
Abstract
Natural products serve as chemical blueprints for most antibiotics in clinical use. The evolutionary process by which these molecules arise is inherently accompanied by the co-evolution of resistance mechanisms that shorten the clinical lifetime of any given class of antibiotics1. Virginiamycin acetyltransferase (Vat) enzymes are resistance proteins that provide protection against streptogramins2, potent antibiotics against Gram-positive bacteria that inhibit the bacterial ribosome3. Owing to the challenge of selectively modifying the chemically complex, 23-membered macrocyclic scaffold of group A streptogramins, analogues that overcome the resistance conferred by Vat enzymes have not been previously developed2. Here we report the design, synthesis, and antibacterial evaluation of group A streptogramin antibiotics with extensive structural variability. Using cryo-electron microscopy and forcefield-based refinement, we characterize the binding of eight analogues to the bacterial ribosome at high resolution, revealing binding interactions that extend into the peptidyl tRNA-binding site and towards synergistic binders that occupy the nascent peptide exit tunnel. One of these analogues has excellent activity against several streptogramin-resistant strains of Staphylococcus aureus, exhibits decreased rates of acetylation in vitro, and is effective at lowering bacterial load in a mouse model of infection. Our results demonstrate that the combination of rational design and modular chemical synthesis can revitalize classes of antibiotics that are limited by naturally arising resistance mechanisms.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
Data availability
Models and maps generated during this study are available in the EMDB and PDB (accessions are listed in Extended Data Tables 4 and 5). Source data are provided with this paper.
Code availability
Forcefield-based refinement is available in PHENIX (versions 1.15 and later) using beta features available in Schrӧdinger 2019-3. Python code for analysing IVT data and VatA kinetics data are available on github: https://github.com/fraser-lab/streptogramin.
References
Wright, P. M., Seiple, I. B. & Myers, A. G. The evolving role of chemical synthesis in antibacterial drug discovery. Angew. Chem. Int. Edn Engl. 53, 8840–8869 (2014).
Stogios, P. J. et al. Potential for reduction of streptogramin A resistance revealed by structural analysis of acetyltransferase VatA. Antimicrob. Agents Chemother. 58, 7083–7092 (2014).
Vazquez, D. in Mechanism of Action (eds Gottlieb, D. & Shaw, P. D.) 387–403 (Springer Berlin Heidelberg, 1967).
Waglechner, N. & Wright, G. D. Antibiotic resistance: it’s bad, but why isn’t it worse? BMC Biol. 15, 84 (2017).
Seiple, I. B. et al. A platform for the discovery of new macrolide antibiotics. Nature 533, 338–345 (2016).
Charest, M. G., Lerner, C. D., Brubaker, J. D., Siegel, D. R. & Myers, A. G. A convergent enantioselective route to structurally diverse 6-deoxytetracycline antibiotics. Science 308, 395–398 (2005).
Vidaillac, C., Parra-Ruiz, J., Winterfield, P. & Rybak, M. J. In vitro pharmacokinetic/pharmacodynamic activity of NXL103 versus clindamycin and linezolid against clinical Staphylococcus aureus and Streptococcus pyogenes isolates. Int. J. Antimicrob. Agents 38, 301–306 (2011).
Wilson, D. N. The A-Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 44, 393–433 (2009).
Noeske, J. et al. Synergy of streptogramin antibiotics occurs independently of their effects on translation. Antimicrob. Agents Chemother. 58, 5269–5279 (2014).
Hershberger, E., Donabedian, S., Konstantinou, K. & Zervos, M. J. Quinupristin-dalfopristin resistance in Gram-positive bacteria: mechanism of resistance and epidemiology. Clin. Infect. Dis. 38, 92–98 (2004).
Sharkey, L. K. R. & O’Neill, A. J. Antibiotic resistance ABC-F proteins: bringing target protection into the limelight. ACS Infect. Dis. 4, 239–246 (2018).
Leclercq, R. & Courvalin, P. Bacterial resistance to macrolide, lincosamide, and streptogramin antibiotics by target modification. Antimicrob. Agents Chemother. 35, 1267–1272 (1991).
Haroche, J. et al. Clonal diversity among streptogramin A-resistant Staphylococcus aureus isolates collected in French hospitals. J. Clin. Microbiol. 41, 586–591 (2003).
Werner, G., Cuny, C., Schmitz, F. J. & Witte, W. Methicillin-resistant, quinupristin-dalfopristin-resistant Staphylococcus aureus with reduced sensitivity to glycopeptides. J. Clin. Microbiol. 39, 3586–3590 (2001).
Valour, F. et al. Pristinamycin in the treatment of MSSA bone and joint infection. J. Antimicrob. Chemother. 71, 1063–1070 (2016).
Delgado, G. Jr, Neuhauser, M. M., Bearden, D. T. & Danziger, L. H. Quinupristin-dalfopristin: an overview. Pharmacotherapy 20, 1469–1485 (2000).
Politano, A. D. & Sawyer, R. G. NXL-103, a combination of flopristin and linopristin, for the potential treatment of bacterial infections including community-acquired pneumonia and MRSA. Curr. Opin. Investig. Drugs 11, 225–236 (2010).
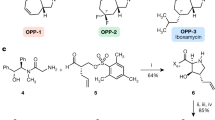
Li, Q. & Seiple, I. B. Modular, scalable synthesis of group A Streptogramin antibiotics. J. Am. Chem. Soc. 139, 13304–13307 (2017).
Li, Q. & Seiple, I. B. A concise route to virginiamycin M2. Tetrahedron 75, 3309–3318 (2019).
Schlessinger, R. H. & Li, Y.-J. Total synthesis of (−)-virginiamycin M2 using second-generation vinylogous urethane chemistry. J. Am. Chem. Soc. 118, 3301–3302 (1996).
Entwistle, D. A., Jordan, S. I., Montgomery, J. & Pattenden, G. Total synthesis of the virginiamycin antibiotic 14,15-anhydropristinamycin IIB. J. Chem. Soc. Perkin Trans. I 1315–1317 (1996).
Tavares, F., Lawson, J. P. & Meyers, A. I. Total synthesis of streptogramin antibiotics. (−)-Madumycin II. J. Am. Chem. Soc. 118, 3303–3304 (1996).
Ghosh, A. K. & Liu, W. A convergent, enantioselective total synthesis of streptogramin antibiotic (−)-madumycin II. J. Org. Chem. 62, 7908–7909 (1997).
Breuilles, P. & Uguen, D. Total synthesis of pristinamycin IIB. Tetrahedr. Lett. 39, 3149–3152 (1998).
Entwistle, D. A. Total synthesis of oxazole-based virginiamycin antibiotics: 14,15-anhydropristinamycin IIB. Synthesis 1998, 603–612 (1998).
Dvorak, C. A. et al. The synthesis of streptogramin antibiotics: (−)-griseoviridin and its C-8 epimer. Angew. Chem. Int. Ed. Engl. 39, 1664–1666 (2000).
Wu, J. & Panek, J. S. Total synthesis of (−)-virginiamycin M2. Angew. Chem. Int. Edn Engl. 49, 6165–6168 (2010).
Wu, J. & Panek, J. S. Total synthesis of (−)-virginiamycin M2: application of crotylsilanes accessed by enantioselective Rh(II) or Cu(I) promoted carbenoid Si–H insertion. J. Org. Chem. 76, 9900–9918 (2011).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018).
Li, J. et al. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins 79, 2794–2812 (2011).
Harms, J. M., Schlünzen, F., Fucini, P., Bartels, H. & Yonath, A. Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin. BMC Biol. 2, 4 (2004).
Osterman, I. A. et al. Madumycin II inhibits peptide bond formation by forcing the peptidyl transferase center into an inactive state. Nucleic Acids Res. 45, 7507–7514 (2017).
Hansen, J. L., Moore, P. B. & Steitz, T. A. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 330, 1061–1075 (2003).
Tu, D., Blaha, G., Moore, P. B. & Steitz, T. A. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell 121, 257–270 (2005).
Hoang, N. H. et al. Regio-selectively reduced streptogramin A analogue, 5,6-dihydrovirginiamycin M1 exhibits improved potency against MRSA. Lett. Appl. Microbiol. 57, 393–398 (2013).
Kingston, D. G. I., Kolpak, M. X., LeFevre, J. W. & Borup-Grochtmann, I. Biosynthesis of antibiotics of the virginiamycin family. 3. Biosynthesis of virginiamycin M1. J. Am. Chem. Soc. 105, 5106–5110 (1983).
Richter, M. F. et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545, 299–304 (2017).
Sharkey, L. K. R., Edwards, T. A. & O’Neill, A. J. ABC-F proteins mediate antibiotic resistance through ribosomal protection. MBio 7, e01975 (2016).
Radika, K. & Northrop, D. B. Correlation of antibiotic resistance with Vmax/Km ratio of enzymatic modification of aminoglycosides by kanamycin acetyltransferase. Antimicrob. Agents Chemother. 25, 479–482 (1984).
Knies, J. L., Cai, F. & Weinreich, D. M. Enzyme efficiency but not thermostability drives cefotaxime resistance evolution in TEM-1 β-lactamase. Mol. Biol. Evol. 34, 1040–1054 (2017).
Polikanov, Y. S., Steitz, T. A. & Innis, C. A. A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nat. Struct. Mol. Biol. 21, 787–793 (2014).
Renaud, J.-P. et al. Cryo-EM in drug discovery: achievements, limitations and prospects. Nat. Rev. Drug Discov. 17, 471–492 (2018).
Wong, W. et al. Mefloquine targets the Plasmodium falciparum 80S ribosome to inhibit protein synthesis. Nat. Microbiol. 2, 17031 (2017).
Llano-Sotelo, B. et al. Binding and action of CEM-101, a new fluoroketolide antibiotic that inhibits protein synthesis. Antimicrob. Agents Chemother. 54, 4961–4970 (2010).
Tropea, J. E., Cherry, S. & Waugh, D. S. Expression and purification of soluble His6-tagged TEV protease. Methods Mol. Biol. 498, 297–307 (2009).
Kuhn, M. L., Majorek, K. A., Minor, W. & Anderson, W. F. Broad-substrate screen as a tool to identify substrates for bacterial Gcn5-related N-acetyltransferases with unknown substrate specificity. Protein Sci. 22, 222–230 (2013).
Winter, G. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 (2010).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Schuwirth, B. S. et al. Structures of the bacterial ribosome at 3.5 A resolution. Science 310, 827–834 (2005).
Passmore, L. A. & Russo, C. J. Specimen preparation for high-resolution cryo-EM. Methods Enzymol. 579, 51–86 (2016).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Cheng, A. et al. High resolution single particle cryo-electron microscopy using beam-image shift. J. Struct. Biol. 204, 270–275 (2018).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Grant, T., Rohou, A. & Grigorieff, N. cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383 (2018).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Noeske, J. et al. High-resolution structure of the Escherichia coli ribosome. Nat. Struct. Mol. Biol. 22, 336–341 (2015).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D 65, 1074–1080 (2009).
Roos, K. et al. OPLS3e: extending force field coverage for drug-like small molecules. J. Chem. Theory Comput. 15, 1863–1874 (2019).
Sindhikara, D. et al. Improving accuracy, diversity, and speed with prime macrocycle conformational sampling. J. Chem. Inf. Model. 57, 1881–1894 (2017).
Bochevarov, A. D. et al. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 113, 2110–2142 (2013).
Acknowledgements
We thank F. Ward and J. Cate for initial advice on ribosome purifications and translation assays, E.Nogales and a UCSF-UCB Sackler Sabbatical Exchange Fellowship (J.S.F.) for initial cryo-EM access and training. A.A.T. and J.P. were supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1650113. D.J.L. was supported by a Postdoctoral Individual National Research Award NIH AI148120. H.A.C. was supported by a National Institute on Minority Health and Health Disparities (NIMHD) research diversity supplement under NIH GM123159. This project was funded by the UCSF Program for Breakthrough Biomedical Research, funded in part by the Sandler Foundation (J.S.F. and I.B.S.), a Sangvhi-Agarwal Innovation Award (J.S.F.), Packard Fellowships from the David and Lucile Packard Foundation (J.S.F. and I.B.S.), NIH GM123159 (J.S.F.), and NIH GM128656 (I.B.S.). We thank G. Meigs and J. Holton at Beamline 8.3.1 at the Advanced Light Source, which is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15-328599, the National Institutes of Health (R01 GM124149 and P30 GM124169), Plexxikon Inc., and the Integrated Diffraction Analysis Technologies program of the US Department of Energy Office of Biological and Environmental Research. The Advanced Light Source (Berkeley, CA) is a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy under contract number DE-AC02-05CH11231, Office of Basic Energy Sciences. We thank M. Thompson for comments on the crystallography methods. We thank A. Myasnikov and D. Bulkley for technical support at the UCSF Center for Advanced CryoEM, which is supported by NIH grants S10OD020054 and S10OD021741 and the Howard Hughes Medical Institute (HHMI). We thank E. Eng and E. Kopylov for technical support at the National Center for CryoEM Access and Training (NCCAT) and the Simons Electron Microscopy Center located at the New York Structural Biology Center, which is supported by the NIH Common Fund Transformative High Resolution Cryo-Electron Microscopy program (U24 GM129539) and by grants from the Simons Foundation (SF349247) and NY State. We thank W. Weiss at the University of North Texas Health Science Center for conducting the animal study.
Author information
Authors and Affiliations
Contributions
Q.L. and I.B.S. determined analogues for synthesis and designed the synthetic routes; Q.L. executed and optimized the syntheses of analogues, with assistance from A.A.T. (analogues 29–32), R.W. (analogue 21), K.J. (analogues 27 and 28), and D.C. (analogue 26); J.P. and D.J.L. prepared samples and collected cryo-EM data; J.P. and A.F.B. calculated cryo-EM reconstructions; J.P. and J.E.P. performed the VatA acetylation assay; J.P. performed the in vitro translation experiments; G.v.Z. and K.B. developed new tools for cryo-EM model refinement; J.P., K.B. and J.T.B. performed cryo-EM model refinements; G.v.Z., J.P., H.A.C., N.Z. and M.P.J. determined relative energies of macrocycle confirmations; H.A.C. collected X-ray crystallographic data and performed X-ray model refinements; D.S., C.W., B.M., E.M, and O.C. designed and executed the MIC assays; Q.L., J.P., I.B.S. and J.S.F. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
K.B. and G.v.Z. are employees of Schrodinger Inc. D.S., C.W. and B.M. are employees of Micromyx.
Additional information
Peer review information Nature thanks Martin Burke, Gerard Wright and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
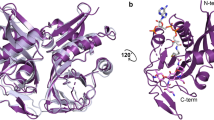
Extended Data Fig. 1 Natural and semisynthetic streptogramins and their molecular mechanisms of action and resistance.
a, Selected natural and semisynthetic streptogramin analogues. Modifications installed by semisynthesis are highlighted in blue. b, 2.5-Å cryo-EM structure of VM2 bound to the 50S subunit of the E. coli ribosome. Coulomb potential density is contoured in dark blue at 4.0σ and light grey at 1.0σ. Atom colouring of VM2 mirrors the building blocks used in its synthesis (see Fig. 2). c, Binding interactions between VM2 and residues in the ribosomal binding site. d, X-ray crystal structure VM1 bound to the resistance protein VatA (PDB ID: 4HUS). e, Binding interactions between VM1 and VatA, highlighting the extensive hydrophobic interactions at C3–C6. Acetylation occurs at the C14 alcohol. f, g, Conformational energy of VM2 showing contributions on a per atom basis when refined with standard CIF-based restraints generated by ‘phenix.eLBOW’ (f) and when refined with OPLS3e/VSGB2.1 force field (g). Colour indicates low strain (green, −14 kcal mol−1) up to high strain (red, 10 kcal mol−1), with total conformational energy of 39.5 kcal mol−1 (f) and −88.3 (g). Hydrogens were added and optimized with fixed heavy atoms for the CIF-based refined conformation using ‘prepwizard’; the PHENIX-OPLS3e/VSGB2.1 refined conformation was taken as is. Energies were calculated using Prime and per atom contribution visualized using Maestro’s prime energy visualization.
Extended Data Fig. 3 Inhibitory activity against Gram-positive organisms.
MIC values for selected analogues against an expanded panel of Gram-positive pathogens.
Extended Data Fig. 4 Inhibitory activity against Gram-negative organisms.
MIC values for selected analogues against an expanded panel of Gram-negative pathogens.
Extended Data Fig. 5 Cryo-EM density for all compounds bound to the E. coli ribosome.
a, 2.6-Å cryo-EM structure of VM2 bound to the 50S subunit of the E. coli ribosome. Coulomb potential density is contoured in dark blue at 4.0σ and light grey at 1.0σ for the entire figure. b, 2.8-Å cryo-EM structure of 21 bound to the 50S subunit of the E. coli ribosome. c, 2.8-Å cryo-EM structure of 40e bound to the 50S subunit of the E. coli ribosome. d, 2.5-Å cryo-EM structure of 40o bound to the 50S subunit of the E. coli ribosome. e, 2.8-Å cryo-EM structure of 40q bound to the 50S subunit of the E. coli ribosome. f, 2.6-Å cryo-EM structure of 41q bound to the 50S subunit of the E. coli ribosome. g, 2.5-Å cryo-EM structure of 46 bound to the 50S subunit of the E. coli ribosome. h, 2.5-Å cryo-EM structure of 47 bound to the 50S subunit of the E. coli ribosome. i, 2.7-Å cryo-EM structure of 46/VS1 bound to the 50S subunit of the E. coli ribosome. j, 2.8-Å cryo-EM structure of 47/VS1 bound to the 50S subunit of the E. coli ribosome.
Extended Data Fig. 6 Gold standard and map to model Fourier shell correlation plots.
a–j, The particle Fourier shell correlation (FSC) curves for reconstructions obtained by cisTEM using a molecular mass of 1.8 MDa are shown in blue with unmasked map–model FSC curves obtained from ‘phenix.mtriage’ shown in orange. Dashed lines indicate FSC of 0.143 for estimating gold standard resolution and FSC of 0.5 for estimating map–model resolution.
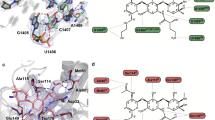
Extended Data Fig. 7 Conformations of 46 and 47 in the ribosome and in VatA.
a, The conformation of 46 minimized by quantum mechanical methods in low dielectric, shows how the isoquinoline side chain packs over the macrocycle. b, By contrast, the ribosome-bound conformations of 46 determined by cryo-EM show that the side chain extends away from the macrocycle due to interactions formed in the binding site. c, Model of 47 in the conformation bound to the ribosome modelled into the active site of VatA (shown in surface). d, Model of 46 in the conformation bound to the ribosome modelled into the active site of VatA. e, Low energy model of 46 modelled into the active site of VatA. f, Overlay of VatA-bound (marine), ribosome-bound (violet), and ribosome with VS1-bound (light pink) conformations of 47. g, X-ray crystal structures of VM1 bound to VatA (PDB code 4HUS; 2.4 Å) and 46 bound to VatA at 2.8-Å resolution.
Supplementary information
Supplementary Information
Procedures for MIC determination and chemical synthesis.
Supplementary Data
Excel file containing MIC data featured in the manuscript and extended data.
Rights and permissions
About this article

Cite this article
Li, Q., Pellegrino, J., Lee, D.J. et al. Synthetic group A streptogramin antibiotics that overcome Vat resistance. Nature 586, 145–150 (2020). https://doi.org/10.1038/s41586-020-2761-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2761-3
This article is cited by
-
Structural basis of Cfr-mediated antimicrobial resistance and mechanisms to evade it
Nature Chemical Biology (2024)
-
Strategies of virginiamycin supplementation in the postweaning phase on growth performance and carcass quality of beef cattle
Tropical Animal Health and Production (2024)
-
Structural insights into the mechanism of overcoming Erm-mediated resistance by macrolides acting together with hygromycin-A
Nature Communications (2023)
-
Molecular mechanism of plasmid-borne resistance to sulfonamide antibiotics
Nature Communications (2023)
-
A large expert-curated cryo-EM image dataset for machine learning protein particle picking
Scientific Data (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
