
Abstract
Cardiovascular disease is the leading cause of death worldwide. Advanced insights into disease mechanisms and therapeutic strategies require a deeper understanding of the molecular processes involved in the healthy heart. Knowledge of the full repertoire of cardiac cells and their gene expression profiles is a fundamental first step in this endeavour. Here, using state-of-the-art analyses of large-scale single-cell and single-nucleus transcriptomes, we characterize six anatomical adult heart regions. Our results highlight the cellular heterogeneity of cardiomyocytes, pericytes and fibroblasts, and reveal distinct atrial and ventricular subsets of cells with diverse developmental origins and specialized properties. We define the complexity of the cardiac vasculature and its changes along the arterio-venous axis. In the immune compartment, we identify cardiac-resident macrophages with inflammatory and protective transcriptional signatures. Furthermore, analyses of cell-to-cell interactions highlight different networks of macrophages, fibroblasts and cardiomyocytes between atria and ventricles that are distinct from those of skeletal muscle. Our human cardiac cell atlas improves our understanding of the human heart and provides a valuable reference for future studies.
Similar content being viewed by others


Main
The heart is a complex organ, composed of four morphologically and functionally distinct chambers (Fig. 1a). Deoxygenated blood from the low-pressure right atrium and ventricle is propelled into the lungs. Oxygenated blood enters the left atrium and ventricle, which propels blood across the body at systemic pressure. Chambers are separated by the interatrial and interventricular septa, and unidirectional flow is established by the atrio-ventricular and ventricular-arterial valves. An intrinsic electrophysiological system rapidly propagates electrical impulses from the sinoatrial node to the atrioventricular node, and along Purkinje fibres to the apex where contraction begins. Cardiac anatomical and functional complexity requires exquisite orchestration of heterogeneous cell populations to enable continuous contraction and relaxation across different pressures, strains and biophysical stimuli in each chamber.

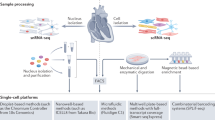
a, Transmural samples were obtained from left and right atrium, left and right ventricles, apex and interventricular septum from 14 individuals. Single nuclei (n = 14) and single cells (n = 7) were processed using Chromium 10x 3′DEG chemistry. b, Infographic shows donors (women, top; men, bottom), age, and contribution to cells and nuclei datasets (orange circle). Data are available in Supplementary Table 1. c, Uniform manifold approximation and projection (UMAP) embedding of 487,106 cells and nuclei delineate 11 cardiac cell types and marker genes. d, Distribution of cell populations, identified from nuclei within atria (left and right) and ventricles (left, right, apex and interventricular septum) after subclustering analysis. Colour code as in c. Data are available in Supplementary Table 2. Adip, adipocytes; Lym, lymphoid; Meso, mesothelial cells; Myel, myeloid; NC, neuronal cells; PC, pericytes. e, Multiplexed smFISH of cell type-specific transcripts in right ventricles (RV; left): TTN (green, cardiomyocytes) and CDH5 (red, EC) right atrium (RA; middle): NPPA (green, aCM) and DCN (red, FB) and left atrium (LA; right): MYH11 (green, SMCs) and KCNJ8 (red, pericytes). Nuclei are counterstained with DAPI (dark blue). Scale bars, 20 μm. For details on statistics and reproducibility, see Methods.
The heart is derived from multipotent progenitor cells that comprise two heart fields. Cells of the first heart field primarily populate the left ventricle; second heart field cells populate the right ventricle, and both fields contribute to the atria. Haemodynamics changes in the postnatal period and the distinct gene regulatory networks that operate in each heart field presumably prime gene expression patterns of adult heart cells1.
Single-cell and single-nucleus RNA sequencing (scRNA-seq and snRNA-seq, respectively) and multiplex single-molecule fluorescence in situ hybridization (smFISH) enable the identification of anatomical specificities, molecular signatures, intercellular networks and spatial relationships by illuminating the coordinated communication of cardiac cells within their microenvironments2.
We present comprehensive transcriptomic data on six distinct cardiac regions, providing, to our knowledge, the largest reference framework so far3,4. We incorporate snRNA-seq to ensure high-throughput capture of large cardiomyocytes (length and width approximately 100 and 25 μm) and scRNA-seq to upsample and enrich endothelial and immune cell populations. Using multiplex smFISH imaging, we describe the spatial distribution of selected cell populations and cell–cell co-localizations. We compare cardiac cell and nuclear transcriptomes with those of skeletal muscle and kidney, highlighting cardiac-specific cell signatures. Our study defines the cellular and molecular signatures of the adult healthy heart, and enables functional plasticity in response to varying physiological conditions and diseases.
Cellular landscape of the adult human heart
We isolate single cells, nuclei and CD45+ enriched cells from the left and right ventricular free walls, left and right atrium, the left ventricular apex, and interventricular septum, from 14 adults (Fig. 1a, b, Supplementary Table 1). After processing with 10X Genomics and a generative deep variational autoencoder, the resulting dataset comprises 45,870 cells, 78,023 CD45+ enriched cells and 363,213 nuclei for 11 major cell types: atrial cardiomyocytes, ventricular cardiomyocytes, fibroblasts (FBs), endothelial cells (ECs), pericytes, smooth muscle cells (SMCs), immune cells (myeloid and lymphoid), adipocytes, mesothelial cells and neuronal cells (Fig. 1c, e, Extended Data Figs. 1, 2).
The distributions of these main cell types, estimated from nuclei data, differ between atrial and ventricular tissues. Atrial tissues contain 30.1% cardiomyocytes, 24.3% FBs, 17.1% mural cells (pericytes and SMCs), 12.2% ECs and 10.4% immune cells (myeloid and lymphoid). By contrast, ventricular regions (apex, interventricular septum, left and ventricle) contain 49.2% ventricular cardiomyocytes, 21.2% mural cells, 15.5% FBs, 7.8% ECs and 5.3% immune cells (Fig. 1d, Supplementary Table 2).
The ventricular proportions of ventricular cardiomyocytes and FBs are negatively correlated, whereas pericytes and SMC proportions are positively correlated, indicating a functional organization (Supplementary Table 3). Cell distributions are generally similar in male and female hearts. However, the mean percentages of ventricular cardiomyocytes from left and right ventricles are higher in female hearts (56 ± 9%; mean ± s.d.) and associated with a stronger negative correlation between ventricular cardiomyocytes and FBs (r = −0.8; slope = −0.9) compared to male hearts (47 ± 11%; P = 0.03; ventricular cardiomyocytes–FBs, r = −0.4; slope = −0.3). Differences in the proportions of cardiomyocytes is unexpected given the average smaller female heart mass, and if confirmed might explain higher cardiac stroke volumes in women5 and lower rates of cardiovascular disease.
Cardiomyocyte heterogeneity
Cardiomyocytes show high-level expression of genes that encode contractile force-generating sarcomere proteins (TTN, MYBPC3 and TNNT2) and calcium-mediated processes (RYR2, PLN and SLC8A1). Consistent with bulk tissue RNA-sequencing (RNA-seq) data6, we observe markedly distinct transcriptional signatures in ventricular and atrial cardiomyocytes, reflecting developmental origins and differences in electrophysiological, contractile and secretory processes (Extended Data Fig. 3, Supplementary Table 4).
Ventricular cardiomyocytes are enriched in genes encoding sarcomere proteins (MYH7 and MYL2), transcription factors (IRX3, IRX5, IRX6, MASP1 and HEY2), and PRDM16, mutated in left ventricular non-compaction cardiomyopathy7. Other abundant transcripts enable tissue integrity despite high ventricular strain: PCDH7 encodes a calcium-dependent strong adhesive molecule8; SMYD2 encodes a lysine methyltransferase that promotes sarcomere formation and stabilization9. Atrial cardiomyocytes abundantly express prototypic genes and also ALDH1A2, an enzyme required for retinoic acid synthesis, ROR2, which participates in Wnt signalling during lineage differentiation10, and SYNPR, which functions in the mechanosensing of TRP channels by atrial volume receptors11.
We identify five ventricular cardiomyocyte (vCM1–vCM5) populations: vCM1 comprise 63.9% of left ventricular cardiomyocytes but only 36.7% of right ventricular cardiomyocytes (Fig. 2a, b, Extended Data Fig. 3a, c–e, Supplementary Tables 5, 6). vCM2 is more enriched in right ventricles (39.9%) than left ventricles (9.1%). However, differences between vCM1 and vCM2 are small, indicating shared gene programs between left (enriched in vCM1) and right (enriched in vCM2) ventricles. vCM2 shows higher expression of PRELID2, a developmental molecule with unknown cardiac function12 (verified by single-molecule fluorescence in situ hybridization (smFISH) (Fig. 2c, Extended Data Fig. 3c). Among vCMs, vCM2 has the highest expression of the myosin gene MYH6 and CDH13, a cell surface T-cadherin receptor for cardioprotective adiponectin and low density lipoproteins, both associated with several cardiometabolic traits13.

a, UMAP embedding of five ventricular cardiomyocyte (vCM) populations. b, Regional distributions of ventricular cardiomyocyte populations. Data are available in Supplementary Table 5. AX, apex; LV, left ventricle; SP, interventricular septum; RV, right ventricle. c, d, Multiplexed smFISH of PRELID2 (red) enriched in vCM2 (c) and of FHL1 (red) enriched in vCM3 (d). e, UMAP embedding of five atrial cardiomyocytes (aCM) populations. f, Regional distributions of atrial cardiomyocyte populations. LA, left atrium; RA, right atrium. g, h, Multiplexed smFISH of HAMP (red) enriched in aCM2 (g) and of CNN1 (red) enriched in aCM3 (h). In c, d, g and h, nuclei are counterstained with DAPI (dark blue). Scale bars, 10 μm. For details on statistics and reproducibility, see Methods.
vCM3 and vCM4 are present across all ventricular regions. The vCM3 transcriptional profile resembles a prominent right atrium population (aCM3, discussed below) with retinoic-acid-responsive SMC gene enrichment (MYH9, NEXN and CNN1)14,15. vCM3 also express stress-response genes including ANKRD116, FHL117 (verified by smFISH) (Fig. 2d, Extended Data Fig. 3c), DUSP2718, and XIRP1 and XIRP2, interacting with intercalated disc ion channel proteins implicated in cardiomyopathy and arrhythmias19. The small population vCM4 is enriched for nuclear-encoded mitochondrial genes (NDUFB11, NDUFA4, COX7C and COX5B) and Gene Ontology terms indicative of a high energetic state (Extended Data Fig. 3f). vCM4 also demonstrates high levels of CRYAB, which encodes a cytoprotective and antioxidant heat shock protein20, of sarcomere protein genes and PLN, indicating that these ventricular cardiomyocytes are outfitted to perform higher workload than other ventricular cardiomyocytes.
vCM5 (approximately 1%) comprises cells with high levels of DLC1 and EBF221, regulating brown adipocyte differentiation and perhaps cardiac pacemaker activity, and transcripts identified in neuronal lineages (SOX5, EBF1 and KCNAB1). As EBF1-depleted mice have a profoundly hypoplastic ventricular conduction system22, vCM5 may participate in electrophysiology.
We identify five atrial cardiomyocyte populations (aCM1–aCM5) (Fig. 2e, f, Extended Data Fig. 3b–d, Supplementary Tables 6, 7). HAMP, a master regulator of iron homeostasis, is considerably enriched in more than 50% of right atrium cardiomyocytes versus 3% left atrium cardiomyocytes (verified by smFISH)23 (Fig. 2g, Extended Data Fig. 3c), indicating energetic differences6. HAMP has unknown roles in cardiac biology, but Hamp-null mice have deficits in the electron transport chain and lethal cardiomyopathy24.
aCM1 shows robust expression of prototypic atrial transcripts, indicative of a basal atrial cardiomyocyte gene program, and lower levels of molecules with neuronal functions (ADGRL2, NFXL1 and ROBO2). aCM2 predominantly expresses HAMP within the right atrium and is enriched for SLIT3, the developmental ligand for cardiac ROBO receptors25, ALDH1A226 and BRINP3, involved in retinoic acid signalling, and GRXCR2, supporting cilia involved in mechanosensing27.
aCM3 and vCM3 share similar transcriptional profiles including enrichment of the SMC gene CNN1 (verified by smFISH) (Fig. 2h, Extended Data Fig. 3c). The molecular signatures of aCM2, aCM3 and vCM3 indicate derivation from the second heart field28. aCM4 transcripts denote high metabolic activity, similar to vCM4, and have the highest expression of NPPA. aCM5 expresses similar transcripts to vCM5.
Vascular, stromal and mesothelial cells
The vascular compartment includes 17 distinct populations of EC, SMC, pericyte, mesothelial cells with anatomical and arterio-venous specificities (Fig. 3a, b, Supplementary Tables 8, 9). Endothelial cells, identified by pan-EC markers PECAM1, CDH5 and VWF, comprise 10 populations (Extended Data Fig. 4a–c, g). Three capillary ECs (EC1-3_cap), which express RGCC and CA429, represent 57.4% of all ECs. Capillary-like EC4_immune ECs express transcripts related to antigen presentation and immune regulation (CX3CL1, CCL2, IL6 and ICAM1)30. Arterial EC5_art ECs are enriched for SEMA3G, EFNB2 and DLL4, whereas EC6_ven ECs express venous markers NR2F231 and ACKR132, which we confirmed by smFISH (Fig. 3c). Mainly atrial EC7_atria ECs express the angiogenesis regulator SMOC133 and NPR3, detected in mouse endocardium34, suggestive of endocardial cells. Lymphatic EC8_ln ECs, enriched for PROX1, TBX1 and PDPN, represent approximately 1% of the captured ECs29.

a, UMAP embedding of 17 vascular and mesothelial populations. EC1/2/3_cap, capillary ECs; EC4_immune, immune-related ECs; EC5_art, arterial ECs; EC6_ven, venous ECs; EC7_atrial, atria-enriched ECs; EC8_ln, lymphatic ECs; EC9_FB-like, ECs with FB features; EC10_CM-like, ECs with cardiomyocyte features; PC1_vent, ventricle-enriched pericytes; PC2_atrial, atria-enriched pericytes; PC3_str, stromal pericytes; PC4_CM-like, pericytes with cardiomyocyte features; SMC1_basic, basic SMCs; SMC2_art, arterial SMCs. b, Schematic of the vascular cells and their placement in the vasculature. c, Multiplexed smFISH of MYH11 (yellow) in SMC (thick in artery and very thin in small vein), CDH5 (red) in the endothelium, and SEMA3G (cyan) and ACKR1 (green) in EC5_art and EC6_ven, respectively in apex. Nuclei are counterstained with DAPI (dark blue). Scale bar, 20 μm. d, Predicted cell–cell interactions in arteries and veins. Data are available in Supplementary Table 10. e, f, Multiplexed smFISH of pan-FB DCN (cyan) and FAP (red) in FB4 in interventricular septum (SP) (e) and DCN (cyan) and LINC001133 (red) in FB5 in the apex (AX) (f). Nuclei are counterstained with DAPI (dark blue). Scale bars, 5 μm. g, UMAP embedding showing six FB populations and their respective marker genes. h, Multiplexed smFISH of C1QA+ macrophages (MP) and PTX3+ FB3, suggesting cross-talk between both cell types. Scale bar, 5 μm. For details on statistics and reproducibility, see Methods.
Pericytes express ABCC9 and KCNJ8 and segregate into four clusters, with PC1_vent cells enriched in ventricles and PC2_atria cells in atria. PC1_vent cells express adhesion molecules (NCAM2 and CD38), and CSPG4, which is involved in microvascular morphogenesis and EC cross-talk35 (Extended Data Fig. 4d–f). PC3_str co-express pericyte markers and very low levels of pan-EC transcripts. RNA velocity analyses suggest a directionality that indicates PC3_str cells as a transitional state between pericytes and ECs (Extended Data Fig. 4h, i). These observations may relate to bidirectional pericyte or endothelial cell (trans)differentiation, which remains controversial36.
Vascular SMCs that express MYH11 split into two populations. SMC1_basic cells express transcripts that indicate immaturity, including the stem-cell marker LGR637 and proliferation-associated RGS538. SMC2_art cells express considerably higher levels of CNN1, ACTA2 and TAGLN, indicating arterial origin, whereas SMC1_basic cells may be venous-derived39 (Extended Data Fig. 4d–f).
Cell–cell interaction analyses indicate connections between ECs and mural cells in different vascular segments (Fig. 3b–d, Extended Data Fig. 4j, k, Supplementary Table 10), including Notch receptor–ligand interactions40 (NOTCH1 or NOTCH4 with JAG1, and NOTCH2 or NOTCH3 with JAG1, JAG2 or DLL4) between EC5_art and SMC2_art cells. Exploratory spatial transcriptomics (Extended Data Fig. 5) shows co-occurrence of EC5_art and SMC2_art markers and JAG1 and NOTCH2, thereby supporting this interaction between ECs and SMCs. A venous-specific DLL1–NOTCH3 interaction is predicted for EC6_ven and SMC1_basic cells. Notably, many of the venous and arterial EC predicted interactions are shared with capillary ECs, which suggests gradual changes along the arterio-venous axis39.
We define a distinct small population as mesothelial cells that enrich for MSLN, WT1 and BNC141 but lack EC, FB or mural genes. smFISH confirms this annotation with localization of BNC1+/CDH5− cells to the epicardium (Extended Data Fig. 4l–n).
Cardiac fibroblasts
Cells of the FB compartment show enriched expression of DCN, GSN and PDGFRA within seven populations (Fig. 3g) with regional enrichment in ventricles (FB1) and atria (FB2). This is consistent with distinctive functional properties, including stronger profibrotic responses, by atrial FBs42. FB1 and FB2 cells express canonical genes and define a basal, chamber-specific FB expression program (Extended Data Fig. 6a, Supplementary Table 11).
FB4 and FB5 cells are less abundant in the right atrium than other regions, whereas FB3 are less abundant in the left ventricle (Extended Data Fig. 6c). FB4 cells express genes responsive to TGFβ signalling (for example, POSTN and TNC) (Fig. 3e). FB5 cells have higher expression of genes involved in the production, remodelling and degradation of extracellular matrix (ECM). By contrast, FB3 cells have lower expression of ECM-related genes but higher expression of cytokine receptors such as OSMR and ILST643 (Fig. 3f, Extended Data Fig. 6b, Supplementary Table 12). These distinctive fibroblast gene programs probably govern stress-responsive cardiac remodelling and contribute to homeostasis.
Separate clustering of atrial and ventricular FBs recapitulated the populations described above, including an OSM-signalling population in each chamber (aFB4 and vFB3). In addition, we identify distinct chamber-specific ECM-producing FBs that differ in the expression of collagen isoforms and other ECM-related (aFB2 versus vFB2) (Extended Data Fig. 6g–m, Supplementary Table 13) or connective tissue-related genes (aFB1 versus vFB4).
Immune cells and cardiac homeostasis
Analysis of cardiac immune cells reveals 21 cell states (Fig. 4a, Extended Data Fig. 7). Myeloid cells comprise 13 populations, including several subtypes of macrophages, monocytes and dendritic cells, whereas the lymphoid compartment comprises 8 populations (Supplementary Tables 14, 15).

a, Manifold of 40,868 myeloid and lymphoid cardiac cells. NP, neutrophils; NK, natural killer; NKT, natural killer T cells; CD4+T_tem, effector-memory CD4+ T cells; CD4+T_cytox, CD4+ cytotoxic T cells; CD8+T_tem, CD8+ effector-memory T cells; CD8+T_cytox, CD8+ cytotoxic T cells; DC, dendritic cells; CD14+Mo, CD14+ monocytes; CD16+Mo, CD16+ monocytes; Mo_pi, pro-inflammatory monocytes; IL17RA+Mo, IL17RA+ monocytes; MP_AgP, HLA class II antigen-presenting macrophages; MP_mod, monocyte-derived macrophages; LYVE1+MP1–3, M2-like, LYVE1+ macrophages sets 1–3; DOCK4+MΦ1–2, DOCK4+ macrophage sets 1–2; B_cells, B cells; plasma, plasma B cells. b, BioRender infographic summarizes predicted cell–cell interaction circuits between atrial and ventricular cardiomyocytes, FB4 and immune cells involved in tissue repair in the heart and SKM. Data are available in Supplementary Table 17. c, Gene expression signature for cardiac-specific LYVE1+ macrophages compared against predicted matched populations in skeletal muscle and kidney.
Macrophages include three LYVE1+ macrophage populations: LYVE1+ MP1–2 enrich for clathrin and cathepsin genes, and LYVE1+ MP3 for HLA-DOA, HLA-DQA1/2 and HLA-DQB1. LYVE1+ macrophages appear related to recently described tissue-resident macrophages associated with cardiovascular remodelling44, although negative for TIMD445 (Extended Data Fig. 8a, b). Monocyte-derived macrophages express LYVE1 and FOLR2, monocyte-like markers CEBPB and S100A8, and chemoattractant cytokine genes CCL13 and CCL18. Antigen-presenting macrophages are FOLR2−, LYVE1− and MERTK−, and enrich for HLA-DRA, HLA-DMA, HLA-DMB, HLA-DPA1 and TREM2 (described in lipid-associated macrophages)46. Although monocytes are abundant in our data and others47, these are likely to be circulating, as supported by computational integration of our data with published peripheral blood mononuclear cell (PBMC) single-cell RNA-seq data (Supplementary Table 16). Two populations of DOCK4+ macrophages differentiated by higher expression of IL4R, STAT3 and ITGAM in DOCK4+ MP1 versus DOCK4+ MP2, do not express C1QA or FOLR2 (Extended Data Fig. 8c).
Predicted cell–cell interactions identify receptor–ligand circuits among immune cells, cardiomyocytes, and FBs. LYVE1+, monocyte-derived and antigen-producing macrophages are predicted to interact with FB4 via CD74–MIF (Fig. 4b, Extended Data Fig. 8d, e, Supplementary Table 17). Inhibition of this interaction leads to fibrosis48 and tissue damage49. FB4 also enrich for FN1, COL4A1 and TNC, facilitating cellular proliferation in the fetal heart50 and predicted to interact with different integrins in atrial and ventricular cardiomyocytes. In skeletal muscle (SKM), predicted cell–cell interactions between PRG4+ FBs (analogous to FB4) and cardiomyocytes involve COL1A2, COL6A2 and α10β1 integrins, whereas SKM FBs and monocytes appear to interact via the ICAM1–AREG and CXCR4–CXCL12 chemokine pairs (Fig. 4b, Supplementary Table 18), indicating tissue-specific homeostatic transcriptional circuits.
Using a logistic regression model, we find that lymphoid cells are more similar across heart, SKM and kidney, whereas there is less concordance for myeloid cells (Extended Data Fig. 8f, Supplementary Tables 16, 19), probably due to tissue-specific adaptability of myeloid cells51. Notably, populations corresponding to cardiac monocyte-derived macrophages, LYVE1+ MP1, DOCK4+ MP1–2 and antigen-presenting macrophages are absent from the SKM and kidney. Cardiac LYVE1+ MP2–3, pro-inflammatory monocytes, classical monocytes and mast cells are more similar to their SKM counterparts, indicating greater similarity of striated muscle and cardiac myeloid populations versus kidney. The transcriptional signature of cardiac LYVE1+ MP2-3 is specific without overlap in SKM and kidney (Fig. 4c).
Conduction system and neuronal cells
Among 3,961 cells expressing prototypic electrophysiologic transcripts (NRXN1, NRXN3 and KCNMB4), we identify six neuronal cell subclusters (Extended Data Fig. 9a–c). NC1 constitutes 75–80% of neuronal cells and exhibits a basal gene program including LGI4, required for glia development and axon myelination52. NC2 and NC4 show strong expression of the central nervous system marker, PRKG153, and co-express typical fibroblast and cardiomyocyte genes, respectively. NC3 has overlapping gene expression signatures with ECs. NC5 expresses LGR5, a Wnt signalling, G-protein-coupled receptor and stem-cell marker that promotes cardiomyocyte differentiation in the outflow tract54, an arrhythmogenic area55. This cluster also expresses the neurodegenerative disease gene PPP2R2B56 (verified by smFISH) (Extended Data Fig. 9d, Supplementary Table 20); LSAMP, which guides the development of specific patterns of neuronal connections57; and the lipoprotein transport enzyme LPL that remyelinates damaged neurons58. NC6 mimics Schwann cells, expressing MBP, PRX and MPZ, which encode myelin constituents59.
Adipocytes
Cardiac adipocytes uniformly express GPAM, FASN and ADIPOQ and at lower levels, LEP60 (Extended Data Fig. 9e–h). ADIP1 expresses genes for PPAR pathways, metabolism of lipids and lipoproteins, and lipolysis. ADIP2 expresses ECM genes such as LAMA2, IGFBP7 and FBN1, which encodes both the glycoprotein fibrillin1 and asprosin, a white adipose tissue secreted hormone involved in glucose homeostasis (Supplementary Table 21). Given a stromal-related molecular signature, ADIP2 cells may represent fibrogenic adipocytes and/or precursors61,62. ADIP3 transcripts encode inflammatory and cytokine responsive molecules.
COVID-19 and GWAS disease relevance
Transcripts encoding the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) receptor ACE263 are highest in pericytes, followed by FBs and lowest in cardiomyocytes, where expression is twofold higher in ventricular than atrial cardiomyocytes. Among proteases priming viral entry63, TMPRSS2 transcripts are absent in pericytes, FBs and cardiomyocytes, whereas CTSB and CTSL are lowly expressed with higher levels in cardiomyocytes. ACE2 expression in pericytes and fibroblasts is depicted by smFISH (Extended Data Fig. 10a–e).
We define cells enriched for genes from 12 cardiovascular genome-wide association studies (GWAS) and involved in SARS-CoV-2 infection using MAGMA64 (Extended Data Fig. 10f). Atrial fibrillation GWAS signals are associated with transcriptional profiles in vCM3, owing to higher mean expression of CAV1, CAV2 and PRRX1. PR interval GWAS signals are associated with vCM3 and aCM5, with high expression of SCN5A, CAV1, ARHGAP24, MEIS1, TBX5 and TTN. GWAS signals for QRS duration are associated with specific gene expression in NC2 (PRKCA, CEP85L, SLC35F1, SIPA1L1, KLF12 and FADS2). Coronary artery disease and hypertension GWAS signals are associated with transcripts from many cell lineages, particularly SMCs, FBs, and ECs, reflecting the relevance of vascular cells in both disorders.
Discussion
Our analyses of approximately half a million single cells and nuclei from six distinct cardiac regions from fourteen donors considerably expand an emerging reference adult heart cell atlas. By combining single-cell and single-nuclear RNA-seq data with machine learning and in situ imaging techniques, we provide detailed insights across the repertoire of cardiac cells, including cardiomyocytes (excluded by single-cell RNA-seq) and ECs (underrepresented in cardiac snRNA-seq). We quantify the cellular composition highlighting chamber-specific features and differences between male and female donors. Within each cell compartment, we identify and validate prototypic lineage-specific genes, and genes with previously unknown cardiac expression. Our results begin to unravel the molecular underpinnings of cardiac physiology and the cellular response to stress and disease.
Cardiomyocytes are the most prevalent cardiac cells and comprise higher percentages in ventricles than atria, and in female versus male ventricular tissues. Transcriptional differences between atrial and ventricular cardiomyocyte populations indicate different developmental origins, distinctive haemodynamic forces and specialized functions in cardiac chambers. Cellular diversity of FBs reveals ECM-producing and ECM-organizing activities that with other cells support cardiomyocytes across varying biophysical stimuli. The vascular compartment contains several ECs and pericyte populations and two SMC subtypes with distinct anatomical and arterio-vascular characteristics. Arterial and venous ECs are predicted to interact with mural cells via Notch signalling pathways involved in regulating vascular homeostasis and development. Immune cells interact with FBs and cardiomyocytes. In addition to confirming previous findings65,66, we show macrophage complexity and infer paracrine circuits for cardiac homeostasis. Cross-tissue analyses delineate cardiac populations distinct from skeletal muscle and kidney.
We illustrate the relevance of cardiac cell atlas by defining cell lineages enriched in cardiovascular GWAS and molecules involved in SARS-CoV-2 infection. High expression of the viral receptor ACE2 in pericytes and its correlation with AGTR1 is consistent with the role of renin–angiotensin–aldosterone system signalling in cardiac haemodynamics67.
We recognize limitations associated with cell capture by different data sources and unintended bias from surgical sampling. However, we expect our results will inform studies of other cardiac regions (valves, papillary muscle and conduction system), propel studies with large cohorts to determine the roles of age, gender and ancestry on normal cardiac physiology and provide crucial insights to enable mechanistic understanding of heart disease.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Research ethics for donor tissues
Heart tissues (donors D1–D7 and D11) were processed at Wellcome Sanger Institute (Hinxton, UK) and obtained from deceased transplant organ donors after Research Ethics Committee approval (ref 15/EE/0152, East of England Cambridge South Research Ethics Committee) and informed consent from the donor families. Heart tissues (donors H2–H7) were processed at Harvard Medical School (Boston, Massachusetts, USA) and obtained from deceased organ donors after Human Research Ethics Board approval Pro00011739 (University of Alberta, Edmonton, Canada). Informed consent from donor families was acquired via the institutional Human Organ Procurement and Exchange Program (HOPE). Cardiovascular history was unremarkable for all donors (Supplementary Table 1).
Tissue acquisition and processing
Tissues were acquired from UK and North American donors (D1–7 and 11, H2–7) after circulatory death (DCD) (D2, D4–D7 and D11) and after brain death (DBD) (D1, D3, H2–H7). For UK DCD donors, after a five-minute stand-off and for DBD, the chest is opened, the aorta is cross-clamped and cardiac samples are acquired. For North American DBD donors, the aorta is cross-clamped, cold cardioplegia (Celsior) is administered under pressure via the aorta to arrest beating, the heart is excised, rinsed in cold saline and samples acquired. All donor samples were full-thickness myocardial biopsies from the left and right atrium, left and right ventricles, interventricular septum and apex, with intentional exclusion of large epicardial fat deposits. Samples used for single nuclei isolation were flash-frozen and stored at −80 °C. Single-cell isolation and CD45+ enrichment was carried out on freshly collected samples. Residual tissue after nuclei and cell isolation procedures was formalin-fixed or frozen in OCT for additional studies.
All tissues were stored and transported on ice at all times until freezing or tissue dissociation to minimise any transcriptional degradation. Previous studies on the post-mortem tissue stability of the GTEx consortium on bulk tissues68 and in single cells69 suggest only minor changes in tissues within the first 24 h post mortem when stored in cold conditions.
Single nuclei isolation
Single nuclei were obtained from flash-frozen tissues using mechanical homogenization as previously described70. Tissues were homogenized using a 7 ml glass Dounce tissue grinder set (Merck) with 8–10 strokes of a loose pestle (A) and 8–10 strokes of a tight pestle (B) in homogenization buffer (250 mM sucrose, 25 mM KCl, 5 mM MgCl2, 10 mM Tris-HCl, 1 mM dithiothreitol (DTT), 1× protease inhibitor, 0.4 U μl−1 RNaseIn, 0.2 U μl−1 SUPERaseIn, 0.1% Triton X-100 in nuclease-free water). Homogenate was filtered through a 40-μm cell strainer (Corning). After centrifugation (500g, 5 min, 4 °C) the supernatant was removed and the pellet was resuspended in storage buffer (1× PBS, 4% bovine serum albumin (BSA), 0.2 U μl−1 Protector RNaseIn). Nuclei were stained with NucBlue Live ReadyProbes Reagents (ThermoFisher) and Hoechst-positive single nuclei were purified by fluorescent activated cell sorting (FACS) using influx, XDP or FACSAria (BD Biosciences) (Supplementary Fig. 1). Nuclei purification and integrity was verified under a microscope, and nuclei were further processed using the Chromium Controller (10X Genomics) according to the manufacturer’s protocol.
Single-cell preparation
Heart tissues (0.2–0.9 g) were transferred from cardioplegic solution into gentleMACS C-tubes (Miltenyi Biotec) containing enzymatic digestion base solution (100 μg ml−1 liberase TH Research grade and 50 μg ml−1 DNase I, HBSS 10 mM HEPES and 30 mM taurine)71. Tissues were minced using scissors (FST) and automatically digested using gentleMACS Octo Dissociator (Miltenyi Biotec) with heaters. Cardiomyocyte-depleted single-cell suspension were washed with base solution containing 20% fetal bovine serum (FBS) (Gibco), filtered through 70-μm nylon strainer (BD Falcon), collected by centrifugation (330g, 10 min, 4 °C) and resuspended in base solution containing 0.2% FBS (Gibco). Cells were manually counted three times by Trypan blue exclusion after each centrifugation and resuspended at a concentration of at least 2 × 106 ml−1. Single cells were processed using Chromium Controller (10X Genomics) according to the manufacturer’s protocol.
CD45+ cell enrichment
Cell suspension was prepared as described above and subsequently labelled using anti-human CD45 monoclonal antibody-conjugated microbeads according to the manufacturer’s protocol (Miltenyi Biotec). In brief, up to 107 cells were incubated for 15 min at 4 °C in 80 μl of PBS, BSA, EDTA buffer (1× PBS pH 7.2, 0.5% BSA, 2 mM EDTA) containing 20 μl CD45 microbeads. Cell suspension was washed in PBS, BSA, EDTA buffer once and collected by centrifugation (330g, 10 min, 4 °C). Resuspended cells were applied to MACS LS columns (Miltenyi Biotec). CD45-depleted cell fraction was discarded after three washes with PBS, BSA and EDTA buffer and the CD45+ cell fraction was collected in PBS, BSA and EDTA buffer by removal of the columns from the magnetic field. CD45+ cells were counted and resuspended in PBS, BSA and EDTA buffer to a concentration of at least 2 × 106 per ml before further processing using a Chromium Controller (10X Genomics) according to the manufacturer’s protocol.
Chromium 10X library preparation
Single cells and nuclei were manually counted by Trypan blue exclusion or automatically using a Countess II (Life Technologies) using at least two separate counts. Cell or nuclei suspension was adjusted to 400–1,000 cells per microlitre and loaded on the Chromium Controller (10X Genomics) with a targeted cell or nuclei recovery of 4,000–10,000 per reaction. 3′ gene expression libraries were prepared according to the manufacturer’s instructions of the v2 or v3 Chromium Single Cell Reagent Kits (10X Genomics). Quality control of cDNA and final libraries was done using Bioanalyzer High Sensitivity DNA Analysis (Agilent) or 4200 TapeStation System (Agilent). Libraries were sequenced using HiSeq 4000 (Illumina) at Wellcome Sanger Institute, and NextSeq 500 (Illumina) at Harvard Medical School with a minimum depth of 20,000–30,000 read pairs per cell or nucleus (Supplementary Table 22).
Spatial validation using smFISH with RNAscope probes
During preparation of formalin-fixed paraffin-embedded (FFPE) samples fresh tissue was fixed in neutral-buffered 10% formalin for 18–36 h and subsequently embedded in paraffin blocks. Fixed-frozen tissue samples were fixed in 4% paraformaldehyde (ThermoFisher). Sections were cut at 5-μm thickness using a microtome and placed onto SuperFrost Plus slides (VWR). FFPE tissue slides were automatically stained using BOND RX (Leica) and the RNAscope Multiplex Fluorescent Reagent Kit v2 Assay (ACDBio) according to the manufacturer’s protocol. Fixed-frozen tissue slides were processed according to the protocol of RNAscope Multiplex Fluorescent Assay v1 (ACDBio). RNAscope ready- or custom-made target probes were run in parallel to multiplex positive and negative controls (Extended Data Fig. 12b, Supplementary Table 23). All nuclei were DAPI-stained. All FFPE tissue slides were imaged using an Opera Phenix High-Content confocal Screening System (Perkin Elmer) with a 1-μm z-step size and 20× water-immersion objective (NA 0.16, 0.299 μm per pixel). Channels: DAPI (excitation 375 nm, emission 435–480 nm), Atto 425 (excitation 425 nm, emission 463–501 nm), opal 520 (excitation 488 nm, emission 500–550 nm), opal 570 (excitation 561 nm, emission 570–630 nm), opal 650 (excitation 640 nm, emission 650–760 nm). Fixed-frozen tissue slides were imaged using a LSM710 confocal microscope (Zeiss) and 40× oil-immersion objective (1.3 oil, DIC III). Channels: DAPI (excitation 375 nm, emission 435–480 nm), Alexa Fluor 488 (excitation 492 nm, emission 517 nm), Atto 550 (excitation 560 nm, emission 575 nm) and Atto 647 (excitation 649 nm, emission 662 nm). Visualization and background removal (rolling ball radius) were done using Fiji/ImageJ72. Pseudocolours were used for better visualization.
Haematoxylin and eosin staining
Tissue samples were fresh-frozen in isopentane (ThermoFisher) at −80 °C and embedded in OCT (VWR). Sections were cut at a thickness of 10 μm using a microtome, placed onto SuperFrostPlus slides (VWR) and further processed according to a standard haematoxylin and eosin staining protocol (Extended Data Fig. 12a).
Acquisition of skeletal muscle tissue
Intercostal muscle samples were obtained from between the second and third rib on the left side. This is typically from the deepest layer of muscle (furthest away from the skin). Samples were collected directly into the cold preservation solution.
Nuclei isolation for skeletal muscle
Muscle tissue was washed in 1× PBS, cleaned of any visible fat depositions and minced to obtain fragments of approximately 1 mm3. Per sample, approximately 0.3 g of minced tissues was homogenized in 3 ml of buffer A (250 mM sucrose, 10 mg ml−1 BSA, 5 mM MgCl2, 0.12 U μl−1 RNaseIn, 0.06 U μl−1 SUPERasIn, 1× protease inhibitor) using Dounce tissue grinder set (Merck) with 50 strokes of the loose pestle (A). The homogenate was filtered through a 100-μm cell strainer (Corning) and the strainer was washed with 1 ml and then 750 μl of buffer A. After the addition of Triton X-100 (final concentration 0.5%), the mixture was further homogenized with 50 strokes of the tight pestle (B). After filtering through a 40-μm strainer, nuclei were centrifuged (3,000g, 5 min, 4 °C), resuspended in 1 ml of buffer B (320 mM sucrose, 10 mg ml−1 BSA, 3 mM CaCl2, 2 mM magnesium acetate, 0.1 mM EDTA, 10 mM Tris-HCl, 1 mM DTT, 1× protease inhibitor, 0.12 U μl−1 RNaseIn, 0.06 U μl−1 SUPERasin) and purified using a 27% Percoll gradient solution. The Percoll mixture was centrifuged at 20,000g (15 min, 4 °C) and the pellet was resuspended in 200 μl of buffer B, followed by centrifugation (20,000g, 3 min, 4 °C). After Trypan Blue staining, the intact nuclei were counted using a haemocytometer. Nuclei were profiled using a Chromium Controller (10X Genomics) according to the manufacturer’s protocol.
Single-cell isolation for skeletal muscle
Muscle tissue was washed in 1× PBS, cleaned of any visible fat depositions and finely minced. Then, 2 g of the minced tissue was transferred to digestion buffer 1 (750 U ml−1 collagenase type 2 in 1× PBS) and incubated at 37 °C in a water bath for 90 min. The partially digested tissue was collected by centrifugation (650g, 5 min, 4 °C) and the pellet was resuspended in digestion buffer 2 (100 U ml−1 collagenase type 2, 2 U ml−1 dispase in PBS). After 30 min incubation at 37 °C in a water bath, the digestion was stopped by the addition of 2% FBS. Cells were filtered through a 100-μm and a 40-μm nylon strainer (BD Falcon), collected by centrifugation (650g, 4 °C, 3 min) and washed with 1× PBS, 2% FBS. Subsequently, a 20% Percoll gradient (15,000g, 4 °C, 20 min) was used for cell purification. The layer containing cells was collected, washed in PBS containing 2% FBS, and viable cells were counted by Trypan Blue exclusion using a haemocytometer. Nuclei were profiled using a Chromium Controller (10X Genomics) according to the manufacturer’s protocol.
The methods key resources table is in Supplementary Table 24.
Transcriptome mapping
After sequencing, samples were demultiplexed and stored as CRAM files. Each sample was mapped to the human reference genome (GRCh38 v.3.0.0) provided by 10X Genomics, and using the CellRanger suite (v.3.0.1) with default parameters. Single-cell samples were mapped against the reference as it was provided. Single-nuclei samples, the reference for pre-mRNA, was created using the 10X Genomics instructions (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references).
Count data processing
After mapping, samples from each data source (single nuclei, single cell and CD45+ cell) were grouped into individual AnnData objects by concatenating the raw_feature_bc_matrix_h5.h5 and adding the appropriate metadata information. For each data source object, the mean of unique molecular identifiers (UMIs) (n_counts) was calculated and used as a threshold for empty droplets.
Doublet detection
After removal of empty droplets, we applied scrublet73 to assign a doublet score (scrublet_score) to each cell. These cells were clustered and visualized using the UMAP method74. In addition, each cell was processed for doublet detection using a percolation method to allow for improved detection of doublets75.
Cell quality control and filtering
Each data source was processed and annotated separately to account for source-specific quality differences. These metrics are included as covariates for further processing. Total cells and CD45+ cells were filtered for counts (500 < n_counts <15,000), genes (200 < n_genes), mitochondrial genes (percent_mito <20%), ribosomal genes (percent_ribo <20%) and scrublet score (scrublet_score <0.3). Single nuclei were filtered for counts (500 < n_counts <15,000), genes (300 < n_genes <6,000), mitochondrial genes (percent_mito <5%), ribosomal genes (percent_ribo <5%) and scrublet score (scrublet_score <0.3). The same filtering thresholds were applied to the skeletal muscle dataset.
Scanpy toolkit 1.576 in Python v.3.7 was used to perform downstream analyses, including normalization (normalize_per_cell: counts_per_cell_after = 10,000), log transformation (log1p), variable gene detection (highly_variable_genes), regressing out unwanted sources of variation (regress_out: n_counts and percent_mito), data feature scaling (scale: max_value = 10) and PCA (pca: using highly variable genes) as previously described77.
Batch alignment using deep variational autoencoder
We built a global manifold by aligning all the data sources and donors in our data. This was done in a three-step procedure: (1) Each source was analysed and annotated separately, aligning only for donors using a pericyte-space linear regression step before batch alignment with bbknn78. Differentially expressed genes (DEGs) were calculated using a Wilcoxon rank sum test with Bonferroni–Hochberg adjustment as implemented in the Scanpy framework. (2) To annotate each cluster, we used an integrative approach by searching the top significant DEGs (P < 1 × 10−5) with a logFC >1 against the ToppFun79 and EnrichR80 databases. Significant hits on pathways, transcriptional regulation and biological processes were prioritized to annotate a given cluster. Each cellular compartment was labelled under the adata.obs[‘cell_type’] slot after grouping source-specific cell states. (3) All sources were combined into a single AnnData object under the label adata.obs[‘cell_sources’]. Batches were aligned using the batch_correction function from the scGen variational autoencoder81. First we align for adata.obs[‘cell_sources’], using adata.obs[‘cell_type’] as an anchor. Next, we aligned for adata.obs[‘donor’], using adata.obs[‘cell_type’] as an anchor. Each batch alignment round was run for 50 epochs.
Manifolds for the adipocytes, vascular and immune cardiac populations, as well as the skeletal muscle analysis, were created using this method and the clustering accuracy was evaluated with SCCAF82 (Extended Data Fig. 12d).
DEGs
To help with the annotation of the subpopulations of each cell compartment, we calculated the DEGs using the Wilcoxon rank sum test as implemented in the scanpy workflow and recommended by recent benchmarking studies83. A gene was considered to be differentially expressed if it has a log2-transformed fold change > 1 and a P < 1 × 10−5, unless stated otherwise in the analysis section.
Cell–cell interactions
Expression matrices of the populations under study were exported from the AnnData, together with a metadata table that contained the cell-barcodes as indices. We then ran CellPhoneDB as follows: cellphonedb method statistical_analysis meta.tsv counts.tsv–counts-data = gene_name–threads = 60. CellPhoneDB raw predictions were filtered by removing those interactions with a P > 1.0 × 10−5. Significant pairs were then submitted for gene set enrichment analysis into ReactomeDB, enrichR and ToppFun for functional classification. The vascular cells were randomly sub-sampled to 39,000 cells before the analysis, and the cardiac repair group (atrial and ventricular cardiomyocytes, FBs, and immune cells) was randomly sub-sampled to 69,295 cells before the analysis.
Visualization of gene expression on 10X Genomics Visium data
We processed the publicly available left ventricular myocardium Visium data from 10X Genomics (https://support.10xgenomics.com/spatial-gene-expression/datasets/1.0.0/V1_Human_Heart) using the Scanpy v.1.5 workflow adapted for the analysis of 10X Genomics Visium data (https://scanpy-tutorials.readthedocs.io/en/latest/spatial/basic-analysis.html). In brief, spots were removed with less than 500 UMIs or more than 20,000 UMIs, and less than 200 genes. Data were log-transformed and normalized before plotting.
Estimation of RNA velocity
To calculate the RNA velocity of the single cells and CD45+ enriched single cells, we used the CellRanger output BAM file and the GENCODE v33 GTF (ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_33/gencode.v33.chr_patch_hapl_scaff.annotation.gtf.gz) file together with the velocyto84 CLI v.0.17.17 to generate a loom file containing the quantification of spliced and unspliced RNA. Next, we built a manifold, cluster the cells and visualize the RNA velocities using scVelo85.
Subpopulation analyses of atrial and ventricular cardiomyocytes, FBs and neuronal cells
All barcodes labelled in the global object as cardiomyocytes, fibroblasts and neural cells were selected for further subpopulation analyses. Additional cell population-specific filtering criteria were applied to nuclei as follows: cardiomyocyte counts (n_counts <12,500), genes (n_genes <4,000), mitochondrial genes (percent_mito <1%), ribosomal genes (percent_ribo <1%) and scrublet score (scrublet_score <0.25); FB mitochondrial genes (percent_mito <1%), ribosomal genes (percent_ribo <1%); neuronal cell genes (n_genes <4000), mitochondrial genes (percent_mito <1%), ribosomal genes (percent_ribo <1%). Total and CD45+ cells were excluded in the atrial and ventricular cardiomyocytes datasets and did not contribute to subpopulation analysis. No further filtering of FBs or neuronal cell total and CD45+ cells was applied. Cardiomyocytes and FBs were then further split into two groupings based on the region of origin: (1) left and right atrium, and (2) left and right ventricles, apex and interventricular septum.
Donor effects were aligned as described in step (1) above. For FB and neuronal cells, sources were aligned as described in step (3) above. Leiden clustering and UMAP visualization were performed for identifying subpopulations and visualization86. Differentially expressed genes were calculated using the Wilcoxon rank sum test. Genes were ranked by score.
Cross-tissue comparison of cardiac immune populations with skeletal muscle, kidney and blood immune populations
We collected single-cell transcriptome data for adult kidneys from ref. 99 (https://www.kidneycellatlas.org/), and subset all immune cells reported in their study. For the SKM we selected the annotated immune cells from the merged manifold. For the human blood, we used the publicly available 10,000 single PBMC cells dataset provided by 10X Genomics (https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/pbmc_10k_v3). As previously described87, we trained a logistic regression model on the cardiac immune cells using 80% of the expression data and tested its accuracy on the remaining 20% to produce a model with an accuracy of 0.6862 (Extended Data Fig. 8f, Supplementary Table 16). We then applied this model to predict analogue cardiac immune populations in the adult kidney, SKM and PBMCs. Predictions with a probability less than 0.8 were excluded from downstream comparative analyses.
Gene Ontology enrichment analysis
For the ventricular cardiomyocyte population, we used the R package gProfileR (https://cran.r-project.org/web/packages/gProfileR/index.html) with the score-ranked gene list of vCM4 as input and the set of genes expressed in ventricular cardiomyocytes as background (those genes having a UMI count >1). To perform the Gene Ontology analysis on the vascular cells, the top 500 significant DEGs (P < 1 × 10−5) with a log-transformed fold change > 1 were searched against the Gene Ontology biological process database using ToppFun79 (Supplementary Table 25). The top five significantly enriched terms (q < 0.05) for each subpopulation were selected and plotted on a heat map. To perform the pathway analysis on the adipocytes, the top 500 significant DEGs (P < 1 × 10−5) with a log-transformed fold change > 0.5 were searched against ToppFun79 pathway databases (Supplementary Table 26). The top five significantly enriched pathways (q < 0.05) for each subpopulation were selected and plotted on a heat map.
Gene set score
We use the score_genes function as implemented in scanpy to calculate the enrichment of genes involved in the Oncostatin M pathway. A list of genes was collected upon literature research88,89. For gene set enrichment, only highly expressed genes were considered to reduce noise (more than 500 UMIs across all cells). The same analysis was performed for comparison of cardiac immune cells in our study with the observations of previous studies on cardiac-resident macrophages51, mouse tissue-remodelling macrophage45 and yolk sac lineage origin90.
Statistics and reproducibility
All analyses were performed using R Software, v.3.6.1. Student’s t-tests were used to compare cell type distributions at each site. P < 0.05 was considered statistically significant. Linear regression models (correlations) were obtained using the R linear model function (lm), which estimates statistical likelihood (P value) of a linear relationship. Bonferroni correction was applied for multiple testing.
The depicted RNAscope micrographs in the figures are representative. The micrographs in Figs. 2g, 3c, h and Extended Data Fig. 3c (HAMP), Extended Data Fig. 3e (CNN1), Extended Data Figs. 4g, m, 6f were repeated with similar results in two individual tissue sections. The micrographs in Figs. 2h, 3f and Extended Data Fig. 3c (CNN1), Extended Data Fig. 3e (PCDH7), Extended Data Figs. 6e, h, 9d were repeated with similar results in three individual tissue sections. The micrographs in Figs. 1e, 2d, 3e and Extended Data Figs. 1f, 3c (FHL1) and Extended Data Fig. 6d were repeated with similar results in four individual tissue sections. The micrographs in Fig. 2c and Extended Data Fig. 3c (PRELID2), Extended Data Figs. 10e, 12a were repeated with similar results in six or more individual tissue sections. Positive and negative controls were done once per used samples.
GWAS enrichment analysis
We downloaded GWAS summary statistics from broad cvdi, EBI GWAS catalogue and GWAS atlas. We selected traits with well-powered GWAS (n > 5,000 and number of significant loci >10). GWAS datasets are summarized in Supplementary Table 27. Gene expression data of protein-coding genes were mapped onto Entrez gene ids and these gene annotations were used on the human genome assembly hg19/37. We only used gene expression data from nuclei. We implemented the analysis previously described64 in python and in R. The log-transformed counts (plus one pseudocount) were used to compute average cell type-specific expression profiles. We performed individual magma analyses for each cell type, always conditioning on default gene level covariates (for example, gene length) and average gene expression across all cells. Subsequently, we applied the Benjamini–Hochberg method and selected cell type trait associations with FDR < 10%. These pairs were then subjected to conditional analysis as previously described64 to define ‘independent’, ‘jointly explained’ and ‘partially jointly explained’ pairs of associations (Supplementary Table 28).
Distributions of dispersed cells and isolated nuclei
The different procedures for obtaining isolated nuclei and dispersed cells resulted in significantly different distributions of cell types (Supplementary Table 29, Extended Data Fig. 2). Notably, 30.1% and 49.2% of isolated nuclei were derived from atrial and ventricular cardiomyocytes in the atrial and ventricular regions, whereas these cells were mostly excluded from preparations of isolated and CD45-selected cells (Supplementary Table 2).
Excluding cardiomyocytes, the distribution of cell types identified from isolated nuclei and dispersed cells remained distinct (Supplementary Table 30). Although 59.0% of dispersed cells were ECs, only 15.7% of nuclei were derived from ECs. By contrast, 64.2% of nuclei were from FBs (31.2%) and pericytes (33.0%), whereas only 17.1% of dispersed cells were FBs (2.3%) and pericytes (14.8%). These differences may reflect sensitivity of EC nuclei to isolation procedures or resistance of pericytes and FBs to cellular enzymatic digestion.
Despite differences in cell distributions between isolated nuclei and dispersed cells, the gene expression profiles of cell lineages were reasonably correlated (r > 0.4 for each cell type). To address the concordance of the genes captured by cells and nuclei, we compared the expression of the major cell type markers from Fig. 1c across the three sources (Extended Data Fig. 1c). As nuclei lack cytoplasmic RNA, the expression of certain genes, especially immune genes NKG7 and C1QA, was lower in nuclei than in cells. Nevertheless, the general trend with respect to marker genes was consistent across the three sources, and the same genes distinguished individual cell types independent of the source.
Further analysis of vascular cells
The PC3_str contained similar contribution of cells and nuclei, and had a scrublet score below the stringent threshold used; nevertheless, the average number of genes and counts in this cluster was higher than average. Thus, despite our stringent quality filtering, we cannot exclude the possibility that there might be doublets in this cluster. EC10_CMC-like and PC4_CMC-like co-express EC or pericyte genes with cardiomyocyte markers and further studies are required to understand whether they represent previously unknown cell states or doublets.
The observations of the arterial and venous SMC are supported by previous studies, which predict that arterial SMCs are more contractile, and venous SMCs are less differentiated91.
EC3_cap enrich for transcripts encoding components of AP1 (JUN and FOS), which mediates multiple EC fate decisions including response to VEGF, inflammatory and stress signals, and ATF3, an adaptive-response gene induced by diverse signals92,93,94.
Skeletal muscle characterization
We collected intercostal skeletal muscle samples from five healthy individuals, including one donor with matched cardiac tissue, and profiled the transcriptome of 35,665 single cells and 39,597 single nuclei. Analogous to the heart, the combination of cells and nuclei allowed us to capture and resolve major cell lineages, including cardiomyocyte, fibroblasts, endothelial cells, smooth muscle cells, pericytes, myeloid and lymphoid immune cells and satellite cells (Extended Data Fig. 11a, b, Supplementary Table 31).
Further analysis of the vascular cells of the skeletal muscle identified ten distinct populations. The endothelial cells showed five clusters separated based on their respective vascular beds with signatures similar to the ones we observe in the heart. The EC_cap expresses VWF and RGCC. Venous EC_ven express ACKR1 and PLVAP, whereas arterial EC_art show SEMA3G and HEY1, in line with our heart data (Extended Data Fig. 11c, d, Supplementary Table 32).
The overall distributions of vascular and stromal cell populations in skeletal and cardiac muscle were similar, including the arterial and venous features of ECs; however; skeletal muscle contained a single SMC cluster, potentially related to the smaller size of the dataset. In skeletal muscle, the predicted cell–cell interactions of the EC_art and SMCs included NOTCH1/4–JAG1 as well as JAG1/JAG2/DLL4–NOTCH3, but not JAG1/JAG2/DLL4–NOTCH2, inferred in the heart (Extended Data Fig. 11e, f, Supplementary Table 33).
Cardiac immune cells
Using the logistic regression model, we did not identify any counterpart of the cardiac IL17RA+ monocytes in SKM or kidney, possibly owing to the small size of this population.
Naive T cells (CD4+T_naive) identified expressed CCR7 and SELL, indicative of their naive and tissue-resident nature95. Memory T cells (CD8+T_tem) expressed BACH2, STAT4 and IL7R, associated with long-term immune memory96,97. We further characterized the lymphoid cells using scNym98, and trained it using published data99,100. The resulting model was applied to our cardiac immune cells and those cells, with a predicted score higher than 0.8 were presumed to be likely candidates for re-annotation. Using this approach, we identified candidates for plasma B cells (109), dendritic cells (645), innate lymphoid cells (89), MAIT T cells (219), T helper cells (80) T regulatory cells (11), T central memory cells (103), γδ T cells (30) and plasmocytoid dendritic cells (27). These annotations can be found in the cardiac immune object annotations under the label ‘scNym’ at www.heartcellatlas.org.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Data objects with the raw counts matrices and annotation are available via the www.heartcellatlas.org webportal. Raw data are available through the Human Cell Atlas (HCA) Data Coordination Platform (DCP) with accession number: ERP123138 (https://www.ebi.ac.uk/ena/browser/view/ERP123138). The 10X Genomics Visium data for the heart left ventricle tissue can be accessed at: https://support.10xgenomics.com/spatial-gene-expression/datasets/1.1.0/V1_Human_Heart. GWAS data used in this study can be found in Supplementary Table 27. All of our data can be explored at www.heartcellatlas.org.
Code availability
All code used for this study can be accessed as Jupyter notebooks in the project GitHub repository: https://github.com/cartal/HCA_Heart.
References
Meilhac, S. M. & Buckingham, M. E. The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol. 15, 705–724 (2018).
Svensson, V., Vento-Tormo, R. & Teichmann, S. A. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protocols 13, 599–604 (2018).
Wang, L. et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 22, 108–119 (2020).
Tucker, N. R. et al. Transcriptional and cellular diversity of the human heart. Circulation 142, 466–482 (2020).
Chung, A. K. et al. Women have higher left ventricular ejection fractions than men independent of differences in left ventricular volume: the Dallas Heart Study. Circulation 113, 1597–1604 (2006).
Johnson, E. K., Matkovich, S. J. & Nerbonne, J. M. Regional differences in mRNA and lncRNA expression profiles in non-failing human atria and ventricles. Sci. Rep. 8, 13919 (2018).
Delplancq, G. et al. Cardiomyopathy due to PRDM16 mutation: first description of a fetal presentation, with possible modifier genes. Am. J. Med. Genet. C. Semin. Med. Genet. 184, 129–135 (2020).
Kim, S.-Y., Yasuda, S., Tanaka, H., Yamagata, K. & Kim, H. Non-clustered protocadherin. Cell Adhes. Migr. 5, 97–105 (2011).
Donlin, L. T. et al. Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organization. Genes Dev. 26, 114–119 (2012).
Mazzotta, S. et al. Distinctive roles of canonical and noncanonical Wnt signaling in human embryonic cardiomyocyte development. Stem Cell Reports 7, 764–776 (2016).
Shenton, F. C. & Pyner, S. Expression of transient receptor potential channels TRPC1 and TRPV4 in venoatrial endocardium of the rat heart. Neuroscience 267, 195–204 (2014).
Gao, M. et al. Conserved expression of the PRELI domain containing 2 gene (Prelid2) during mid-later-gestation mouse embryogenesis. J. Mol. Histol. 40, 227–233 (2009).
Putku, M. et al. CDH13 promoter SNPs with pleiotropic effect on cardiometabolic parameters represent methylation QTLs. Hum. Genet. 134, 291–303 (2015).
Liu, R. & Jin, J.-P. Calponin isoforms CNN1, CNN2 and CNN3: regulators for actin cytoskeleton functions in smooth muscle and non-muscle cells. Gene 585, 143–153 (2016).
Besnard, S. et al. Smooth muscle dysfunction in resistance arteries of the staggerer mouse, a mutant of the nuclear receptor RORα. Circ. Res. 90, 820–825 (2002).
Mikhailov, A. T. & Torrado, M. The enigmatic role of the ankyrin repeat domain 1 gene in heart development and disease. Int. J. Dev. Biol. 52, 811–821 (2008).
Kwapiszewska, G. et al. Fhl-1, a new key protein in pulmonary hypertension. Circulation 118, 1183–1194 (2008).
Geng, T. et al. H19 lncRNA promotes skeletal muscle insulin sensitivity in part by targeting AMPK. Diabetes 67, 2183–2198 (2018).
Huang, L. et al. Critical roles of Xirp proteins in cardiac conduction and their rare variants identified in sudden unexplained nocturnal death syndrome and Brugada syndrome in Chinese Han population. J. Am. Heart Assoc. 7, e006320 (2018).
Fittipaldi, S. et al. Alpha B-crystallin induction in skeletal muscle cells under redox imbalance is mediated by a JNK-dependent regulatory mechanism. Free Radic. Biol. Med. 86, 331–342 (2015).
Rajakumari, S. et al. EBF2 determines and maintains brown adipocyte identity. Cell Metab. 17, 562–574 (2013).
2016 AHA late-breaking basic science abstracts. Circ. Res. 119, e160–e171 (2016).
Bolotta, A. et al. New insights into the hepcidin-ferroportin axis and iron homeostasis in iPSC-derived cardiomyocytes from Friedreich’s ataxia patient. Oxid. Med. Cell. Longev. 2019, 7623023 (2019).
Lakhal-Littleton, S. et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. eLife 5, e19804 (2016).
Medioni, C. et al. Expression of Slit and Robo genes in the developing mouse heart. Dev. Dyn. 239, 3303–3311 (2010).
Lee, J. H., Protze, S. I., Laksman, Z., Backx, P. H. & Keller, G. M. Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell 21, 179–194.e4 (2017).
Liu, C., Luo, N., Tung, C.-Y., Perrin, B. J. & Zhao, B. GRXCR2 regulates taperin localization critical for stereocilia morphology and hearing. Cell Rep. 25, 1268–1280.e4 (2018).
Liu, X. et al. Single-cell RNA-Seq of the developing cardiac outflow tract reveals convergent development of the vascular smooth muscle cells. Cell Rep. 28, 1346–1361.e4 (2019).
Kalucka, J. et al. Single-cell transcriptome atlas of murine endothelial cells. Cell 180, 764–779.e20 (2020).
Pober, J. S., Merola, J., Liu, R. & Manes, T. D. Antigen presentation by vascular cells. Front. Immunol. 8, 1907 (2017).
Corada, M., Morini, M. F. & Dejana, E. Signaling pathways in the specification of arteries and veins. Arterioscler. Thromb. Vasc. Biol. 34, 2372–2377 (2014).
Kashiwazaki, M. et al. A high endothelial venule-expressing promiscuous chemokine receptor DARC can bind inflammatory, but not lymphoid, chemokines and is dispensable for lymphocyte homing under physiological conditions. Int. Immunol. 15, 1219–1227 (2003).
Awwad, K. et al. Role of secreted modular calcium-binding protein 1 (SMOC1) in transforming growth factor β signalling and angiogenesis. Cardiovasc. Res. 106, 284–294 (2015).
Tang, J. et al. Genetic fate mapping defines the vascular potential of endocardial cells in the adult heart. Circ. Res. 122, 984–993 (2018).
Stallcup, W. B. The NG2 proteoglycan in pericyte biology. Adv. Exp. Med. Biol. 1109, 5–19 (2018).
Chen, Q. et al. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat. Commun. 7, 12422 (2016).
Barker, N. & Clevers, H. Leucine-rich repeat-containing G-protein-coupled receptors as markers of adult stem cells. Gastroenterology 138, 1681–1696 (2010).
Daniel, J.-M. et al. Regulator of G-protein signaling 5 prevents smooth muscle cell proliferation and attenuates neointima formation. Arterioscler. Thromb. Vasc. Biol. 36, 317–327 (2016).
Vanlandewijck, M. et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480 (2018).
Sweeney, M. & Foldes, G. It takes two: endothelial-perivascular cell cross-talk in vascular development and disease. Front. Cardiovasc. Med. 5, 154 (2018).
Zhou, B. et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Invest. 121, 1894–1904 (2011).
Burstein, B., Libby, E., Calderone, A. & Nattel, S. Differential behaviors of atrial versus ventricular fibroblasts: a potential role for platelet-derived growth factor in atrial-ventricular remodeling differences. Circulation 117, 1630–1641 (2008).
Zhang, F., Li, C., Halfter, H. & Liu, J. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene 22, 894–905 (2003).
Lim, H. Y. et al. Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49, 326–341.e7 (2018).
Dick, S. A. et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29–39 (2019).
Jaitin, D. A. et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178, 686–698.e14 (2019).
Nahrendorf, M., Pittet, M. J. & Swirski, F. K. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 121, 2437–2445 (2010).
Heinrichs, D. et al. Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc. Natl Acad. Sci. USA 108, 17444–17449 (2011).
Valiño-Rivas, L. et al. CD74 in kidney disease. Front. Immunol. 6, 483 (2015).
Hortells, L., Johansen, A. K. Z. & Yutzey, K. E. Cardiac fibroblasts and the extracellular matrix in regenerative and nonregenerative hearts. J. Cardiovasc. Dev. Dis. 6, 29 (2019).
Bajpai, G. et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 24, 1234–1245 (2018).
Kegel, L., Aunin, E., Meijer, D. & Bermingham, J. R. LGI proteins in the nervous system. ASN Neuro 5, 167–181 (2013).
Struk, A. A. et al. Self-regulation and the foraging gene (PRKG1) in humans. Proc. Natl Acad. Sci. USA 116, 4434–4439 (2019).
Sahara, M. et al. Population and single-cell analysis of human cardiogenesis reveals unique LGR5 ventricular progenitors in embryonic outflow tract. Dev. Cell 48, 475–490.e7 (2019).
Lerman, B. B. Outflow tract ventricular arrhythmias: an update. Trends Cardiovasc. Med. 25, 550–558 (2015).
Dagda, R. K. et al. The spinocerebellar ataxia 12 gene product and protein phosphatase 2A regulatory subunit Bβ2 antagonizes neuronal survival by promoting mitochondrial fission. J. Biol. Chem. 283, 36241–36248 (2008).
Wang, L. et al. Polymorphisms of the tumor suppressor gene LSAMP are associated with left main coronary artery disease. Ann. Hum. Genet. 72, 443–453 (2008).
Bruce, K. D. et al. Lipoprotein lipase is a feature of alternatively-activated microglia and may facilitate lipid uptake in the CNS during demyelination. Front. Mol. Neurosci. 11, 57 (2018).
Kim, H.-S. et al. Schwann cell precursors from human pluripotent stem cells as a potential therapeutic target for myelin repair. Stem Cell Reports 8, 1714–1726 (2017).
Antonopoulos, A. S. & Antoniades, C. The role of epicardial adipose tissue in cardiac biology: classic concepts and emerging roles. J. Physiol. (Lond.) 595, 3907–3917 (2017).
Vijay, J. et al. Single-cell analysis of human adipose tissue identifies depot and disease specific cell types. Nat. Metab. 2, 97–109 (2020).
Hepler, C. et al. Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. eLife 7, e39636 (2018).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Watanabe, K., Umićević Mirkov, M., de Leeuw, C. A., van den Heuvel, M. P. & Posthuma, D. Genetic mapping of cell type specificity for complex traits. Nat. Commun. 10, 3222 (2019).
Leid, J. et al. Primitive embryonic macrophages are required for coronary development and maturation. Circ. Res. 118, 1498–1511 (2016).
Chen, B., Brickshawana, A. & Frangogiannis, N. G. The functional heterogeneity of resident cardiac macrophages in myocardial injury CCR2+ cells promote inflammation, whereas CCR2− cells protect. Circ. Res. 124, 183–185 (2019).
Vaduganathan, M. et al. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N. Engl. J. Med. 382, 1653–1659 (2020).
Ferreira, P. G. et al. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nat. Commun. 9, 490 (2018).
Madissoon, E. et al. scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 21, 1 (2019).
Litvinukova, M. et al. Single cell and single nuclei analysis human heart tissue. https://doi.org/10.17504/protocols.io.veae3ae (2018).
Noseda, M. et al. PDGFRα demarcates the cardiogenic clonogenic Sca1+ stem/progenitor cell in adult murine myocardium. Nat. Commun. 6, 6930 (2015).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019).
McInnes, L. & Healy, J. UMAP: Uniform manifold approximation and projection for dimension reduction. Preprint at https://arxiv.org/abs/1802.03426 (2018).
Popescu, D.-M. et al. Decoding human fetal liver haematopoiesis. Nature 574, 365–371 (2019).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
Polański, K. et al. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965 (2019).
Chen, J., Bardes, E. E., Aronow, B. J. & Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–W311 (2009).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–W97 (2016).
Lotfollahi, M., Wolf, F. A. & Theis, F. J. scGen predicts single-cell perturbation responses. Nat. Methods 16, 715–721 (2019).
Miao, Z. et al. Putative cell type discovery from single-cell gene expression data. Nat. Methods 17, 621–628 (2020).
Soneson, C. & Robinson, M. D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 15, 255–261 (2018).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Bergen, V., Lange, M., Peidli, S., Alexander Wolf, F. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. https://doi.org/10.1038/s41587-020-0591-3 (2020).
Traag, V. A., Waltman, L. & van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019).
Miragaia, R. J. et al. Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity 50, 493–504.e7 (2019).
Dey, G. et al. Signaling network of oncostatin M pathway. J. Cell Commun. Signal. 7, 103–108 (2013).
Abe, H. et al. Macrophage hypoxia signaling regulates cardiac fibrosis via oncostatin M. Nat. Commun. 10, 2824 (2019).
Bian, Z. et al. Deciphering human macrophage development at single-cell resolution. Nature 582, 571–576 (2020).
Wong, A. P., Nili, N. & Strauss, B. H. In vitro differences between venous and arterial-derived smooth muscle cells: potential modulatory role of decorin. Cardiovasc. Res. 65, 702–710 (2005).
Pober, J. S. & Sessa, W. C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 7, 803–815 (2007).
McDonald, A. I. et al. Endothelial regeneration of large vessels is a biphasic process driven by local cells with distinct proliferative capacities. Cell Stem Cell 23, 210–225.e6 (2018).
Chen, S.-C., Liu, Y.-C., Shyu, K.-G. & Wang, D. L. Acute hypoxia to endothelial cells induces activating transcription factor 3 (ATF3) expression that is mediated via nitric oxide. Atherosclerosis 201, 281–288 (2008).
Hashimoto, K. et al. Single-cell transcriptomics reveals expansion of cytotoxic CD4 T cells in supercentenarians. Proc. Natl Acad. Sci. USA 116, 24242–24251 (2019).
Roychoudhuri, R. et al. BACH2 regulates CD8+ T cell differentiation by controlling access of AP-1 factors to enhancers. Nat. Immunol. 17, 851–860 (2016).
Ahrends, T. et al. CD4+ T cell help creates memory CD8+ T cells with innate and help-independent recall capacities. Nat. Commun. 10, 5531 (2019).
Kimmel, J. C. & Kelley, D. R. scNym: Semi-supervised adversarial neural networks for single cell classification. Preprint at https://doi.org/10.1101/2020.06.04.132324 (2020).
Stewart, B. J. et al. Spatiotemporal immune zonation of the human kidney. Science 365, 1461–1466 (2019).
James, K. R. et al. Distinct microbial and immune niches of the human colon. Nat. Immunol. 21, 343–353 (2020).
Acknowledgements
This publication is part of the Human Cell Atlas (www.humancellatlas.org/publications). We acknowledge the Sanger Flow Cytometry Facility, Sanger Cellular Generation and Phenotyping (CGaP) Core Facility, and Sanger Core Sequencing pipeline for support with sample processing and sequencing library preparation. We acknowledge the HMS MicRoN Core for their support and assistance in this work. We thank J. Eliasova for graphical images; M. Prete, V. Kiselev and O. Tarkowska for IT support; S. Aldridge for editing the manuscript; C. Domínguez-Conde, J. Park, K. James, K. Tuong, R. Elmentaite and M. Haniffa for discussions about immune cell annotation, and M. Luecken, M. Lotfollahi, D. Fischer and F. Theis for discussions on computational analyses. We thank T. R. W. Oliver and L. Campos for their assistance with orientation of basic cardiac histology. M.N. is the recipient of a British Heart Foundation (BHF) grant (PG/16/47/32156) and J.J.B. is recipient of a BHF grant (FS/13/12/30037, PG/15/57/31580, PG/17/71/33242). M.N., N.H. and S.A.T. are funded by a BHF/DZHK grant (SP/19/1/34461). N.H. is the recipient of an ERC Advanced Grant under the European Union Horizon 2020 Research and Innovation Program (grant agreement AdG788970) and the Federal Ministry of Education and Research of Germany in the framework of CaRNAtion (031L0075A). N.H., C.E.S. and J.G.S. are supported by grants from the Leducq Fondation (16CVD03). D.R. is supported by the German Research Foundation (DFG). H.Z. is supported by the long-term Chinese Council Scholarship (CSC). M.K. received funding provided by the Alexander von Humboldt Foundation. H.Z. was supported by an EMBO long-term postdoctoral fellowship. G.O. received funding provided by the CIHR Canadian Institutes for Health Research, HSF Heart and Stroke Foundation and AI Alberta Innovates. This project has been made possible in part by grant number 2019-202666 from the Chan Zuckerberg Initiative (C.E.S., J.G.S., M.N., M.H., N.H. and S.A.T.). C.T.-L., K.P., E.F., E.T., K.R., O.B., H.Z. and S.A.T. are supported by the Wellcome Sanger Institute grant (WT206194) and the Wellcome Science Strategic Support for a Pilot for the Human Cell Atlas (WT211276/Z/18/Z)). J.G.S. and C.E.S. are supported by NIH grants (2R01HL080494, 2R01HL080494, 1UM1HL098166). J.G.S. and C.E.S. are supported by Engineering Research Centers Program of the National Science Foundation under NSF Cooperative Agreement no. EEC-1647837. C.E.S. and B.M. are supported by the Howard Hughes Medical Institute. We are grateful to the deceased donors and their families, the Human Organ Procurement and Exchange Program (HOPE) and to the Cambridge Biorepository for Translational Medicine (CBTM) for access to human tissue. E.R.N. was supported by a fellowship from the Sarnoff Cardiovascular Research Foundation.
Author information
Authors and Affiliations
Contributions
Conceived the study: N.H., S.A.T., M.N., C.E.S., J.G.S., H.M. Acquired tissue: K.M., K.S.P., G.O., H.Z., A.V. Processed tissue: M. Litviňuková, D.R., S.S., E.F., L.T., H.W., J.M.G., B.M., D.M.D., K.R., M.N. Computational methods development: C.T.-L., K.P., M.H., J.G.S. Cell biology methods development: M. Litviňuková, E.L.L., M.N., H.M., D.R. Histopathological evaluation: J.J.B., M.N. Data processing: C.T.-L., K.P., G.P. Global analysis: C.T.-L., D.R., M. Litviňuková, C.L.W., E.L.L., J.G.S. Cardiomyocyte analysis: D.R., C.L.W., E.R.N., H.M., D.M.D., N.H., J.G.S., C.E.S. Vascular analysis: M. Litviňuková, M.N., C.T.-L., M.K., S.A.T. Fibroblast analysis: E.L.L., H.M., D.R., M. Litviňuková, M.K., M.N., J.G.S., C.E.S., N.H. Immune analysis: C.T.-L., S.A.T. Neuronal analysis: E.R.N., D.R., C.L.W., J.G.S., C.E.S. Adipocyte analysis: M. Lee, M.N. Comparative analysis: C.T.-L., M. Litviňuková, M.N., S.A.T. GWAS analysis: M.H., C.T.-L., N.H., S.A.T. Interpreted results: M. Litviňuková, C.T.-L., D.R., H.M., C.L.W, E.L.L., M.H., J.B., M.N., C.E.S., J.G.S., S.A.T., N.H. Wrote the manuscript: M. Litviňuková, C.T.-L., H.M., D.R., C.L.W., E.L.L., M.K., M.H., J.G.S., C.E.S., M.N., N.H., S.A.T.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Expression of the canonical markers.
a, UMAP embedding of selected canonical markers shown in Fig. 1c. b, Scaled expression (log2-transformed fold change, log2FC) of selected canonical markers shown in Fig. 1c. c, Expression (log2FC) of marker genes from Fig. 1c in each source highlighting that the same marker genes are used for identification of the same cell types in both cells and nuclei. d, Multiplexed smFISH staining of cell type-specific transcripts from Fig. 1e in right ventricles (top): TTN (green, cardiomyocytes) and CDH5 (red, EC) right atrium (middle): NPPA (green, aCM) and DCN (red, FB) and LA (bottom): MYH11 (green, SMC) and KCNJ8 (red, pericytes), nuclei are DAPI-stained (dark blue). Scale bars, 20 μm. For details on statistics and reproducibility, see Methods.
Extended Data Fig. 2 Source and region covariates of the global dataset.
a, UMAP embedding of the major cell types coloured by source. b, UMAP embedding highlighting the individual sources c, Distribution of cell types obtained by each source. Data are available in Supplementary Table 29. Further analyses and descriptions are available in the Methods and Supplementary Table 30. d, UMAP embedding of the major cell types coloured by region. e, UMAP embedding highlighting the individual regions f, Distribution of cell types across the six sampled regions (nuclei only). Data are available in Supplementary Table 2.
Extended Data Fig. 3 Ventricular and atrial cardiomyocytes.
a, Expression (log2FC) of selected marker genes in ventricular cardiomyocyte subpopulations. b, Expression (log2FC) of selected marker genes in atrial cardiomyocyte subpopulations c, Single channel multiplexed smFISH images of overlay shown in Fig. 2c, d, g, h. d, Expression (log2FC) of specific markers in cardiomyocyte subpopulations. I and II, PCDH7 expression in ventricular and atrial cardiomyocytes, respectively. III, PRELID2 expression is highest in vCM2 and is enriched in right ventricles. IV and V, CNN1 expression is enriched in both vCM3 and aCM3. VI and VII, HAMP expression is enriched in the right atrium. e, Multiplexed smFISH of transcripts enriched in cardiomyocyte subpopulations. Left, expression of TNNT2 (green) and PCDH7 (red) in left ventricles. Right, expression of TNNT2 (green) and CNN1 (red) in right ventricles, nuclei are DAPI-stained (dark blue). Scale bars, 10 μm. f, Gene Ontology analysis results for vCM4 showing significant terms related to energy metabolism and muscle contraction. Data are available in Supplementary Table 6. g, Multiplexed smFISH of positive and negative RNAscope control probes. Scale bars, 5 μm. For details on statistics and reproducibility, see Methods.
Extended Data Fig. 4 Vascular and mesothelial populations.
a, Scaled expression (log2FC) of selected marker genes for EC subpopulations. b, c, Distribution of the EC subpopulations across the sources (b) and the regions (c) (nuclei only). Data are available in Supplementary Table 9. d, Scaled expression (log2FC) of selected marker genes of pericytes and smooth muscle cell subpopulations. e, f, Distribution of the mural subpopulations across the sources (e) and the regions (f) (nuclei only). Data are available in Supplementary Table 9. g, Multiplexed smFISH in apex section shows MYH11 (yellow) expression in vascular SMC (thick in artery and very thin in nearby small calibre vein), CDH5 (red) in the endothelium, and SEMA3G (cyan) and ACKR1 (green) expression respectively in arterial and venous ECs, nuclei are DAPI-stained (dark blue). Scale bars, 20 μm. h, UMAP embedding of vascular and mesothelial cells with stochastic representation of the RNA velocity. i, Latent time of the vascular cells showing predicted directionalities of the cell populations based on the RNA splicing dynamics. The analysis uses only cells, nuclei are omitted. EC_cap, capillary ECs; EC_art, arterial ECs; EC_ven, venous ECs; EC atrial, atrial endothelial cells; EC_ln, lymphatic endothelial cells; PC, pericytes; PC_str, stromal pericytes; SMC_basic, smooth muscle cells; SMC_art, arterial smooth muscle cells. j, Predicted cell–cell interactions using the CellphoneDB statistical inference framework on 39,000 cells from 14 biologically independent individuals (n = 14). Selected ligand–receptor interactions show specificity of NOTCH ligands-receptors pairing in defined vasculature beds. Mean of combined gene expression of interacting pairs (log2FC). CellPhoneDB P value of the specificity of the interactions = 10 × 10−5. The red rectangles highlight the arterial interactions and the blue rectangle highlights venous interactions depicted in Fig. 3d. Notably, even though the EC6_ven and SMC2_art interaction is unexpected, we cannot exclude that those cell states are restricted to their respective vascular beds. Further validation is needed to determine the exact spatial distribution of EC6_ven and SMC2_art and subsequently verify whether the interaction is plausible in vivo. Data are available in Supplementary Table 10. k, Scaled expression (log2FC) of the ligands and receptors from g across the vascular populations described in Fig. 3a. l, Scaled expression (log2FC) of selected marker genes of mesothelial cells. m, Multiplexed smFISH localizes the mesothelial cells expressing BNC1 into the epicardium of the left atria. CDH5 shows endothelial cells in the tissue and is absent from the mesothelial cells, nuclei are DAPI-stained (dark blue). Scale bars, 20 μm. n, Distribution of the mural subpopulations across the sampled regions (nuclei only). Data are available in Supplementary Table 9. For details on statistics and reproducibility, see Methods.
Extended Data Fig. 5 Vascular markers visualized on 10X Genomics Visium data.
a–d, Spatial expression (log2FC) of CDH5 (pan-EC marker), SEMA3G and GJA5 (arterial EC markers) (a), ACKR1 and PLVAP (venous EC markers) (b), MYH11 and ACTA2 (pan-SMC markers) (c), and JAG1 and NOTCH2 (d) on publicly available 10X Visium section of human left ventricle. JAG1 and NOTCH2 are the predicted interaction partners for arterial ECs and SMCs, respectively.
Extended Data Fig. 6 Fibroblasts.
a, Scaled expression (log2FC) of selected marker genes of identified FB populations. b, Enrichment for oncostatin M pathway for FB populations showing enriched activity in FB3. A list of genes with which the score was calculated is in Supplementary Table 12. c, Regional distribution per FB population. Some FB populations show enrichment in the atria (left and right), such as FB2 and FB3. FB1, FB4–FB6 are enriched in the ventricles (left, right, apex and interventricular septum). Data are available in Supplementary Table 35. d–f, Multiplexed smFISH for probes targeting FAP, LINC01133 and PTX3 confirming FB4, FB5 and FB3 subpopulations. FAP (red) is imaged in interventricular septum, LINC01133 (red) in apex and PTX3 (red) in right atrium tissue section. DCN (cyan) is used as a pan-FB marker, C1QA (green) as a pan-macrophage marker, nuclei are DAPI-stained (dark blue). Scale bars, 5 μm. g. Scaled expression (log2FC) of APOD and CFH genes, which represent differences between ventricular and atrial fibroblasts. h, Multiplexed smFISH of apex section representing DCN (cyan), APOD (red) and CFH (green), nuclei are DAPI-stained (dark blue). Although the APOD signal colocalized with DCN, expression of CFH was absent. Scale bars, 5 μm. i, UMAP embedding of the ventricular fibroblast cell-states. j, UMAP embedding of atrial fibroblasts cell types. k, Scaled expression (log2FC) of marker genes for ventricular fibroblast subpopulations. l, Scaled expression (log2FC) of marker genes for atrial fibroblast subpopulations. m, Scaled expression (log2FC) of ECM genes differentiating atrial (aFB1, aFB2) and ventricular (vFB2, vFB4) clusters which suggest different ECM mechanisms. For details on statistics and reproducibility, see Methods.
Extended Data Fig. 7 Covariates of immune cardiac populations.
a–j, UMAP embedding of cell source (a), donor (b), gender (c), type (d), number of genes (e), number of counts (f), percentage of mitochondrial genes (g), percentage of ribosomal genes (h), scrublet score (i) and annotation of the cell populations of the immune cells (j).
Extended Data Fig. 8 Immune cardiac populations.
a, Visualization of transcriptional signatures from published studies. The score values represent the likelihood of the external transcriptional signature to be present when comparing it against the transcriptional background of a cardiac immune population. Bajpai_2018 = CCR2-MERTK+ tissue-resident macrophages from ref. 51. Dick_2019 = self-renewing tissue macrophages from ref. 45. Bian_2020 = yolk sac-derived macrophages from ref. 90. The complete signature can be found in Supplementary Table 19. b, Expression (log2FC) of LYVE1, FOLR2 and TIMD4 characteristic of the self-renewing tissue-resident murine macrophages previously described45, as well as MERTK as previously described51 and the TREM2 expression associated to lipid-associated macrophages (LAM) previously described46. Complete signatures can be found in Supplementary Table 19. c, Scaled expression (log2FC) of genes differentiating DOCK4+ MP1 from DOCK4+ MP2: IL4R, ITGAM, STAT3, DOCK1, HIF1A and RASA2. d, Predicted cell–cell interactions calculated for 69,295 cardiomyocytes, fibroblasts and myeloid cells from 14 donors (n = 14) and enriched for ‘extracellular matrix organization’. Mean of combined gene expression of interacting pairs (log2FC). Data are available in Supplementary Table 17. e, Spatial mapping of the CD74–MIF interaction between LYVE1+MP and FB4 on a publicly available 10X Genomics Visium dataset for left ventricular myocardium. We identified four spots where we observe co-expression of FN1, LYVE1, CD74 and MIF, as predicted from the cell–cell interactions. The bar represents the log2FC. f, Confusion matrix for the logistic regression model trained on cardiac immune cells. This model reached an accuracy score of 0.6862, showing a stronger accuracy with lymphoid cells, compared with the myeloid ones.
Extended Data Fig. 9 Neuronal and adipocyte populations.
a, UMAP embedding identifies six neuronal subpopulations. b, Regional distribution of neuronal cell subpopulations identified in a. Data are available in Supplementary Table 20. c, Expression (log2FC) dot plot of selected marker genes in neuronal cell subpopulations. d, Multiplexed smFISH of NRXN1 (green) and PPP2R2B (red), nuclei were DAPI-stained (dark blue). Scale bars, 5 μm. For details on statistics and reproducibility, see Methods. e, UMAP embedding showing four adipocyte subpopulations. f, UMAP embedding of expression of gene markers associated with adipocytes (GPAM, FASN, ADIPOQ, LEP). g, Top five significantly enriched pathways for each adipocyte subpopulation, using differentially expressed genes calculated using the Wilcoxon rank sum test with Benjamini–Hochberg correction (logFC >0.5, P < 1.0 × 10−5) and tested using a hypergeometric distribution with Bonferroni correction as implemented in ToppFun. Data are available in Supplementary Table 21. h, Expression (log2FC) of adipocyte associated genes and select marker genes from the top enriched pathway for each adipocyte subpopulation.
Extended Data Fig. 10 Relevance for COVID-19 and GWAS studies.
a, Global expression (log2FC) of ACE2 in all cardiac cells. b–d, Gene expression of ACE2, TMPRSS2, CTSB and CTSL in cardiomyocytes (b), FBs (c) and vascular cells (d). e, Multiplexed smFISH expression of DCN (cyan), KCNJ8 (green) and ACE2 (red), nuclei are DAPI-stained (dark blue) marking fibroblasts (#; expression of DCN) and pericytes (*; co-expression of DCN and KCNJ8) in right ventricular tissue section. Scale bars, 5 μm. For statistics and reproducibility, see Methods. f, The colour coding of the heat map shows the −log10(P value) of the MAGMA GWAS enrichment analysis for the association between cell type-specific expression (y axis) and GWAS signals (x axis). The cell types refer to the subcluster annotations and GWAS studies refer to Supplementary Table 27. AF, atrial fibrillation; CAD, coronary artery disease; HF, heart failure; HR, heart rate; HT, hypertension; LVD, left ventricular diameter; NICM, non-ischaemic cardiomyopathy; PR, PR interval; PWAVE, P-wave duration; T2D, type 2 diabetes; QRS, QRS complex duration; QT, QT interval. Dots mark significant associations (FDR < 10%). The colour of the dots indicates the type of association as determined by pairwise conditional analysis (green: independent association, blue: partially jointly explained with other cell types, grey: explained away by other cell types). Data are available in Supplementary Table 28.
Extended Data Fig. 11 Skeletal muscle populations.
a, UMAP embedding of transcriptional data from skeletal muscle using cells and nuclei. Mural, pericytes and smooth muscle cells. b, Scaled expression (log2FC) of selected markers for the major skeletal muscle populations. c, UMAP embedding of vascular and stromal populations of skeletal muscle. d, Scaled expression (log2FC) of marker genes used in Extended Data Fig. 3 for identification of vascular cell states. e, Predicted cell–cell interactions inferred using CellphoneDB statistical inference framework in skeletal muscle cells with 9,220 cells from five donors (n = 5) depicting cell states from c, Selected ligand–receptor interactions show specificity of NOTCH ligand–receptor pairing in defined vasculature beds. The interactions of EC_art-SMC are highlighted by a red rectangle and EC_ven-SMC are highlighted by a blue rectangle. Colour of the dots indicates the mean expression level of interacting molecule in partner 1 and interacting molecule partner 2. Mean of combined gene expression of interacting pairs (log2FC). CellPhoneDB P value of the specificity of the interactions = 10 × 10−5. Data are available in Supplementary Table 10. f, Scaled expression (log2FC) of the ligands and receptors from Extended Data Fig. 3 depicted on vascular populations of skeletal muscle.
Extended Data Fig. 12 Analysis technical information.
a, Locations and representative histology section of six cardiac regions sampled, including right and left atrium, right and left ventricular free wall and left ventricular apex and interventricular septum. H&E, magnification ×10; scale bars, 500 μm. b, Spatial visualization of positive and negative RNAscope control probes. Scale bars, 5 μm. For statistics and reproducibility, see Methods. c, Heat map of top five significantly enriched Gene Ontology Biological Processes term for each of the vascular subpopulations from Fig. 3a. Data are available in Supplementary Table 25. d, SCCAF scores for each batch aligned manifold. For each population, we plotted the true positive (TPR) versus false positive (FPR) learning ratios from the subpopulation in each manifold. Next, we plotted how accurately the manifold represents each learned subpopulation based on the test training set and the CV cross-validation set. The closer the CV value to the test value, the better the manifold is at representing the subpopulations.
Supplementary information
Supplementary Figure 1
FACS gating strategy for removal of cell debris and purification of nuclei. a. Size gating to remove doublets and aggregates was applied (SSC-A, FSC-A, SSC-W, SSC-H, FSC-H, FSC-W), followed by sorts for Hoechst-positive nuclei (purple population). Used for all nuclei processed in North America b. Example of negative control (no NucBlue Live ReadyProbes Reagent applied, ThermoFisher) with the same parameters as a. c. Gating strategy used for all nuclei processed in the United Kingdom. The Hoechst-positive nuclei were selected in P1. Further gating for size was applied (P2, P3) to remove doublets and aggregates. d. Example of a negative control with the same parameters as c.
Supplementary Tables
This zipped file contains Supplementary Tables 1-35 and their legends.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Litviňuková, M., Talavera-López, C., Maatz, H. et al. Cells of the adult human heart. Nature 588, 466–472 (2020). https://doi.org/10.1038/s41586-020-2797-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2797-4
This article is cited by
-
Catecholamine treatment induces reversible heart injury and cardiomyocyte gene expression
Intensive Care Medicine Experimental (2024)
-
Macrophage and fibroblast trajectory inference and crosstalk analysis during myocardial infarction using integrated single-cell transcriptomic datasets
Journal of Translational Medicine (2024)
-
Identification of porcine fast/slow myogenic exosomes and their regulatory effects on lipid accumulation in intramuscular adipocytes
Journal of Animal Science and Biotechnology (2024)
-
The Abl1 tyrosine kinase is a key player in doxorubicin-induced cardiomyopathy and its p53/p73 cell death mediated signaling differs in atrial and ventricular cardiomyocytes
Journal of Translational Medicine (2024)
-
Spatial multi-omics: novel tools to study the complexity of cardiovascular diseases
Genome Medicine (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
