped lentiviruses.
a, Infection of target HEK293T cells expressing TMPRSS2 and ACE2 receptors using equal amounts of virus as determined by reverse transcriptase activity. Data points are mean ± s.e.m . of technical replicates (n = 2); data are representative of n = 2 independent experiments. b, Representative inverse dilution plots for spike variants against convalescent plasma units 1–3. Data are mean ±s.e.m. of the neutralization of technical replicates (n = 2). Data are representative of two independent experiments (n = 2).
Extended Data Fig. 8 Neutralizing activity of a panel of monoclonal antibodies against SARS-CoV-2 spike-pseudotyped viruses.
a, Lentiviruses pseudotyped with the spike protein (wild type (WT) (D614G background), D796H, ΔH69/ΔV70, D796H + ΔH69/ΔV70) of SARS-CoV-2 were produced in HEK293T cells and used to infect target Hela cells stably expressing ACE2 in the presence of serial dilutions of the indicated monoclonal antibodies (mAb). Data are mean ± s.d. of n = 2 technical replicates. Data are representative of at least two independent experiments. b, Classes of RBD-binding antibodies and fold changes for spike mutations D796H or ΔH69/ΔV70 are indicated on a previously published study50. Clusters II and V contain only non-neutralizing monoclonal antibodies; the smaller neutralizing monoclonal antibody clusters IV (n = 2) and X (n = 1) were not tested. Red indicates significant fold changes.
Extended Data Fig. 9 Location of spike mutations ΔH69/Y70 and D796H.
a, The SARS-CoV-2 spike trimer (PDB ID: 6XR8) with two protomers represented as surfaces and one protomer represented as a ribbon. The NTD is coloured in light blue, the RBD in light pink, the fusion peptide in dark pink, the HR1 domain in yellow, the CH domain in pale green and the CD domain in brown. The location of D796 and H69 are indicated by red spheres. The loop connecting D796 to the fusion peptide is coloured magenta to improve visibility. The double grey lines provide orientation relative to the membrane. b, Magnification of the region defined by the box around H69 in a. H69 is highlighted in yellow. Residues containing atoms that are within 6 Å of H69 are highlighted in cyan. c, Magnification of the region defined by the box around D796 in a. D796 is highlighted in yellow. Residues containing atoms that are within 6 Å of D796 are highlighted in cyan. Hydrogen bonds are indicated by dashed yellow lines. Hydrophobic residues in the vicinity of D796 have been labelled. Y707 is from the neighbouring protomer. d, Global prevalence of selected spike mutations described in this paper. All high-coverage sequences were downloaded from the GISAID database on 6 January 2021 and aligned using MAFFT; as of this date there were 298,254 sequences available. The global prevalence of each of the six spike variants (W64G, ΔH69/ΔV70, Y200H, T240I, P330S and D796H) were assessed by viewing the multiple sequence alignment in AliView, sorting by the column of interest and counting the number of mutations.
Supplementary information
Supplementary Information
A full list of members of the CITIID-NIHR BioResource COVID-19 Collaboration.
Supplementary Information
This file contains Supplementary Clinical Case details, Supplementary Tables 1-4 and Supplementary Figs 1-2.
Rights and permissions
About this article

Cite this article
Kemp, S.A., Collier, D.A., Datir, R.P. et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 592, 277–282 (2021). https://doi.org/10.1038/s41586-021-03291-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03291-y
This article is cited by
-
Evidence of SARS-CoV-2 convergent evolution in immunosuppressed patients treated with antiviral therapies
Virology Journal (2024)
-
Impact of plasma Epstein–Barr virus DNA in posttreatment nasopharyngeal carcinoma patients after SARS-CoV-2 infection
Infectious Agents and Cancer (2024)
-
Chronic shedding of a SARS-CoV-2 Alpha variant in wastewater
BMC Genomics (2024)
-
Chronic COVID-19 infection in an immunosuppressed patient shows changes in lineage over time: a case report
Virology Journal (2024)
-
Clinical course and management of COVID-19 in the era of widespread population immunity
Nature Reviews Microbiology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
