
Abstract
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) pandemic continues, with devasting consequences for human lives and the global economy1,2. The discovery and development of virus-neutralizing monoclonal antibodies could be one approach to treat or prevent infection by this coronavirus. Here we report the isolation of sixty-one SARS-CoV-2-neutralizing monoclonal antibodies from five patients infected with SARS-CoV-2 and admitted to hospital with severe coronavirus disease 2019 (COVID-19). Among these are nineteen antibodies that potently neutralized authentic SARS-CoV-2 in vitro, nine of which exhibited very high potency, with 50% virus-inhibitory concentrations of 0.7 to 9 ng ml−1. Epitope mapping showed that this collection of nineteen antibodies was about equally divided between those directed against the receptor-binding domain (RBD) and those directed against the N-terminal domain (NTD), indicating that both of these regions at the top of the viral spike are immunogenic. In addition, two other powerful neutralizing antibodies recognized quaternary epitopes that overlap with the domains at the top of the spike. Cryo-electron microscopy reconstructions of one antibody that targets the RBD, a second that targets the NTD, and a third that bridges two separate RBDs showed that the antibodies recognize the closed, ‘all RBD-down’ conformation of the spike. Several of these monoclonal antibodies are promising candidates for clinical development as potential therapeutic and/or prophylactic agents against SARS-CoV-2.
Similar content being viewed by others

Main
The novel coronavirus SARS-CoV-21,2 has caused more than 14 million confirmed infections globally, and has caused more than 600,000 deaths. This pandemic has also put much of the world on pause, with unprecedented disruption of lives and unparalleled damage to the economy. A return to some semblance of normality will depend on the ability of science to deliver an effective solution, and the scientific community has responded admirably. Drug development is well underway, and vaccine candidates have entered clinical trials. Another promising approach is the isolation of SARS-CoV-2-neutralizing monoclonal antibodies (mAbs) that could be used as therapeutic or prophylactic agents. The primary target for such antibodies is the viral spike, a trimeric protein3,4 that is responsible for binding of the virus to the ACE2 receptor on the host cell1,3,5,6. The spike protein is comprised of two subunits. The S1 subunit has two major structural elements, RBD and NTD; the S2 subunit mediates virus–cell membrane fusion after the RBD has engaged ACE2. Reports of the discovery of neutralizing mAbs that target the RBD have been published recently7,8,9,10,11. We now describe our efforts in isolating and characterizing a collection of mAbs that not only target multiple epitopes on the viral spike but also show very high potency in neutralizing SARS-CoV-2.
Patient selection
Forty patients with PCR-confirmed SARS-CoV-2 infection were enrolled in a cohort study on virus-neutralizing antibodies. Plasma samples from all participants were first tested for neutralizing activity against SARS-CoV-2 pseudovirus (Wuhan-Hu-1 spike pseudotyped with vesicular stomatitis virus). Neutralizing titres varied widely, with half-maximal inhibitory concentrations (IC50s) ranging from a reciprocal plasma dilution of less than 100 to roughly 13,000 (Fig. 1a). We selected five patients for isolation of mAbs because their plasma virus-neutralizing titres were among the highest. The clinical characteristics of these five patients are summarized in Extended Data Table 1. All were severely ill with acute respiratory distress syndrome requiring mechanical ventilation.

a, Plasma neutralization profile of 40 patients against SARS-CoV-2 pseudovirus (highlighted are five top neutralizers chosen for further study). b, All 252 transfection supernatants were screened for binding to the S trimer and RBD, as well as for neutralization against SARS-CoV-2 pseudovirus and live virus. For pseudovirus neutralization, the 50% inhibitory dilutions (IC50) of each supernatant are plotted. For live virus, semiquantitative representation of the inhibition at a dilution of 1:50, with neutralization levels ranging from (−) for none to (+++) for complete neutralization, is plotted. Potent antibodies later identified are marked by vertical lines and labelled at the bottom. The antibodies from each patient are coloured as in a.
Isolation and construction of mAbs
Peripheral blood mononuclear cells from each patient were processed as shown in Extended Data Fig. 1a, starting with cell sorting by flow cytometry. The sorting strategy focused on live memory B lymphocytes that were CD3−, CD19+, and CD27+ (Extended Data Fig. 1b). The final step focused on those cells that bound the SARS-CoV-2 spike trimer (S trimer)4. A total of 602, 325, 14, 147, and 145 such B cells from patients 1, 2, 3, 4, and 5, respectively, were labelled with unique hashtags (Extended Data Fig. 1c). The cells were then placed into the 10X Chromium (10X Genomics) for single-cell 5′-mRNA and V(D)J sequencing to obtain paired heavy (H) and light (L) chain sequences. A careful bioinformatic analysis was carried out on 1,145 paired sequences to downselect ‘high-confidence’ antigen-specific mAbs. We recovered 331 mAb sequences, representing 252 individual clones. Only six mAbs were from patient 3, whereas 44 to 100 mAbs were identified from each of the other patients (Extended Data Fig. 2a). The VH and VL sequences of 252 antibodies (one per clone) were codon-optimized and synthesized, and each VH and VL gene was then cloned into an expression plasmid with corresponding constant regions of H chain and L chain of human IgG1. Monoclonal antibodies were then expressed by co-transfection of paired full-length H chain and L chain genes into Expi293 cells.
Monoclonal antibody screening
All 252 transfection supernatants were screened for binding to the S trimer and RBD by enzyme-linked immunosorbent assays (ELISAs), as well as for their ability to neutralize SARS-CoV-2 pseudovirus and live virus (Fig. 1b, Extended Data Fig. 2). A substantial percentage of the mAbs in the supernatants bound S trimer, and a subset of those bound RBD. Specifically, 121 supernatants were scored as positive for S trimer binding, yielding an overall hit rate of 48%. Of these, 38 were positive for RBD binding while the remaining 83 were negative. None of the 13 trimer-specific mAbs from patient 5 recognized RBD. In the pseudovirus neutralization screen, 61 supernatants were scored as positive, indicating that half of the trimer-specific mAbs were virus-neutralizing. In the screen for neutralization against SARS-CoV-2 (strain USA-WA1/2020), 41 supernatants were scored as positive. Overall, this screening strategy was quite effective in identifying neutralizing mAbs (vertical lines and labelled antibodies at the bottom of Fig. 1b) that were later identified as potent.
Sequence analysis of S trimer-specific mAbs
Of the 121 mAbs that bound the S trimer, 88% were IgG isotype, with IgG1 being predominant (Extended Data Fig. 3a). Comparison to the IgG repertoires of three healthy human donors12 revealed a statistically significant over-representation of IGHV3-30, IGKV3-20, and IGHJ6 genes for this collection of SARS-CoV-2 mAbs (Extended Data Figs. 3b, c). In addition, the average CDRH3 length was also longer (Extended Data Fig. 3d). Notably, the average percentages of somatic hypermutation in VH and VL were 2.1 and 2.5, respectively, which were significantly lower than those found in healthy individuals (Extended Data Fig. 3e) and remarkably close to those of germline sequences.
Antigen binding and virus neutralization
Since the screening for pseudovirus neutralization was performed quantitatively with serial dilutions of the transfection supernatants, we plotted in Extended Data Fig. 2b the best-fit neutralization curves for 130 samples that were positive in at least one of the screens shown in Fig. 1b. Most were non-neutralizing or weakly neutralizing, but 18 showed better potency. One additional supernatant was initially missed in the pseudovirus screen (patient 1 in Extended Data Fig. 2b) but was later found to be a potent neutralizing mAb. Together, these 19 mAbs were purified from transfection supernatants and further characterized for their binding and neutralization properties. As shown in Fig. 2a, quantitative ELISA showed that all but one (2-43) of the mAbs bound the S trimer. Nine of the antibodies clearly bound RBD, with little or no binding to NTD. Eight antibodies bound NTD to varying degrees, with no binding to RBD. Two mAbs bound neither RBD nor NTD, and were therefore categorized as ‘other’.

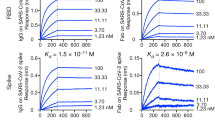
a, Binding profiles of 19 purified potent neutralizing mAbs against the S trimer (left), RBD (middle), and NTD (right) of SARS-CoV-2. Note that mAb 2-30 bound multiple proteins at high concentrations. b, Neutralization profiles of the pseudovirus (top) and live virus (bottom) for the 19 purified mAbs. Epitope classifications are listed above plots. A single replicate of the binding experiment and triplicates of neutralization are presented as mean ± s.e.m.
The pseudovirus neutralization profiles for these purified 19 mAbs are shown in Fig. 2b (top). The RBD-directed antibodies neutralized the pseudovirus with IC50 values of 0.005 to 0.512 μg ml−1; the NTD-directed antibodies were slightly less potent, with IC50 values ranging from 0.013 to 0.767 μg ml−1. A common feature of the NTD mAbs was the plateauing of virus neutralization at levels short of 100%. Two antibodies, categorized as ‘other’, neutralized with IC50 values of 0.071 and 0.652 μg ml−1. Antibody neutralization of the authentic or live SARS-CoV-2 (strain USA-WA1/2020) was carried out using Vero cells inoculated with a multiplicity of infection of 0.1. As shown in the bottom portion of Fig. 2b, the RBD-directed antibodies again neutralized the virus but with IC50 values of 0.0007 to 0.209 μg ml−1; the NTD-directed antibodies showed similar potency, with IC50 values ranging from 0.007 to 0.109 μg ml−1. Here, the plateauing effect seen in the pseudovirus neutralization assay was less apparent. Antibodies 2-43 and 2-51 neutralized the live virus with IC50 values of 0.003 and 0.007 μg ml−1, respectively. Overall, nine mAbs exhibited high potency in neutralizing authentic SARS-CoV-2 in vitro with IC50 values of 0.009 μg ml−1 or less, including four against RBD (2-15, 2-7, 1-57, and 1-20), three against NTD (2-17, 5-24, and 4-8), and two against undetermined regions on the S trimer (2-43 and 2-51). Patient 2 alone contributed five of the top nine SARS-CoV-2 neutralizing mAbs. A correlation of the results of the two virus-neutralizing assays is shown in Extended Data Fig. 4.
Epitope mapping
All 19 potent neutralizing mAbs (Fig. 2) were further studied in antibody competition experiments to gain insight into their epitopes. We also chose 12 mAbs that bound the S trimer strongly during the initial supernatant screen, including 5 that bound RBD (1-97, 2-26, 4-13, 4-24, and 4-29) and 7 that did not bind RBD (1-21, 2-29, 4-15, 4-32, 4-33, 4-41, and 5-45). Four of these mAbs were weak in neutralizing SARS-CoV-2 pseudovirus, and the remaining eight were non-neutralizing (Extended Data Fig. 5). We used ELISA to evaluate 16 non-RBD mAbs for competition in binding to the S trimer in a ‘checkerboard’ experiment. The extent of the antibody competition is reflected by the intensity of the heatmap in Fig. 3a. There is one large cluster (A) of mAbs that competed with one another, which partially overlaps with a small cluster (B). A third cluster (C) does not overlap at all. Note that all but one of the antibodies in cluster A recognized NTD. Antibody 2-51 is clearly directed against the NTD region even though it could not bind NTD. Moreover, one mAb from each of clusters B and C also recognized NTD, thereby indicating that all three clusters are within or near the NTD. One mAb, 1-21, appears to have a unique non-overlapping epitope (epitope region D).

a, Competition results of non-RBD binders (left) and RBD binders (right) in blocking binding of ACE2 or biotinylated mAb to the S trimer. In addition, the ability of each mAb to bind NTD and RBDmut is shown. The numbers in each box show the area under each competition curve (AUC) as tested by ELISA. Plus and minus signs indicate binding and no binding, respectively, of the mAb to the protein. The letters A to H at the bottom denote clusters of antibody epitopes defined by the competition experiments. b, Venn diagram interpretation of results from a and Extended Data Fig. 6b.
We carried out a similar ‘checkerboard’ competition experiment by ELISA for 14 of our RBD-directed mAbs plus CR302213,14. Here, the heatmap shows four epitope clusters that are serially overlapping (Fig. 3a). There is one large cluster (E) that contains mAbs that can largely block ACE2 binding. Furthermore, four antibodies in this cluster lost the ability to bind to a mutant RBD (L455R, A475R, G502R) that could no longer bind ACE2 (unpublished data). Together, these results suggest that most of the mAbs in cluster E are directed against the ACE2-binding interface of RBD. The next cluster (F) connects to both cluster E and cluster G, the location of which is defined by its member CR302215. Last, cluster G overlaps another cluster (H), which includes 1-97, which strongly inhibited the binding of 2-30 to the S trimer. This finding suggests that cluster H may be proximal to one edge of cluster E.
One potent neutralizing mAb, 2-43, did not bind the S trimer in ELISA (Fig. 2a) and thus could not be tested in the above competition experiments. However, 2-43 did strongly bind the S trimer when expressed on the cell surface, as determined by flow cytometry (Extended Data Fig. 6a), and this binding was competed out by itself but not by RBD, NTD, ACE2, or the soluble S trimer4 (Extended Data Fig. 6b). NTD-directed mAbs had only a modest effect on its binding to cell-surface S trimer, but numerous RBD-directed mAbs in cluster E potently blocked the binding of 2-43, demonstrating that this antibody is likely to target a quaternary epitope on the top of RBD.
These mapping results could be represented by two sets of Venn diagrams shown in Fig. 3b. In the non-RBD region, the potent neutralizing mAbs reside exclusively in cluster A and bind a patch on the NTD. Weaker neutralizing mAbs recognize a region at the interface between clusters A and B. In the RBD region, the most potent neutralizing mAbs also group together within one cluster (E). Given that all block ACE2 binding, it is likely they recognize the top of RBD and neutralize the virus by competitive inhibition of receptor binding. Cluster G contains CR3022, a mAb known to be directed against an epitope on a cryptic site on the side of RBD when it is in the ‘up’ position15. Cluster F is therefore likely situated between the top and this ‘cryptic’ site. The Venn diagram also suggests that cluster H may occupy a different side surface of RBD, perhaps in the region recognized by S309, a mAb isolated from a patient with SARS-CoV-18.
Cryo-electron microscopy
We produced cryo-electron microscopy (cryo-EM) reconstructions of antigen-binding fragments (Fabs) from three mAbs in complex with the S trimer4. First, single-particle analysis of the complex with the Fab of mAb 2-4 (RBD-directed) yielded maps of high quality (Fig. 4a; Extended Data Table 2; Extended Data Fig. 7a–d), with the most abundant particle class representing a 3-Fab-per-trimer complex, refined to an overall resolution of 3.2 Å. While density for the constant portion of the Fabs was visible, it was blurred as a result of molecular motion, and thus only the variable domains were included in the molecular model. Fab 2-4 bound the spike protein near the apex, with all RBDs in the ‘down’ orientation, and the structure of the antibody-bound spike protein was highly similar to previously published unliganded spike structures in the ‘all-down’ conformation3,4. Detailed interactions between mAb 2-4 and RBD are shown in Extended Data Fig. 7e–i. Overall, the structure of the 2-4 Fab–spike complex shows that neutralization of SARS-CoV-2 by this mAb is likely to result from locking the RBD in the down conformation while also occluding access to ACE2.

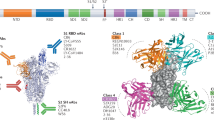
a, Cryo-EM reconstruction of 2-4 Fab in complex with the S trimer at 3.2 Å overall resolution. Density is coloured with RBD in green, NTD in orange, and other regions in grey. b, Cryo-EM reconstruction of 4-8 Fab in complex with the S trimer (ribbon diagram, coloured as in a) at 3.9 Å overall resolution, with RBDs in the ‘all-down’ configuration. c, Cryo-EM reconstruction of the 2-43 Fab in complex with the S trimer at 5.8 Å resolution reveals a quaternary epitope involving RBD from one subunit and another RBD from the next. d, Mapping of the Venn diagrams from Fig. 3b onto the surface of the viral spike.
We also produced 3D cryo-EM reconstructions of 4-8 Fab (NTD-directed) in complex with the S trimer (Extended Data Table 2, Extended Data Fig. 8a–f). Two main particle classes were observed—one for a 3-Fab-bound complex with all RBDs ‘down’ at 3.9 Å resolution (Fig. 4b), and another a 3-Fab-bound complex with one RBD ‘up’ at 4.0 Å resolution (Extended Data Fig. 8g). However, molecular motion prevented visualization of the interaction at high resolution. Nevertheless, the density in the 4-8 map reveals the overall positions of the antibody chains that target the NTD. It is unclear how binding to the tip of the NTD results in neutralization of SARS-CoV-2.
Third, a 5.8 Å resolution reconstruction of 2-43 Fab in complex with the S trimer (Extended Data Table 2, Extended Data Fig. 8h–k) revealed three bound Fabs, each targeting a quaternary epitope on the top of the spike that included elements of the RBDs from two adjacent S1 protomers (Fig. 4c), consistent with the epitope mapping results (Fig. 3b, Extended Data Fig. 6b), including the lack of binding to isolated RBD (Fig. 2a). Given these findings, the inability of 2-43 to bind the S trimer in ELISA studies is likely to be an artefact of the assay format, as this mAb did bind the spike expressed on the cell surface and in the cryo-EM study.
Armed with these three cryo-EM reconstructions, we used the Venn diagrams from Fig. 3b to map the epitopes of many of our SARS-CoV-2-neutralizing mAbs onto the surface of the spike (Fig. 4d). This is obviously a rough approximation because antibody footprints are much larger than the area occupied by the mAb number. However, the spatial relationship of the antibody epitopes should be reasonably represented by Fig. 4d.
mAb 2-15 protects hamsters against SARS-CoV-2
To assess the in vivo potency of mAb 2-15, we performed a protection experiment in a golden Syrian hamster model of SARS-CoV-2 infection. The hamsters were first given an intraperitoneal injection of the antibody at a dose of 1.5 mg kg−1 or 0.3 mg kg−1, or PBS alone. Intranasal inoculations of 105 plaque-forming units (PFU) of the HKU-001a strain of SARS-CoV-2 were carried out 24 h later. Four days after virus challenge, lung tissues were removed to quantify the viral load. As shown in Fig. 5, both viral RNA copy numbers and infectious virus titres were reduced by 4 logs or more in hamsters given 1.5 mg kg−1 of mAb 2-15. The protection at 0.3 mg kg−1 was borderline, as we had estimated. This pilot animal study demonstrates that the potency of mAb 2-15 in vitro is reflected in vivo, with complete elimination of infectious SARS-CoV-2 at a relatively modest antibody dose.

One day before intranasal challenge with SARS-CoV-2, each group of hamsters was given a single intraperitoneal dose of 1.5 mg kg−1 of mAb 2-15 (n = 4), 0.3 mg kg−1 of mAb 2-15 (n = 4), or saline as control (n = 4). The viral loads in the lung tissues on day 4 after viral challenge were determined by quantitative PCR with reverse transcription (qRT–PCR; red), as well as by an assay to quantify PFUs of infectious SARS-CoV-2 (blue). All data points are shown, along with the mean ± s.d. The differences between the 1.5 mg kg−1 group and the control group are statistically significant at P < 0.05.
Discussion
We have identified a collection of SARS-CoV-2-neutralizing mAbs that are not only potent but also diverse. Nine of these antibodies can neutralize the authentic virus in vitro at concentrations of 9 ng ml−1 or less (Fig. 2b), including four directed against the RBD, three directed against the NTD, and two directed against nearby quaternary epitopes. Unexpectedly, many of the these mAbs have V(D)J sequences close to germline sequences, without extensive somatic hypermutations (Extended Data Fig. 3e), a finding that bodes well for vaccine development. Our most potent RBD-specific mAbs (for example, 2-15, 2-7, 1-57, and 1-20) compare favourably with such antibodies recently reported7,8,10,16,17,18,19,20, including those with high potency9,11,21,22. The in vitro potency of 2-15 is well reflected in vivo in the hamster protection experiment (Fig. 5). It appears from the epitope-mapping studies that mAbs directed against the top of the RBD compete strongly with ACE2 binding and potently neutralize the virus, whereas those directed against the side surfaces of the RBD do not compete with ACE2 and neutralize less potently (Figs. 3b, 4d). Our collection of non-RBD neutralizing mAbs is unprecedented, to our knowledge, in that such antibodies have been reported only sporadically and only with substantially lower potencies22,23,24. The most potent of these mAbs are directed against (for example, 2-17, 5-24, and 4-8) or overlapping with (2-51) a patch on the NTD (Figs. 3b, 4d). It is unclear how NTD-directed mAbs block SARS-CoV-2 infection and why their neutralization profiles are different from those of RBD-directed antibodies (Fig. 2b). Nevertheless, vaccine strategies that do not include the NTD will be unable to induce an important class of virus-neutralizing antibodies.
The isolation of two mAbs (2-43 and 2-51) directed against epitopes that do not map to the RBD or NTD is also unprecedented, to our knowledge. Cryo-EM of 2-43 Fab bound to the S trimer has confirmed its epitope as quaternary in nature, crossing from the top of one RBD to the top of another RBD (Fig. 4c). It will be equally informative to understand the epitope of 2-51. We have also shown cryo-EM evidence for a neutralizing mAb (4-8) bound to the NTD of the viral spike (Fig. 4b), as well as another high-resolution structure of an mAb (2-4) bound to the RBD (Fig. 4a).
The potency and diversity of our SARS-CoV-2-neutralizing mAbs are probably attributable to patient selection. Infected individuals with severe disease develop a more robust virus-neutralizing antibody response25. If patient 2 had not been included, five of the top neutralizing mAbs would have been lost. The diversity of our antibodies is also attributable, in part, to the choice of using the S trimer to sort from memory B cells, while most groups have used the RBD7,9,10,11,16,17,18,19,21. The characterization of this diverse collection of mAbs has allowed us to observe that all potent SARS-CoV-2-neutralizing antibodies described to date are directed against the top of the viral spike. RBD and NTD are, undoubtedly, quite immunogenic. Neutralizing antibodies to the stem region of the S trimer remain to be discovered. In conclusion, we believe that several of our monoclonal antibodies with strong virus-neutralizing activity are promising candidates for development as modalities to treat or prevent SARS-CoV-2 infection.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment, except where stated.
Expression and purification of SARS-CoV-2 proteins
The mammalian expression vector that encodes the ectodomain of the SARS-CoV-2 S trimer and the vector encoding RBD fused with SD1 at the N terminus and an HRV-3C protease cleavage site followed by a mFc tag and an 8 × His tag at the C terminus were kindly provided by Jason McLellan4. SARS-CoV-2 NTD (aa1-290) with an HRV-3C protease cleavage site, a mFc tag, and an 8 × His tag at the C terminus was also cloned into mammalian expression vector pCAGGS. Each expression vector was transiently transfected into Expi293 cells using 1 mg/ml polyethylenimine (Polysciences). Five days after transfection, the S trimer was purified using Strep-Tactin XT Resin (Zymo Research), and the RBD-mFc and NTD-mFc were purified using protein A agarose (ThermoFisher Scientific). In order to obtain RBD-SD1 and NTD, the mFc and 8 × His tags at the C terminus were removed by HRV-3C protease (Millipore-Sigma) and then purified using Ni-NTA resin (Invitrogen) followed by protein A agarose.
Sorting for S trimer-specific B cells and single-cell B cell receptor sequencing
Peripheral blood mononuclear cells from five patients and one healthy donor were stained with LIVE/DEAD Fixable Yellow Dead Cell Stain Kit (Invitrogen) at ambient temperature for 20 min, followed by washing with RPMI-1640 complete medium and incubation with 10 μg/ml S trimer at 4 °C for 45 min. Afterwards, the cells were washed again and incubated with a cocktail of flow cytometry and hashtag antibodies, containing CD3 PE-CF594 (BD Biosciences), CD19 PE-Cy7 (Biolegend), CD20 APC-Cy7 (Biolegend), IgM V450 (BD Biosciences), CD27 PerCP-Cy5.5 (BD Biosciences), anti-His PE (Biolegend), and human Hashtag 3 (Biolegend) at 4 °C for 1 h. Stained cells were then washed, resuspended in RPMI-1640 complete medium and sorted for S trimer-specific memory B cells (CD3−CD19+CD27+S trimer+ live single lymphocytes). The sorted cells were mixed with mononuclear cells from the same donor, labelled with Hashtag 1, and loaded into the 10X Chromium chip of the 5′ Single Cell Immune Profiling Assay (10X Genomics) at the Columbia University Human Immune Monitoring Core (HIMC; RRID:SCR_016740). The library preparation and quality control were performed according to the manufacturer’s protocol and sequenced on a NextSeq 500 sequencer (Illumina).
Identification of S trimer-specific antibody transcripts
For each sample, full-length antibody transcripts were assembled using the VDJ module in Cell Ranger (version 3.1.0, 10X Genomics) with default parameters and the GRCh38 genome as reference. To identify cells from the antigen sort, we first used the count module in Cell Ranger to calculate copies of all hashtags in each cell from the Illumina NGS raw reads. High-confidence antigen-specific cells were identified as follows. In brief, based on the copy numbers of the hashtags observed, a cell must contain more than 100 copies of the antigen sort-specific hashtag to qualify as an antigen-specific cell. Because hashtags can fall off cells and bind to cells from a different population in the sample mixture, each cell usually has both sorted and spiked-in-specific hashtags. To enrich for true antigen-specific cells, the copy number of the specific hashtag has to be at least 1.5× higher than that of the non-specific hashtag. Low-quality cells were identified and removed using the cell-calling algorithm in Cell Ranger. Cells that did not have productive H and L chain pairs were excluded. If a cell contained more than two H or/and L chain transcripts, the transcripts with fewer than three unique molecular identifiers were removed. Cells with identical H and L chain sequences, which may have resulted from mRNA leakage, were merged into one cell. Additional filters were applied to remove low-quality cells and/or transcripts in the antibody gene annotation process.
Antibody transcript annotation and selection criteria
Antigen-specific antibody transcripts were processed using our bioinformatics pipeline SONAR for quality control and annotation26. In brief, V(D)J genes were assigned for each transcript using BLAST27 with customized parameters against a germline gene database obtained from the international ImMunoGeneTics information system (IMGT) database26,28. On the basis of BLAST alignments of V and J regions, CDR3 was identified using the conserved second cysteine in the V region and WGXG (H chain) or FGXG (L chain) motifs in the J region (X represents any amino acid). For H chain transcripts, the constant domain 1 (CH1) sequences were used to assign isotype using BLAST with default parameters against a database of human CH1 genes obtained from IMGT. A BLAST E-value threshold of 10−6 was used to find significant isotype assignments, and the CH1 allele with the lowest E-value was used. Sequences other than the V(D)J region were removed and transcripts containing incomplete V(D)J or/and frame shift were excluded. We then aligned each of the remaining transcripts to the assigned germline V gene using CLUSTALO29 and calculated the somatic hypermutation level.
To select representative antibodies for functional characterization, we first clustered all antibodies using USEARCH30 with the following criteria: identical heavy chain V and J gene assignments, the same length of CDRH3, and CDRH3 identity higher than 0.9. For each cluster, cells with the same light chain V and J gene assignments were grouped into a clone. All clone assignments were manually checked. We then calculated the clonal size for each clone, and one H and L chain pair per clone was chosen for antibody synthesis. For clones with multiple members, the member with the highest somatic hypermutation level was chosen for synthesis. For cells having multiple high quality H or L chains, which may be from doublets, we synthesized all H and L chain combinations.
Analysis of S trimer-specific antibody repertoire
Because 88% of the S trimer-specific antibodies were IgG isotype, we compared the repertoire features to IgG repertoires from three healthy donors12 (17,243 H chains, 27,575 kappa L chains, 20,889 lambda L chains). The repertoire data from the three healthy donors were combined and annotated using SONAR with the same process as above.
Antibody expression and purification
For each antibody, variable genes were optimized for human cell expression and synthesized by GenScript. VH and VL were inserted separately into plasmids (gWiz or pcDNA3.4) that encode the constant region for H chain and L chain. Monoclonal antibodies were expressed in Expi293 (ThermoFisher, A14527) by co-transfection of H chain and L chain expressing plasmids using polyethylenimine and culture at 37 °C with shaking at 125 rpm and 8% CO2. On day 3 after transfection, 400 μl supernatant were collected for screening for binding to the S trimer and RBD by ELISA, and for neutralization of SARS-CoV-2 pseudovirus and authentic virus. Supernatants were also collected on day 5 for antibody purification using rProtein A Sepharose (GE, 17-1279-01) affinity chromatography.
Production of pseudoviruses
Recombinant Indiana vesicular stomatitis virus (rVSV) expressing the SARS-CoV-2 spike was generated as previously described31,32. HEK293T cells were grown to 80% confluency before transfection with pCMV3-SARS-CoV-2-spike (Sino Biological) using FuGENE 6 (Promega). Cells were cultured overnight at 37 °C with 5% CO2. The next day, medium was removed and VSV-G pseudotyped ΔG-luciferase (G*ΔG-luciferase, Kerafast) was used to infect the cells in DMEM at a MOI of 3 for 1 h before the cells were washed three times with 1× DPBS. DMEM supplemented with 2% fetal bovine serum, 100 IU/ml of penicillin and 100 μg/ml of streptomycin were added to the inoculated cells, which were cultured overnight as described above. The supernatant was removed the following day and clarified by centrifugation at 300g for 10 min before aliquoting and storing at −80 °C.
Pseudovirus neutralization
Neutralization assays were performed by incubating pseudoviruses with serial dilutions of heat-inactivated plasma together with supernatant or purified antibodies, and scored by the reduction in luciferase gene expression. In brief, Vero E6 cells (ATCC) were seeded in a 96-well plate at a concentration of 2 × 104 cells per well. Pseudoviruses were incubated the next day with serial dilutions of the test samples in duplicate or triplicate for 30 min at 37 °C. The mixture was added to cultured cells and incubated for an additional 24 h. The luminescence was measured using a Britelite plus Reporter Gene Assay System (PerkinElmer). IC50 was defined as the dilution at which the relative light units were reduced by 50% compared with the virus control wells (virus + cells) after subtraction of the background in the control groups with cells only. The IC50 values were calculated using nonlinear regression in GraphPad Prism 8.0.
Authentic SARS-CoV-2 neutralization
Supernatants containing expressed mAbs were diluted 1:10 and 1:50 in EMEM with 7.5% inactivated fetal calf serum and incubated with authentic SARS-CoV-2 (strain USA-WA1/2020; MOI 0.1) for 1 h at 37 °C. After incubation, the mixture was transferred onto a monolayer of Vero-E6 cells that was cultured overnight. After incubation of the cells with the mixture for 70 h at 37 °C, cytopathic effects (CPEs) caused by the infection were scored for each well from 0 to 4 to indicate the degree of virus inhibition. Semi-quantitative representation of the inhibition for each antibody-containing supernatant at a dilution of 1:50 is shown in the lowest panel of Fig. 1b with neutralization levels ranging from (−) for none to (+++) for complete neutralization.
An end-point dilution assay in a 96-well plate format was performed to measure the neutralization activity of select purified mAbs. In brief, each antibody was serially diluted (fivefold dilutions) starting at 20 μg/ml. Triplicates of each mAb dilution were incubated with SARS-CoV-2 at a MOI of 0.1 in EMEM with 7.5% inactivated fetal calf serum for 1 h at 37 °C. After incubation, the virus–antibody mixture was transferred onto a monolayer of Vero-E6 cells grown overnight. The cells were incubated with the mixture for 70 h. CPEs were visually scored for each well in a blinded fashion by two independent observers. The results were then converted into percentage neutralization at a given mAb concentration, and the averages ± s.e.m. were plotted using a five-parameter dose–response curve in GraphPad Prism 8.0.
Epitope mapping by ELISA
We coated 50 ng per well of S trimer, 50 ng per well of RBD, and 100 ng per well of NTD onto ELISA plates at 4 °C overnight. The ELISA plates were then blocked with 300 μl blocking buffer (1% BSA and 10% bovine calf serum (BCS) (Sigma)) in PBS at 37 °C for 2 h. Afterwards, supernatants from the antibody transfection or purified antibodies were serially diluted using dilution buffer (1% BSA and 20% BCS in PBS), incubated at 37 °C for 1 h. Next, 100 μl of 10,000-fold diluted Peroxidase AffiniPure goat anti-human IgG (H+L) antibody (Jackson ImmunoResearch) was added into each well and incubated for 1 h at 37 °C. The plates were washed between each step with PBST (0.5% Tween-20 in PBS). Finally, the TMB substrate (Sigma) was added and incubated before the reaction was stopped using 1 M sulfuric acid. Absorbance was measured at 450 nm.
For the competition ELISA, purified mAbs were biotin-labelled using One-Step Antibody Biotinylation Kit (Miltenyi Biotec) following the manufacturer’s recommendations and purified using 40K MWCO Desalting Column (ThermoFisher Scientific). Serially diluted competitor antibodies (50 μl) were added into S trimer-precoated ELISA plates, followed by 50 μl of biotinylated antibodies at a concentration that achieves an OD450 reading of 1.5 in the absence of competitor antibodies. Plates were incubated at 37 °C for 1 h, and 100 μl of 500-fold diluted Avidin-HRP (ThermoFisher Scientific) was added into each well and incubated for another 1 h at 37 °C. The plates were washed with PBST between each of the previous steps. The plates were developed afterwards with TMB and absorbance was read at 450 nm after the reaction was stopped.
For the ACE2 competition ELISA, 100 ng of ACE2 protein (Abcam) was immobilized on the plates at 4 °C overnight. The unbound ACE2 was washed away by PBST and then the plates were blocked. After washing, 100 ng of S trimer in 50 μl dilution buffer was added into each well, followed by addition of another 50 μl of serially diluted competitor antibodies and then incubation at 37 °C for 1 h. The ELISA plates were washed four times with PBST and then 100 μl of 2,000-fold diluted anti-strep-HRP (Millipore Sigma) was added into each well for another 1 h at 37 °C. The plates were then washed and developed with TMB, and absorbance was read at 450 nm after the reaction was stopped.
For all the competition ELISA experiments, the relative binding of biotinylated antibodies or ACE2 to the S trimer in the presence of competitors was normalized by comparing to competitor-free controls. Relative binding curve and the area under curve (AUC) were generated by fitting the nonlinear five-parameter dose–response curve in GraphPad Prism 8.0.
Cell-surface competition binding assay
Expi293 cells were co-transfected with vectors encoding pRRL-cPPT-PGK-GFP (Addgene) and pCMV3-SARS-CoV-2 (2019-nCoV) Spike (Sino Biological) at a ratio of 1:1. Two days after transfection, cells were incubated with a mixture of biotinylated mAb 2-43 (0.25 μg/ml) and serially diluted competitor antibodies at 4 °C for 1 h. Then 100 μl of diluted APC-streptavidin (Biolegend) was added to the cells and incubated at 4 °C for 45 min. Cells were washed three times with FACS buffer before each step. Finally, cells were resuspended and binding of 2-43 to cell-surface S trimer was quantified on an LSRII flow cytometer (BD Biosciences). The mean fluorescence intensity of APC in GFP-positive cells was analysed using FlowJo and the relative binding of 2-43 to the S trimer in the presence of competitors was calculated as the percentage of the mean fluorescence intensity compared to that of the competitor-free controls.
Cryo-EM data collection and processing
SARS-CoV-2 S trimer at a final concentration of 2 mg/ml was incubated with sixfold molar excess per spike monomer of the antibody Fab fragments for 30 min in 10 mM sodium acetate pH 5.5, 150 mM NaCl, and 0.005% n-dodecyl-β-d-maltoside (DDM). Sample (2 μl) was incubated on C-flat 1.2/1.3 carbon grids for 30 s and vitrified using a Leica EM GP Plunge Freezer. Data were collected on a Titan Krios electron microscope operating at 300 kV equipped with a Gatan K3 direct detector and energy filter using the Leginon software package33. A total electron fluence of 51.3 e/Å2 was fractionated over 40 frames, with a total exposure time of 2 s. A magnification of 81,000× resulted in a pixel size of 1.058 Å, and a defocus range of −0.4 to −3.5 μm was used. All processing was done using cryoSPARC v2.14.234. Raw movies were aligned and dose-weighted using patch motion correction, and the CTF was estimated using patch CTF estimation. A small subset of approximately 200 micrographs were picked using blob picker, followed by 2D classification and manual curation of particle picks, and used to train a Topaz neural network35. This network was then used to pick particles from the remaining micrographs, which were extracted with a box size of 384 pixels.
For the 2-4 Fab dataset, 2D classification followed by ab initio modelling and 3D heterogeneous refinement revealed 83,927 particles with three 2-4 Fabs bound, one to each RBD. A reconstruction of these particles using non-uniform refinement with imposed C3 symmetry resulted in a 3.6 Å map, as determined by the gold standard Fourier shell correlation (FSC). Given the relatively low resolution of the RBD–Fab interface, masked local refinement was used to obtain a 3.5 Å map with improved density. A masked local refinement of the remainder of the S trimer resulted in a 3.5 Å reconstruction. These two local refinements were aligned and combined using the vop maximum function in UCSF Chimera36. This was repeated for the half maps, which were used, along with the refinement mask from the global non-uniform refinement, to calculate the 3D FSC37 and obtain an estimated resolution of 3.2 Å. All maps have been submitted to the EMDB with the ID EMD-22156.
For the 4-8 Fab dataset, image preprocessing and particle picking were performed as above. 2D classification, ab initio modelling, and 3D heterogeneous classification revealed 47,555 particles with 3 Fabs bound, one to each NTD and with all 3 RBDs in the down conformation. While this particle stack was refined to 3.9 Å using non-uniform refinement with imposed C3 symmetry, substantial molecular motion prevented the visualization of the Fab epitope at high resolution (EMD-22159). In addition, 105,278 particles were shown to have 3 Fabs bound, but with 1 RBD in the up conformation. These particles were refined to 4.0 Å using non-uniform refinement with C1 symmetry (EMD-22158), and suffered from the same conformational flexibility as the all-RBD-down particles. This flexibility was visualized using 3D variability analysis in cryoSPARC.
For the 2-43 Fab dataset, which was collected at an electron fluence of 51.69 e/Å2, image preprocessing was performed as above, and particle picking was performed using blob picker. 2D classification, ab initio modelling, and 3D heterogeneous classification revealed 10,068 particles with 3 Fabs bound, which was refined to 5.8 Å resolution (EMD-22157).
Cryo-EM model fitting
An initial homology model of the 2-4 Fab was built using Schrodinger Release 2020-2: BioLuminate38. The RBD was initially modelled using the coordinates from PDB ID 6W41. The remainder of the S trimer was modelled using the coordinates from PDB ID 6VSB. These models were docked into the consensus map using Chimera. The model was then fitted interactively using ISOLDE 1.0b539 and COOT 0.8.9.240, and using real space refinement in Phenix 1.1841. In cases where side chains were not visible in the experimental data, they were truncated to alanine. Validation was performed using Molprobity42 and EMRinger43. The model was submitted to the PDB with the ID 6XEY. Figures were prepared using ChimeraX44.
Hamster protection experiment
In vivo evaluation of mAb 2-15 in an established golden Syrian hamster model of SARS-CoV-2 infection was performed as described previously with slight modifications45. Approval was obtained from the University of Hong Kong (HKU) Committee on the Use of Live Animals in Teaching and Research. In brief, 6–8-week-old male and female hamsters were obtained from the Chinese University of Hong Kong Laboratory Animal Service Centre through the HKU Laboratory Animal Unit and kept in biosafety level-2 (BSL-2) housing with access to standard pellet feed and water ad libitum until virus challenge in the BSL-3 animal facility. Each hamster (n = 4 per group) was intraperitoneally administered one dose of 1.5 mg/kg of mAb 2-15 in phosphate-buffered saline (PBS), 0.3 mg/kg of mAb 2-15 in PBS, or PBS alone as control. Twenty-four hours later, each hamster was intranasally inoculated with a challenge dose of 100 μl Dulbecco’s modified Eagle medium containing 105 PFU of SARS-CoV-2 (HKU-001a strain, GenBank accession no: MT230904.1) under intraperitoneal ketamine (200 mg/kg) and xylazine (10 mg/kg) anaesthesia. The hamsters were monitored twice daily for clinical signs of disease and killed on the fourth day after the challenge. Half of each hamster’s lung tissue was used for viral load determination by a quantitative SARS-CoV-2 RdRp/Hel RT–PCR assay46 and an infectious virus titration using a plaque assay described previously45. Student’s t-test was used to determine significant differences among the groups, and P < 0.05 was considered statistically significant.
Ethics statement
The acquisition of samples from recovering patients for isolation and identification of potent monoclonal antibodies against COVID-19 (AAAS9517) was approved by the Columbia University Institutional Review Board. Informed consent was obtained from all participants or surrogates.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The 19 neutralizing antibodies have been deposited in GenBank (https://www.ncbi.nlm.nih.gov/genbank/) with accession numbers from MT712278 to MT712315. Coordinates for the antibody 2-4 complex have been deposited in the Protein Data Bank as PDB 6XEY. Cryo-EM maps and data have been deposited in EMDB with deposition codes EMDB-22156 for antibody 2-4, EMDB-22158 and EMDB-22159 for antibody 4-8, and EMDB-22275 for antibody 2-43. These data are used in Fig. 4 and Extended Data Figs. 7, 8.
Code availability
Next-generation sequencing data of antibody repertoires were processed using Cell ranger v3.1.0, SONAR V1, BLAST v2.2.25, CLUSTALO1.2.3, and USEARCH v9.2.64. Cryo-EM data was collected using Leginon 3.4.beta. Cryo-EM data was processed using cryoSPARC v2.14.2, MotionCor2, Topaz v0.2.4, 3DFSC v3.0, UCSF Chimera v1.13.1, ChimeraX v0.93, ISOLDE v1.0b5, Phenix v1.18, and COOT v0.8.9.2.
References
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Wang, C., Horby, P. W., Hayden, F. G. & Gao, G. F. A novel coronavirus outbreak of global health concern. Lancet 395, 470–473 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e286 (2020).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e278 (2020).
Wang, Q. et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181, 894–904.e899 (2020).
Ju, B. et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature https://doi.org/10.1038/s41586-020-2380-z (2020).
Pinto, D. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295 (2020).
Cao, Y. et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 182, 73–84.e16 (2020).
Wu, Y. et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 368, 1274–1278 (2020).
Hansen, J. et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science https://doi.org/10.1126/science.abd0827 (2020).
Sheng, Z. et al. Gene-specific substitution profiles describe the types and frequencies of amino acid changes during antibody somatic hypermutation. Front. Immunol. 8, 537 (2017).
ter Meulen, J. et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 3, e237 (2006).
Tian, X. et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 9, 382–385 (2020).
Yuan, M. et al. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 368, 630–633 (2020).
Rogers, T. F. et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science https://doi.org/10.1126/science.abc7520 (2020).
Chen, X. et al. Human monoclonal antibodies block the binding of SARS-CoV-2 spike protein to angiotensin converting enzyme 2 receptor. Cell. Mol. Immunol. 17, 647–649 (2020).
Zeng, X. et al. Isolation of a human monoclonal antibody specific for the receptor binding domain of SARS-CoV-2 using a competitive phage biopanning strategy. Antib. Ther. 3, 95–100 (2020).
Liu, X. et al. Neutralizing antibodies isolated by a site-directed screening have potent protection on SARS-CoV-2 infection. Preprint at https://doi.org/10.1101/2020.05.03.074914 (2020).
Zost, S. J. et al. Rapid isolation and profiling of a diverse panel of human monoclonal antibodies targeting the SARS-CoV-2 spike protein. Nat. Med. https://doi.org/10.1038/s41591-020-0998-x (2020).
Robbiani, D. F. et al. Convergent antibody responses to SARS-CoV-2 infection in convalescent individuals. Nature https://doi.org/10.1038/s41586-020-2456-9 (2020).
Brouwer, P. J. M. et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science https://doi.org/10.1126/science.abc5902 (2020).
Chi, X. et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science https://doi.org/10.1126/science.abc6952 (2020).
Wang, C. et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 11, 2251 (2020).
Wang, P. et al. SARS-CoV-2 neutralizing antibody responses are more robust in patients with severe disease. Preprint at https://doi.org/10.1101/2020.06.13.150250 (2020).
Schramm, C. A. et al. SONAR: a high-throughput pipeline for inferring antibody ontogenies from longitudinal sequencing of B cell transcripts. Front. Immunol. 7, 372 (2016).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Lefranc, M. P. et al. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 37, D1006–D1012 (2009).
Sievers, F. & Higgins, D. G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 1079, 105–116 (2014).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Nie, J. et al. Establishment and validation of a pseudovirus neutralization assay for SARS-CoV-2. Emerg. Microbes Infect. 9, 680–686 (2020).
Whitt, M. A. Generation of VSV pseudotypes using recombinant ΔG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 169, 365–374 (2010).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Zhu, K. et al. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins 82, 1646–1655 (2014).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 74, 519–530 (2018).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron Radiat. 11, 53–55 (2004).
Davis, I. W., Murray, L. W., Richardson, J. S. & Richardson, D. C. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–W619 (2004).
Barad, B. A. et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Chan, J. F. et al. Simulation of the clinical and pathological manifestations of Coronavirus Disease 2019 (COVID-19) in golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa325 (2020).
Chan, J. F. et al. Improved molecular diagnosis of COVID-19 by the novel, highly sensitive and specific COVID-19-RdRp/Hel real-time reverse transcription-PCR assay validated in vitro and with clinical specimens. J. Clin. Microbiol. 58, e00310-20 (2020).
Acknowledgements
We thank N. Wang and J. McLellan for providing reagents for generating the SARS-CoV-2 S trimer and RBD-SD1 with mFc tag; W. Chen for assistance with generating the Venn diagrams; and B. DeKosky and X. Wu for helpful input. This study was supported by funding to D.D.H. from the Jack Ma Foundation, the JPB Foundation, Samuel Yin, Brii Biosciences, Tencent Charity Foundation, Roger Wu, Carol Ludwig, and Peggy and Andrew Cherng. Cryo-EM data collection was performed at the National Center for CryoEM Access and Training and the Simons Electron Microscopy Center located at the New York Structural Biology Center, supported by the NIH Common Fund Transformative High Resolution Cryo-Electron Microscopy program (U24 GM129539) and by grants from the Simons Foundation (SF349247) and NY State Assembly. Data analysis was performed at the National Resource for Automated Molecular Microscopy (NRAMM), supported by the NIH National Institute of General Medical Sciences (GM103310). Hamster experiments were conducted with support from the Health@InnoHK (Centre for Virology, Vaccinology and Therapeutics), Innovation and Technology Commission, The Government of the Hong Kong Special Administrative Region.
Author information
Authors and Affiliations
Contributions
D.D.H. conceived the project. L.L., P.W., M.S.N, J.Y., Q.W. and Y.H. performed many of the experiments. M.T.Y. was responsible for recruiting patients, obtaining clinical specimens, and summarizing clinical data. L.L., V.S., A.F. and X.V.G. performed and analysed the B cell sorting, 10X Genomics, sequencing and analysis of the clones. Z.S. performed bioinformatic analyses on 10X next-generation sequencing data and antibody repertoire. J.Y. cloned, expressed, and purified the mAbs. L.L. and Q.W. performed the epitope mapping and binding experiments. P.W. conducted the pseudovirus neutralization assays and M.S.N and Y.H.performed infectious SARS-CoV-2 neutralization assays. M.R., G.C., J.B., J.G., and L.S. carried out the cryo-EM studies. J.F.-W.C., Z.C., and K.-Y.Y. were responsible for the hamster experiment. Y.L. helped with project management. T.Z. and P.D.K. provided key reagents for the study, and P.D.K. contributed to the analysis and discussion of the data. L.L., P.W., M.S.N., J.Y., Y.H., Z.S., M.R., Q.W., L.S., and D.D.H. analysed the results, and D.D.H. wrote the manuscript, with contributions from each author. J.G.S. provided valuable suggestions.
Corresponding authors
Ethics declarations
Competing interests
A provisional patent application has been filed for the monoclonal antibodies described in the manuscript. L.L. and D.D.H. are inventors.
Additional information
Peer review information Nature thanks Antonio Lanzavecchia and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 SARS-CoV-2 S trimer-specific antibody isolation strategy.
a, Schema for isolating of S trimer-specific mAbs from memory B cells in the blood of infected patients. b, Sorting results on the isolation of S trimer-specific memory B cells using flow cytometry. c, Magnified representation of the panel of S trimer-positive memory B cells for each patient. Inset numbers indicate the absolute number and the percentage of S trimer-specific memory B cells isolated from each case.
Extended Data Fig. 2 Summary of mAb screening of transfection supernatants.
a, Numbers of binding and neutralizing antibodies from patients 1 to 5. b, The best-fit pseudovirus neutralization curves for 130 samples that were positive in at least one of the screens shown in Fig. 1b. The 18 transfection supernatants that showed evidently better potency are highlighted in colours, while others with non-neutralizing or weakly neutralizing activities are shown in grey. One additional supernatant (Patient 1) that was initially missed in the pseudovirus screen but later found to be a potent neutralizing mAb (1-87) is also highlighted.
Extended Data Fig. 3 Genetic features of SARS-CoV-2-specific antibody repertoire.
a, 108 of the 123 antigen-specific antibodies are from IgG isotype. The kappa and lambda light chains are comparably used. b, Compared to IgG repertoires of healthy human donors (17,243, 27,575, and 20,889 transcripts for heavy, kappa, and lambda chains respectively), IGHV3-30 (antigen-specific n = 26 and healthy donor n = 1117) and IGKV3-20 genes (antigen-specific n = 15 and healthy donor n = 4,071) are over-represented in heavy and light chain repertoires respectively (P values are 6.415 × 10−11 and 0.04332 respectively, χ2-test with 1 degree of freedom). We did not test the enrichment of other genes because the numbers of antigen-specific antibodies are less than 15. c, The usage of IGHJ6 gene (antigen-specific n = 36 and healthy donor n = 3646) was significantly higher in antigen-specific antibodies (χ2-test with 1 degree of freedom, P = 0.02807). d, The CDRH3 length of antigen-specific antibodies is significantly longer than in healthy donors (two-sided Kolmogorov–Smirnov test, P = 0.014). e, For both heavy and light chains, the V region nucleotide somatic hypermutation levels are significantly lower than in antibodies of healthy donors (two-sided Kolmogorov–Smirnov test, P < 2.2 × 10−16 for both heavy and light chains). For the boxplots, the middle lines are medians. The lower and upper hinges correspond to the first and third quartiles respectively. The upper whisker extends to values no larger than 1.5× IQR (the interquartile range or distance between the first and third quartiles) from the hinge. The lower whisker extends to values no smaller than 1.5× IQR from the hinge. Data points beyond the whiskers were plotted as outliers using dots.
Extended Data Fig. 4 Correlation of neutralizing antibody titres of the top 19 mAbs in the live SARS-CoV-2 assay versus the pseudovirus assay.
Green circles represent RBD-directed antibodies; orange circles represent NTD-directed antibodies; and black circles represent antibodies in the ‘Others’ category. The Pearson correlation coefficient (R) and the p value were calculated using GraphPad Prism. Experiments were performed in triplicates for all mAbs tested.
Extended Data Fig. 5 The pseudovirus neutralization profiles for 12 purified mAbs that strongly bound the S trimer but with weak or no virus-neutralizing activities.
The four mAbs with weak neutralizing activities against SARS-CoV-2 pseudovirus are shown in sold lines, and the remaining 8 non-neutralizing mAbs are shown in dashed lines.
Extended Data Fig. 6 Cell-surface staining with antibodies.
a, Antibody binding to the SARS-CoV-1 (blue) and SARS-CoV-2 (red) spike proteins expressed on the cell surface. Expi293 cells were co-transfected with GFP and full-length SARS-CoV-1 or SARS-CoV-2 spike genes. After 48 h, antibody binding to spike protein in the GFP-positive cells was detected by flow cytometer. The data show all antibodies tested were able to recognize the wildtype SARS-CoV-2 spike protein but not SARS-CoV-1 spike protein. b, Monoclonal Ab 2-43 bound to S trimer expressed on Expi293 cell surface can be competed out by mAbs directed against RBD but only minimally by mAbs to the NTD region. Shown are representative data from three independent experiments.
Extended Data Fig. 7 Cryo-EM analysis of antibody 2-4 in complex with the S trimer.
a, Representative micrograph and CTF of the micrograph. 8,324 micrographs were collected in total. b, Representative 2D class averages. c, Resolution of the consensus map with C3 symmetry as calculated by 3DFSC. d, The local resolution of the full map as calculated by cryoSPARC at an FSC cutoff of 0.5. e, Representative density of the Fab 2-4 (blue) and RBD (green) interface, showing interactions of CDR H3 in red, L1 in magenta, and L3 in light magenta (left), along with CDR H2 and the N-linked glycosylation added by SHM at ASN58 (right). f, Fab 2-4 binding interface with RBD. VH is shown in blue, VL in light blue, with CDRs H1 in orange, H2 in yellow, H3 in red, L1 in magenta, and L3 in light magenta. g, Positions of antibodies 2-4, S3098, and BD-239 on the trimeric CoV-2 spike. h, Antibody BD-239 in complex with S trimer. i, Somatic hypermutations found only in the antibody 2-4 heavy chain, shown in brown. The mutation A60T creates an NxT sequence leading to N58 glycosylation.
Extended Data Fig. 8 Cryo-EM data processing for antibodies 4-8 and 2-43 in complex with S trimer.
a, Representative 4-8 micrograph and CTF of the micrograph. 3,153 micrographs were collected in total. b, Representative 2D class averages. c, Resolution of the spike in the RBD down conformation in complex with Fab 4-8. d, Resolution of the spike in the RBD up conformation in complex with Fab 4-8. e, Local resolution of the spike in the RBD down conformation in complex with Fab 4-8 at an FSC cutoff of 0.5, with two thresholds shown. f, Local resolution of the spike in the RBD up conformation in complex with Fab 4-8 at an FSC cutoff of 0.5, with two thresholds shown. g, Although the map was reconstructed at 4.0Å resolution, density for 4-8 Fab is poor due to molecular motion. A rigid body fit with SARS-CoV-2 spike and an antibody variable domain model is shown. h–k, Cryo-EM data processing for antibody 2-43 in complex with the S trimer. h, Representative 2-43 micrograph and CTF of the micrograph. i, Representative 2D class averages. j, Resolution of Fab 2-43 in complex with S trimer. k, The local resolution of the full map as calculated by cryoSPARC at an FSC cutoff of 0.5.
Supplementary information
Rights and permissions
About this article

Cite this article
Liu, L., Wang, P., Nair, M.S. et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 584, 450–456 (2020). https://doi.org/10.1038/s41586-020-2571-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2571-7
This article is cited by
-
Novel neutralizing SARS-CoV-2-specific mAbs offer detection of RBD linear epitopes
Virology Journal (2024)
-
The role of nutritional support with probiotics in outpatients with symptomatic acute respiratory tract infections: a multicenter, randomized, double-blind, placebo-controlled dietary study
BMC Nutrition (2024)
-
An ancestral SARS-CoV-2 vaccine induces anti-Omicron variants antibodies by hypermutation
Nature Communications (2024)
-
Simulation-driven design of stabilized SARS-CoV-2 spike S2 immunogens
Nature Communications (2024)
-
An integrated technology for quantitative wide mutational scanning of human antibody Fab libraries
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
