
Abstract
Endothelial cells adopt tissue-specific characteristics to instruct organ development and regeneration1,2. This adaptability is lost in cultured adult endothelial cells, which do not vascularize tissues in an organotypic manner. Here, we show that transient reactivation of the embryonic-restricted ETS variant transcription factor 2 (ETV2)3 in mature human endothelial cells cultured in a serum-free three-dimensional matrix composed of a mixture of laminin, entactin and type-IV collagen (LEC matrix) ‘resets’ these endothelial cells to adaptable, vasculogenic cells, which form perfusable and plastic vascular plexi. Through chromatin remodelling, ETV2 induces tubulogenic pathways, including the activation of RAP1, which promotes the formation of durable lumens4,5. In three-dimensional matrices—which do not have the constraints of bioprinted scaffolds—the ‘reset’ vascular endothelial cells (R-VECs) self-assemble into stable, multilayered and branching vascular networks within scalable microfluidic chambers, which are capable of transporting human blood. In vivo, R-VECs implanted subcutaneously in mice self-organize into durable pericyte-coated vessels that functionally anastomose to the host circulation and exhibit long-lasting patterning, with no evidence of malformations or angiomas. R-VECs directly interact with cells within three-dimensional co-cultured organoids, removing the need for the restrictive synthetic semipermeable membranes that are required for organ-on-chip systems, therefore providing a physiological platform for vascularization, which we call ‘Organ-On-VascularNet’. R-VECs enable perfusion of glucose-responsive insulin-secreting human pancreatic islets, vascularize decellularized rat intestines and arborize healthy or cancerous human colon organoids. Using single-cell RNA sequencing and epigenetic profiling, we demonstrate that R-VECs establish an adaptive vascular niche that differentially adjusts and conforms to organoids and tumoroids in a tissue-specific manner. Our Organ-On-VascularNet model will permit metabolic, immunological and physiochemical studies and screens to decipher the crosstalk between organotypic endothelial cells and parenchymal cells for identification of determinants of endothelial cell heterogeneity, and could lead to advances in therapeutic organ repair and tumour targeting.
Similar content being viewed by others
Main
Endothelial cells (ECs) in zonated capillaries sustain tissue-specific homeostasis and supply angiocrine factors to guide organ regeneration1,2. By contrast, maladaptation of ECs contributes to fibrosis and tumour progression6,7. The mechanism(s) by which ECs acquire adaptive tissue-specific heterogeneity or maladapt within the scarred tissues or tumour microenvironment are unknown. Identifying the molecular determinants of vascular heterogeneity requires the generation of malleable and perfusable vascular networks that are responsive and can conform to microenvironmental and biophysical signals8.
Attempts to uncover the crosstalk between adult ECs and non-vascular cells—for example, through the generation of decellularized scaffolds9,10, organ-on-chip models11,12 and three-dimensional (3D) bioprinting, as well as the culturing of normal13 and malignant organoids14—have met with hurdles. In these approaches, ECs do not have the cellular freedom to directly interact with parenchymal and tumour cells, owing to the physical constraints that are imposed by artificial semipermeable biomaterials used in organ-on-chip systems and low-volume microfluidic devices, and the lack of adaptive ECs11. Moreover, the use of non-physiological matrices such as Matrigel, poses challenges for translation to the clinic. Thus, transcriptional resetting of adult human ECs to generate adaptable tubulogenic and perfusable ECs in defined matrices will provide insights into vascular diversity and therapeutic organ regeneration.
During development, ETV2 functions as a pioneer transcription factor that induces vascular cell fate and lumen morphogenesis3,15. ETV2 is expressed in ECs during vasculogenesis, but is turned off mid-gestation, when the primitive capillary networks are established15, and is not expressed in adult ECs. Transient reintroduction of ETV2 into parenchymal cells induces a stable EC fate16. Here, we show that in addition to specifying vascular cell fate, transient reactivation of ETV2 resets mature adult human vascular ECs (VECs) to embryonic-like malleable vasculogenic ECs, hereafter referred to as ‘reset VECs’ (R-VECs). R-VECs self-organize into adaptable, large-volume 3D lumenized vascular networks that can transport human blood and physiologically arborize decellularized tissues, islets and normal and malignant organoids, and that can build durable capillaries in vivo.
R-VECs form stable vessels in vitro
Human ECs transduced with lentivirus to express ETV2, in serum-free medium form functional, durable and adaptable 3D vessels by transitioning through three stages (Fig. 1a, Extended Data Fig. 1a). In the first (induction) stage, ETV2 upregulates vasculogenic and tubulogenic factors in flat EC cultures. During the second (remodelling) stage, R-VECs that are placed in matrices, self-assemble into sprouting and lumenized 3D vessels. At the third stage, R-VECs are non-proliferative, maintaining stabilized and adaptive 3D patterned capillaries (Extended Data Fig. 1b).

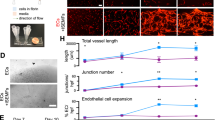
a, Experimental set-up for vessel formation. A total of 105 control ECs or R-VECs were plated on Matrigel in serum-free StemSpan tube-formation medium (Supplementary Data 2). Lenti-EVT2, lentiviral ETV2 expression construct. b, Z-stack of GFP+ R-VEC vessels at week 16. Scale bar, 1,000 μm. c, Quantification of tube formation in control ECs (HUVECs) and R-VECs (HUVEC-ETV2). d, Quantification of R-VEC vessels on Matrigel or LEC matrix. e, Electron microscopy images of stage-3 vessels on Matrigel and LEC matrix. L, lumen. Scale bars, 5 μm. f, Top, schematic of in vivo plug experiment in which control ECs or R-VECs fluorescently labelled with GFP were subcutaneously injected as a single-cell LEC suspension into SCID-beige mice. Bottom, whole-mount confocal images of R-VEC plugs and control EC plugs at five months. A fluorescently labelled antibody against human VEcadherin (hVEcad) was injected retro-orbitally before mice were euthanized. Scale bars, 200 μm. g, Orthogonal projection showing the anastomosis of mouse vessels and human VEcad+ vessels. Sections were post-stained for mouse endomucin (mEndomucin). Scale bar, 10 μm. h, Quantification of the density of human vessels in the plugs, defined as the percentage of GFP positive vessels of the scanned area. i, j, Experimental procedure for the decellularized intestine cultures (i, left). R-VECs repopulated the vasculature, lining blood vessels including the distal capillaries. At day 7 the bioreactors were stained for human CD31, imaged (i, right) and quantified (j). Scale bars, 500 μm. Data are mean ± s.e.m. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001. For statistics, see Supplementary Data 1.
Human umbilical vein ECs (HUVECs) transduced with ETV2 showed a 50-fold increase in the area of vessels formed over 8 weeks compared to naive HUVECs, which did not form durable vessels in any of the enriched angiogenic media that we tested (Fig. 1b, c, Extended Data Fig. 1c, d, Supplementary Video 1a). In addition, mature adult human EC populations isolated from adipose, cardiac, aortic and dermal tissues and transduced with ETV2 formed long-lasting and patterned R-VEC plexi (Extended Data Fig. 1e–g). Next, we investigated whether R-VEC vessel formation could be achieved without Matrigel. We identified a stoichiometrically defined ratio of laminin, entactin and type-IV collagen that is sufficient for the self-assembly of R-VECs into lumenized vessels similar to those formed in Matrigel (Fig. 1d, Extended Data Fig. 1h). This composite matrix of laminin, entactin and type-IV collagen is hereafter referred to as LEC matrix. Confocal and electron microscopy showed that R-VECs organized into vessels that exhibit a continuous, patent (open) lumen with the apicobasal polarity on both Matrigel and LEC matrix (Fig. 1e, Extended Data Fig. 1i). Moreover, transduction of ETV2 reduces stiffness in adult ECs (as measured by atomic force microscopy; AFM), which facilitates lumen formation (Extended Data Fig. 1j). To assess whether this tubulogenesis-promoting activity is specific to ETV2, we transduced HUVECs with a lentiviral vector expressing another ETS transcription factor, ETS1; and to test whether the survival of ECs could drive tubulogenesis, HUVECs were also transduced with a lentiviral construct of constitutively active myristoylated AKT1 (myrAKT1). Neither ETS1 nor myrAKT1 reset ECs to form stable vessels (Extended Data Fig. 1k–m).
We quantified the mRNA and protein levels of ETV2 in R-VECs from stages 1 to 3 (Extended Data Fig. 2a–d). ETV2 protein levels peaked during stage 2 but were downregulated by more than 90% at stage 3, which could not be accounted for by the minor drop in ETV2 mRNA levels (Extended Data Fig. 2a–d). Treatment with the proteasome inhibitor MG132 at stage 3 restored ETV2 protein levels by sixfold—approaching its original expression levels—which indicates that proteasomal proteolysis regulates ETV2 expression (Extended Data Fig. 2e, f). To examine whether short-term induction of ETV2 is sufficient to generate R-VECs, we used a reverse tetracycline-controlled transactivator (rtTA) doxycycline-inducible system, in which doxycycline induces the expression of ETV2 (induced R-VECs; iR-VECs) (Extended Data Fig. 2g, h). Induction of ETV2 was transiently required until the first week of stage 2; after that, iR-VEC vessels sustain their stability without continuous ETV2 induction (Extended Data Fig. 2i–k).
Thus, short-term expression of ETV2 confers adult ECs with the capacity to self-assemble into stable and durable patterned vessels, without affecting cell survival and proliferation and without the physical constraints of artificial bioprinted scaffolds and restrictive synthetic barriers.
R-VECs form durable vessels in vivo
SCID-beige mice were implanted subcutaneously with mCherry- or GFP-labelled control human ECs or R-VECs suspended in LEC matrix. One to five-months after implantation, R-VECs—but not control ECs—self-organized into long-lasting, branching and patterned vessels in vivo. Injection of R-VEC-implanted mice with an antibody directed against human vascular endothelial cadherin (VEcad) showed that R-VEC vessels anastomose to the endomucin-positive mouse vasculature, establishing a mosaic of functional perfused vessels throughout the plug (Fig. 1f–h, Extended Data Fig. 3a). Mouse perivascular cells wrap around R-VEC vessels, with larger arterioles covered with a thicker layer of smooth muscle cells and less coverage in smaller capillaries (Extended Data Fig. 3b, c). iR-VECs also assembled into stable vessels in LEC matrix, and one week of doxycycline treatment in vivo was sufficient to retain vascular stability (Extended Data Fig. 3d, e). The lack of extravasation of intravenously injected 70-kDa dextran in mice indicated that R-VEC and iR-VEC vessels in in vivo plugs were non-leaky and patent. By contrast, human ECs that were transduced with KRAS formed leaky and disorganized vessels, reminiscent to those of haemangiomas (Extended Data Fig. 3f). Unlike implants of KRAS-transduced endothelial cells, R-VEC implants did not exhibit aberrant growth, haemangiomas or tumours, and they retained perfused and organized vessels for 10 months (Extended Data Fig. 4a–e). In summary, R-VECs build durable, anastomosed and pericyte-covered capillaries that are structurally normal and show no signs of vascular anomalies or tumours.
R-VECs arborize decellularized scaffolds
We next examined whether R-VECs can functionally populate the denuded vascular lining of decellularized tissues. Although large vessels in decellularized scaffolds can be colonized with ECs, it is challenging to vascularize the abundant smaller capillaries9. Stage-1 R-VECs, but not control ECs, fully populated the narrow small capillaries evenly throughout the decellularized rat intestine scaffolds ex vivo (Fig. 1i, j, Extended Data Fig. 5a–d). After one week of ex vivo culture, the revascularized intestinal explants were implanted in the omentum of immunocompromised mice. Intravital anti-human VEcad staining at one and four weeks showed that R-VEC-vascularized scaffolds retained their patency and anastomosed to the mouse vasculature (Extended Data Fig. 5e). At four weeks, R-VEC vessels persisted at a higher rate in vivo compared to naive control ECs, owing to their integrity and low rate of apoptosis (Extended Data Fig. 5f, g). Thus, R-VECs enable the functional arborization of decellularized tissues for therapeutic regeneration.
ETV2 remodels ECs to primitive plexi
To uncover the mechanism by which ETV2 drives vascular resetting, we performed RNA sequencing (RNA-seq) in stage-1 R-VECs and control ECs (Fig. 2a–c). Gene Ontology (GO) analyses revealed the upregulation of genes in pathways that regulate vasculogenesis, angiogenesis, GTPase activity, extracellular matrix remodelling and the response to mechanical stimuli (Fig. 2b, c, Extended Data Fig. 6a, b). At stage 1, R-VECs maintain their vascular identity by sustaining the expression of EC-specific genes (Extended Data Fig. 6c). After ETV2 induction, a group of 490 genes was differentially expressed among various tissue-specific adult human ECs, including cardiac, dermal, aortic, pulmonary and adipose-derived R-VECs (Fig. 2d, Extended Data Fig. 6d). Chromatin immunoprecipitation followed by sequencing (ChIP–seq) analysis of K4me3, K27ac and K27me3 histone modifications in both R-VECs and control ECs showed that ETV2 bound to the promoters of several differentially expressed vascular-specific genes—and to the promoters of pro-tubulogenesis genes, which are silenced in mature ECs (Fig. 2c, e, Extended Data Fig. 6e–h). Therefore, ETV2 resets the chromatin and transcriptome of mature ECs through the direct reactivation of suppressed tubulogenic and vasculogenic genes.

a, Schematic of RNA-seq and ChIP–seq performed in the induction phase (day 14) on R-VECs and control ECs. b, RNA-seq of R-VECs or control HUVECs in stage 1 (2D monolayers). GO term analysis was performed on differentially expressed genes. GO categories are ordered on the basis of the number of differentially expressed genes. Heat maps for GO categories in red are presented in Fig. 2c and Extended Fig. 6b. ECM, extracellular matrix; Pos. reg., positive regulation. c, Heat map of genes in one top GO category. Values are log2-normalized counts per million (CPM), centred and scaled by row. ETV2 binding from ChIP–seq at the promoter of each differentially expressed gene is shown in the yellow-and-green heat map (left). d, Heat map of 490 differentially expressed genes across ECs of different tissues (stage 1, induction phase) upon ETV2 expression. Tissue-adjusted log2-transformed CPM, centred and scaled by row. e, ETV2 ChIP–seq in R-VECs during the induction phase (stage 1; 2D) using an anti-Flag antibody or mouse IgG as control. ChIP for H3K4me3, H3K27ac and H3K27me3 was performed in both control ECs and R-VECs at stage 1. Enriched regions were analysed by ChIP–seq. Horizontal bars underneath peaks represent significantly changed regions. Promoter regions bound by ETV2 are highlighted in cream. Track range ETV2/K27me3/K27ac, 0–0.3; K4me3/input/IgG, 0–1. f, Western blot for active RAP1–GTP compared to total RAP1 input for stage 1 2D control ECs (HUVECs) and R-VECs (HUVEC-ETV2). The quantification of RAP1–GTP compared to total RAP1 is shown below the blot and presented as mean ± s.e.m. g, Quantification of R-VEC vessel formation after treatment with RAP1 inhibitor or dimethyl sulfoxide (DMSO). h, Z-stack confocal images and electron microscopy images of R-VEC vessels treated with RAP1 inhibitor or DMSO at four weeks. Red circles indicate orthogonal cross-sections. Scale bars, 5 μm (top); 2 μm (bottom). Data are mean ± s.e.m. NS, not significant; *P < 0.05, **P < 0.01. For statistics, see Supplementary Data 1.
After ETV2 transduction, genes encoding Ras-interacting protein 1 (RASIP1) and three guanine nucleotide exchange factors (GEFs) that are involved in the activation of the small GTPase RAP1 (RASGRP2, RASGRP3 and RAPGEF5)—all of which are crucial for lumen formation4,5—were upregulated in all tissue-specific ECs (Fig. 2c, d). Similarly, differential expression of genes in the RAP1 pathway was found in ETV2-positive ECs isolated from ETV2–Venus reporter mouse embryos at embryonic stage 9.5 (E9.5) (Extended Data Fig. 7a, b). ChIP–seq analysis of stage-1 R-VECs confirmed the direct binding of ETV2 to RASGRP3 and RASIP1 promoters and a subsequent increase in K4me3 and K27ac histone marks at these genes (Fig. 2e). A pull-down of active RAP1–GTP in stage-1 R-VECs showed that the levels of active RAP1–GTP were higher in R-VECs than in naive ECs (Fig. 2f). Vessel formation was reduced and no lumen was present after treatment with the RAP1 inhibitor GGTI-298 (Fig. 2g, h). Similarly, knockdown of RASGRP3 by short hairpin RNA (shRNA) disrupted R-VEC-mediated tubulogenesis (Extended Data Fig. 7c). Therefore, ETV2 potentiates lumen formation in part through the upregulation of RAP1 GEFs.
In vitro, stage-3 R-VECs upregulate the expression of genes that are involved in mechanosensing (PIEZO2, KLF2 and KLF4) and EC remodelling (ATF3), which are not expressed in cultured mature ECs (Extended Data Fig. 7d). We confirmed this result by isolating R-VECs from in vivo plugs and comparing their transcriptome to that of freshly isolated HUVECs and stage-3 R-VEC stable vessels (Extended Data Fig. 7d). Notably, the genes upregulated in stage-3 R-VECs (PIEZO2, KLF2 and KLF4) were bound by ETV2 and epigenetically primed for expression in stage-1 two-dimensional (2D) R-VECs (Extended Data Fig. 7e). Thus, ETV2 resets the chromatin landscape of mature ECs to an in vivo physiological configuration that is responsive and conforms to microenvironmental cues—reminiscent of generic vasculogenic ECs.
R-VECs build haemodynamic vessels
We tested the capacity of R-VEC vessels to self-congregate into sprouting vascular networks in the absence of pre-patterned scaffolds and synthetic barriers and to sustain a laminar flow in vitro in large-volume microfluidic devices. R-VECs or control ECs were seeded in a 5 × 3 × 1-mm microfluidic device that can accommodate more than 45,000 stage-1 ECs within a 15-μl volume of fibrin gel17 (Fig. 3a). Within three days, R-VECs self-organized into a multilayered, branching and interconnected vascular plexus, maintaining their 3D lumenized stability (Fig. 3b–d). Notably, R-VEC vessels allowed the gravity-driven transport of heparinized human whole peripheral blood with a full complement of plasma, platelets, white and red blood cells (Fig. 3e, Supplementary Video 1b, c). During the transport of blood, R-VEC capillary networks sustained their vascular integrity and were haemodynamically stable from the inlet to the outlet chambers of the microfluidic device, enduring the force of blood flow without collapse, regression or thrombosis. R-VECs therefore maintain the haemodynamic vascularization of tissues, and pave the way for an Organ-On-VascularNet platform (Supplementary Video 1d).

a, Overview of microfluidic device measuring 5 × 3 × 1 mm and holding 15 μl fibrin gel. b, Representative images of devices with control ECs or R-VECs stained with human VEcad antibody at day 7. Scale bars, 3 mm. c, Orthogonal representation of intact lumen formation in R-VECs. Scale bar, 50 μm. d, Quantification of vessel area in devices with control ECs versus R-VECs. e, Intact heparinized human peripheral blood (100 μl) composed of a full complement of red blood cells, white blood cells, platelets and unperturbed plasma was injected and perfused through the R-VEC vessels. Right, representative live image of blood flow through R-VECs (see also Supplementary Video 1b, c). Scale bar, 25 μm. f, Experimental set-up for co-seeding human islets with control ECs or R-VECs in microfluidic devices. g, Fluorescently labelled human heparinized whole blood (red, PKH26 red fluorescent dye) was perfused through the microfluidic devices (day 4) (see also Supplementary Video 2b–d). Z-stack projections of whole devices of islet explants post-stained with EpCAM and VEcad (day 4). Scale bars, 3 mm. h, Magnified area of direct interaction of R-VECs with co-cultured islets in a microfluidic device. Scale bar, 100 μm. i, Single section and orthogonal projection of human islets vascularized by R-VECs in a microfluidic device. Scale bars, 50 μm. j, Experimental set-up for the glucose-stimulation test in microfluidic devices. k, Insulin levels were measured at 2 mM glucose (t = −10 and 0 min, basal level) and 9 and 24 min after stimulation with 16.7 mM glucose. * represents statistical tests versus islets alone; # represents statistical tests versus islets + control ECs. l, Fold change in insulin levels at the outlet (insulin levels at 16.7 mM/insulin levels at 2 mM), 9 min after high-glucose stimulation. Data are mean ± s.e.m. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ###P < 0.001. For statistics, see Supplementary Data 1.
R-VECs physiologically vascularize islets
We assessed the potential of R-VECs to functionally vascularize human islets in the perfusable microfluidic devices. Currently, organ-on-chip devices11,12 segregate ECs from parenchymal cells with physical barriers, and are thus unsuitable for studying islets, which require active interaction with ECs to maintain their function18. We seeded around 40 human islets, alone or in the presence of control ECs or R-VECs, in 15-μl microfluidic devices (Fig. 3f). Within three days, R-VECs—but not control ECs—arborized islets with continuous 3D vascular networks, which extended deep into islets and metabolically irrigated insulin-secreting β-cells (Fig. 3g–i, Supplementary Video 2a–f). Heparinized human blood travelled through the R-VEC-co-opted islets, with intact haematopoietic cells perfusing the vascularized islets (Fig. 3g, Supplementary Video 2b–e).
We used a glucose-stimulation test to assess islet function (Fig. 3j), and found that islets arborized with R-VECs responded to high glucose by secreting insulin, as measured at the device outlet at 9 and 24 minutes of stimulation (Fig. 3k). There was a sevenfold increase in insulin secretion in glucose-stimulated R-VEC-co-opted islets, but not in control ECs or islet-alone cultures (Fig. 3l). Similar results were observed in co-cultured islet explants arborized by R-VECs in static Matrigel droplets (Extended Data Fig. 8a–e). Thus, R-VECs self-congregate in large-volume microfluidic devices into haemodynamically stable vessels that physiologically perfuse and sustain glucose-sensing human β-cells.
R-VECs vascularize organoids and tumoroids
We next assessed the capacity of R-VECs to functionally arborize organoids composed of healthy or malignant human cells, in order to model tissue- and tumour-specific adaptive responses of ECs and set the stage for organ regeneration. Normal colon organoids (COs) were established and maintained from healthy human colon crypts19,20 (Extended Data Fig. 8f). Next, the COs were mixed with either control ECs or stage-1 R-VECs in static 50-μl droplets of Matrigel or LEC matrix (Fig. 4a). R-VECs sustained the arborization of the COs throughout the matrix droplet, with a higher vessel area than control ECs (Fig. 4b, c, Supplementary Video 3a). Moreover, as tracked in a 72-h time-lapse video, R-VECs interacted and engaged significantly more with the cells within the COs, as compared to control ECs (Fig. 4d, Supplementary Video 3b). The surface area of COs was larger in the presence of R-VECs, with no change in the differentiation of COs as assessed by the expression of stem and progenitor cell markers (Fig. 4e, Extended Data Fig. 8g). R-VECs also arborized mouse small intestinal organoids, with an increase in the vessel area and the number of R-VEC sprouts per organoid (Extended Data Fig. 8h–j). Thus, R-VECs instructively sustain the proliferation and integrity of COs, while preserving their differentiation status.

a, Control ECs or R-VECs were seeded with human normal colon organoids (COs) or colorectal cancer organoids (CRCOs) in Matrigel droplets. b, c, Confocal Z-projections (b) and magnified images (c) of human COs alone or co-cultured with control ECs or R-VECs on day 8. Scale bars, 1 mm (b); 100 μm (c, left image); 50 μm (c, right images). d, Surface area of control ECs or R-VECs interacting with human COs in LEC matrix, quantified over a 72-h time lapse. e, Quantification of colon area (as stained by EpCAM) per field in COs alone or co-cultured with control ECs or R-VECs on day 8. f, Human CRCOs were seeded with control ECs or R-VECs in Matrigel droplets. Confocal images of CRCOs alone or co-cultured with control ECs or R-VECs, post-stained for KRT20 at day 8 after a 4.5-h EdU pulse. Scale bars, 100 μm. g, Surface area of control ECs or R-VECs interacting with human CRCOs in Matrigel, quantified over a 78-h time lapse. h, Quantification of EdU+ CRCO cells (in f) in CRCOs alone or CRCOs co-cultured with control ECs or R-VECs on day 8. i, j, R-VECs in single-cell suspension were cultured either alone or mixed with human COs and subjected to scRNA-seq on day 7. i, Uniform manifold approximation and projection (UMAP) plot for the EC fractions of R-VECs alone and R-VECs co-cultured with COs. j, UMAP plot of 9 unique clusters, in ECs from both the R-VECs alone group and the R-VECs +COs group. k, Heat map of differentially expressed genes from cluster 5, enriched among R-VECs but absent in R-VEC-alone cultures. l, m, R-VECs in single-cell suspension were cultured either alone or mixed with human CRCOs and subjected to scRNA sequencing on day 7. l, UMAP plot for the EC fractions of R-VECs alone and R-VECs co-cultured with CRCOs. m, UMAP plot of 8 unique clusters, in ECs from both the R-VECs alone group and the R-VECs + CRCOs group. n, Heat map of differentially expressed genes from cluster 8, enriched among R-VECs in culture with CRCOs. o, Schematic of the adaptation and maladaptation of R-VECs. Data are mean ± s.e.m. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001. For statistics, see Supplementary Data 1.
Tumour vasculature is composed of abnormal capillaries that supply aberrant factors that instigate tumour growth7. To determine whether R-VECs can acquire and report on the maladapted features of tumour vessels, we mixed stage-1 R-VECs with patient-derived colorectal cancer organoids (CRCOs) (Fig. 4f, Supplementary Video 3c). Within 24 hours, R-VECs, but not control ECs, migrated to and erratically infiltrated tumour organoids (Supplementary Video 3c). Similar to human COs, the vessel area in CRCOs mixed with R-VECs, and the interaction of R-VECs with CRCOs—as tracked in a 72-h time-lapse video—were increased compared to control ECs (Fig. 4f, g, Extended Data Fig. 8k, Supplementary Video 3c). Staining for the epithelial marker EpCAM revealed intimate cell–cell interactions between the tumoroids and the R-VECs, with a higher percentage of EdU-positive proliferating tumour cells in the R-VEC than the control EC co-cultures (Fig. 4f, h, Extended Data Fig. 8l). Hence, R-VECs establish an adaptive 3D vascular niche that can be used to decipher the crosstalk between ECs and normal or tumour organoids.
R-VECs adapt to organoids and tumoroids
We performed single-cell RNA-seq (scRNA-seq) on the 3D R-VEC-vascularized human COs or CRCOs to assess the adaptability of R-VECs. R-VECs were cultured alone or co-cultured with human COs or CRCOs for seven days, isolated and subjected to scRNA-seq using the 10X Genomics Chromium platform (Extended Data Figs. 9a, 10a). ECs were identified by their expression of VEcad (also known as CDH5) CD31 (PECAM1) and VEGFR2 (KDR) and epithelial cells by their expression of EPCAM, CDH1 and KRT19 (Extended Data Figs. 9b–e, 10b–e). The identity of the COs was validated by the expression of SATB2, CA4 and CA2, among other genes (Extended Data Fig. 9f).
R-VECs that were co-cultured with malignant or normal organoids showed changes in their clustering patterns and gene expression when compared to R-VECs that were cultured alone (Fig. 4i–n). R-VECs that interacted with COs were enriched in EC organotypic marker genes, including PLVAP and TFF3 (cluster 5, absent in R-VECs alone)1,21 (Fig. 4i–k). By contrast, R-VECs that arborized CRCOs were enriched in clusters of genes with typical attributes of tumour ECs, including ID1, JUNB and ADAMTS4 (cluster 8), whereas genes responsible for junctional integrity—such as CLDN5 (cluster 5, cluster 7)—were selected against22 (Fig. 4l–n). In response to association with R-VECs, colon tumour cells upregulated their expression of marker genes that are linked to poor prognosis and high rates of metastasis, including higher levels of MSLN23, and downregulated their expression of MT1G, MT1X and MT2A24 (Extended Data Fig. 10f–h). These data provide further evidence that R-VECs model an adaptable 3D vascular niche that responds to microenvironmental stimuli (Fig. 4o).
Discussion
We have created haemodynamic, self-organizing, large-volume 3D R-VEC vascular plexi in a Matrigel-free LEC matrix, which mimic primitive pliable blood vessels. R-VECs sustain their tubulogenic potential in diverse serum-free media compositions enabling the functional vascularization of organoids and tissue explants, notably islets. These networks do not have the constraints of synthetic scaffolds and semipermeable membranes, and allow the direct cellular interaction of ECs with parenchymal and tumour cells. Transient reintroduction of ETV2—which is silenced during fetal development—into adult human ECs induces a molecular reset of cell tubulogenic and adaptability attributes that are lost in cultured mature ECs4,5. In R-VECs, the RAP1 pathway is activated through RAP1 GEFs and the RASIP1 effector, allowing lumen formation in a flow- and pericyte-independent manner. ETV2 resets the vasculogenic memory to an early embryonic stage, and thereby renders R-VECs receptive to microenvironmental cues1,2. In stabilized R-VEC vessels, the expression of ETV2 was spontaneously reduced through proteasomal proteolysis, suggesting that transient expression of ETV2 is sufficient to reset ECs into a plastic and adaptive state.
The capacity of R-VECs to self-assemble into perfusable vascular networks that can transport human blood enables the 3D physiological vascularization of scalable and organ-level micro- and macrofluidic devices. This licenses R-VECs to recapitulate the physiochemical and multicellular geometry of blood-perfusable vascular niches that, by deploying angiocrine factors, directly enhance the frequency of co-cultured organoids. In addition, R-VECs conform to signals that are produced by organoids or tumoroids and, reciprocally, tumour cells upregulate markers that are associated with poor outcomes in response to signals induced by subverted R-VECs. Our R-VEC Organ-On-VascularNet platform therefore overcomes the constraints of costly, technically challenging and non-physiological organ-on-chip models, the design of which prevents the direct cellular interaction of ECs with non-vascular cells.
Co-cultures of organoids with blood-perfusable pericyte-coated R-VECs could serve as a tissue-specific biological platform for the delivery of engineered immune cells (such as CAR-T cells) and chemotherapeutic agents, and could also be used to unravel the pathogenesis of microangiopathy in diseases such as coronavirus disease 2019 (COVID-19). The durable tubulogenic capacity, scalability, haemodynamic blood perfusibility, geometrical malleability, medium compatibility and cellular adaptability of R-VECs—which are capable of vascularizing normal and malignant organoids, as well as decellularized scaffolds—will lay the foundation for physiological, metabolic and immunological studies and pharmaceutical screening. The R-VEC Organ-On-VascularNet model permits the construction of functional and perfused implantable tissues ex vivo, opening a new chapter in translational vascular medicine for tissue-specific regeneration and for targeting the corrupted vascular niches of tumours.
Methods
Cell culture of ECs
Approval for the use of discarded left-over HUVECs and human adipose tissue ECs was obtained through the Weill Cornell Medicine Institutional Review Board (IRB). The ECs were isolated in the laboratory as previously described, using a collagenase-based digestion approach25,26. The cells were then grown in tissue culture dishes coated with 0.2% gelatin in complete EC medium. Complete EC medium is composed of 400 ml M199, 100 ml heat-inactivated fetal bovine serum (FBS), 7.5 ml HEPES, 5 ml antibiotics (Thermo Fisher Scientific, 15070063), 5 ml glutamax (Thermo Fisher Scientific, 35050061), 5 ml lipid mixture (Thermo Fisher Scientific, 11905031), and 25 mg EC growth supplement (Alpha Aesar, J64516-MF) (Supplementary Data 2). The cells were transduced with lenti-PGK-ETV2 or an empty lentiviral vector at passage 1–2. In some instances, the cells were also labelled by using PGK-mCherry or PGK-GFP lenti-viral vectors. The cells were split 1:2 using accutase and passaged on gelatinized plates. As required, cells in 2D (stage 1, induction) were frozen down to be used in future experiments. Comparisons for all assays and co-cultures were performed using the same parental EC line lentivirally transduced with and without ETV2. Overall, HUVECs from more than 10 different isolations were used for the experiments. R-VECs used for tube-formation assays were of passage 5–10.
Human adipose-derived ECs were isolated by mechanical fragmentation followed by collagenase digestion for 30 min. After plating the crude population of cells on the plastic dish and expansion for 5 to 7 days, the cells were then sorted to purify VEcad+CD31+ ECs and expanded as described above. Human adipose ECs were cultured in the same medium as that described above for HUVECs. At least three different isolations of adipose ECs were used in our experiments. Human microvascular cardiac (PromoCell, C12286), aortic (PromoCell, C12272), pulmonary (PromoCell, C-12282) and microvascular dermal (PromoCell, C12265) ECs were acquired from PromoCell and cultured in EC growth medium MV (PromoCell, C22020).
Lentiviral transduction of ECs
ECs were transduced with ETV2 lenti-particles or empty vector lenti-particles. ETV2 cDNA (NM_014209.3) was introduced into the pCCL-PGK lentivirus vector (Genecopeia). For ChIP analysis, a triple Flag tag was subcloned in the ETV2 construct at the amino terminus27. After one week of transduction, ECs were collected for mRNA isolation and quantitative PCR with reverse transcription (qRT–PCR) analysis. The relative ETV2 RNA unit was determined by calculating the relative ETV2 mRNA expression compared to GAPDH using the following formula: (2−[Ct(ETV2) − Ct(GAPDH)]) × 1,000. Primers are found in Supplementary Data 4). Cells with a relative ETV2 RNA unit within the range of 60–100 were used for all experiments. A multiplicity of infection (MOI) of 3 gave us relative expression levels of 60–80 as calculated by mRNA expression. MOI was calculated by converting particles of antigen P24 to infectious units per ml (IFU) and then to MOI based on cell number (kit: Katara, 632200). An MOI of 3 was found to be adequate for cardiac and aortic ECs; an MOI of 6 was required for adipose and dermal ECs. Polybrene at 2 μg ml−1 was used for all transductions. ETS1, myrAKT, mCherry and GFP were also introduced into the pCCL-PGK lentivirus vector and an MOI of 3 was used for all transductions.
For inducible expression of ETV2, ECs were transduced with doxycycline-inducible ETV2 lenti-viruses (pLV[Exp]-Puro-TRE > hETV2 (NM_014209.3), VectorBuilder VB170514-1062dfs and pLV[Exp]-Neo-CMV > tTS/rtTA_M2, VectorBuilder VB160419-1020mes) in which the presence of doxycycline turns on ETV2 expression. After 1 week of doxycycline (1 μg ml−1) induction of ETV2, cells were collected to determine the relative ETV2 mRNA unit. Cells with a relative ETV2 RNA unit within 60–100 were used for all experiments. An MOI of 50 was required for the inducible ETV2 lentiviral particles and rtTA lentiviral particles.
Lentivirus production
All lentiviral plasmids were prepared with a DNA Midiprep kit (Qiagen, 12145). Viruses were packaged in 293T cells by co-transduction with a second or third generation of packaging plasmids. Culture media were collected 48 h after transduction and virus particles were concentrated using a Lenti-X concentrator (Katara, 631232), resuspended in phosphate-buffered saline (PBS) without calcium or magnesium (Corning, 21040CV) and stored at −80 °C in small aliquots. Virus titres were determined with a Lenti-X p24 titre kit (Katara, 632200).
Tube-formation assays
Twenty-four-well plates were coated with 300 μl of Matrigel (Corning) for 30 min in a 37 °C incubator. Meanwhile, cells with or without ETV2 were accutased and counted. Cells were then resuspended in StemSpan (Stem Cell Technologies) supplemented with 10% knockout serum (Thermo Fisher Scientific, 10828028) and cytokines: 10 ng ml−1 FGF2 (bFGF) (Peprotech, 1000-18B), 10 ng ml−1 IGF1 (Peprotech, 100-11), 20 ng ml−1 EGF (Peprotech, AF-100-15), 20 ng ml−1 SCF (Peprotech, 300-07) and 10 ng ml−1 IL-6 (Peprotech, 200-06). One hundred thousand cells either with or without ETV2 were then dispersed in each well in 1 ml of medium. Cultures were placed in a 37 °C incubator with 5% oxygen for the remainder of the tube-formation experiments. The medium was changed every other day, by replacing 750 μl of medium with fresh medium. Care was taken to not disrupt the tubes during all medium changes. In several cases, a mixture of defined matrices comprising a mixture of laminin and entactin (Corning, 354259) and collagen IV (Corning, 354245) (LEC matrix) was used instead of Matrigel as indicated in the text. We combined these defined matrices at different ratios of laminin, entactin and collagen IV (LEC) components and ultimately found the most effective combination of these gel mixtures for tube-formation assays, which was: 200 μl of laminin and entactin (note that concentrations slightly vary for each lot; always diluted to 16.5 mg ml−1 in PBS first) and 100 μl of collagen IV (concentrations slightly vary for each lot; first diluted to 0.6 mg ml−1 in PBS), mixed together on the ice and stored at 4 °C overnight before use. The final format of the LEC matrix consisted of 11 mg ml−1 of the laminin and entactin mixture, and 0.2 mg ml−1 collagen IV. The volume of LEC was increased as needed, as long as the ratios and final concentrations were maintained. Vessel area was measured over the course of 24 h to 12 weeks for stage-2 (remodelling) and stage-3 (stabilization) phases. An EVOS inverted microscope with a 4× objective was used to capture images in their different (randomized) locations in each well for each condition and time point. All of the images were then analysed for the lumenized vessel area using ImageJ to trace the vessel area. The same procedure was used for cells transduced with ETS1 or myrAKT128, and for KRAS-transduced ECs26.
Tube-formation assay in different medium formulations
ECs were accutased and plated on Matrigel at 100,000 cells per well in 24-well plates as described above. To assess the tube-formation assays of ETV2 ECs versus control ECs, we compared their capacity to form a tubular network in three different enriched pro-angiogenic medium formulations (Extended Data Fig. 1d). Medium formulation 1 (MF1) is a StemSpan tube-formation medium (Supplementary Data 2)—a serum-free medium containing StemSpan supplemented with knockout serum and cytokines (StemSpan (Stem Cell Technologies) supplemented with 10% knockout serum (Thermo Fisher Scientific, 10828028) and cytokines: 10 ng ml−1 FGF (Peprotech, 1000-18B), 10 ng ml−1 IGF1 (Peprotech, 100-11), 20 ng ml−1 EGF (Peprotech, AF-100-15), 20 ng ml−1 SCF (Peprotech, 300-07) and 10 ng ml−1 IL-6 (Peprotech, 200-06)). Medium formulation 2 (MF2, EGM-2) is an EC growth medium (Supplementary Data 2) (PromoCell, C22111). Medium formulation 3 (MF3) is the complete EC medium (Supplementary Data 2) with serum that was used to maintain and propagate ECs (400 ml M199, 100 ml heat-inactivated FBS, 7.5 ml HEPES, 5 ml antibiotics (Thermo Fisher Scientific, 15070063), 5 ml glutamax (Thermo Fisher Scientific, 35050061), 5 ml lipid mixture (Thermo Fisher Scientific, 11905031) and 25 mg endothelial cell growth supplement (Alpha Aesar, J64516-MF). Media were changed every other day. Images were acquired at different time points. ImageJ was used to measure vessel area over time.
Video set-up for HUVECs cultured in 3D matrices in different medium formulations
GFP-labelled control HUVECs and R-VECs were embedded inside LEC matrix at 5 million cells per ml. Gels were polymerized on glass-bottomed culture dishes at 37 °C incubator for 15 min. Subsequently, either EGM-2 or StemSpan tube-formation medium (Supplementary Data 2) was added into the cell culture as described above. The medium was also supplemented with Trolox, a vitamin E analogue (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) (Sigma) at 100 μM to enable long-term imaging. The cultures were mounted in a temperature- and gas-controlled chamber for live-cell imaging. Time-lapse videos were acquired with a Zeiss Cell Observer confocal spinning disk microscope (Zeiss) equipped with a Photometrics Evolve 512 EMCCD camera at an interval of 40 min over 3 days. The medium was refreshed every two days.
Immunofluorescent staining of tubes in vitro
At 8 to 12 weeks all medium was removed from the wells. The tubes were washed once with PBS and fixed for 30 min in 4% paraformaldehyde (PFA) at room temperature. Then, the wells were rewashed with PBS and put in blocking buffer (containing 0.1% Triton-X) for 1 h at room temperature. For proliferation studies, a 16-h pulse of EdU (Click-iT EdU kit, Thermo Fisher Scientific, C10337) was used for all three stages of vessel formation.
Electron microscopy
Tissues were washed with serum-free medium or PBS then fixed with a modified Karmovsky’s fix of 2.5% glutaraldehyde, 4% PFA and 0.02% picric acid in 0.1 M sodium cacodylate buffer at pH 7.2. After a secondary fixation in 1% osmium tetroxide and 1.5% potassium ferricyanide, samples were dehydrated through a graded ethanol series and embedded in an Epon analogue resin. Ultrathin sections were cut using a Diatome diamond knife (Diatome) on a Leica Ultracut S ultramicrotome (Leica). Sections were collected on copper grids, further contrasted with lead and viewed on a JEM 1400 electron microscope (JEOL) operated at 100 kV. Images were recorded with a Veleta 2k × 2k digital camera (Olympus SIS).
AFM measurements
AFM was used to examine the stiffness of HUVECs and adult human adipose ECs. Bright-field images of cells, for determination of the location of stiffness measurements, were acquired using an inverted microscope (Zeiss Axio Observer Z1) as the AFM base (20× 0.8 NA objective). An MFP-3D-BIO Atomic Force Microscope (Asylum Research) was used to collect force maps. A 5-μm borosilicate glass beaded probe (Novascan) with a nominal spring constant of 0.12 N m−1 was used for all measurements. Each force map sampled a 60 μm × 60-μm region, in a 20 × 20 grid of force curves (400 force curves total) under fluid conditions which covered an area of 360 μm2. The trigger point was set to 2 nN with an approach velocity of 5 μm s−1. The force-indentation curves were fit to the Hertz model for spherical tips using the Asylum Research software to determine Young’s modulus, with an assumed Poisson’s ratio value of 0.45 for the sample. Force maps of stiffness along with individual stiffness values for each measured point were then exported from the Asylum Research software for further analysis. A custom-made MATLAB (MathWorks) script was written to correctly analyse the data for the stiffness of the cells and filter measurements such that only data 1 μm from the glass bottom dish were analysed (to remove any substrate effect from the measurements).
RNA and protein collection from endothelial cell tubular capillaries
At indicated time points, capillaries of ECs from tube-formation assays were collected for RNA sequencing and western blotting. Before the cells were collected, the medium was completely removed from the well. Two millilitres of 2 mg ml−1 dispase (Roche 38621000) was added into each well to dissociate the EC tubes for 45 min at 37 °C with gentle shaking. Dissociated cells were pelleted, washed once in PBS and subsequently collected for either mRNA or protein isolation. On several occasions, dissociated ECs from tubes were pooled from multiple wells of the same EC line and experiment to allow sufficient isolation of mRNA and protein for downstream analysis.
Western immunoblot
Cells were lysed into 1× SDS loading buffer (50 mM Tris-HCl pH 6.8, 5% β-mercaptoethanol, 2% SDS, 0.01% bromophenol blue, 10% glycerol) followed by sonication (Bioruptor, 2 × 30 s at high setting). Proteins were solved on a 5–15% gradient Tris–glycine SDS–PAGE gel and semi-dry-transferred to nitrocellulose membranes. The following primary antibodies were used at the indicated dilutions: RAP1 (CST, 2399, 1:1,000); RASGRP3 (CST, 3334, 1:1,000), GAPDH (CST, 5174, 1:10,000); AKT (CST, 34685, 1:5,000); p-S473-AKT (CST, 4060, 1:2,000); ETS1 (CST, 14069, 1,000) and ETV2 (Abcam, ab181847, 1:1,000). All antibody information can be found in the Reporting Summary. Horseradish peroxidase (HRP)-conjugated secondary antibodies and the ECL prime western blotting system (GE Healthcare, RPN2232) were then used. Chemiluminescent signals were captured with a digital camera (Kindle Biosciences) and images of protein bands were taken for quantification using ImageJ.
In vivo experiments
All animal experiments were performed under the approval of the Weill Cornell Medicine Institutional Animal Care and Use Committee (IACUC). HUVECs transduced with an empty lentiviral vector or lentiviral vectors carrying ETV2 construct, and labelled with GFP or mCherry (2 million cells per plug), were injected subcutaneously into male or female 8–12-week-old SCID-beige mice (Taconic). The cells were first resuspended in PBS (50 μl) and then mixed with Matrigel (Corning, 356237) or LEC matrix as described above to a final volume of 350 μl. The gels were also supplied with FGF2 (10 ng ml−1) (Peprotech, 1000-18B), VEGF-A (20 ng ml−1) (Peprotech, 100-20) and heparin (100 μg ml−1) (Sigma H3149-100KU). Each mouse received two plugs: one with control cells and the other with cells transduced with ETV2. Mice implanted with plugs were injected retro-orbitally with anti-human VEcad (clone BV9- Biolegend) conjugated to Alexa-647 (25 μg in 100 μl of PBS) or 70-kDa fluorescently labelled lysine-fixable dextran (Thermo Fisher Scientific) and euthanized 8 min after injection. Whole-mount images were taken directly on a Zeiss 710 confocal microscope using a well containing a coverslip bottom. The plugs were fixed in 4% PFA overnight and then dehydrated in ethanol or put in sucrose for further immunostaining. The dehydrated plugs were sent to Histoserv for further processing, sectioning and haematoxylin and eosin (H&E), picrosirius or Masson staining. The sections were processed for immunostaining as described below. GFP-labelled lentiviral KRAS-transduced cells were injected in mice as described above, but owing to a rapid increase in size, mice bearing plugs with KRAS-transduced cells were euthanized at 2 weeks.
Immunostaining of sections
Optimal cutting temperature compound (OCT) -frozen sections (20 μm), previously fixed in 4% PFA and treated in sucrose, were washed once with PBS. The slides were then incubated in blocking buffer (0.1% Triton-X, 5% normal donkey serum, 0.1% bovine serum albumin (BSA), for 30 min at room temperature and overnight in primary antibodies at the appropriate dilution (listed in the Reporting Summary) at 4 °C in blocking buffer. For thicker sections (50 μm) tissues were blocked overnight in blocking buffer at 4 °C (0.3% Triton-X, 5% normal donkey serum, 0.1% BSA) and then for two days in primary antibody in blocking buffer at 4 °C (0.3% Triton-X, 5% normal donkey serum, 0.1% BSA). The next day, the slides were washed 3 times for 10 min at room temperature and then incubated for three hours in fluorescently conjugated secondary antibodies (1:1,000). Finally, the slides were washed 3 times for 10 min and counterstained with DAPI. The sections were mounted on coverslips. A Zeiss 710 confocal or Zeiss Cell Observer confocal spinning disk microscope was used to acquire images. For stroma staining, a mouse anti-PDGFRβ antibody (1:500, Biolegend) or an anti-mouse SMA antibody (1:200, Abcam) was used. Mouse ECs were counterstained with mouse anti-endomucin antibody (1:100, Santa Cruz). Several images were taken from sections from different layers of each plug. At least 12 pictures (4 per mouse) from different slides were taken for each condition and time point. Images were processed using ImageJ and the percentage of vessel area within the area of each image field was quantified using the threshold feature in ImageJ.
RAP1 pull-down and western blots
A 10-cm plate of either HUVECs or ETV2-transduced HUVECs (flat 2D induction stage) was used for the active RAP1 assay (Cell Signaling, 8818S) according to the manufacturer’s guidelines for the kit. In brief, the cells were washed once with PBS and then starved for three hours in M199 medium with 0.5% BSA. The cells were then scraped in the lysis buffer supplied with the kit and resuspended at around 1 mg ml−1. A fraction was saved as input and the rest of the cells were used for RAP1–GTP pull-down. Positive and negative controls, as well as a beads-only control, were performed according to the manufacturer’s guidelines. Proteins were solved on a 5–15% gradient Tris–glycine SDS–PAGE gel and semi-dry-transferred to nitrocellulose membranes. The membranes were then blocked in 5% milk in PBST and incubated in the provided RAP1 (1:1,000) antibody, GAPDH and/or ETV2 antibody for 48 h. After 48 h, the membranes were washed 3 times for 5 min and incubated in HRP-conjugated secondary antibody. Finally, after secondary washings, the membrane was blotted in ECL, chemiluminescent signals were captured with a digital camera (Kindle Biosciences) and images of protein bands were taken for densitometric quantification using ImageJ.
RAP1 inhibition experiment
Tube-formation assays for ECs with or without ETV2 were set up in 24 wells as described above. The next day, RAP1 inhibitor (GGTI-298, Tocris) resuspended in DMSO was added to the wells at a 1:1,000 dilution at the final concentration of 10 μM, and the same amount of DMSO was added to the control wells. The inhibitor and medium were changed every other day for 4 weeks. Images were obtained and the vessel area was calculated as described above at one-week and four-week time points.
RASGRP3 knockdown experiments
shERWOOD-UltramiR RASGRP3 shRNA lentiviral constructs (in pZIP-TRE3G) were purchased from TransOMIC Technologies. The clone number and targeted RASGRP3 sequences are as follows: ULTRA-3265848, AAGGGCAGAAGTCATCACAAA;ULTRA-3265850, CCTTGGAGTACACTTGAAAGA. The control shRNA (ULTRA-NT, ATGCTTTGCATACTTCTGCCT) targets a fly luciferase RNA sequence. Lentivirus was prepared as described above, using second-generation packaging plasmids. R-VECs (stage 1) were transduced with either shRNA virus or control shRNA virus (MOI = 3). Doxycycline was added at day 1 of the remodelling stage (stage 2) and the medium with doxycycline was replaced every other day for 4 weeks. Images were obtained and the vessel area was calculated as described above at two-week and four-week time points. To confirm RASGRP3 knockdown, doxycycline was added to stage-1 R-VEC cells for one week and then the cells were collected for western blot analysis.
Proteasome inhibition experiment
R-VEC vessels were prepared on Matrigel as described above. At the stabilization stage (4 weeks), R-VEC tubes were treated with either 20 μM of MG132 (Selleck Chemicals) or DMSO for 6 h. The medium was removed and the wells were washed once with PBS. R-VEC tubes were then incubated in a solution of 2 mg ml−1 Dispase (Roche) for 45 min at 37 °C to dissociate the tubes. 20μM of MG132 (Selleck Chemicals) or DMSO was continuously provided during the dissociation period. Dissociated cells were collected and further processed for western blotting as described above.
Isolation of ECs from ETV2 reporter mice
ETV2-Venus reporter mice were a gift from V. Kouskoff29. In brief, embryos were isolated at E9.5 from pooled litters of ETV2-Venus reporter mice. For each independent biological replicate, five litters of mice at E9.5 were pooled together. All embryos were accutased for 20 min at 37 °C and then triturated several times with a pipette. The cells were post-stained for anti-mouse CD31 and anti-mouse CD45 antibodies, and then sorted as either ETV2Venus+CD31+CD45− or CD31+CD45− (ARIAII, BD). Cells were sorted straight into Trizol-LS and the RNA was further purified using a Qiagen RNA-easy isolation kit.
Intestinal tissue collection and decellularization
Intestines were collected from Sprague Dawley rats ranging from 250–350 g in weight. In brief, under aseptic conditions a midline laparotomy was performed and the intestine exposed. A 5-cm-long intestinal segment was isolated, preserving the mesenteric artery and the mesenteric vein that perfuse the isolated segment. Both vessels were cannulated with a 26G cannula, and the intestinal lumen was cannulated using 1/4-inch barbed connectors. The isolated segments were decellularized, with perfusion through vasculature and lumen provided at 1 ml min−1 using a peristaltic pump (iPump). The decellularization process consisted of Milli-Q water for 24 h, sodium deoxycholate (Sigma) for 4h and DNase I (Sigma) for 3 h. Decellularized intestines were sterilized with gamma radiation before use.
Bioreactor culture
Decellularized intestines were seeded either with 5 million GFP+ETV2+ human ECs or with 5 million GFP+ control ECs. Cells were seeded through the mesenteric artery and mesenteric vein. Seeded intestines were mounted inside a custom-made bioreactor under sterile conditions. After 24 h, perfusion was started through the mesenteric artery at 1 ml min−1 using a peristaltic pump (iPump). Cells were grown in complete EC medium (M199/EBSS (HyClone, SH302503.01) supplemented with 20% heat-inactivated FBS, 1% penicillin–streptomycin, 1.5% HEPES (Corning, 25-060-Cl), 1% glutamax (Gibco 35050-061), 1% lipid mixture (Gibco, 11905-031), 1% heparin (Sigma, H3149-100KU) and 15 μg ml−1 endothelial cell growth supplement (Merck, 324845)) for the first 5 days, and then cells were grown for 2 days in StemSpan (Stem Cell Technologies) supplemented with 10% knockout serum (Thermo Fisher Scientific, 10828028), 1% penicillin–streptomycin, 1% glutatamax, 10 ng ml−1 FGF2 (Peprotech100018B), 20 ng ml−1 EGF (InvitrogenPHG0311), 10ng ml−1 IGF2 (Peprotech 100-12), 20 ng ml−1 SCF (Peprotech 300-07) and 10 ng ml−1 IL-6 (Peprotech 200-06). After 7 days, re-endothelialized intestines were collected under sterile conditions and segments of 5 × 7 mm were excised for heterotopic implantation. The remaining intestinal tissue was then fixed in 4% PFA, mounted and prepared for imaging by fluorescent microscopy. To assess the patency of the vessels, some re-endothelialized intestines were perfused with fluorescently labelled LDL.
Heterotopic graft implantation
Mice used for these studies were maintained and experiments performed in accordance with the UK Animals (Scientific Procedures) Act 1986 and approved by the University College London Biological Services Ethical Review Process (PPL 70/7622). Animal husbandry at UCL Biological Services was in accordance with the UK Home Office certificate of designation. NOD-SCID-gamma (NSG) mice, aged between 8 and 12 weeks, were anaesthetized with a 2–5% isoflurane–oxygen gas mix for induction and maintenance. Buprenorphine (0.1 mg kg−1) was administered at the induction of analgaesia. A midline laparotomy was performed under aseptic conditions. The stomach was externalized from the incision and the omentum stretched from the great curvature. A segment of the engineered intestine was then enveloped in the omentum, using 8/0 prolene sutures to secure the closure of the omental wrap. The stomach and the omentum were placed back in the abdomen and the laparotomy closed using 6/0 vicryl sutures. Mice were allowed to eat and drink normally immediately after surgery and no further medications were administered during the post-operative periods. After one week or four weeks, the mice were intravenously injected with fluorescently labelled anti-human VEcad (BV9 Biolegend) as described in ’In vivo experiments’, and then euthanized. Grafts were retrieved together with the omental envelope and fixed in 4% PFA, mounted and prepared for imaging by fluorescent microscopy.
Analysis of vascular parameters for decellularized intestine experiments
Images for in vitro EC revascularization were processed using ImageJ by setting a threshold and quantifying the area covered by the CD31 signal with respect to the intestine area. In vivo quantification of cells positive for GFP and VEcad was performed on images acquired with a confocal microscope (Zeiss LSM710) and evaluation of vascular parameters was performed using Angiotool software (National Cancer Institute)30.
Quantification of proliferating cells and apoptotic cells in decellularized scaffolds
Explanted intestinal grafts were fixed in 4% PFA, embedded in OCT and sectioned. Sections were stained for cleaved caspase 3 (Cell Signaling, 9661S) and for Ki67 (Abcam, AB15580). First, the sections were blocked for 1 h in PBS with 10% donkey serum. Then, primary antibodies were incubated overnight at 4 °C in blocking solution with the addition of 0.5% Triton-X. Primary antibodies were washed 3 times with PBS before the secondary antibody was added. Secondary antibody for donkey anti-mouse or rabbit (Alexa Fluor 547 or 647; Life Tech) was used at a dilution of 1:500 in blocking solution with 0.5% Triton X-100 and incubated at room temperature for 1 hour. Secondary antibody buffer was washed off with PBS 3 times and the slides mounted in a solution containing DAPI. Images were acquired with a confocal microscope (Zeiss LSM710). Three fields of view (425.10 μm × 425.10 μm in size) were evaluated per animal and the ratio between human VEcad (injected intra-vitally before euthanasia) and cleaved caspase 3- or Ki67-positive cells quantified.
Primary human pancreatic islets in static co-culture with ECs
Primary human islets were purchased from Prodo Laboratories. Twenty-five human islets were cultured alone, co-cultured with control ECs or co-cultured with R-VECs. Control ECs and R-VECs were used at 5 million cells per ml. The human islets with and without ECs were mixed in 40 μl Matrigel and plated into wells of a Nunc IVF 4-well dish (Thermo Fisher Scientific, 144444). Islets and ECs were co-cultured with serum-free islet medium (SFIM, Supplementary Data 2). The medium was composed of glucose-free RPMI 1640 supplemented with 0.1% human serum albumin, 10 μg ml−1 human transferrin, 50 μM ethanolamine, 50 μM phosphoethanolamine, 6.7 μg ml−1 sodium selenite, 10 ng ml−1 FGF2, 100 μg ml−1 heparin and 5.5 mM glucose. After two weeks of co-culture, samples were prepared for glucose-stimulated insulin secretion (GSIS). Samples were starved in Krebs-Ringer bicarbonate HEPES (KRBH) buffer containing 2 mM glucose for 2 h, followed by 45 min in 2 mM glucose as the basal insulin secretion and 45 min in 16.7 mM glucose as the stimulated insulin secretion. Insulin concentrations at the end of basal and stimulated phases were determined using the STELLUX Chemi Human Insulin ELISA (ALPCO). For each group, there were 11 replicates, with islets derived from 4 different donors. In other experiments, 200 human islets were cultured alone or mixed with 250,000 control ECs or 250,000 R-VECs in 50-μl Matrigel droplets. Human islet explants in co-culture were stained for EpCAM and VEcad and imaged at one and two weeks. In brief, the growth medium was removed and the cells were fixed in 4% PFA for 20 min. They were then permeabilized in 0.5% Triton-X for 20 min and blocked in IF Buffer (PBS, 0.2% Triton-X, 0.05% Tween, 1% BSA) for 1 h. Then, the cells were incubated in primary antibodies overnight in IF buffer: anti-EpCAM (1:100, Biolegend), VEcad (1:100, R&D). They were then washed 3 times with PBS 0.1% Tween. The wells were then incubated with secondary antibodies (1:1,000) in the IF buffer for 3 h. The solution was removed, DAPI in PBS was added for 5 min and cells were washed twice with PBS 0.1% Tween.
To quantify the interacting vessels with human pancreatic islets, co-cultures were imaged using a 10× objective to capture both GFP-labelled vessels and human pancreatic islets in the bright field. Using the custom MATLAB code, we traced the area of GFP-labelled vessels that surrounded and wrapped the human pancreatic islets for co-cultures with control ECs and with R-VECs.
Vascular network formation in microfluidic devices
We produced a more substantial scale device using photo-lithography as previously described17. The distance between the two fluidic channels or the width of the device is 3 mm (increased from 1 mm). The length of the device or the length of the fluidic channels is 5 mm. The height of the device is 1 mm. The total volume of the device is 15 μl. In brief, each device comprises two layers of poly(dimethylsiloxane) (PDMS; Sylgard 184; Dow-Corning), which are cast from silicon wafer masters. The devices are plasma-treated with plasma etcher (Plasma Etch) and subsequently treated with (3-glycidyloxypropyl)trimethoxysilane (Sigma, 440167) overnight. The next day, they are submerged in water to wash overnight before use. All devices are kept in a 37 °C incubator with 20% oxygen.
A mixture of 3 million ml−1 ETV2 HUVECs or control HUVECs in 5 mg ml−1 bovine fibrinogen (Sigma) and 3 U ml−1 bovine thrombin (Sigma) was injected into the devices with two 400-μm acupuncture needles (Hwato). After the cell and gel mixture polymerized, the acupuncture needles were pulled out leaving two hollow channels. HUVECs were seeded into the hollow channels to form two parent vessels on the next day. The devices were placed on a platform rocker for the entire experiment (Benchmark). Cells were cultured in the medium for vessel formation in microfluidic devices (Supplementary Data 2) and refreshed daily until day 7, when the devices were fixed and imaged.
For human pancreatic islet culture experiments, devices were set up similarly to experiments with ECs alone. Approximately 75 human pancreatic islets were mixed either alone or with control ECs or R-VECs (4 million cells per ml) cells in 5 mg ml−1 bovine fibrinogen and 3 U ml−1 bovine thrombin to a total volume of 30 μl and injected into the devices. The needles were removed after fibrin gel polymerization, and 200 μl of human pancreatic islet co-culture medium (Supplementary Data 2) was added into each of the fluidic channels. The devices were placed on a platform rocker (Benchmark 2000) during the entire experiment.
GSIS assay for human pancreatic islets in the devices
Human pancreatic islets were placed in the devices as described above either alone or in co-culture with control ECs or R-VECs. Human cadaveric islets (Prodo Labs) were procured from three healthy separate donors, with a total of n = 4 devices for no ECs, n = 4 devices for control ECs and n = 8 devices for R-VECs. After 4 days, the medium was removed in all the devices. The devices were then starved with 2 mM glucose for 2 h in the incubator. At the end of starvation, 300 μl of 2 mM glucose KRBH buffer was added at the inlet of the device, and devices were incubated at 37 °C for 3 min. Driven by gravity, KRBH buffer perfused through to the other side (outlet) of the device during the incubation. After the 3-min incubation, fluid from the outlets was collected for insulin measurement through ELISA. The inlets were also emptied of any remaining fluid. Then, another 300 μl KRBH buffer was added to inlets, leaving the outlets empty. In R-VEC co-culture devices, 30–150 μl fluid collected in the outlets, owing to high perfusion rates. In islets-alone and control-EC co-culture devices, only a small amount of fluid (less than 10 μl) was found in the outlets. To enable sample collection, we rinsed the outlets of islets alone and control-EC co-culture devices with 150 μl KRBH buffer and collected all outlet liquid for insulin measurement using ELISA. Sample collection was repeated for a total of 8 times using 2 mM glucose KRBH buffer, and another 8 times using 16.7 mM glucose KRBH buffer. In the end, we acquired a series of semi-dynamic GSIS samples. We examined the insulin concentration at the outlet of the device at the third (at t = 9 min) and eighth (t = 24 min) collections at both the 2 mM and the 16.7 mM glucose phases. The insulin level per device was calculated as: insulin per device = insulin concentration × collected volume. Basal insulin levels were determined as the average of the third and eighth collections at 2 mM glucose. Insulin concentration was determined using the STELLUX Chemi Human Insulin ELISA (ALPCO).
Staining protocol for experiments in devices
To stain for ECs in the devices, immediately before the experiment was terminated, all medium was aspirated in both fluidic channels in the devices. VEcad antibody (200 μl) conjugated with Alexa 647 at 10 μg ml−1 (Biolegend) was placed in one of the fluidic channels and allowed to slowly perfuse through the lumenized R-VEC vessels for 15–20 min in the incubator from one fluidic channel to the other fluidic channel. The device was then washed 3 times with basal medium and fixed with PFA for 45 min.
When co-culture experiments were set up with human pancreatic islets, the same protocol was used to stain for R-VEC lumenized vessels with VEcad-conjugated antibody. Post-fixation, the device was permeabilized with 0.1% Triton-X for 45 min and further stained with either EpCAM for human COs or EpCAM and insulin for human pancreatic islets. To stain for EpCAM (Biolegend) the conjugated antibodies were added to both fluidic channels at 10 μg ml−1 for 48 h on a rocker at 4 °C. The devices were washed 3 times with 1× PBS and subsequently washed and submerged into 1× PBS for 24 h on a rocker at 4 °C. A similar staining procedure was used for insulin and post-VEcad staining, except that permeabilization was carried out overnight, followed by primary antibody staining as described above and secondary staining for 24 h on a rocker at 4 °C. The devices went through washing for another 24 h with 1× PBS on a rocker at 4 °C and were then imaged using a Zeiss 710 confocal microscope.
Whole-blood perfusion in vascularized microfluidic devices
For blood perfusion videos, vessels were prepared with 3 million R-VEC cells per ml, as described above. Medium (400 μl) (Promocell) was refreshed. On day 7, blood was collected from a donor following IRB protocol in a heparinized tube. We sealed one end of both of the fluidic channels leaving two reservoirs diagonal to one another open for the perfusion experiment. Human heparinized whole peripheral blood (BD Vacutainer) was obtained from consented healthy individuals by phlebotomy. Then 200 microlitres of whole blood were immediately pipetted into one of the fluidic channels at the open reservoir. The blood cells along with intact plasma entered the fluidic channel, traversed through the lumenized R-VEC vessels and exited to the reservoir diagonal to the reservoir in which blood entered. In experiments to perfuse blood in devices with R-VECs in co-culture with human pancreatic islets, we stained blood cells with PKH26 red fluorescent dye (Sigma, MMIDI26-1KT) according to the manufacturer’s protocol for 5 min on ice. Fluorescently labelled blood cells were pipetted into the reservoir, traversed through the lumenized R-VEC vessels and exited to the diagonal reservoir. In other devices (control ECs + human pancreatic islets, and human pancreatic islets alone), fluorescently labelled blood cells were not able to traverse from one fluidic channel to the other fluidic channel. Images were taken with an Axio Observer Z1 equipped with Hamamatsu Flash 4.0 v2, sCMOS camera and 10×/0.45 objective.
Isolation and culture of mouse small intestine organoids
Mouse small intestine organoids were isolated as previously described31. Fifteen centimetres of the proximal small intestine was removed and flushed with cold PBS. After opening longitudinally, it was washed in cold PBS until the supernatant was clear. The intestine was then cut into 5-mm pieces and placed into 10 ml cold 5 mM EDTA-PBS and vigorously resuspended using a 10-ml pipette. The supernatant was aspirated and replaced with 10 ml EDTA and placed at 4 °C on a benchtop roller for 10 min. This was then repeated for a second time for 30 min. The supernatant was aspirated and then 10 ml of cold PBS was added to the intestine and resuspended with a 10 ml pipette. After collecting this 10 ml fraction of PBS containing crypts, this was repeated and each successive fraction was collected and examined underneath the microscope for the presence of intact intestinal crypts and lack of villi. The 10-ml fraction was then mixed with 10 ml DMEM basal medium (Advanced DMEM F12 containing penicillin–streptomycin, glutamine, HEPES (10 mM) and 1mM N-acetylcysteine (Sigma Aldrich A9165-SG) containing 10 U ml−1 DNase I (Roche, 04716728001), and filtered through a 100-μm filter into a BSA (1%)-coated tube. It was then filtered through a 70-μm filter into a BSA (1%)-coated tube and spun at 1,200 rpm for 3 min. The supernatant was aspirated and the cell pellet mixed with 5 ml basal medium containing 5% FBS and centrifuged at 200g for 5 min. The purified crypts were then resuspended in basal medium and mixed 1:10 with Growth Factor Reduced (GFR) Matrigel (Corning, 354230). A 40-μl sample of the resuspension fluid was plated in a 48 well plate and allowed to polymerize. Mouse small intestine organoid growth medium composed of basal medium containing 40 ng ml−1 EGF (Invitrogen PMG8043), 100 ng ml−1 Noggin (Peprotech 250-38) and 500 ng ml−1 R-spondin1 (R&D Systems, 3474-RS-050), were then laid on top of the Matrigel. In some experiments, small intestinal organoid growth medium was made with R-spondin1 from conditioned medium, collected from HEK293 cell lines expressing recombinant R-spondin1 (provided by C. Kuo).
Maintenance of mouse small intestine organoids
The medium of the organoids was changed every two days and they were passaged 1:4 every 5–7 days. To passage, the growth medium was removed and the Matrigel was resuspended in cold PBS and transferred to a 15-ml falcon tube. The organoids were mechanically disassociated using a p1000 or a p200 pipette and pipetting 50–100 times. Seven ml of cold PBS was added to the tube and pipetted 20 times to fully wash the cells. The cells were then centrifuged at 1,000 rpm for 5 min and the supernatant was aspirated. They were then resuspended in GFR Matrigel and replated as above. For freezing, after spinning the cells were resuspended in basal medium containing 10% FBS and 10% DMSO and stored in liquid nitrogen indefinitely.
Mouse small intestine organoid co-culture and staining
Mouse small intestine organoids were co-cultured for 4–7 days either alone or with control ECs or R-VECs, at a final concentration of 5 million cell per ml of Matrigel. Organoids were mechanically dissociated as described above and mixed with the ECs, spun down and resuspended in GFR Matrigel. The mixture was then dispersed in 30-μl droplets in 8-well chamber slides (Lab-Tek II, 154534) or 50-μl droplets in a Nunc IVF 4-well dish (Thermo Fisher Scientific, 144444). Cells were cultured in mouse small intestine organoid medium (Supplementary Data 2) as described above (EGF 40 ng ml−1, Noggin 50 ng ml−1, R-spondin1 conditioned medium (10%) + FGF-2 (10 ng ml−1) (Peprotech, 1000-18B) and heparin (100 μg ml−1) (Sigma, H3149-100KU). Vessel area was quantified by the threshold function in ImageJ and individual sprouts in contact with the mouse small intestine organoids were counted and reported as vessel sprouts per organoid. Where indicated, 10 μM EdU was added to the growth medium for 6 h before fixing. The growth medium was removed and the cells were fixed in 4% PFA for 20 min. They were then permeabilized in 0.5% Triton-X for 20 min and blocked in IF buffer (PBS, 0.2% Triton-X, 0.05% Tween, 1% BSA) for 1 h or immediately processed for EdU staining according to directions provided with the Click-iT Edu Imaging Kit (Invitrogen C10340). For immunofluorescent staining, cells were incubated in primary antibodies overnight in IF buffer: anti-KRT20 (1:200, Cell Signaling Technologies, 13063). They were then washed 3 times with PBS 0.1% Tween. The wells were then incubated with secondary antibodies (1:1,000) in the IF buffer for 3 h. The solution was removed, DAPI in PBS was added for 5 min and cells were washed twice with PBS 0.1% Tween. The chambers were then removed and cover slips were mounted using Prolong Gold antifade medium (Invitrogen P36930).
Isolation and culture of human normal and tumour colon organoids
Isolation of human colonic crypts and adenoma, and culture and maintenance of organoid cultures, were performed as previously described32. Normal and adenoma tissues were collected from colonic resections according to protocols approved by the Weill Cornell Medicine IRB. In brief, human colonic mucosa samples were obtained by trimming surgically resected specimens. The underlying muscle layer was removed using fine scissors under a stereomicroscope, leaving the mucosa, which was cut into 5-mm pieces on a Petri dish, placed into a 15-ml centrifuge tube containing 10 ml of cold DPBS and washed 3 times. Ten millilitres of cold DPBS supplemented with 2.5 mM EDTA was added to the tube and the tube was incubated for 1 h at room temperature with gentle shaking. Isolated crypts were mixed with Matrigel (Corning, 354230), dispensed in the centre of each well of a 6-well plate using a 200-μl pipette and placed at 37 °C for 10 min to solidify the Matrigel.
Normal COs were procured from Jason Spence’s (J.S.) laboratory at the University of Michigan as previously described33,34 (specifically, human CO lines 87 and 89). Healthy human COs were passaged 1:3 every 7 days by mechanical dissociation (pipetting) and grown in 12-well low-attachment plates in 30-μl Matrigel droplets. Normal COs were cultured in human CO medium (Supplementary Data 2) comprising Advanced DMEM F12, penicillin–streptomycin, 4 mM glutamax, 1% HEPES, primocin (100 μg ml−1), 50% L-WRN (WNT3a, R-spondin1, Noggin)-conditioned medium, N2, B27 without vitamin A, N-acetylcysteine (1 mM), human recombinant EGF (50 ng ml−1), Y-27632 (10 μM), A-83-01 (500 nM) and SB202190 (10 μM). The L-WRN-conditioned medium was generated using L-WRN cells. Conditioned medium was collected for 4 days, pooled, sterile-filtered and frozen into aliquots until use.
Human CRCOs were procured through the Institute for Precision Medicine at Weill Cornell Medicine35. The CRCOs were split 1:3 every 7 days by digesting in TrypLE Select (Thermo Fisher Scientific) supplemented with 10 μM Y27632 (Tocris Bioscience), and were maintained in human CRCO medium and propagated in GFR Matrigel. Human CRCO medium (Supplementary Data 2) comprises Advanced DMEM F12, 1% penicillin–streptomycin, 1% glutamax, 1% HEPES, R-spondin1-conditioned medium (5%), N-acetylcysteine (1.25 mM), human recombinant EGF (50 ng ml−1), human recombinant FGF-10 (20 ng ml−1), FGF-2 (1 ng ml−1), Y-27632 (10 μM), A-83-01 (500 nM), SB202190 (10 μM), nicotinamide (10 mM), PGE2 (1 μM), NRG (10 ng ml−1) and human gastrin 1 (10 nM).
Co-cultures of normal and tumour organoids with ECs
R-VECs or control ECs (at a final concentration of 5 million cells per ml) were mixed with healthy human COs or patient-derived CRCOs, spun down and resuspended in Matrigel (Corning, 354230) or LEC matrix as described above. The cells were then dispersed in 30–70-μl Matrigel or LEC droplets in 8-well chamber slides (Lab-Tek II, 154534) or a Nunc IVF 4-well dish (Thermo Fisher Scientific, 144444) and cultured in the respective organoid medium with the addition of FGF-2 (10 ng ml−1) (Peprotech, 1000-18B) and heparin (100 μg ml−1) (Sigma H3149-100KU). The medium was changed every other day. A 4.5-h pulse of EdU was used for all tumour organoid co-culture experiments (Click-iT EdU kit, Invitrogen C10340). The co-cultures were maintained in a 37 °C incubator with 20% oxygen. Human COs and CRCOs were stained similarly to mouse small intestinal organoid co-cultures. Antibodies against human EpCAM (Biolegend) and VEcad (R&D) were added and co-cultures were incubated overnight, followed by secondary antibody staining.
Preparation of normal and tumour organoids cultured with ECs for molecular profiling and single-cell sequencing
For single-cell sequencing, co-cultures were maintained for seven days. To collect cells in co-culture for single-cell sequencing, the medium was removed from the culture and the organoid–endothelial cell droplets were incubated in 2 mg ml−1 dispase (Roche) for 20 min at 37 °C with shaking. The cells were then spun down and incubated for an additional 15 min at 37 °C in accutase. At this point, the endothelial cells were mostly released from the co-cultures and collected by filtering through a 40-μm mesh. The rest of the undigested cells (mainly organoid clusters) were further dissociated into single cells by incubating with TryplE for an additional 45 min at 37 °C until the cells were completely separated as single cells. This two-step digestion allowed for increased viability and efficient dissociation of both endothelial cells and organoids. Both the first and the second fraction were further processed for single-cell analysis. Single cells were collected and filtered through a 35-μm nylon mesh and processed for single-cell sequencing.
For qRT–PCR experiments, co-cultures were maintained for seven days in Matrigel. To collect cells and dissociate organoids in co-cultures, we incubated the Matrigel droplets with TrypLE-Express enzyme (Thermo Fisher Scientific, 3 ml per 50-μl Matrigel droplet) for 45 min at 37 °C with vigorous shaking. The dissociated cells were then washed twice, once with organoid culture medium and once with MACs buffer. Dissociated cells were resuspended in 100 μl MACS buffer and anti-human CD31 (Biolegend, 10 μg ml−1) was used to stain for endothelial cells for 30 min on ice. The cell suspension was washed with MACS buffer and resuspended in MACS buffer with DAPI (1μg ml−1). Subsequently, cells were sorted to purify the DAPI−CD31− population. An Accurus PicoPure RNA isolation kit (Thermo Fisher Scientific) was used to isolate RNA from the collected cells.
Quantification of vessels that interact with human COs and CRCOs in serial confocal videos
Human CRCOs and COs were stained with CellTracker (Invitrogen, C34565) as per the instruction manual of the manufacturer. CRCOs and COs were embedded inside Matrigel or LEC matrix with either control ECs or R-VECs at 5 million cells per ml. A mixture of gel and cells was pipetted onto a glass-bottomed dish and polymerized inside a 37 °C incubator for 15 min. The culture was then fed with organoid medium supplemented with 10 ng ml−1 bFGF (Peprotech) and 100 μg ml−1 heparin (Sigma H3149-100KU). To enable long-term imaging, 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Sigma), as an antioxidant, was also added into the medium at 100 μM. The culture was immediately mounted onto a temperature- and gas-controlled chamber. Time-lapse videos were acquired with a Zeiss Cell Observer confocal spinning disk microscope (Zeiss) equipped with a Photometrics Evolve 512 EMCCD camera at an interval of 40 min over 3–4 days. Medium was refreshed every two days.
To quantify the vessels interacting with normal and tumour colon organoids, Z-projection images of time-lapse videos from several time points were obtained using ImageJ. Custom MATLAB codes were written to quantify the interacting vessel areas with all individual organoids. The custom MATLAB codes are provided in Supplementary Data 3. In brief, the code was used to manually trace the perimeter of all vessels around which ECs were wrapping and tapping the organoids. The area of the manually traced interacting vessels was quantified and reported.
RNA library preparation and analysis of sequencing data
RNA was isolated and purified using the Rneasy Mini Kit (Qiagen) or Accurus PicoPure RNA isolation kit (Thermo Fisher Scientific). RNA quality was verified using an Agilent Technologies 2100 Bioanalyzer. RNA libraries were prepared and multiplexed using the Illumina TruSeq RNA Library Preparation Kit v.2 (non-stranded and poly-A selection) and 10 nM of cDNA was used as input for high-throughput sequencing with Illumina’s HiSeq 2500 or HiSeq 4000, producing 51-bp paired-end reads. Sequencing reads were de-multiplexed (bcl2fastq) and mapped with STAR v.2.6.0c (ref. 36) with default parameters to the appropriate NCBI reference genome (GRCh38.p12 for human samples and GRCm38.p6 for mouse samples). Fragments per gene were counted with featureCounts v.1.6.2 (ref. 37) with respect to Gencode comprehensive gene annotations (release 28 for human samples and M17 for mouse samples).
Transcriptome data analysis
Differential gene expression analysis was performed using DESeq2 v.1.18.1 (ref. 38), and only false discovery rate (FDR)-adjusted P values of less than 0.05 were considered statistically significant. Before differential gene expression analysis, genes expressed at low levels were filtered out by only retaining genes that have more than 1CPM in the condition with the least number of replicates. Base-2 log-transformed CPM values were used for heat map plots, which were centred and scaled by row. Before visualization, tissue-specific effects were removed using the removeBatchEffect function from limma v.3.34.9 (ref. 39). GO analysis was performed using DAVID Bioinformatics Resource Tools v.6.8 (ref. 40).
ChIP and antibodies
To identify the genome-wide localization of ETV2, K4me3, K27me3 and K27ac modifications in R-VECs or control ECs, ChIP assays were performed with approximately 1 × 107 cells per experiment, as previously described41. Cells introduced with triple Flag-tagged ETV2 lentivirus (as described above) were used for the ETV2 ChIP. In brief, cells were cross-linked in 1%PFA for 10 min at 37 °C, then quenched by 0.125 M glycine. Chromatin was sheared using a Bioruptor (Diagenode) to create fragments of 200–400 bp, immunoprecipitated by 2–5 μg of antibody or mouse IgG bound to 75 μl Dynabeads M-280 (Invitrogen) and incubated overnight at 4 °C. Magnetic beads were washed and chromatin was eluted. The ChIP DNA was reverse-cross-linked and column-purified. All ChIP antibodies are identified in the Reporting Summary.
ChIP–seq library construction and sequencing
ChIP–seq libraries were prepared with the Illumina TruSeq ChIP Library Preparation Kit for DNA from ETV2 ChIP, and K4me3, K27me3 and K27ac modification ChIP. ChIP–seq libraries were sequenced with the Illumina HiSeq 4000 system.
ChIP–seq data processing and analysis
ChIP–seq reads were aligned to the reference human genome (hg19, GRCh37) using the BWA alignment software (v.0.5.9)42. Unique reads mapped to a single best-matching location with no more than 4% of the read length of mismatches were kept for peak identification and profile generation. Sequence data were visualized with IGV by normalizing to 1 million reads43. The software MACS2 (ref. 44) was applied to the ChIP–seq data with sequencing data from input DNA as control to identify genomic enrichment (peak) of ETV2. The SICER (v.1.1) (ref. 45) algorithm was applied to the ChIP–seq data with sequencing data from input DNA as a control to identify genomic regions with significant enrichment differences in different cell types. The resulting peaks were filtered by P < 0.05 for ETV2 and FDR < 0.01 for histone modifications. We computed the read counts in individual promoters using HOMER46. Each identified peak was annotated to promoters ( ±2 kb from transcription start site), gene body or intergenic region by HOMER. Summary and peak call information for all ChIP–seq data processing and analysis is provided in Supplementary Table 1.
10X Chromium single-cell transcriptomics and analysis
The following two experiments were performed for single-cell library preparation to establish the adaptation of R-VECs when co-cultured with normal or malignant organoids.
Experiment 1: R-VECs were co-cultured alone or together with human COs for seven days in human CO medium supplemented with 10 ng ml−1 FGF2 (Promocell) and 100 μg ml−1 heparin. COs were also cultured alone in CO medium supplemented with 10 ng ml−1 FGF and 100 ng ml−1 heparin for seven days. After seven days, all three conditions (R-VECs alone, R-VECs + COs, or COs alone) were dissociated with dispase and TryplE (Thermo Fisher Scientific) as described above, and submitted for 10X Chromium single-cell analysis. All three samples were processed and run at the same time.
Experiment 2: R-VECs were co-cultured alone or together with human CRCOs for seven days in CRCO medium supplemented with 10 ng ml−1 FGF2 and 100 μg ml−1 heparin. The CRCOs were also cultured alone in CRCO medium with 10 ng ml−1 FGF and 100 μg ml−1 heparin for 7 days. After 7 days, all three conditions (R-VECs alone, R-VECs + CRCOs, or CRCOs alone) were dissociated with collagenase, dispase and TryplE as described above, and submitted for 10 Chromium single-cell analysis. All three samples were processed and run at the same time.
The single-cell suspension was loaded onto a well on a 10X Chromium Single Cell instrument (10X Genomics). Barcoding and cDNA synthesis were performed according to the manufacturer’s instructions. In brief, the 10X GemCode Technology partitions thousands of cells into nanolitre-scale gel bead-in-emulsions (GEMs), in which all the cDNA generated from an individual cell share a common 10X barcode. To identify the PCR duplicates, a unique molecular identifier (UMI) was also added. The GEMs were incubated with enzymes to produce full length cDNA, which was then amplified by PCR to generate enough quantity for library construction. Qualitative analysis was performed using the Agilent Bioanalyzer High Sensitivity assay. The cDNA libraries were constructed using the 10X Chromium single-cell 3′ Library Kit according to the manufacturer’s original protocol. In brief, after the cDNA amplification, enzymatic fragmentation and size selection were performed using SPRI select reagent (Beckman Coulter, B23317) to optimize the cDNA size. P5, P7, a sample index and read 2 (R2) primer sequence were added by end repair, A-tailing, adaptor ligation and sample-index PCR. The final single-cell 3′ library contains standard Illumina paired-end constructs (P5 and P7), Read 1 (R1) primer sequence, 16-bp 10X barcode, 10-bp randomer, 98-bp cDNA fragments, R2 primer sequence and 8-bp sample index. For quality control after library construction, 1 μl of the sample was diluted 1:10 and run on the Agilent Bioanalyzer High Sensitivity chip for qualitative analysis. For quantification, the Illumina Library Quantification Kit (KAPA Biosystems, KK4824) was used.
Libraries were sequenced on an Illumina NextSeq500 with a 150-cycle kit using the following read length: 26-bp Read 1 for cell barcode and UMI, 8-bp I7 index for sample index and 132-bp Read 2 for transcript. Cell Ranger 2.2.0 (http://10xgenomics.com) was used to process Chromium single-cell 3′ RNA-seq output. First, ‘cellranger mkfastq’ demultiplexed the sequencing samples based on the 8-bp sample index read to generate fastq files for the Read 1 and Read 2, followed by extraction of 16-bp cell barcode and 10-bp UMI. Second, ‘cellranger count’ aligned the Read 2 to the human reference genome (GRCh38) using STAR36. Then aligned reads were used to generate the data matrix only when they have valid barcodes and UMI and map to exons (Ensembl GRCh38) without PCR duplicates. Valid cell barcodes were defined on the basis of UMI distribution.
All single-cell analyses were performed using the Seurat package in R (v.2.3.4) (ref. 47). Once the gene–cell data matrix was generated, poor-quality cells were excluded, including cells with more than 6,000 uniquely expressed genes (as they are potentially cell doublets). Only genes expressed in three or more cells in a sample were used for further analysis. Cells were also discarded if their mitochondrial gene percentages were over 10% or if they expressed fewer than 600 unique genes, resulting in 20,778 genes across 24,478 cells, with the median UMI count for each cell across the entire dataset being 7,845 and the median number of unique genes per cell being 2,397. Further information on each sample that passed the quality filters is available in Supplementary Table 2. Following best practices in the package suggestions, UMI counts were log-normalized and after the most highly variable genes were selected the data matrices were scaled using a linear model, with variation arising from UMI counts and mitochondrial gene expression mitigated for. Principal component analysis was subsequently performed on this matrix and after reviewing principal component heat maps and jackstraw plots, UMAP visualization was performed on the top 29 components and the clustering resolution was set at 1.0 for visualizations. Differential gene expression for gene-marker discovery across the clusters was performed using the Wilcoxon rank-sum test in the Seurat package.
Epithelial cells were identified by the epithelial cell markers EPCAM, CDH1 and KRT19 and ECs were identified by the EC markers VEcad, CD31 and VEGFR2. Epithelial cells were filtered out from the next analysis to identify heterogeneity amongst the EC populations of the co-cultured normal and tumour cell populations. The epithelial cell fraction was also analysed on its own in the tumour and co-cultured samples. In both of these analyses best practices were again followed for cluster discovery using the top 20 components and cluster resolution 0.6 in the matched tumour and normal sample sets and differential gene expression for gene-marker discovery across the clusters were performed using the Wilcoxon rank-sum test in the Seurat package.
Statistical analysis and data reporting
Data were assessed and analysed using appropriate statistical methods. The normality of data was assessed using the Kolmogorov–Smirnov test. Sample sizes and statistics for each experiment are provided in Supplementary Data 1. GraphPad Prism v.7 was used for all statistical analysis, unless otherwise indicated. No statistical methods were used to determine sample size. Unless otherwise stated, the experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Source ChIP–seq data are provided in Supplementary Table 1 and source scRNA-seq data are provided in Supplementary Table 2. The RNA-seq data can be viewed at the Gene Expression Omnibus (GEO) under accession number GSE131039. The ChIP–seq data and scRNA-seq data can be viewed at the GEO under accession numbers GSE147746 and GSE148996, respectively. Source data are provided with this paper.
Code availability
All of the code used in this paper is available from the authors on request.
References
Augustin, H. G. & Koh, G. Y. Organotypic vasculature: from descriptive heterogeneity to functional pathophysiology. Science 357, eaal2379 (2017).
Rafii, S., Butler, J. M. & Ding, B. S. Angiocrine functions of organ-specific endothelial cells. Nature 529, 316–325 (2016).
Lee, D. et al. ER71 acts downstream of BMP, Notch, and Wnt signaling in blood and vessel progenitor specification. Cell Stem Cell 2, 497–507 (2008).
Barry, D. M. et al. Rasip1-mediated Rho GTPase signaling regulates blood vessel tubulogenesis via nonmuscle myosin II. Circ. Res. 119, 810–826 (2016).
Strilić, B. et al. The molecular basis of vascular lumen formation in the developing mouse aorta. Dev. Cell 17, 505–515 (2009).
Carmeliet, P. & Jain, R. K. Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307 (2011).
Cao, Z. et al. Molecular checkpoint decisions made by subverted vascular niche transform indolent tumor cells into chemoresistant cancer stem cells. Cancer Cell 31, 110–126 (2017).
Nolan, D. J. et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 26, 204–219 (2013).
Pellegata, A. F., Tedeschi, A. M. & De Coppi, P. Whole organ tissue vascularization: engineering the tree to develop the fruits. Front. Bioeng. Biotechnol. 6, 56 (2018).
Giobbe, G. G. et al. Extracellular matrix hydrogel derived from decellularized tissues enables endodermal organoid culture. Nat. Commun. 10, 5658 (2019).
Ronaldson-Bouchard, K. & Vunjak-Novakovic, G. Organs-on-a-chip: a fast track for engineered human tissues in drug development. Cell Stem Cell 22, 310–324 (2018).
Bhatia, S. N. & Ingber, D. E. Microfluidic organs-on-chips. Nat. Biotechnol. 32, 760–772 (2014).
Lancaster, M. A. & Knoblich, J. A. Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125 (2014).
Tuveson, D. & Clevers, H. Cancer modeling meets human organoid technology. Science 364, 952–955 (2019).
Koyano-Nakagawa, N. & Garry, D. J. Etv2 as an essential regulator of mesodermal lineage development. Cardiovasc. Res. 113, 1294–1306 (2017).
Ginsberg, M. et al. Efficient direct reprogramming of mature amniotic cells into endothelial cells by ETS factors and TGFβ suppression. Cell 151, 559–575 (2012).
Nguyen, D. H. et al. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc. Natl Acad. Sci. USA 110, 6712–6717 (2013).
Eberhard, D., Kragl, M. & Lammert, E. ‘Giving and taking’: endothelial and beta-cells in the islets of Langerhans. Trends Endocrinol. Metab. 21, 457–463 (2010).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011).
Miyoshi, H. & Stappenbeck, T. S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protocols 8, 2471–2482 (2013).
Stan, R. V. et al. The diaphragms of fenestrated endothelia: gatekeepers of vascular permeability and blood composition. Dev. Cell 23, 1203–1218 (2012).
Lyden, D. et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature 401, 670–677 (1999).
Li, S. et al. Plasma mesothelin as a novel diagnostic and prognostic biomarker in colorectal cancer. J. Cancer 8, 1355–1361 (2017).
Si, M. & Lang, J. The roles of metallothioneins in carcinogenesis. J. Hematol. Oncol. 11, 107 (2018).
Baudin, B., Bruneel, A., Bosselut, N. & Vaubourdolle, M. A protocol for isolation and culture of human umbilical vein endothelial cells. Nat. Protocols 2, 481–485 (2007).
Seandel, M. et al. Generation of a functional and durable vascular niche by the adenoviral E4ORF1 gene. Proc. Natl Acad. Sci. USA 105, 19288–19293 (2008).
Ginsberg, M., Schachterle, W., Shido, K. & Rafii, S. Direct conversion of human amniotic cells into endothelial cells without transitioning through a pluripotent state. Nat. Protocols 10, 1975–1985 (2015).
Schachterle, W. et al. Sox17 drives functional engraftment of endothelium converted from non-vascular cells. Nat. Commun. 8, 13963 (2017).
Wareing, S., Eliades, A., Lacaud, G. & Kouskoff, V. ETV2 expression marks blood and endothelium precursors, including hemogenic endothelium, at the onset of blood development. Dev. Dyn. 241, 1454–1464 (2012).
Zudaire, E., Gambardella, L., Kurcz, C. & Vermeren, S. A computational tool for quantitative analysis of vascular networks. PLoS One 6, e27385 (2011).
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Sugimoto, S. & Sato, T. Establishment of 3D intestinal organoid cultures from intestinal stem cells. Methods Mol. Biol. 1612, 97–105 (2017).
Dame, M. K. et al. Identification, isolation and characterization of human LGR5-positive colon adenoma cells. Development 145, dev153049 (2018).
Tsai, Y. H. et al. A method for cryogenic preservation of human biopsy specimens and subsequent organoid culture. Cell. Mol. Gastroenterol. Hepatol. 6, 218–222.e7 (2018).
Puca, L. et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 9, 2404 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols 4, 44–57 (2009).
Liu, Y. et al. Epigenetic profiles signify cell fate plasticity in unipotent spermatogonial stem and progenitor cells. Nat. Commun. 7, 11275 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Thorvaldsdóttir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013).
Zhang, Y. et al. Model-based analysis of ChIP–seq (MACS). Genome Biol. 9, R137 (2008).
Zang, C. et al. A clustering approach for identification of enriched domains from histone modification ChIP–seq data. Bioinformatics 25, 1952–1958 (2009).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Acknowledgements
S.R. is supported by the Ansary Stem Cell Institute; grants from the National Institutes of Health (NIH) (R35 HL150809, R01s DK095039, HL119872, HL128158, HL115128, HL139056, RC2 DK114777 and U01AI138329); the Empire State Stem Cell Board; grants from New York State Stem Cell Science (NYSTEM) (C026878, C028117, C029156, C030160); the Daedalus Fund for Innovation from Weill Cornell Medicine; and the Starr Foundation stem cell core project; and Qatar National Priorities Research Program (NPRP 8-1898-3-392); and the Tri-Institutional Stem Cell Initiatives (TRI-SCI 2013-032, 2014-023, 2016-013 and 2019-029). R.E.S. is supported by NIH grants R01CA234614, 2R01AI107301, R01DK121072,1R21AI117213, RO3DK117252, R01AA027327 and K08DK101754 and Department of Defense CA170574. R.E.S. is a Irma Hirschl Trust Research Award Scholars. R.S. is supported by NYSTEM contract C32596GG; P.D.C. by NIHR Research (NIHR-RP-2014-04-046); P.D.C., A.F.P., A.M.T. and F.S. by the OAK Foundation (W1095/OCAY-14-191), H2020 grant INTENS 668294 and the NIHR Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust; A.M.T. by the BBSRC ICASE studentship 167881; Y. Liu and J.M.G.-S. by a New York Stem Cell Foundation Druckenmiller fellowship; B.K. by a T32 fellowship; O.E. by NIH UL1TR002384, R01CA194547, LLS SCOR 180078-02, 7021-20 and Janssen and Eli Lilly research grants; J.S. by HL119215; and Q.J.Z. by NIH R01DK106253, UC4DK116280, and the Neuroendocrine Tumor Research Foundation grant NTRF192269; J.R.S. by NIH R01HL119215. We thank R. Chavez for technical assistance and animal maintenance, the Visual Function Core at Weill Cornell Medicine for live-imaging resources, J. Shieh for HUVEC isolations, C. Wasserstein, M. Mishaan, M. Bell, C. Cheung and W. Gu for support with organoid cultures and A. Gjinovci for surgical help with the isolation of mouse small bowels.
Author information
Authors and Affiliations
Contributions
B.P. and S.R. conceived the study and wrote the manuscript. B.P., D.-H.T.N., R.S., K.S., R.E.S., S.Y.R. and S.R. discussed and analysed data. R.S. provided microscopy expertise. B.P., D.-H.T.N., G.L., R.S., Y. Liu, F.G., Y. Lin, J.M.G.-S., M.Y., S.Y.R. and S.R. performed experiments and analysed data. A.F.P., A.M.T., F.S. and P.D.C. carried out and analysed experiments on the decellularized intestines. Y. Liu, D.R., P.Z., T.Z., B.K., O.E. and J.Z.X. analysed ChIP–seq, RNA-seq and single-cell sequencing. M.W., T.H., S.L., L.D., J.S. and Q.J.Z. assisted with organoid cultures. All the authors read and provided feedback on the figures and manuscript.
Corresponding author
Ethics declarations
Competing interests
S.R. is the founder and a non-paid consultant to Angiocrine Bioscience. O.E. is a scientific advisor and equity holder in Freenome, Owkin, Volastra Therapeutics and One Three Biotech. R.E.S. is a scientific advisor and member of the SAB for Miromatrix Inc.
Additional information
Peer review information Nature thanks Hans Clevers, Harald Ott and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 ETV2 confers mature human ECs with the ability to autonomously self-assemble into lumenized, durable, branching and patterned vessels in vitro without the constraints of bioprinted scaffolds.
a, Overview of experimental set-up for vessel formation in vitro for screen of different media, extracellular matrix components and tissue-specific ECs. b, The proliferation of GFP-transduced R-VECs and control ECs (CTRL-ECs) at each stage of vessel formation. EdU+ cells were quantified after a 16-hour EdU pulse. c, Time course of vessel formation on Matrigel for GFP+ CTRL-EC and R-VECs over 8 weeks. d, Vessel formation using R-VEC or CTRL-EC in three different enriched pro-angiogenic media (Supplementary Data 2): Serum-free StemSpan with Knockout serum replacement and Cytokines, EGM-2 and complete EC media on Matrigel. R-VEC formed the most robust lumenized vessels in serum-free StemSpan with knockout serum replacement medium and cytokines, as compared to other media with serum. CTRL-EC failed to form durable stable vessels. e, f, Time course (e) and quantification (f) of tube formation for GFP+ human Adipose CTRL-EC and human Adipose R-VEC on Matrigel. g, Representative images of tissue-specific GFP+ R-VEC and CTRL-EC isolated from adult human heart (cardiac EC), aorta (aortic EC) and skin (dermal EC) demonstrated robust and stable vessels at 4 weeks on Matrigel. h, Representative images of GFP+ R-VEC vessels formed on Matrigel or a pre-defined matrix of laminin/entactin and collagen IV (LEC). i, Immunostaining of R-VEC-tubes displayed apicobasal proper polarity with podocalyxin, apical (in red) and laminin, basal (in green). The right image is an orthogonal projection. j, Stiffness measurements by atomic force microscopy (AFM) of adult Adipose and HUVEC ECs with and without ETV2. In both cases, ETV2-transduced ECs are significantly less stiff than their counterparts. The abbreviated box plots indicate the interquartile range and median for each condition. k, HUVECs were transduced with either an empty lentiviral vector or lentiviral vectors with ETV2, myrAKT or ETS1 constructs, and used in a vessel formation assay. Western Blot analysis for expression of ETV2, p-AKT, total AKT and ETS1 in those cells. l, Representative images for ETS1 or myrAKT1 transduced GFP+ HUVECs in a vessel formation assay on Matrigel. m, Quantification of vessel area for ETS1, myrAKT1 and ETV2 (R-VEC) cells indicated that ETS1-EC and myrAKT1-EC fail to form robust vessel formation as compared to R-VEC. Data are mean ± s.e.m. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001. For statistics, see Supplementary Data 1. For medium formulations, see Supplementary Data 2.
Extended Data Fig. 2 Transient ETV2 expression in adult human ECs is sufficient for the generation and maintenance of durable long-lasting R-VEC vessels in vitro.
a, Schematic for ETV2 mRNA and protein levels assessment at each of the three stages of R-VEC vessel formation. b, Quantification of ETV2 mRNA levels at each stage of vessel formation. c, d, Western blot analysis (c) and densitometric quantification (d) of ETV2 protein levels at each stage of vessel formation. GAPDH was used as a loading control. e, A proteasome inhibitor (MG132) restored ETV2 levels by ~sixfold when added to R-VECs during the stabilization stage. f, Densitometric quantification of western blots in e. g, qRT–PCR (g) and western blot (h) assessment of ETV2 levels after doxycycline removal. i, Representative images of GFP+ iR-VECs on Matrigel with inducible ETV2 expression at 2 months. ETV2 was turned off at day 0, day 7 and at 4-weeks post start of the remodelling stage 2. j, Quantification of iR-VEC vessels at 2 months. k, Electron microscopy pictures of a lumen present both in vessels in which doxycycline was continuously present and in vessels in which doxycycline was removed after 1 month. Data are mean ± s.e.m. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001. For statistics, see Supplementary Data 1. For medium formulations, see Supplementary Data 2.
Extended Data Fig. 3 R-VEC vessels are functionally anastomosed to host vessels and not leaky in vivo.
a, Fluorescently labelled R-VEC or CTRL-EC cells in LEC matrix were subcutaneously injected in the flank of SCID beige mice and retrieved at 2 months. Human-specific VEcad antibody (hVEcad) was injected intravitally right before euthanasia. Sections of the plugs were stained for mouse ECs with an anti-mouse endomucin antibody (mEndomucin), identifying properly organized human R-VECs anastomosing with mouse vessels (thickness = 50 μm). Sections were also stained with the nuclear stain DAPI. b, c, Plugs in a were post-stained with hVEcad and a mouse PDGFRβ antibody (b) or mouse SMA antibody (c) (thickness = 50 μm). d, In vivo plug assay, in which mice were subcutaneously injected with either control ECs (HUVECs transduced only with rtTA lentivirus) or stage 1 doxycycline-inducible-ETV2 ECs (iR-VECs: HUVECs transduced with both rtTA and inducible ETV2 lentivirus) in LEC matrix. One group of mice was on doxycycline (ETV2 continuously on) and another group of mice was on doxycycline food diet for 1 week (ETV2 on) and then switched to regular food (ETV2 off). All mice were euthanized 2 months post-implantation. Red indicates the GFP labelled human ECs, white: Anti-VEcad antibody that was retro-orbitally injected before euthanizing the mice. e, Quantification of vessel area for rtTA only plugs, mice on doxycycline for 1 week, and mice continuously on doxycycline diet (ETV2 on). All mice were euthanized 2 months post-implantation. f, 70 kDa fluorescent dextran (in blue) and human VEcad (in white) were injected in mice implanted with fluorescently labelled R-VECs (in red, 5-months post-implantation), iR-VECs (in red, 1 week on doxycycline food and euthanized at 2 months) or K-RAS-HUVECs (K-RAS-EC) (in red, 2-weeks post-implantation) to assess anastomosis and leakiness of vessels. K-RAS-EC vessels showed dextran leakiness, whereas R-VEC and iR-VEC vessels exhibited patency and non-leakiness. Green arrows point at perfused mouse vessels that were also perfused with dextran. Data are mean ± s.e.m. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001. For statistics, see Supplementary Data 1. For medium formulations, see Supplementary Data 2.
Extended Data Fig. 4 Implanted R-VECs form stable, patterned, branching and durable vessels in vivo without features of vascular malformations, cysts, adenomas, haemangiomas or metastasis.
a, Representative images of non-haemorrhagic R-VEC plugs at 10 months. b, Whole-mount microscopy of R-VEC plugs at 10-months post perfusion with anti-human VEcad antibody (hVEcad). c, d, Representative H&E and Masson staining of R-VEC plugs at 10 months (c). There were no features of cysts or haemangiomas present, in contrast to KRAS-EC plugs (at 4 weeks) that formed an EC tumour (d). e, There was no metastasis of R-VECs to other tissues 10 months after plug implantation and the tissues were assessed to be normal without fibrosis and architectural disruption or tumorigenesis as evaluated by H&E, Masson and picrosirius staining.
Extended Data Fig. 5 Decellularized intestinal scaffolds re-endothelialized with R-VECs engraft in vivo after omental implantation.
a, Schematic of experimental procedure for heterotopic implantation of decellularized intestinal scaffold vascularized using R-VECs. b, Rat intestines were cannulated through lumen, mesenteric artery and mesenteric vein. c, Decellularized intestine preserves native vasculature (green = GFP+ R-VECs). d, Seeded GFP labelled R-VECs spread evenly and reach distal capillaries. e, Heterotopic implantation of re-endothelialized intestines in immunodeficient mice omentum shows engraftment after 1 and 4 weeks of GFP+ R-VECs and anastomosis to the host vasculature as indicated by intravital intravenous injection of anti-human VEcad antibody (hVEcad). Representative H&E stainings show anatomical normal perfused vessels. f, Quantification of the area covered by R-VEC compared to CTRL-EC in implanted re-endothelialized intestines at 1 week and 4 weeks. g, Quantification of R-VEC and CTRL-EC proliferation and apoptosis in implanted re-endothelialized intestines at 1 and 4 weeks. Data are mean ± s.e.m. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001. For statistics, see Supplementary Data 1.
Extended Data Fig. 6 ETV2, by directly binding to promoters and enhancers of target genes, regulates differentially expressed genes in R-VECs.
a, Schematic of RNA-sequencing performed on R-VECs and CTRL-ECs derived from different tissue-specific ECs during stage 1 induction phase (2D monolayers). b, R-VECs or CRTL-ECs were analysed by RNA sequencing. Heat maps of selected genes within top enriched GO categories. Values are log2-normalized CPM, centred and scaled by row. ETV2 binding from ChIP–seq at the promoter of each differentially expressed gene is shown in the yellow-green heat map. c, R-VECs retain essential EC fate genes at stage 1 induction phase across all tissue-specific ECs. The data are presented as log2(CPM) with no scaling by row or column. d, PCA plot based on the top 1,000 most variable genes across ECs with and without ETV2 from different tissues during stage 1 induction, using log2-normalized CPM after subtracting tissue-specific effects using limma’s removeBatchEffect function. e, ETV2 ChIP was performed on R-VECs using an anti-flag antibody at the induction stage 1 (2D), along with histone modification ChIP for H3K4me3 and H3K27ac. Enriched regions were analysed by ChIP–seq. f, Genomic distribution of ETV2 peaks in R-VEC (Stage 1). The number in brackets is the number of ETV2 peaks in each region. g, Promoters bound by ETV2 have an increase in both K4me3 and K27ac. h, GO enrichment in genes with ETV2 binding at promoters. For statistics, see Supplementary Data 1.
Extended Data Fig. 7 ETV2 in R-VECs endows ECs with transcriptional adaptability and plasticity.
a, Diagram of EC sample preparation from ETV2 Venus reporter mice by FACS sorting. ETV2+ and ETV2− ECs were sorted at day E9.5. ECs were sorted as non-haematopoietic CD31+CD45neg cells. b, Heat map of overlap of differentially expressed genes in ETV2+ vs. ETV2− ECs at E9.5 and R-VECs (stage 1) vs. CTRL-EC from different tissues, using tissue-adjusted log2(CPM), centred and scaled by row. c, Knockdown of RASGRP3 by two different shRNAs in R-VECs, shRNA against Luciferase was used as control. Vessel quantification upon RASGRP3 knockdown. d, Heat map displaying overlapping differentially expressed genes from R-VEC at stabilization stage 3 (4 weeks) vs. R-VEC at induction stage 1, R-VECs in vitro pre-plug (stage 1 induction stage) vs. R-VECs in vivo in plugs (1 month), and freshly isolated vs. cultured HUVECs. Values represent tissue-adjusted log2(CPM), centred and scaled by row. e, ChIP–seq depicting genes that are differentially expressed in the stabilization stage 3 phase, but that are already directly bound by ETV2 and epigenetically primed for expression at induction stage 1 (2D monolayers). ETV2 ChIP–sequencing was performed on R-VECs using an anti-flag antibody. Mouse IgG was used as a control for ETV2 ChIP. Histone modification ChIP for H3K4me3, H3K27ac and H3K27me3 was performed on both CTRL-EC and R-VEC at the induction stage 1 (2D monolayers). Enriched regions were analysed by ChIP–sequencing. Black bar, ETV2 enriched regions. Green bar, the region with increased K4me3 modification. Blue bar, the region with increased K27ac modification. Promoter regions bound by ETV2 are highlighted in cream. Track range ETV2/K27me3/K27ac/, 0–0.3; K4me3/input/IgG, 0–1. For statistics, see Supplementary Data 1. For medium formulations, see Supplementary Data 2.
Extended Data Fig. 8 R-VECs physiologically arborize human pancreatic islet explants and organoids.
a, Human islet explants were cultured in Matrigel droplets (volume 50 μl) either with GFP labelled CTRL-EC or R-VEC (day 4). b, Insulin secretion fold change after glucose stimulation at 16.7 mM vs. 2mM glucose (2-week time point). c, Vessel area of ECs directly interacting with islets at week 2. d, EpCAM and VEcad staining of islets co-cultured in a Matrigel droplets at 2 weeks. e, Orthogonal projections of R-VECs in co-culture with human islets at two weeks, demonstrating strong interaction of the sprouting R-VEC vessels with islets. f, Human COs were derived from isolated crypts from colon biopsies of healthy human donors. Colon organoids were confirmed to express proper markers by quantitative RT–PCR. g, Quantitative RT–PCR of various colon markers for human COs, co-cultured with CTRL-EC or co-cultured with R-VEC for 8 days. Epithelial cells were sorted out as live CD31neg non-vascular cells. h, Mouse small intestine organoids were cultured alone, or in the presence of CTRL-EC or R-VEC (day 8). Confocal representative images of EdU+ (proliferating cells), KRT20+ (differentiated epithelial cells in blue) and ECs (mCherry - red) of co-culture experiment with mouse intestinal organoids. i, Quantification of vessel area over the course of 7 days in co-cultures of mouse intestine organoids with CTRL-EC or R-VEC. j, Vessel arborization quantified as EC sprouts in direct contact/organoid in CTRL-EC versus R-VEC wells. k, Time-lapse representative images show the progression of interacting ECs with CRCOs. CTRL-EC (in green) did not interact with CRCOs (in red) (top panel), whereas R-VEC (in green) form robust EC tubes to tap and wrap CRCOs (in red) (bottom panel). l, Orthogonal projections of CRCOs co-cultured with R-VECs (day 8). Data are mean ± s.e.m. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001. For statistics, see Supplementary Data 1. For medium formulations, see Supplementary Data 2.
Extended Data Fig. 9 Endothelial and epithelial cell identification by scRNA-seq from co-cultures of normal COs with R-VECs.
a, Schematic of 10X Chromium scRNA-seq experiments of R-VECs alone, R-VECs co-cultured with human COs, or COs alone. Samples were analysed 7 days post co-culture. The same compatible medium was used across all three conditions. b, UMAP of cells from each condition alone and the three conditions merged. c, Endothelial cells were identified as cells expressing either VEcad, CD31 or VEGFR2 and negative for the epithelial marker EPCAM. Epithelial cells were defined as positive for EPCAM and negative for any of the EC markers VEcad, CD31 or VEGFR2. d, UMAP of the 9 unique clusters identified in the merged samples. e, Endothelial and epithelial cell specific markers were used to confirm the EC clusters (clusters 1 to 7) vs. epithelial cell clusters (clusters 8 and 9). f, The identity of epithelial cells in clusters 8 and 9 was confirmed as colon-specific by expression of marker genes including SATB2, CA4, CA2 and others. For statistics, see Supplementary Data 1. For medium formulations, see Supplementary Data 2.
Extended Data Fig. 10 Endothelial and epithelial cell identification by scRNA-seq from co-cultures of CRCOs with R-VECs.
a, Schematic of 10X Chromium scRNA-seq experiments of R-VECs alone, R-VECs co-cultured with human CRCOs or CRCOs alone. Samples were analysed 7 days after co-culture. The same compatible medium was used across all three conditions. b, UMAP of cells from each condition alone and the three conditions merged. c, Endothelial cells were identified as cells expressing either VEcad, CD31 or VEGFR2 and negative for the epithelial marker EPCAM. Epithelial cells were defined as positive for EPCAM and negative for any EC markers VEcad, CD31 or VEGFR2. d, UMAP of the 9 unique clusters identified in the merged samples. e, Endothelial and epithelial cell-specific markers were used to confirm the endothelial cell clusters (clusters 6, 7, 8) vs. epithelial cell clusters (clusters 1, 2, 3, 4, 5, 9). f, UMAP of merged epithelial cell fractions from hCRCO cultured alone or co-cultured with R-VECs. Six unique clusters were identified. g, h, Heat map (g) and dot plot (h) of differentially expressed genes in tumour epithelial cells in cluster 2 and cluster 5 that are enriched in co-culture with R-VECs. Differential expression was performed using the Wilcoxon rank-sum test; FDR-adjusted P < 0.05. For statistics, see Supplementary Data 1. For medium formulations, see Supplementary Data 2.
Supplementary information
Supplementary Data
Supplementary Data 1: All statistics and replicates for Figures 1–4 and Extended Figures 1–10.
Supplementary Data
Supplementary Data 2: Summary of all medium components.
Supplementary Data
Supplementary Data 3: MATLAB code for calculation of EC area interacting with organoids (pertaining to Figure 4d and Figure 4g).
Supplementary Data
Supplementary Data 4: Summary of quantitative PCR primers.
Supplementary Information
This file contains Supplementary Video Legends.
Supplementary Table
Supplementary Table 1: Summary and source data for ChIP analysis.
Supplementary Table
Supplementary Table 2: Summary and source data for single-cell RNA-sequencing experiments.
Supplementary Figure
Supplementary Figure 1a: Original Western Blots.
Supplementary Figure
Supplementary Figure 1b: Original Western Blots.
Video 1
CTRL-EC and R-VEC in 3D gel matrices and and in large-volume perfusable in microfluidic devices.
Video 2
Interaction of R-VECs with human pancreatic islets.
Video 3
Time-lapse videos of R-VEC or CTRL-EC in co-culture with either normal human colon organoids (hCO) or human colorectal cancer organoids (hCRCO) in 3D matrix.
Rights and permissions
About this article

Cite this article
Palikuqi, B., Nguyen, DH.T., Li, G. et al. Adaptable haemodynamic endothelial cells for organogenesis and tumorigenesis. Nature 585, 426–432 (2020). https://doi.org/10.1038/s41586-020-2712-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2712-z
This article is cited by
-
Advances in secondary prevention mechanisms of macrovascular complications in type 2 diabetes mellitus patients: a comprehensive review
European Journal of Medical Research (2024)
-
Harnessing 3D in vitro systems to model immune responses to solid tumours: a step towards improving and creating personalized immunotherapies
Nature Reviews Immunology (2024)
-
HIF-1α promotes kidney organoid vascularization and applications in disease modeling
Stem Cell Research & Therapy (2023)
-
ETV2/ER71, the key factor leading the paths to vascular regeneration and angiogenic reprogramming
Stem Cell Research & Therapy (2023)
-
Bioengineering human intestinal mucosal grafts using patient-derived organoids, fibroblasts and scaffolds
Nature Protocols (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
