
Abstract
The expanding pandemic of coronavirus disease 2019 (COVID-19) requires the development of safe, efficacious and fast-acting vaccines. Several vaccine platforms are being leveraged for a rapid emergency response1. Here we describe the development of a candidate vaccine (YF-S0) for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that uses live-attenuated yellow fever 17D (YF17D) vaccine as a vector to express a noncleavable prefusion form of the SARS-CoV-2 spike antigen. We assess vaccine safety, immunogenicity and efficacy in several animal models. YF-S0 has an excellent safety profile and induces high levels of SARS-CoV-2 neutralizing antibodies in hamsters (Mesocricetus auratus), mice (Mus musculus) and cynomolgus macaques (Macaca fascicularis), and—concomitantly—protective immunity against yellow fever virus. Humoral immunity is complemented by a cellular immune response with favourable T helper 1 polarization, as profiled in mice. In a hamster model2 and in macaques, YF-S0 prevents infection with SARS-CoV-2. Moreover, a single dose conferred protection from lung disease in most of the vaccinated hamsters within as little as 10 days. Taken together, the quality of the immune responses triggered and the rapid kinetics by which protective immunity can be attained after a single dose warrant further development of this potent SARS-CoV-2 vaccine candidate.
Similar content being viewed by others

Main
Protective immunity against SARS-CoV-2 and other coronaviruses is believed to depend on neutralizing antibodies (NAbs) that target the viral spike (S) protein. In particular, NAbs specific for the N-terminal S1 domain—which contains the ACE2 receptor-binding domain—have previously been shown to prevent viral infection in several animal models3.
The YF17D vaccine is known to rapidly induce broad multifunctional innate, humoral and cell-mediated immune responses that may result in lifelong protection after a single vaccine dose in nearly all vaccinees4. These favourable characteristics also translate to vectored vaccines that are based on the YF17D backbone5. YF17D is used as a vector in two licensed human vaccines, which were generated by swapping genes that encode the YF17D surface antigens for those of the Japanese encephalitis virus (for the Imojev vaccine) or dengue virus (for the Dengvaxia vaccine).
Vaccine design and rationale
YF17D is a small positive-sense single-stranded RNA live-attenuated virus with limited vector capacity, which tolerates some insertion of foreign antigens in the viral polyprotein6. Such insertions are constrained by (1) the topology and post-translational processing of the YF17D polyprotein; and (2) the need to express the antigen of interest in an immunogenic, probably natively folded form to yield a potent recombinant vaccine.
Using an advanced reverse genetics system, we generated a panel of YF17D-based candidate vaccines (YF-S) that express the S protein of SARS-CoV-2 in its native cleavable S1 and S2 subunits (hereafter, S1/2) form, its noncleavable S0 version or its S1 subdomain as in-frame fusions within the E and NS1 intergenic region of YF17D (which we termed YF-S1/2, YF-S0 and YF-S1, respectively) (Fig. 1a, Extended Data Fig. 1a). As outlined in ‘Full protection in hamsters’, we finally selected the YF-S0 variant as the lead vaccine candidate on the basis of its superior immunogenicity, efficacy and favourable safety profile.

a, Schematic of YF17D-based vaccine candidates (Extended Data Fig. 1). C, core or capsid protein. b, Syrian hamsters were immunized twice intraperitoneally with 103 PFU of each vaccine construct and inoculated with 2 × 105 median tissue-culture infectious dose (TCID50) SARS-CoV-2 (n = 12 from 2 independent experiments for all groups). μCT, micro-computed tomography. c, d, NAb (c) and total binding IgG (binding antibody (bAb)) (sera of three hamsters were pooled) (d) on day 21 after vaccination. Dashed line represents lower limit of quantification (LLOQ) (c) or lower limit of detection (LLOD) (d). e, Seroconversion rates at the indicated days. f, g, Viral loads in hamster lungs four days after infection quantified by quantitative PCR with reverse transcription (RT–qPCR) (f) and virus titration (g). h, Box plot showing NAbs before, and four days after, challenge; centre line represents the median, the lower and upper hinges represent the first and third quartiles, and the whiskers represent the minimum and maximum. i, Representative haematoxylin and eosin-stained images of sham- or YF-S0-vaccinated hamster lungs after challenge. Perivascular oedema (blue arrow); peribronchial inflammation (red arrows); perivascular inflammation (green arrow); bronchopneumonia (circle), apoptotic bodies in bronchial wall (red arrowhead). Scale bars, 100 μm. j, Spider web plot showing histopathological score for lung damage, normalized to sham treatment (grey). k, Heat map showing normalized RNA levels of antiviral and pro-inflammatory cytokine genes in lungs after challenge as determined by RT–qPCR relative to non-treated and non-infected controls (n = 4). IP-10 is also known as CXCL10. Data are median ± IQR. Two-tailed uncorrected Kruskal–Wallis test (c–g), or a two-tailed Wilcoxon matched-pairs rank test (h) was applied. NS, not significant.
Live attenuated viruses can be rescued by plasmid transfection into BHK-21 cells, which are an established substrate for the production of biological agents7 and suitable for vaccine production at industrial scale (Extended Data Fig. 2) when following the guidelines of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH)8. Infectious progeny from each recombinant construct showed a unique small-plaque phenotype as compared to parental YF17D (Extended Data Fig. 1b), consistent with some replicative trade-off posed by the inserted foreign sequences. We visualized the S or S1 and YF17D antigens intracellularly in YF-S-infected cells by staining with SARS-CoV-2 S- and YF17D-specific antibodies (Extended Data Fig. 1c). We further confirmed the proper expression and N-glycosylation9 of S or S1 by immunoblotting, with or without prior treatment with PNGase F (Extended Data Fig. 1d). The full-length S1/2 and S0 antigens that contain the original S2 subunit (stalk and cytoplasmic domains) of S may be expected to (1) form spontaneously trimers10,11,12 and (2) to be intracellularly retained (reinforced by C-terminal fusion to an extra transmembrane domain known to function as endoplasmic reticulum retention signal) (Extended Data Fig. 1a).
Consistent with a smaller plaque phenotype, intracranial inoculation in suckling mice13 confirmed the attenuation of the YF-S variants (Extended Data Fig. 3a, b). Mouse pups inoculated with 100 plaque-forming units (PFU) of parental YF17D stopped growing and succumbed to infection within seven days (median day of euthanasia), whereas pups inoculated with the YF-S variants continued to grow to a median day of euthanasia of 10, 12 and 17.5 days for YF-S1, YF-S1/2 and YF-S0, respectively. Thus, YF-S0 in particular has markedly reduced neurovirulence relative to YF17D. Akin classical YF17D potency testing13, the presence of some mortality confirms the viability and replication competence of the YF-S variants in vivo.
Likewise, YF-S0 is also highly attenuated in AG129 mice, which are highly susceptible to neuroinvasive YF17D infection14,15. Whereas intraperitoneal inoculation with 1 PFU of YF17D was uniformly lethal (a median day of euthanasia of 16 days), a 10,000× higher inoculum of YF-S0 resulted in only 1 out of 12 mice displaying disease (Extended Data Fig. 3c).
In humans, YF17D itself has an excellent safety profile4,16,17. However, in very rare cases, YF17D vaccination may result in viscerotropic disease16,17. These events are poorly understood4, but are probably linked to functional immune deficiencies (including deficiency in antiviral interferon responses)18. STAT2−/− hamsters2, which are deficient in type I and type III interferons, are particularly susceptible to infection with YF17D. When inoculated with 104 PFU of YF17D, 12 out of 14 STAT2−/− hamsters rapidly succumbed (a median day of euthanasia of 7 days; <15% survival) and presented with severe viscerotropic (haemorrhages in the liver) and neurotropic disease (paralysis) (Extended Data Fig. 3d). By contrast, >90% (12 out of 13) of STAT2−/− hamsters inoculated with the same dose of YF-S0 survived, without obvious viscerotropic disease and with some signs of neurological involvement in only 1 out of 13 hamsters18. As expected, in wild-type hamsters18 (Extended Data Fig. 3e, f) and in Ifnar−/− (Ifnar is also known as Ifnar1) mice19,20 (Extended Data Fig. 3g) YF-S0 was very well-tolerated at all of the doses we tested. Viraemia, a hallmark of viscerotropic disease in humans4, was observed in most of the hamsters vaccinated with YF17D and was often prolonged over several days; by contrast, inoculation with YF-S0 resulted only in 1 out of 6 hamsters at a single time point with any detectable vaccine virus RNA, which provides further evidence for YF-S0 being much less viscerotropic than YF17D (Extended Data Fig. 3e).
In summary, we generated a set of recombinant replication-competent YF17D variants (YF-S) that express different SARS-CoV-2 S antigens and that are notably less neurovirulent, neurotropic and viscerotropic than YF17D.
Full protection in hamsters
We assessed vaccine potency in a stringent hamster challenge model2. Hamsters were vaccinated at day 0 with a low dose13,21 of 103 PFU (via the intraperitoneal route18,22) of the different constructs or YF17D and sham (as negative controls), and boosted after 7 days (Fig. 1b). At day 21, all hamsters vaccinated with YF-S1/2 and YF-S0 had seroconverted to high levels of S-specific IgG and virus NAbs (Fig. 1c, d, Extended Data Fig. 4a) with log10-transformed geometric mean titres for YF-S1/2 of 3.2 (95% confidence interval of 2.9–3.5) for IgG and 1.4 (95% confidence interval of 1.1–1.9) for NAbs; and for YF-S0 of 3.5 (95% confidence interval of 3.3–3.8) for IgG and 2.2 (95% confidence interval of 1.9–2.6) for NAbs, with rapid seroconversion kinetics (Fig. 1e). Thus, in case of YF-S0, NAb levels exceeded those reported for hamsters after experimental infection with SARS-CoV-2 (by 2.5- to 7.5-fold)23,24,25. By contrast, only 1 out of 12 YF-S1-vaccinated hamsters seroconverted, and then only to low levels of NAbs. An adequate humoral immune response apparently depends on encountering the full-length S antigen.
After 23 or 28 days, hamsters were challenged intranasally with 2 × 105 PFU of SARS-CoV-2. Four days after infection, we detected high viral loads in the lungs of sham-vaccinated controls and hamsters vaccinated with YF17D as a matched placebo (Fig. 1f, g). Infection was characterized by severe lung pathology with multifocal necrotizing bronchiolitis, leukocyte infiltration and oedema, resembling findings in patients with COVID-19 who display severe bronchopneumonia (Fig. 1i, j, Extended Data Fig. 5a). By contrast, hamsters vaccinated with YF-S0 were protected against this aggressive challenge (Fig. 1f, g), with a median reduction of 5 log10-transformed viral RNA loads (interquartile range (IQR) of 4.5–5.4) in viral RNA loads (Fig. 1f), and of 5.3 log10-transformed virus titre (IQR of 3.9–6.3) for infectious virus in the lungs as compared to sham (Fig. 1g). Moreover, infectious virus was no longer detectable in 10 of 12 hamsters and viral RNA was reduced to nonquantifiable levels; RNA measured in the two remaining hamsters is equally likely to have represented residues of inoculum as viral RNA, as has previously been observed in nonhuman primate models26,27,28. Vaccination with YF-S0 also efficiently prevented systemic viral dissemination. In most hamsters, no or only very low levels of viral RNA were detectable in spleen, liver, kidney and heart four days after infection (Extended Data Fig. 4b). Similarly, a slightly different dose and schedule used for vaccination resulted in all vaccinated hamsters in, respectively, a 6 log10-transformed (IQR of 4.6–6.6) and 5.7 log10-transformed (IQR of 5.7–6.6) reduction of viral RNA and infectious virus titres as compared to sham (Extended Data Fig. 4f–i). Finally, vaccination with YF-S0 may induce saturating levels of NAbs, as in at least 4 out of 12 YF-S0-vaccinated hamsters no anamnestic NAb response (<2× increase) was observed after challenge (Fig. 1h, Extended Data Fig. 4c–e), similar to the original YF17D vaccination29,30. By contrast, in hamsters vaccinated with the second-best vaccine candidate (YF-S1/2), NAb levels further increased after SARS-CoV-2 infection (in 11 out of 12 hamsters) and approached a plateau only after challenge.
The lungs of YF-S0-vaccinated hamsters remained normal or near to normal with no further signs of bronchopneumonia, including (1) a reduction or lack of detectable lung pathology by histological inspection (Fig. 1i, Extended Data Fig. 5a); and (2) a considerable improvement of individual lung scores (Fig. 1j, Extended Data Fig. 5b–d) and marked reduction in nonaerated lung volume (Extended Data Fig. 5e), derived by micro-computed tomography of the chest relative to sham. In addition, immunization with YF-S0 resulted in an almost complete (in most cases full) normalization of cytokines—for example, IL-6, IL-10 and IFNγ—that are linked to disease exacerbation in COVID-19 (Fig. 1k, Extended Data Fig. 5f). Even the most sensitive markers of infection, such as the induction of antiviral type-III interferons (IFNλ) or of IFN-stimulated genes such as MX2 and IP-10, showed no elevation in YF-S0-vaccinated hamsters and remained similar to noninfected healthy controls.
Overall, YF-S0, which expresses a noncleavable S, outcompeted the YF-S1/2 construct that expresses the cleavable version of S; this argues for the stabilized form of S, probably presenting its trimeric prefusion conformation11,12,31, serving as the relevant protective antigen. Moreover, consistent with its failure to induce NAbs (Fig. 1c), the YF-S1 construct that expresses only the human ACE2 receptor-binding S1 domain (Extended Data Fig. 1d) did not confer any protection (Fig. 1f–j).
T helper 1 cell polarization of immunity in mice
As the tools do not exist for hamsters, we studied humoral and cell-mediated immune responses elicited by the different YF-S constructs in parallel in mice. Because YF17D does not readily replicate in wild-type mice32,33, we used Ifnar−/− mice, which are susceptible to vaccination with YF17D20,33,34.
We vaccinated mice with 400 PFU (of the YF-S variants, YF17D or sham) at day 0 and boosted 7 days later. S-specific IgG was detectable as early as 7 days after the first immunization (Fig. 2c). At day 21, all YF-S1/2- and YF-S0-vaccinated mice had seroconverted to high levels of S-specific IgG and NAbs with log10-transformed geometric mean titres of 3.5 (95% confidence interval of 3.1–3.9) for IgG and 2.2 (95% confidence interval of 1.7–2.7) for NAbs in the case of YF-S1/2, or 4.0 (95% confidence interval of 3.7–4.2) for IgG and 3.0 (95% confidence interval of 2.8–3.1) for NAbs for YF-S0 (Fig. 2a, b). The excess of IgG2b or IgG2c over IgG1 indicated a dominant pro-inflammatory and therefore antiviral T helper 1 (TH1) cell polarization of the immune response (Fig. 2d), as is desirable for protection against SARS-CoV-235,36,37. Similar to the situation in hamsters, YF-S1 did not induce SARS-CoV-2 NAbs (Fig. 2a, b). However, high and potentially protective immune responses to yellow fever virus (Extended Data Figs. 8a–d, g, 9) were induced by all constructs, confirming a consistent immunization.

Ifnar−/− mice were vaccinated twice (at day 0 and day 7) intraperitoneally with 400 PFU of each construct. a, b, NAbs (a) and binding antibodies (b) on day 21 after vaccination. c, Seroconversion rates at the indicated days. d, Ratios of IgG2b or IgG2c to IgG1, compared to a theoretical TH1 or TH2 cell response. In a–d, mice were vaccinated with YF-S1/2 (n = 11), YF-S0 (n = 11), YF-S1 (n = 13), sham (n = 9) or YF17D (n = 9); data from 3 independent experiments. For binding antibody quantification (b) and IgG subtyping (d), serum minipools from two or three mice were used. e, Number of IFNγ-secreting cells after SARS-CoV-2 S-peptide pool stimulation. Mice were vaccinated with YF-S1/2 (n = 11), YF-S0 (n = 10), YF-S1 (n = 7), sham (n = 9) or YF17D (n = 5). YF-S1 and YF17D from two, and YF-S1/2, YF-S0 and sham from three, independent experiments. f, Normalized mRNA expression levels of Tbx21, Gata3, Rorc and Foxp3 quantified by RT–qPCR in S-peptide-stimulated splenocytes. Data are expressed as fold change over median of uninfected controls (n = 5 YF-S1/2, YF-S0 and YF-S1, n = 7 sham and n = 3 uninfected controls from a single experiment). g, Percentage of IFNγ- and TNF-expressing CD8 cells, and IFNγ-expressing CD4 and γδ T cells, after S peptide stimulation. Mice were vaccinated with YF-S1/2 (n = 11), YF-S0 (n = 10), YF-S1 (n = 5), YF17D (n = 5) or sham (n = 8); YF-S1 and YF17D from a single experiment; YF-S1/2, YF-S0 and sham from three independent experiments. h, i, t-SNE of S-specific CD8 T cells positive for at least one intracellular marker (IFNγ, TNF or IL-4) after S-peptide stimulation (n = 6 for YF-S1/2 and YF-S0). i, Heat map of IFNγ expression density of S-specific CD8 T cells after YF-S1/2 and YF-S0 vaccination. Scale bar represents density of IFNγ-expressing cells; blue, low expression; red, high expression. Data in b–k are median ± IQR. Two-tailed uncorrected Kruskal–Wallis test (b, c, f–k) or a one-sample t-test (e) was applied.
To assess SARS-CoV-2-specific cell-mediated immune responses (which have a pivotal role in shaping and in the longevity of vaccine-induced immunity, as well as in the pathogenesis of COVID-1938,39), we analysed recall responses in splenocytes from vaccinated mice. In general, vaccination with any of the YF-S variants resulted in marked S-specific T cell responses with a favourable TH1 cell polarization, as detected by IFNγ ELISpot (Fig. 2e); this was further supported by an upregulation of T-BET (Tbx21), in particular in cells isolated from YF-S0-vaccinated mice. This cell-mediated immune profile was balanced by a concomitant elevation of GATA3 levels (Gata3) (which drives T helper 2 (TH2) cells), but no marked overexpression of RORγt (Rorc) (which drives T helper 17 (TH17)) or FOXP3 (Foxp3) (which drives regulatory T (Treg) cells) (Fig. 2f). In stark contrast to its failure to induce NAbs (Fig. 2a) or to protect (Fig. 1), YF-S1-vaccinated mice had a greater number of S-specific splenocytes than those vaccinated with YF-S1/2 or YF-S0 (Fig. 2e). Thus, even a vigorous cell-mediated immune response may not be sufficient for vaccine efficacy. In-depth profiling of the T cell compartment by flow cytometry confirmed the presence of S-specific IFNγ- and TNF-expressing CD8+ T lymphocytes, and of IFNγ-expressing CD4+ and γδ T lymphocytes (Fig. 2g, Extended Data Fig. 6a). A specific elevation of other markers such as IL-4, IL-17A or FOXP3 (reflecting TH2, TH17 and Treg cell polarization, respectively) was not observed, supported by t-distributed stochastic neighbour embedding (t-SNE) analysis of the respective T cell populations in YF-S1/2- and YF-S0-vaccinated mice (Fig. 2h; Extended Data Fig. 6b). Our analyses further revealed a similar composition for both of the CD4+ cell sets, comprising an equally balanced mixture of TH1 (IFNγ+ and/or TNF+) and TH2 (IL-4+) cells, and possibly a slight raise in TH17 cells for YF-S0. For YF-S1/2 and YF-S0, CD8+ T lymphocyte populations were dominated by IFNγ- or TNF-expressing cells, consistent with their transcriptional profiles (Fig. 2f). Of note, although similar in number, the YF-S0 and YF-S1/2 vaccines showed non-overlapping profiles regarding their CD8+ T lymphocyte populations: YF-S0 induced stronger IFNγ expression (Fig. 2h, i, Extended Data Fig. 6b). In summary, YF-S0 induces a vigorous and balanced cell-mediated immune response with a favourable TH1 cell polarization, dominated by SARS-CoV-2-specific CD8+ T cells that express high levels of IFNγ when encountering SARS-CoV-2 S antigen.
High levels of NAb in macaques
Six cynomolgus macaques were vaccinated with 105 PFU of YF-S0 (similar to a human dose for YF17D13,21 or YF17D-based recombinant vaccines40,41) via the subcutaneous route40,41 using the same schedule as in mice and hamsters. Six macaques received recombinant YF17D expressing an irrelevant control antigen as a matched placebo. No adverse signs or symptoms were observed. Macaques were bled weekly and assessed for seroconversion to NAb. At day 14 and day 21, all macaques vaccinated with YF-S0 had seroconverted to consistently high levels of virus NAbs (Fig. 3a), with geometric mean titres 2.6 (95% confidence interval of 2.4–2.8) and 2.5 (95% confidence interval of 2.3–2.7) respectively. These levels reach—if not exceed—those reported for other vaccine candidates12,27,28,42,43,44,45,46 (range of 0.3 to 2.6 log10-transformed geometric mean titres), and correlate with protection as confirmed by a reduction in SARS-CoV-2 RNA levels in YF-S0-vaccinated macaques upon challenge (Fig. 3b). Seroconversion occurred rapidly: at day 7 (following a single dose) 2 out of 6 macaques receiving YF-S0 already had SARS-CoV-2 NAbs. In addition, YF-S0 induced protective levels of NAbs against yellow fever virus4,29 (Extended Data Fig. 8e, f).

Twelve cynomolgus macaques (M. fascicularis) were immunized twice (at day 0 and day 7) subcutaneously with 105 PFU of YF-S0 (n = 6) or matched placebo (n = 6). On day 21 after vaccination, all macaques were challenged with 1.5 × 104 TCID50 SARS-CoV-2. a, NAbs on indicated days after first vaccination. Data are median ± IQR. b, Virus RNA loads in throat swabs at indicated time points, quantified by RT–qPCR. Different symbols (squares, triangle and diamond) indicate values for individual macaques followed over time with virus RNA loads above the lower limit of quantification. Histological examination of the lungs (day 21 after challenge) revealed no evidence of any SARS-CoV-2-induced pathology in macaques vaccinated with either YF-S0 or placebo. Two-tailed uncorrected Kruskal–Wallis test was applied.
Rapid protection after a single dose
Vaccination of hamsters using a single dose of YF-S0 induced high levels of NAbs and binding antibodies (Fig. 4a, b) in a dose- and time-dependent manner (for mice, see Extended Data Fig. 7a–d). This single-dose regimen resulted in protection against challenge with SARS-CoV-2, as demonstrated by absence of infectious virus in the lungs in 8 out of 8 hamsters (Fig. 4d) and a marked reduction in lung pathology scores (Fig. 4e, Extended Data Fig. 7e). Viral RNA at quantifiable levels was present in only 1 out of 8 hamsters (Fig. 4c). Protective immunity mounted rapidly. At 10 days after vaccination, 5 out of 8 hamsters that received 104 PFU of YF-S0 were already protected against challenge (Fig. 4c–e, Extended Data Fig. 7e). Notably, the persistence of NAbs and binding antibodies during long-term follow-up hints at a considerable longevity of immunity induced by this single-dose vaccination (Extended Data Fig. 7f, g).

Three groups of hamsters were vaccinated once intraperitoneally with sham (white) or YF-S0 at two different doses; 103 PFU (low) (green circles) and 104 PFU (high) (green triangles) of YF-S0 at 21 days before challenge. A fourth group was vaccinated with the high 104 PFU dose of YF-S0 at 10 days before challenge (green squares). Data are from n = 8 hamsters per group from a single experiment a, b, NAbs (a) and binding antibodies (n = 3; sera from 2 or 3 hamsters were pooled) (b) before challenge. c, d, Viral loads in lungs four days after infection, quantified by RT–qPCR (c) and virus titration (d). e, Spider web plot showing histopathological score for lung damage normalized to sham treatment (white circles). Data are median ± IQR. Two-tailed uncorrected Kruskal–Wallis test was applied.
Discussion
Vaccines against SARS-CoV-2 need to be safe and to result rapidly (ideally after one single dose) in long-lasting protective immunity. We report encouraging results from YF-S0, a YF17D-vectored SARS-CoV-2 vaccine candidate that induces robust immune responses in hamsters, mice and macaques. Because SARS-CoV-2 replicates extensively in the lungs of infected hamsters and results in major lung pathology2,23,24,25, we selected this model to assess vaccine efficacy. YF-S0 resulted in protection against stringent SARS-CoV-2 challenge that was comparable—if not more vigorous—to that of other vaccine candidates in nonhuman primate models12,27,28,42,43,44,45,46. At least in some of the YF-S0-vaccinated hamsters (4 out of 12), no anamnestic response in NAb levels after SARS-CoV-2 challenge was observed—this suggests that sterilizing immunity, similar to that conferred by YF17D vaccination29,30, can be achieved. In hamsters challenged 3 weeks after a single 104-PFU dose vaccination, no infectious virus was detected in the lungs. Considering the severity of the model, it is also notable that no infectious virus was recovered in several hamsters that were challenged 10 days after vaccination. A reduction of viral replication mitigated lung pathology, with normalization of biomarkers—such as IL-6—that are associated with infection and disease (Fig. 1, Extended Data Fig. 5). The vaccination of macaques with a relatively low subcutaneous dose of YF-S0 led to rapid seroconversion to high NAb titres (Fig. 3). It is tempting to speculate that this encouraging potency may translate into a simple one-shot dosing regimen for clinical use in humans.
Moreover, YF-S0 has a markedly improved safety profile over YF17D in several models (Extended Data Fig. 3), and is well-tolerated in hamsters (Extended Data Fig. 3) and nonhuman primates. Notably, updated WHO (World Health Organization) recommendations endorse the general use of YF17D in all people aged nine months or older who live in areas at risk47—including elderly individuals and persons with underlying medical conditions16,47. Our data therefore suggest that YF-S0 might also be safe for those persons who are most vulnerable to COVID-19. Cell-mediated immune responses studied in mice further revealed that YF-S0 favours a TH1 cell response, which is relevant as a skewed TH2 cell polarization may cause an induction and dysregulation of alternatively activated wound-healing monocytes and macrophages35,36,37 that results in an overshooting inflammatory response (the cytokine storm), which leads to acute lung injury38,48. No indication of such a disease enhancement or of antibody-dependent enhancement49 via Fcγ-receptor-mediated mechanisms50 was observed in any of our models.
In conclusion, YF-S0 confers vigorous protective immunity against infection with SARS-CoV-2. This immunity can be achieved within 10 days of a single-dose vaccination. In light of the threat that SARS-CoV-2 will remain endemic with spikes of reinfection, vaccines with this profile may be ideally suited for population-wide immunization programmes.
Methods
Cells and viruses
BHK-21J (baby hamster kidney fibroblasts) cells51 were provided by P. Bredenbeek and maintained in minimum essential medium (Gibco), Vero E6 (African green monkey kidney, ATCC CRL-1586) and HEK293T (human embryonic kidney cells, ATCC CRL-3216) cells were maintained in Dulbecco’s modified Eagle medium (DMEM) (Gibco). All media were supplemented with 10% fetal bovine serum (Hyclone), 2 mM l-glutamine (Gibco), 1% sodium bicarbonate (Gibco). BSR-T7/5 (T7 RNA polymerase expressing BHK-21)52 cells were provided by I. Goodfellow and kept in DMEM supplemented with 0.5 mg ml−1 geneticin (Gibco).
For all challenge experiments in hamsters, SARS-CoV-2 strain BetaCov/Belgium/GHB-03021/2020 (EPI ISL 407976|2020-02-03) was used from passage 4, grown on Vero E6 cells as previously described2. YF17D (Stamaril, Sanofi-Pasteur) was passaged twice in Vero E6 cells before use. All cells were regularly monitored for the absence of mycoplasma contamination.
Vaccine design and construction
Different vaccine constructs were generated using an infectious cDNA clone of YF17D (in an inducible BAC expression vector pShuttle-YF17D, patent number WO2014174078 A1)34,53,54. A panel of several SARS-CoV-2 vaccine candidates was engineered by inserting a codon-optimized sequence of either the SARS-CoV-2 S protein (GenBank: MN908947.3) or variants thereof into the full-length genome of YF17D (GenBank: X03700) as translational in-frame fusion within the yellow fever virus E/NS1 intergenic region6,55. The variants generated contained (i) either the S protein sequence from amino acids 14–1273, expressing S in its cleavable and/or non-cleavable form or version (YF-S1/2 and YF-S0, respectively), or (ii) its subunit S1 (amino acids 14–722) (YF-S1). To ensure a proper yellow fever virus topology and correct expression of different S antigens in the yellow fever virus backbone, transmembrane domains derived from West Nile virus were inserted.
The SARS2-CoV-2 vaccine candidates were cloned by combining the S cDNA (obtained after PCR on overlapping synthetic cDNA fragments; IDT) by a NEB Builder Cloning kit (New England Biolabs) into the pShuttle-YF17D backbone. NEB Builder reaction mixtures were transformed into Escherichia coli EPI300 cells (Lucigen) and correct integration of the S protein cDNA was confirmed by Sanger sequencing. Recombinant plasmids were purified by column chromatography (Nucleobond Maxi Kit, Machery-Nagel) after growth overnight, followed by an additional amplification of the BAC vector for 6 h by addition of 2 mM l-arabinose, as previously described34.
Infectious vaccine viruses were generated from plasmid constructs by transfection into BHK-21J cells using standard protocols (TransIT-LT1, Mirus Bio). The supernatant was collected four days after transfection, when most of the cells showed signs of cytopathic effect. Infectious virus titres (PFU ml−1) were determined by a plaque assay on BHK-21J cells, as previously described15,34. The presence of inserted sequences in generated vaccine virus stocks was confirmed by RNA extraction and DNase I treatment (Direct-zol RNA kit, Zymo Research) followed by RT–PCR (qScript XLT, Quanta) and Sanger sequencing, and by immunoblotting of extracts of freshly infected cells (as described in ‘Immunoblot analysis’). YF-S0 vaccine virus stock used for macaque studies was concentrated by tangential flow filtration (Minimate, Pall), and subjected to deep sequencing on a MiSeq platform (Illumina) following an established metagenomics pipeline56,57 or quality control (identity, consistency and genetic homogenicity) and to confirm the absence of adventitious agents, as previously described2.
Analysis of genetic stability of the YF-S0 vaccine virus
To test the genetic stability of the YF-S0 vaccine virus, virus supernatants recovered from transfected BHK-21J cells (P0) were plaque-purified once (P1) and serially passaged on BHK-21J cells (P3–P6). Furthermore, the genetic stability of 25 plaque isolates from a second round of plaque purification were analysed after amplification (P4*).
For all passages, fresh BHK-21J cells were infected for 1 h with a 1:2 dilution of the virus supernatant from the respective previous passage. After infection, the cells were washed twice with PBS. Supernatants of the infected cells were routinely collected 72 h after infection. The presence of inserted sequences in generated passages was confirmed by RNA extraction and Dnase I treatment (Direct-zol RNA kit, Zymo Research) followed by RT–PCR (qScript XLT, Quanta) and Sanger sequencing, and by immunoblotting of freshly infected cells (as described in ‘Immunoblot analysis’).
Immunofluorescent staining
In vitro antigen expression of vaccine candidates was verified by immunofluorescent staining as previously described34. In brief, BHK-21J cells were infected with 100 PFU of the YF-S vaccine candidates. Infected cells were stained three days after infection. For detection of yellow fever virus antigens, polyclonal mouse anti-YF17D antiserum was used. For detection of SARS-CoV-2 S antigen, rabbit SARS-CoV S1 antibody (40150-RP01, Sino Biological; 1:250 dilution) and rabbit SARS-CoV S primary antibody (40150-T62-COV2, Sino Biological; 1:250 dilution) were used. Secondary antibodies were goat anti-mouse Alexa Fluor 594 and goat anti-rabbit Alexa Fluor 488 (Life Technologies; 1:500 dilution). Cells were counterstained with DAPI (Sigma). All confocal fluorescent images were acquired using the same settings on a Leica TCS SP5 confocal microscope, using a HCX PL APO 63× (NA 1.2) water immersion objective.
Immunoblot analysis
Infected BHK-21J cells were collected and washed once with ice-cold phosphate buffered saline, and lysed in radio-immunoprecipitation assay buffer (Thermo Fisher Scientific) containing 1× protease inhibitor and phosphatase inhibitor cocktail (Thermo Fisher Scientific). After centrifugation at 15,000 rpm at 4 °C for 10 min, protein concentrations in the cleared lysates were measured using BCA (Thermo Fisher Scientific). Immunoblot analysis was performed by a simple western size-based protein assay (Protein Simple) following manufactures instructions. In brief, after loading of 400 ng of total protein onto each capillary, specific S protein levels were identified using specific primary antibodies (NB100-56578, Novus Biologicals and 40150-T62-CoV2, Sino Biological; both 1:100 diluted), and HRP-conjugated secondary antibody (Protein Simple; 1:100 dilution). Chemiluminescence signals were analysed using Compass software (Protein Simple). To evaluate the removal of N-linked oligosaccharides from the glycoprotein, protein extracts were treated with PNGase F according to manufactures instructions (New England Biolabs).
Animals
Hamsters and mice
Wild-type Syrian hamsters (M. auratus) and BALB/c mice and pups were purchased from Janvier Laboratories. Type-I-interferon-receptor-deficient (Ifnar1−/−) mice58, type-I-and-II-interferon-receptor-deficient AG12959 and STAT2−/− hamster were bred in-house. The generation of STAT2−/− hamsters (gene identifier: 101830537) has previously been described60. Functional ablation of type I and III signalling has been confirmed2. STAT2−/− hamsters have previously been demonstrated to be particularly susceptible to flavivirus infection61.
Six- to ten-week-old male and female Ifnar−/− mice, six- to eight-week-old male and female AG129 mice and six- to eight-week-old female wild-type hamsters were used throughout the study. For vaccine safety studies, equal amounts of age-matched male and female wild-type, as well as STAT2−/− hamsters, were used.
Macaques
Twelve outbred mature male cynomolgus macaques (M. fascicularis) were used in this study. Macaques were purpose-bred and housed at the Biomedical Primate Research Centre (BPRC). All macaques selected for the study were in good physical health with normal baseline biochemical and haematological values.
Animal experiments
Hamsters and mice were housed in individually ventilated cages (Sealsafe Plus, Tecniplast), per 5 (for mice) (cage type GM500) or per 2 (for hamsters) (cage type GR900), at 21 °C, 55% humidity and 12:12 light:dark cycles. Hamsters and mice were provided with food and water ad libitum, as well as cotton and cardboard play tunnels (mice) or extra bedding material and wooden gnawing blocks (hamsters). This project was approved by the KU Leuven ethical committee (P015-2020), following institutional guidelines approved by the Federation of European Laboratory Animal Science Associations (FELASA). Hamsters and mice were euthanized by intraperitoneal administration of 100 μl (mice) or 500 μl (hamsters) Dolethal (200 mg ml−1 sodium pentobarbital, Vétoquinol SA).
For mice and hamsters, sample sizes were chosen based on the results of pilot experiments. Pivotal studies have been performed in at least two independent biological repeats. In the case of macaques, statistical power calculations considered the number of macaques required to detect significant induction of immune responses compared to nonvaccinated controls. With groups of n = 6, a vaccine efficacy >85% can be demonstrated (α = 0.05, β = 0.2 (power = 80%), normal distribution). Allocation of experimental groups was done randomly. Analysis of animal samples was performed blinded, except for ELISpot (machine-based counting), flow cytometry (gates defined on negative control samples, identical gates for all groups) and survival data (predefined humane end points).
Immunization and infection of hamsters
Hamsters were intraperitoneally vaccinated with the indicated amount of PFUs of the vaccine constructs using a prime and boost regimen (at day 0 and day 7). As a control, two groups were vaccinated at day 0 and day 7 with either 103 PFU of YF17D or with MEM containing 2% FBS (sham). All hamsters were bled at day 21 to analyse serum for binding and neutralizing antibodies against SARS-CoV-2. At the indicated time after vaccination and before challenge, hamsters were anaesthetized by intraperitoneal injection of a xylazine (16 mg kg−1, XYL-M, V.M.D.), ketamine (40 mg kg−1, Nimatek, EuroVet) and atropine (0.2 mg kg−1, Sterop) solution. Each hamster was inoculated intranasally by gently adding 50 μl droplets of virus stock containing 2 × 105 TCID50 of SARS-CoV-2 in both nostrils. Hamsters were monitored daily for signs of disease (lethargy, heavy breathing or ruffled fur). Four days after challenge, all hamsters were euthanized to collect end sera and lung tissue in RNAlater, MEM or formalin for gene-expression profiling, virus titration or histopathological analysis, respectively. To determine the viraemia profile of the vaccine virus, a subset of hamsters was vaccinated with 104 PFU YF17D or YF-S0 intraperitoneally and bled on days 1, 2, 3, 4, 5, 7, 9, 11, 15 and 29 to measure serum viral load.
Immunization and infection of mice
Ifnar1−/− mice were intraperitoneally vaccinated with vaccine constructs by using a prime and boost of each 4 × 102 PFU (at day 0 and day 7). As a control, two groups were vaccinated (at day 0 and day 7) with either YF17D or sham. All mice were bled weekly and serum was separated by centrifugation for indirect immunofluorescence assay (IIFA) and serum neutralization test (SNT). Three weeks after the first vaccination, mice were euthanized, spleens were collected for ELISpot cytokine detection, transcription-factor analysis by RT–qPCR and intracellular cytokine staining (ICS). For yellow fever challenge, mice were inoculated with a lethal dose of YF17D via the intracranial route, as previously described14,34. In brief, three weeks after intraperitoneal vaccination with 4 × 102 PFU of YF-S0, YF17D or sham, mice were anaesthetized and inoculated intracranially with 30 μl containing 3 × 103 PFU of YF17D, and then monitored daily for signs of disease and weight change for 4 weeks. Sick mice were euthanized on the basis of morbidity (hind limb paralysis, weakness and ruffled fur) or weight loss of more than 25%.
Vaccine safety testing in suckling mice, AG129 mice and STAT2 −/− hamsters
To evaluate neurovirulence and neurotropism, BALB/c mice pups and AG129 mice were, respectively, intracranially or intraperitoneally inoculated with the indicated PFU amount of YF17D and YF-S vaccine constructs and monitored daily for morbidity and mortality for 21 days after inoculation. To study viscerotropic disease, STAT2−/− hamsters were inoculated for highest exposure via the intraperitoneal route with 104 PFU of YF17D or YF-S0 and followed daily for 21 days for well-being. Euthanasia was performed if the following occurred: total weight loss of >20%; day-to-day weight loss of >10%; clear signs of paralysis; signs of distress (laboured breathing, ruffled hair, abnormal or hunched posture, severe agitation or depression).
Immunization and infection challenge of cynomolgus macaques
All housing and animal procedures took place at the BPRC, upon positive advice by the independent ethics committee (DEC-BPRC), under project licence AVD5020020209404 issued by the Central Committee for Animal Experiments, and following approval of the detailed study protocol by the institutional animal welfare body. All animal handlings were performed within the Department of Animal Science according to Dutch law, regularly inspected by the responsible national authority (Nederlandse Voedsel- en Warenautoriteit, NVWA), and the animal welfare body.
Macaques were pair-housed with a socially compatible cage-mate and randomly assigned to two groups. Six (n = 6) cynomolgus macaques vaccinated subcutaneously in the inner upper limbs using a dose of 105 PFU of YF-S0 at days 0 (prime) and 7 (boost). As a control, n = 6 macaques were vaccinated twice with 105 PFU of a matched placebo vaccine, consisting of recombinant YF17D with an irrelevant control antigen with no sequence homology to SARS-CoV-2 inserted in the same location (E/NS1 junction).
A temperature monitor was implanted in the abdominal cavity of each macaque three weeks before the start of the study (Anapill DSI) providing continuous real-time measurement of body temperature and activity. Health was checked daily and macaques monitored for appetite, general behaviour and stool consistency. Blood was collected for regular assessment of whole blood counts and clinical chemistry with no changes out of normal ranges detected.
On day 21 after vaccination, all macaques were challenged by a combined intranasal–intratracheal inoculation with nominally 1.5 × 104 TCID50 of SARS-CoV-2 (as determined by back titration on Vero cells) in total volume 5 ml; split over the trachea (4 ml) and nares (0.25 ml each). The resulting virus RNA loads were quantified in throat swabs using RT–qPCR as described with a lower limit of detection of 200 RNA copies per ml62. After a follow-up for 21 days, macaques were euthanized for histological analysis of their lungs26.
SARS-CoV-2 and yellow fever virus RT–qPCR
The presence of infectious SARS-CoV-2 particles in hamster lung homogenates was quantified by RT–qPCR2. In brief, for quantification of viral RNA levels and gene expression after challenge, RNA was extracted from homogenized organs using the NucleoSpin Kit Plus (Macherey-Nagel), following the manufacturer’s instructions. Reactions were performed using the iTaq Universal Probes One-Step RT–qPCR kit (BioRad), with primers and probes (Integrated DNA Technologies) listed in Supplementary Table 1. The relative RNA fold-change was calculated with the 2−ΔΔCq method63 using housekeeping gene β-actin for normalization. For detection of vaccine virus in blood, RNA from 50 μl of serum was extracted with the NucleoSpin RNA virus kit (Macherey-Nagel). Primers and probe were derived from the yellow fever virus nonstructural gene 3, and are shown in Supplementary Table 1. RT–qPCR was performed using the ABI 7500 Fast Real-Time PCR System (Applied Biosystems). For absolute quantification, standard curves were generated using fivefold dilutions of a cDNA plasmid template (plasmid pShuttle/YFV-17D14,34) of known concentration. On the basis of repeated standard curves, the lower limit of detection was established at 8,900 copies per ml, corresponding to a Ct value of 35. Ct values above 35 were considered below the limit of detection and represented as the square root of the lower limit of detection.
End-point virus titrations
To quantify infectious SARS-CoV-2 particles, end-point titrations were performed on confluent Vero E6 cells in 96-well plates. Lung tissues were homogenized using bead disruption (Precellys) in 250 μl minimal essential medium and centrifuged (10,000 rpm, 5 min, 4 °C) to pellet the cell debris. Viral titres were calculated by the Reed and Muench method64 and expressed as TCID50 per mg tissue.
Histology
For histological examination, lung tissues (for macaques, after inflation with 10% neutral-buffered formalin) were fixed in 4% paraformaldehyde (hamsters) or 10% neutral-buffered formalin (macaques) and embedded in paraffin. Tissue sections (5 μm for hamsters, 3 μm for macaques) were stained with haematoxylin and eosin and analysed for lung damage by an expert pathologist.
Micro-computed tomography and image analysis
To monitor the development of lung pathology after challenge with SARS-CoV-2, hamsters were imaged using an X-cube micro-computed tomography scanner (Molecubes) as previously described2. Quantification of reconstructed micro-computed tomography data was performed with DataViewer and Ctan software (Bruker Belgium). A semiquantitative scoring of micro-computed tomography data was performed as primary outcome measure and imaging-derived biomarkers (nonaerated lung volume) as secondary measures, as previously described2,65,66,67,68.
Detection of total binding IgG and IgG isotyping by IIFA
To detect SARS-CoV-2-specific antibodies in hamster and mouse serum, an in-house-developed IIFA was used. Using CRISPR–Cas9, a CMV-SARS-CoV-2-Spike-Flag-IRES-mCherry-P2A-BlastiR cassette was stably integrated into the ROSA26 safe harbour locus of HEK293T cells69. To determine SARS-CoV-2 S binding antibody end titres, 1/2 serial serum dilutions were made in 96-well plates on HEK293T-spike stable cells and HEK293T wild-type cells in parallel. Goat-anti-mouse IgG Alexa Fluor 488 (A11001, Life Technologies; 1:250 dilution), goat-anti-mouse IgG1, IgG2b and IgG2c Alexa Fluor 488 (respectively 115-545-205, 115-545-207 and 115-545-208 from Jackson ImmunoResearch; all 1:250 diluted) were used as secondary antibody. After counterstaining with DAPI, fluorescence in the blue channel (excitation at 386 nm) and the green channel (excitation at 485 nm) was measured with a Cell Insight CX5 High Content Screening platform (Thermo Fischer Scientific). Specific SARS2-CoV-2 S staining is characterized by cytoplasmic (endoplasmic reticulum) enrichment in the green channel. To quantify this specific SARS-CoV-2 S staining, the difference in cytoplasmic and nuclear signal for the HEK293T wild-type conditions was subtracted from the difference in cytoplasmic and nuclear signal for the HEK293T SARS-CoV-2 S conditions. All positive values were considered as specific SARS-CoV-2 staining. The IIFA end titre of a sample is defined as the highest dilution that scored positive in this way. Because of the limited volume of serum, IIFA end titres for most conditions (unless indicated otherwise) were determined on minipools of two or three samples.
SARS-CoV-2 pseudotyped virus and YF17D virus SNT
SARS-CoV-2 VSV pseudotypes were generated as previously described70,71,72. In brief, HEK-293T cells were transfected with a pCAGGS-SARS-CoV-2Δ18-Flag expression plasmid encoding SARS-CoV-2 S protein carrying a C-terminal 18-amino-acid deletion73,74. One day after transfection, cells were infected with VSVΔG expressing a green fluorescent protein (GFP) reporter gene (multiplicity of infection of 2) for 2 h. The medium was changed with medium containing anti-VSV-G antibody (I1, mouse hybridoma supernatant from CRL-2700; ATCC) to neutralize any residual VSV-G virus input75. Twenty-four hours later, supernatant containing SARS-CoV-2 VSV pseudotypes was collected.
To quantify SARS-CoV-2 NAbs, serial dilutions of serum samples were incubated for 1 h at 37 °C with an equal volume of SARS-CoV-2 pseudotyped VSV particles and inoculated on Vero E6 cells for 18 h.
Neutralizing titres (SNT50) for yellow fever virus were determined with an in-house-developed fluorescence-based assay using a mCherry-tagged variant of YF17D virus34,54. To this end, serum dilutions were incubated in 96-well plates with the YF17D–mCherry virus for 1 h at 37 °C, after which serum–virus complexes were transferred for 72 h to BHK-21J cells. The percentage of GFP- or mCherry-expressing cells was quantified on a Cell Insight CX5/7 High Content Screening platform (Thermo Fischer Scientific) and neutralization half-maximal inhibitory concentration values were determined by fitting the serum neutralization dilution curve that is normalized to a virus (100%) and cell control (0%) in Graphpad Prism (GraphPad Software).
SARS-CoV-2 and YF17D plaque reduction neutralization test
Sera were serially diluted with an equal volume of 70 PFU of SARS-CoV-2 or 40 PFU of YF17D before incubation at room temperature for 1 h. Serum-virus complexes were added to Vero E6 (SARS-CoV-2) or BHK-21J (YF17D) cell monolayers in 24-well plates and incubated at 37 °C for 1 h. Three days later, overlays were removed and stained with 0.5% crystal violet after fixation with 3.7% PFA. Neutralization titres (PRNT50) of the test serum samples were defined as the reciprocal of the highest test serum dilution resulting in a plaque reduction of at least 50%.
Antigens for T cell assays
PepMix yellow fever (NS4B) (JPT-PM-YF-NS4B) and subpool 1 (158 overlapping 15-mers) of PepMix SARS-CoV-2 S (JPT-PM-WCPV-S-2) were used as recall antigens for ELISpot and ICS. Diluted Vero E6 cell lysate (50 μg ml−1) and a combination of PMA (50 ng ml−1) (Sigma-Aldrich) and ionomycin (250 ng ml−1) (Sigma-Aldrich) served as negative and positive control, respectively.
ICS and flow cytometry
Fresh mouse splenocytes were incubated with 1.6 μg ml−1 yellow fever NS4B peptide mixture; 1.6 μg ml−1 S peptide subpool 1; PMA (50 ng ml−1); ionomycin (250 ng ml−1) or 50 μg ml−1 Vero E6 cells for 18 h at 37 °C. After treatment with brefeldin A (Biolegend) for 4 h, the splenocytes were stained for viability with Zombie Aqua Fixable Viability Kit (Biolegend) and Fc receptors were blocked by the mouse FcR blocking reagent (Miltenyi Biotec)(0.5 μl per well) for 15 min in the dark at room temperature. Cells were then stained with extracellular markers BUV395 anti-CD3 (17A2; 1:167 dilution) (BD), BV785 anti-CD4 (GK1.5; 1:100 dilution) (Biolegend), APC/cyanine7 anti-CD8 (53-6.7; 1:100 dilution) (Biolegend) and PerCP/cyanine5.5 anti-TCR γδ (GL3; 1:67 dilution) (Biolegend) in brilliant stain buffer (BD) before incubation on ice for 25 min. Cells were washed once with PBS and fixed and permeabilized for 30 min by using the FOXP3 transcription factor buffer kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Finally, cells were stained with following antibodies: PE anti-IL-4 (11B11; 1:50 dilution), APC anti-IFNγ (XMG1.2; 1:100 dilution), PE/Dazzle 594 anti-TNF (MP6-XT22; 1:50 dilution), Alexa Fluor 488 anti-FOXP3 (MF-14; 1:20 dilution) and Brilliant Violet 421 anti-IL-17A (TC11-18H10.1; 1:50 dilution) (all from Biolegend) and acquired on a BD LSRFortessa X-20 (BD). All measurements were calculated by subtracting from nonstimulated samples (incubated with noninfected Vero E6 cell lysates) from corresponding stimulated samples. The gating strategy used for ICS analysis is depicted in Extended Data Fig. 6a. The strategy used for comparative expression profiling of vaccine-induced T cell populations by t-SNE analysis is outlined in Extended Data Fig. 6b. All flow cytometry data were analysed using FlowJo Version 10.6.2 (LLC)). t-SNE plot was generated in Flowjo after concatenating spike-specific CD4 and CD8 T cell separately based on gated splenocyte samples.
ELISpot
ELISpot assays for the detection of IFNγ-secreting mouse splenocytes were performed with mouse IFNγ kit (ImmunoSpot MIFNG-1M/5, CTL Europe). IFNγ spots were visualized by stepwise addition of a biotinylated detection antibody, a streptavidin-enzyme conjugate and the substrate. Spots were counted using an ImmunoSpot S6 Universal Reader (CTL Europe) and normalized by subtracting spots numbers from control samples (incubated with non-infected Vero E6 cell lysates) from the spot numbers of corresponding stimulated samples. Negative values were corrected to zero.
RT–qPCR for transcription factor profile
S-peptide-stimulated splenocytes were used for RNA extraction by using the NucleoSpin Kit Plus kit (Macherey-Nagel). cDNA was generated by using a high-capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Real-time PCR was performed using the TaqMan gene expression assay (Applied Biosystems) on an ABI 7500 fast platform. Expression levels of Tbx21, Gata3, Rorc and Foxp3 (all from Integrated DNA Technologies (IDT)) were normalized to the expression of GAPDH (IDT). Relative gene expression was assessed by using the 2−ΔΔCq method.
Statistical analysis
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment. GraphPad Prism (GraphPad Software) was used for all statistical evaluations. The number of animals and independent experiments that were performed is indicated in the figure legends. Statistical significance was determined using the non-parametric Mann–Whitney U-test and Kruskal–Wallis test if not otherwise stated. Values were considered significantly different at P ≤ 0.05.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The SARS-CoV-2 strain BetaCov/Belgium/GHB-03021/2020 sequence is available from GISAID (EPI ISL 407976|2020-02-03) (https://www.gisaid.org). The prototypic Wuhan-Hu-1 2019-nCoV sequence is available from GenBank (accession number MN908947.3). The full-length YF17D sequence is available from GenBank (accession number X03700). Flow cytometry data that support the findings in this study are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
WHO. Draft landscape of COVID-19 candidate vaccines, https://www.who.int/who-documents-detail/draft-landscape-of-covid-19-candidate-vaccines (accessed 4 November 2020).
Boudewijns, R. et al. STAT2 signaling restricts viral dissemination but drives severe pneumonia in SARS-CoV-2 infected hamsters. Nat. Commun. 11, 5838 (2020).
Cao, Y. et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 182, 73–84.e16 (2020).
Barrett, A. D. & Teuwen, D. E. Yellow fever vaccine – how does it work and why do rare cases of serious adverse events take place? Curr. Opin. Immunol. 21, 308–313 (2009).
Draper, S. J. & Heeney, J. L. Viruses as vaccine vectors for infectious diseases and cancer. Nat. Rev. Microbiol. 8, 62–73 (2010).
Bonaldo, M. C., Sequeira, P. C. & Galler, R. The yellow fever 17D virus as a platform for new live attenuated vaccines. Hum. Vaccin. Immunother. 10, 1256–1265 (2014).
Afonja, O. et al. Baby hamster kidney cell-derived recombinant factor VIII: a quarter century of learning and clinical experience. Expert Rev. Hematol. 9, 1151–1164 (2016).
ICH. Quality guidelines for pharmaceutical quality based on Good Manufacturing Practice (GMP); Quality of Biotechnological Products (Q5A–Q5E), https://www.ich.org/page/quality-guidelines (accessed 4 November 2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e6 (2020).
Du, L. et al. The spike protein of SARS-CoV—a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 7, 226–236 (2009).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Mercado, N. B. et al. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature 586, 583–588 (2020).
Ferguson, M., Shin, J., Knezevic, I., Minor, P. & Barrett, A. WHO Working Group on technical specifications for manufacture and evaluation of yellow fever vaccines, Geneva, Switzerland, 13–14 May 2009. Vaccine 28, 8236–8245 (2010).
Kum, D. B. et al. Limited evolution of the yellow fever virus 17d in a mouse infection model. Emerg. Microbes Infect. 8, 1734–1746 (2019).
Mishra, N. et al. A chimeric Japanese encephalitis vaccine protects against lethal yellow fever virus infection without inducing neutralizing antibodies. MBio 11, e02494-19 (2020).
Thomas, R. E., Lorenzetti, D. L., Spragins, W., Jackson, D. & Williamson, T. The safety of yellow fever vaccine 17D or 17DD in children, pregnant women, HIV+ individuals, and older persons: systematic review. Am. J. Trop. Med. Hyg. 86, 359–372 (2012).
Rafferty, E., Duclos, P., Yactayo, S. & Schuster, M. Risk of yellow fever vaccine-associated viscerotropic disease among the elderly: a systematic review. Vaccine 31, 5798–5805 (2013).
Mateo, R. I. et al. Yellow fever 17-D vaccine is neurotropic and produces encephalitis in immunosuppressed hamsters. Am. J. Trop. Med. Hyg. 77, 919–924 (2007).
Erickson, A. K. & Pfeiffer, J. K. Spectrum of disease outcomes in mice infected with YFV-17D. J. Gen. Virol. 96, 1328–1339 (2015).
Watson, A. M., Lam, L. K., Klimstra, W. B. & Ryman, K. D. The 17D–204 vaccine strain-induced protection against virulent yellow fever virus is mediated by humoral immunity and CD4+ but not CD8+ T cells. PLoS Pathog. 12, e1005786 (2016).
Martins, R. M. et al. 17DD yellow fever vaccine: a double blind, randomized clinical trial of immunogenicity and safety on a dose-response study. Hum. Vaccin. Immunother. 9, 879–888 (2013).
Tesh, R. B., Travassos da Rosa, A. P., Guzman, H., Araujo, T. P. & Xiao, S. Y. Immunization with heterologous flaviviruses protective against fatal West Nile encephalitis. Emerg. Infect. Dis. 8, 245–251 (2002).
Chan, J. F.-W. et al. Simulation of the clinical and pathological manifestations of coronavirus disease 2019 (COVID-19) in golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin. Infect. Dis. 71, 2428–2446 (2020).
Sia, S. F. et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 583, 834–838 (2020).
Imai, M. et al. Syrian hamsters as a small animal model for SARS-CoV-2 infection and countermeasure development. Proc. Natl Acad. Sci. USA 117, 16587–16595 (2020).
Rockx, B. et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 368, 1012–1015 (2020).
van Doremalen, N. et al. ChAdOx1 nCoV-19 vaccine prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature 586, 578–582 (2020).
Yu, J. et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science 369, 806–811 (2020).
Staples, J. E., Barrett, A. D. T., Wilder-Smith, A. & Hombach, J. Review of data and knowledge gaps regarding yellow fever vaccine-induced immunity and duration of protection. npj Vaccines 5, 54 (2020).
Casey, R. M. et al. Immunogenicity of fractional-dose vaccine during a yellow fever outbreak - final report. N. Engl. J. Med. 381, 444–454 (2019).
Barnes, C. O. et al. Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell 182, 828–842.e16 (2020).
Meier, K. C., Gardner, C. L., Khoretonenko, M. V., Klimstra, W. B. & Ryman, K. D. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 5, e1000614 (2009).
Erickson, A. K. & Pfeiffer, J. K. Dynamic viral dissemination in mice infected with yellow fever virus strain 17D. J. Virol. 87, 12392–12397 (2013).
Kum, D. B. et al. A yellow fever–Zika chimeric virus vaccine candidate protects against Zika infection and congenital malformations in mice. npj Vaccines 3, 56 (2018).
Channappanavar, R. et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 19, 181–193 (2016).
Channappanavar, R. & Perlman, S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 39, 529–539 (2017).
Page, C. et al. Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. J. Virol. 86, 13334–13349 (2012).
Grifoni, A. et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 181, 1489–1501.e15 (2020).
Ni, L. et al. Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity 52, 971–977.e3 (2020).
Appaiahgari, M. B. & Vrati, S. IMOJEV®: a yellow fever virus-based novel Japanese encephalitis vaccine. Expert Rev. Vaccines 9, 1371–1384 (2010).
Guy, B. et al. A recombinant live attenuated tetravalent vaccine for the prevention of dengue. Expert Rev. Vaccines 16, 671–684 (2017).
Gao, Q. et al. Development of an inactivated vaccine candidate for SARS-CoV-2. Science 369, 77–81 (2020).
Wang, H. et al. Development of an inactivated vaccine candidate, BBIBP-CorV, with potent protection against SARS-CoV-2. Cell 182, 713–721.e9 (2020).
Yang, J. et al. A vaccine targeting the RBD of the S protein of SARS-CoV-2 induces protective immunity. Nature 586, 572–577 (2020).
Feng, L. et al. An adenovirus-vectored COVID-19 vaccine confers protection from SARS-COV-2 challenge in rhesus macaques. Nat. Commun. 11, 4207 (2020).
Erasmus, J. H. et al. An Alphavirus-derived replicon RNA vaccine induces SARS-CoV-2 neutralizing antibody and T cell responses in mice and nonhuman primates. Sci. Transl. Med. 12, eabc9396 (2020).
WHO. International Travel and Health. Yellow fever, https://www.who.int/ith/vaccines/yf/en/ (accessed 4 November 2020).
Lambert, P. H. et al. Consensus summary report for CEPI/BC March 12-13, 2020 meeting: assessment of risk of disease enhancement with COVID-19 vaccines. Vaccine 38, 4783–4791 (2020).
Liu, L. et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 4, e123158 (2019).
Smatti, M. K., Al Thani, A. A. & Yassine, H. M. Viral-induced enhanced disease illness. Front. Microbiol. 9, 2991 (2018).
Lindenbach, B. D. & Rice, C. M. trans-Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J. Virol. 71, 9608–9617 (1997).
Buchholz, U. J., Finke, S. & Conzelmann, K. K. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73, 251–259 (1999).
Dallmeier, K. & Neyts, J. Simple and inexpensive three-step rapid amplification of cDNA 5′ ends using 5′ phosphorylated primers. Anal. Biochem. 434, 1–3 (2013).
Sharma, S. et al. Small-molecule inhibitors of TBK1 serve as an adjuvant for a plasmid-launched live-attenuated yellow fever vaccine. Hum. Vaccin. Immunother. 16, 2196–2203 (2020).
Bredenbeek, P. J. et al. A recombinant yellow fever 17D vaccine expressing Lassa virus glycoproteins. Virology 345, 299–304 (2006).
Conceição-Neto, N. et al. Modular approach to customise sample preparation procedures for viral metagenomics: a reproducible protocol for virome analysis. Sci. Rep. 5, 16532 (2015).
Conceição-Neto, N., Yinda, K. C., Van Ranst, M. & Matthijnssens, J. NetoVIR: modular approach to customize sample preparation procedures for viral metagenomics. Methods Mol. Biol. 1838, 85–95 (2018).
Müller, U. et al. Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921 (1994).
van den Broek, M. F., Müller, U., Huang, S., Zinkernagel, R. M. & Aguet, M. Immune defence in mice lacking type I and/or type II interferon receptors. Immunol. Rev. 148, 5–18 (1995).
Fan, Z. et al. Efficient gene targeting in golden Syrian hamsters by the CRISPR/Cas9 system. PLoS ONE 9, e109755 (2014).
Siddharthan, V. et al. Zika virus infection of adult and fetal STAT2 knock-out hamsters. Virology 507, 89–95 (2017).
Corman, V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 25, 2000045 (2020).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25, 402–408 (2001).
Reed, L. J. & Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27, 493–497 (1938).
Vandeghinste, B. et al. Iterative CT reconstruction using shearlet-based regularization. IEEE Trans. Nucl. Sci. 60, 3305–3317 (2012).
Vande Velde, G. et al. Longitudinal micro-CT provides biomarkers of lung disease that can be used to assess the effect of therapy in preclinical mouse models, and reveal compensatory changes in lung volume. Dis. Model. Mech. 9, 91–98 (2016).
Berghen, N. et al. Radiosafe micro-computed tomography for longitudinal evaluation of murine disease models. Sci. Rep. 9, 17598 (2019).
Kaptein, S. J. et al. Antiviral treatment of SARS-CoV-2-infected hamsters reveals a weak effect of favipiravir and a complete lack of effect for hydroxychloroquine. Preprint at https://doi.org/10.1101/2020.06.19.159053 (2020).
Geisinger, J. M., Turan, S., Hernandez, S., Spector, L. P. & Calos, M. P. In vivo blunt-end cloning through CRISPR/Cas9-facilitated non-homologous end-joining. Nucleic Acids Res. 44, e76 (2016).
Whitt, M. A. Generation of VSV pseudotypes using recombinant ΔG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 169, 365–374 (2010).
Berger Rentsch, M. & Zimmer, G. A vesicular stomatitis virus replicon-based bioassay for the rapid and sensitive determination of multi-species type I interferon. PLoS ONE 6, e25858 (2011).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020).
Fukushi, S. et al. Vesicular stomatitis virus pseudotyped with severe acute respiratory syndrome coronavirus spike protein. J. Gen. Virol. 86, 2269–2274 (2005).
Wang, C. et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 11, 2511 (2020).
Kleine-Weber, H. et al. Mutations in the spike protein of Middle East respiratory syndrome coronavirus transmitted in Korea increase resistance to antibody-mediated neutralization. J. Virol. 93, e01381-18 (2019).
Op De Beeck, A., Rouillé, Y., Caron, M., Duvet, S. & Dubuisson, J. The transmembrane domains of the prM and E proteins of yellow fever virus are endoplasmic reticulum localization signals. J. Virol. 78, 12591–12602 (2004).
Bonaldo, M. C. et al. Construction and characterization of recombinant flaviviruses bearing insertions between E and NS1 genes. Virol. J. 4, 115 (2007).
Barban, V. et al. High stability of yellow fever 17D-204 vaccine: a 12-year restrospective analysis of large-scale production. Vaccine 25, 2941–2950 (2007).
Acknowledgements
We thank E. Maas, J. Rymenants, T. Van Buyten, C. Collard, B. Voeten, D. Buyst and N. Cremers for the in vitro cell and virus culture and purification; K. Van den Eynde, E. Allegaert, S. Cumps and W. Versin for technical support with the preparation of specimens for histology; C. Coun, J. Paulissen, C. Sablon and N. Thys for technical assistance in cloning the different vaccine constructs and for generating serology data; J. Wouters and J. Nuyts for help with micro-computed tomography image analysis and support with imaging file processing; E. Martens for help with biomarker analysis; N. Berghmans, S. Knoops, T.-T. Pham, H. Crijns, M. Lox and N. Ongenae for help with hamster husbandry and bleeding; J. Vercruysse, C. Vansalen and N. Goris for help with large scale plasmid production; L. Close for next-generation sequencing analysis of vaccine virus stocks; D. Daelemans for access and W. Chiu for technical assistance with the high content screening platform; R. Gijsbers for helping with generation of pseudotyped viral vectors; H. Serroyen for assisting in figure design; M. A. Whitt for providing plasmids to rescue VSV-dG-GFP pseudoviruses; H. Kleine-Weber, M. Hoffmann and S. Pöhlmann for sharing L1-hybridoma supernatants and protocols for the generation of VSV pseudovirions; B. J. Bosch and W. Li for sharing SARS-CoV-2 S expression plasmids; I. Goodfellow for providing BSR-T7 cells; E. Brouwers for assistance in culturing hybridoma cells; P. Bredenbeek for providing BHK-21J and Vero E6 cells; and C. Libert for providing Ifnar1−/− mouse breeding couples. This list of people who have provided help during these exceptional times may not be complete, and the corresponding authors apologize for any accidental omissions. This project received funding from the European Union’s Horizon 2020 research and innovation programme (no. 101003627 (SCORE project) and no. 733176 (RABYD-VAX consortium)), funding from the Bill and Melinda Gates Foundation (INV-00636), the Research Foundation Flanders (FWO) under the Excellence of Science (EOS) program (VirEOS project 30981113), the FWO Hercules Foundation (Caps-It infrastructure). the KU Leuven Rega Foundation, the FWO (no. G0G4820N) and the KU Leuven/UZ Leuven COVID-19 Fund (COVAX-PREC project). J.M. and X.Z. were supported by grants from the China Scholarship Council (CSC). C.C. was supported by the FWO (FWO 1001719N). L.-H.L. was supported by a KU Leuven DBOF PhD scholarship. G.V.V. acknowledges grant support from KU Leuven Internal Funds (C24/17/061) and K.D. acknowledges grant support from KU Leuven Internal Funds (C3/19/057 Laboratory of Excellence). G.O. and P.M. received funding from KU Leuven (C16/17/010) and from FWO–Vlaanderen. K.B. was supported by the European Union’s Marie Skłodowska-Curie Innovative Training Network HONOURs (no. 721367). We appreciate the in-kind contribution of UCB Pharma (Brussels). We thank everyone who has supported this research by donating money, organizing fundraising campaigns or helping to spread the word. Our efforts to develop a vaccine were strengthened by support from the KU Leuven COVID-19 Fund. We thank all donors and volunteers for their continued support, generosity and optimism.
Author information
Authors and Affiliations
Contributions
L.S.-F., T.V., S.D., N.M. and K.D. designed and constructed vaccine candidates; V.L., M.P.A.J., W.D. and J. Matthijnssens analysed genetic stability and antigenicity; S.S., J. Ma, M.P.A.J., R.B., L.L. and L.-H.L. performed vaccine safety and biodistribution experiments; Z.W. provided the STAT2−/− hamsters; T.V., D.V.L. and M.R. performed and analysed serological assays; S.S., R.B., L.L., S.J.F.K., C.D.K. and L.B. performed vaccination, infection and dissection of hamsters; K.P.B., G.K.-K. and N.v.D. performed immunization and infection of macaques; B.E.V. and E.V. supervised the macaque study; S.S., J. Ma, R.B., L.L., S.J.F.K., C.D.K., L.B., L.-H.L., S.J., M.B.Y., K.P.B., G.K.-K., N.v.D., O.Q., X.Z. and S.t.H. performed and analysed RT–qPCR, titration and plaque reduction neutralization test (PRNT) experiments; L.S., B.W., T.K. and G.V.V. performed histological and micro-computed tomography analysis; J. Ma and M.P.A.J. performed vaccination of mice; J. Ma and B.M.-D. performed flow and ELISpot assays; J. Ma and M.P.A.J. performed the YF17D-challenge experiment; L.C., C.V., C.C., K.V.L., G.O., D.E.T. and P.M. provided advice on data interpretation and critical edits to the text; E.H., V.V. and D.S. provided and facilitated access to essential infrastructure; L.C., C.V., W.B., O.Q., J.N. and K.D. acquired funding; L.S.-F., T.V., S.S., J. Ma, V.L., D.V.L., M.P.A.J., R.B., B.M.-D., L.C., E.V., D.E.T, J.N., H.J.T. and K.D. wrote the original draft with input from co-authors; L.S.-F., T.V., D.E.T., J.N., H.J.T. and K.D. wrote the final draft; J.N., H.J.T. and K.D. supervised the study, performed experimental design and provided scientific direction; all authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
L.S.-F., J.N. and K.D. are named as inventors on a patent describing the invention and use of coronavirus vaccines. All other authors have nothing to disclose.
Additional information
Peer review information Nature thanks Alan Barrett, Bali Pulendran and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Vaccine design and antigenicity.
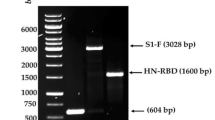
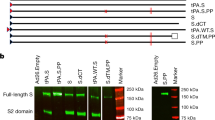
a, SARS-CoV-2 S (S1/2, S0 or S1) antigens were inserted into the E/NS1 intergenic region as translational fusion within the YF17D polyprotein (dark grey) inserted in the endoplasmic reticulum (pale blue). To cope with topological constraints of the fold of both SARS-CoV-2 S antigens and the polyprotein of the YF17D vector, one extra transmembrane domain76,77 (derived from the West Nile virus E protein; light grey) was added to the C-terminal cytoplasmic domain of the full-length S proteins (S1/2 and S0). Likewise, two transmembrane domains were fused to the endoplasmic-reticulum-resident C terminus of the S1 subunit in construct YF-S1. Scissors indicate proposed maturation cleavage sites, including the S1/2 furin-cleavage site mutated in YF-S0. b, Representative images of plaque phenotypes from YF17D and different YF-S vaccine constructs on BHK-21 cells. c, Confocal immunofluorescent images of BHK-21 cells three days after infection with different YF-S vaccine constructs staining for SARS-CoV-2 S antigen (green) and YF17D (red) (nuclei stained with DAPI, blue). Scale bar, 25 μm. Similar results were obtained from two independent experiments. d, Immunoblot analysis of SARS-CoV-2 S (S1/2, S0 and S1) expression after transduction of BHK-21 cells with different YF-S vaccine candidates. Before analysis, cell lysates were treated with PNGase F to remove their N-linked oligosaccharides or left untreated (black arrows denote glycosylated forms of S; white arrow heads denote deglycosylated proteoforms without N-linked oligosaccharides). Similar results were obtained from two technical repeats from the same experiment. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 2 Genetic stability of YF-S0 during passaging in BHK-21 cells.
a, Schematic of YF-S0 passaging in BHK-21 cells. YF-S0 vaccine virus recovered from transfected BHK-21 cells (P0) was plaque-purified once (P1) (n = 5 plaque isolates), amplified (P2) and serially passaged on BHK-21 cells (P3–P6). In parallel, each amplified plaque isolate (P2) (n = 5) from the first plaque purification was subjected to a second round of plaque purification (P3*) (n = 25 plaque isolates) and amplification (P4*). b, Schematic of tiled RT–PCR amplicons from three different primer pairs used for detection of the inserted SARS-CoV-2 S viral RNA sequence present in supernatants of different passages. All data are from a single representative experiment. c, RT–PCR fingerprinting performed on the virus supernatant collected from serial passage 3 (P3) and 6 (P6) of plaque-purified YF-S0. d, Immunoblot analysis of S expression by P3 and P6 of YF-S0. e, RT–PCR fingerprinting on amplified plaque isolates from the second round of plaque purification (P4*), 20 individual amplified plaque isolates are shown here (1–20). c, e, Control, YF-S0 cDNA (0.5 ng); ladder, 1-kb DNA ladder. Direct Sanger sequencing confirmed maintenance of full-length S inserts for 25 out of 25 plaques (100%). After two rounds of plaque purification and amplification, only in three isolates a single point mutation was found (two silent mutations and one missense mutation resulting in a S47P amino acid change in the N terminus of S1); at a low <10−4 mutation frequency (that is, 3 nt changes observed in a total of 25 × 4,196 nt = 104,900 nt sequenced; of which 25 × 3,780 nt = 94,500 nt were of S transgene sequence). This mutation rate is similar to that of parental YF17D under current vaccine manufacturing conditions14,78. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 3 Attenuation of YF-S vaccine candidates.
a, Survival and weight curve of suckling BALB/c mice inoculated intracranially with 100 PFU of the different vaccine constructs. Mice were inoculated with YF-S1/2 (n = 8), YF-S0 (n = 8), YF-S1 (n = 10), sham (n = 10) or YF17D (n = 9). b, Representative images of BALB/c mice at seven days after intracranial inoculation with sham, or 102 PFU of either YF-S0 or YF17D. c, Survival and weight curve of AG129 mice (n = 8 for each group) after intraperitoneal inoculation with a dose range of YF-S0 (102, 103 and 104 PFU) (green) in comparison to YF17D (1, 10 and 102 PFU) (black and grey). d, Survival curve of wild-type (WT) and STAT2-knockout (STAT2−/−) hamsters inoculated intraperitoneally with 104 PFU of YF17D or YF-S0. Wild-type hamsters inoculated with YF17D (n = 6) and YF-S0 (n = 6); STAT2−/− hamsters inoculated with YF17D (n = 14) and YF-S0 (n = 13). The number of surviving hamsters at study end point is indicated (a, c, d). e, f, Vaccine virus RNA (viraemia) in the serum (e) and weight evolution (f) of wild-type hamsters after intraperitoneal inoculation with 104 PFU YF17D (n = 6) or YF-S0 (n = 6). The number of hamsters that showed viraemia on each day after inoculation is indicated below (e). g, Weight evolution of Ifnar−/− mice after intraperitoneal inoculation with 400 PFU each at day 0 and 7 of YF-S0, YF17D and sham. Mice were inoculated with YF17D (n = 5), YF-S0 (n = 5) or sham (n = 5). Data in d are from two independent experiments, data in other panels are from a single experiment.
Extended Data Fig. 4 Immunogenicity and protective efficacy in hamsters.
a, Left, Correlation analysis of NAb titres using SARS-CoV-2 (PRNT) and rVSV-ΔG-S (SNT) for a panel of seven sera. SNT50 and PRNT50 values were plotted to determine the correlation between the neutralization assays with a Pearson regression coefficient of 0.77 (P = 0.04). a, Right, NAbs in sera from four convalescent patients as determined by SNT. Hamsters were vaccinated and challenged as depicted in Fig. 1b. b, Viral RNA in spleen, liver, kidney, heart and ileum of hamsters (n = 6 for each group from a single experiment) vaccinated with YF-S1/2, YF-S0 or sham, and challenged by infection with SARS-CoV-2. Viral RNA levels were determined by RT–qPCR, normalized against β-actin mRNA levels, and calculated relative to the median of sham-vaccinated hamsters. c–e, NAbs (c, d) and binding antibodies (e) in vaccinated hamsters four days after challenge with SARS-CoV-2 (n = 12 hamsters per group from 2 independent experiments; for binding antibody quantification, sera of 3 hamsters were pooled). d, Pair-wise comparison of NAbs at day 21 after immunization (circles), and 4 days after challenge (squares). f, Syrian hamsters were immunized twice intraperitoneally with 1.5 × 103 PFU each of vaccine construct YF-S0 produced on Vero E6 cells (n = 7 hamsters), or sham (n = 3 hamsters). At day 23 after vaccination, hamsters were inoculated with 2 × 105 TCID50 SARS-CoV-2 and followed up for four days. g, NAbs 21 days after vaccination. h, i, Viral loads in lungs of hamsters four days after infection quantified by RT–qPCR (h) and virus titration (i). Data in g–i are from a single experiment. Data are median ± IQR. Two-tailed uncorrected Kruskal–Wallis test (b, c, e) or a two-tailed Mann–Whitney U-test was applied (g–i). ns, not significant.
Extended Data Fig. 5 Protection from lung pathology.
Hamsters (n = 12 from 2 independent experiments for all groups) were vaccinated and challenged as depicted in Fig. 1b. a, Representative haematoxylin and eosin (H&E) images of a diseased lung (sham-vaccinated and infected). Top, area of bronchopneumonia (circle). Scale bar, 1 mm. Bottom, magnification of inflamed tissue showing a mixture of histiocytes (yellow arrows), neutrophils (green arrows) and lymphocytes (blue arrows). Scale bar, 50 μm. b, Cumulative histopathology score for signs of lung damage in H&E-stained lung sections (dotted line denotes the maximum score in the sham-vaccinated group). c, Representative micro-computed tomography images of the lungs of sham- and YF-S0-vaccinated hamsters four days after SARS-CoV-2 infection. Arrows indicate examples of pulmonary infiltrates seen as consolidation of lung parenchyma (cyan). d, e, Five transverse cross sections at different positions in the lung were selected for each hamster and scored to quantify lung consolidations (d) or used to quantify the nonaerated lung volume (NALV) (e), as functional biomarker reflecting lung consolidation. f, Individual expression profiles for 10 genes in lungs of vaccinated hamsters four days after SARS-CoV-2 infection (as in Fig. 1k, presented as log10-transformed fold change relative to uninfected controls (n = 4)). Levels of individual mRNAs were determined by RT–qPCR and normalized for β-actin mRNA. Changes are reported as values over the median of uninfected controls. Only for IFNλ (for which all control hamsters had undetectable RNA levels), fold changes were calculated over the lowest detectable value. Data are median ± IQR. Two-tailed uncorrected Kruskal–Wallis test was applied.
Extended Data Fig. 6 Gating strategy and profiling of CD8 and CD4 T cells.
a, First, live cells were selected by gating out Zombie Aqua (ZA)-positive and low forward scatter (FSC) events. Then, doublets were eliminated in an FSC-H versus FSC-A plot. T cells (CD3+) were stratified into γδ T cells (γδTCR+), CD4 T cells (γδTCR−CD4+) and CD8 T cells (γδTCR−CD8+). Boundaries defining positive and negative populations for intracellular markers were set on the basis of non-stimulated control samples. b, Full representation of t-SNE analysis of S-specific CD4 and CD8 T cells positive for at least one intracellular marker (IFNγ, TNF, IL-4 or IL17A) from splenocytes of YF-S1/2-, YF-S0- and sham-vaccinated Ifnar−/− mice (n = 6 per group) after overnight stimulation with SARS-CoV-2 S peptide pool (blue, IFNγ-expressing T cells; red, TNF-expressing T cells; green, IL-4-expressing T cells; yellow, IL17A-expressing T cells). t-SNE plots were generated using FlowJo by first concatenating S-specific CD8 (top panels) or CD4 T cells (bottom panels) from all mice.
Extended Data Fig. 7 Immunogenicity and protective efficacy in mice and hamsters after single-dose vaccination.
a, Ifnar−/− mice were vaccinated once intraperitoneally with 400 PFU YF-S0 (n = 9), sham (n = 6) or YF17D (n = 6). b, c, NAbs (b) and binding antibodies (c) at day 21 after vaccination; minipools of sera of 2–3 mice analysed for quantification of binding antibodies. d, Spot counts for IFNγ-secreting cells per 106 splenocytes after stimulation with SARS-CoV-2 S peptide pool. e, Hamsters (n = 8 from a single experiment for all groups) were vaccinated with a single dose of YF-S0 and challenged as outlined in Fig. 4. Cumulative histopathology score for signs of lung damage after SARS-CoV-2 infection in H&E-stained lung sections (dotted line denotes the maximum score in sham-vaccinated group). f, g, Hamsters (n = 6 per group from a single experiment) were vaccinated with a single dose of YF-S0 (104 PFU intraperitoneally) and sera were collected at 3, 10 and 12 weeks after vaccination. NAbs (f) and binding antibodies (g) at the indicated weeks post vaccination. Data are median ± IQR. Two-tailed uncorrected Kruskal–Wallis test was applied.
Extended Data Fig. 8 YF17D-specific immune responses in hamsters, mice and macaques.
a, Correlation analysis of NAb titres determined by using YF17D (PRNT) and YF17D–mCherry (SNT) for a panel of 21 antisera (historical samples from previous vaccination studies in mice and hamsters). SNT50 and PRNT50 values were plotted to determine the correlation between both neutralization assays with a Pearson regression coefficient of 0.68 (P = 0.0006). b, c, NAb titres in hamsters (n = 12 from 2 independent experiments for all groups) (b) and Ifnar−/− mice (YF-S1/2 (n = 11), YF-S0 (n = 11), YF-S1 (n = 8), sham (n = 9) and YF17D (n = 8) from 2–3 independent experiments) (c); sera collected at day 21 after vaccination in both experiments (two-dose vaccination schedule; Figs. 1, 2). d, Ifnar−/− mice vaccinated according to a two-dose vaccination schedule (YF-S1/2 (n = 11), YF-S0 (n = 10), YF-S1 (n = 7), sham (n = 9) and YF17D (n = 8) from 3 independent experiments). Spot counts for IFNγ-secreting cells per 106 splenocytes after stimulation with a YF17D NS4B peptide mixture. e, f, NAb titres after vaccination in macaques with YF-S0 (e) or placebo (f) (6 macaques per group from a single experiment); sera collected at indicated times after vaccination (two-dose vaccination schedule; Fig. 3). g, Ifnar−/− mice vaccinated according to a single-dose vaccination schedule (YF-S0 (n = 8), sham (n = 5) and YF17D (n = 5) from 2 independent experiments). Spot counts for IFNγ-secreting cells per 106 splenocytes after stimulation with a YF17D NS4B peptide mixture. Data are median ± IQR. Two-tailed uncorrected Kruskal–Wallis test was applied.
Extended Data Fig. 9 Protection from lethal YF17D.
a, Ifnar−/− mice were vaccinated with either a single 400 PFU intraperitoneal (i.p.) dose of YF17D (black) (n = 7) or YF-S0 (green) (n = 10), or sham (grey, n = 9). After 21 days, mice were challenged by intracranial (i.c.) inoculation with a uniformly lethal dose of 3 × 103 PFU of YF17D and monitored for weight evolution (b) and survival (c). The number of surviving mice at study end point (day 15) is indicated. Data are from two independent experiments.
Supplementary information
Supplementary Table 1
A list of primers and probes used for RT-qPCR.
Supplementary Figure 1
Original WES immunoblots with respective chromatographs, and uncropped RT-PCR gels. Dotted lines specify each that area which was cropped to generate respective panels in Extended Data Fig. 1d, and Extended Data Fig. 2c, 2d and 2e as indicated.
Rights and permissions
About this article

Cite this article
Sanchez-Felipe, L., Vercruysse, T., Sharma, S. et al. A single-dose live-attenuated YF17D-vectored SARS-CoV-2 vaccine candidate. Nature 590, 320–325 (2021). https://doi.org/10.1038/s41586-020-3035-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-3035-9
This article is cited by
-
The SARS-CoV-2 nucleocapsid protein: its role in the viral life cycle, structure and functions, and use as a potential target in the development of vaccines and diagnostics
Virology Journal (2023)
-
Revolutionizing viral disease vaccination: the promising clinical advancements of non-replicating mRNA vaccines
Virology Journal (2023)
-
YF17D-vectored Ebola vaccine candidate protects mice against lethal surrogate Ebola and yellow fever virus challenge
npj Vaccines (2023)
-
Vaccines platforms and COVID-19: what you need to know
Tropical Diseases, Travel Medicine and Vaccines (2022)
-
Synthetic multiantigen MVA vaccine COH04S1 protects against SARS-CoV-2 in Syrian hamsters and non-human primates
npj Vaccines (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
