
Abstract
The ongoing pandemic of coronavirus disease 2019 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is a major threat to global health1 and the medical countermeasures available so far are limited2,3. Moreover, we currently lack a thorough understanding of the mechanisms of humoral immunity to SARS-CoV-24. Here we analyse a large panel of human monoclonal antibodies that target the spike (S) glycoprotein5, and identify several that exhibit potent neutralizing activity and fully block the receptor-binding domain of the S protein (SRBD) from interacting with human angiotensin-converting enzyme 2 (ACE2). Using competition-binding, structural and functional studies, we show that the monoclonal antibodies can be clustered into classes that recognize distinct epitopes on the SRBD, as well as distinct conformational states of the S trimer. Two potently neutralizing monoclonal antibodies, COV2-2196 and COV2-2130, which recognize non-overlapping sites, bound simultaneously to the S protein and neutralized wild-type SARS-CoV-2 virus in a synergistic manner. In two mouse models of SARS-CoV-2 infection, passive transfer of COV2-2196, COV2-2130 or a combination of both of these antibodies protected mice from weight loss and reduced the viral burden and levels of inflammation in the lungs. In addition, passive transfer of either of two of the most potent ACE2-blocking monoclonal antibodies (COV2-2196 or COV2-2381) as monotherapy protected rhesus macaques from SARS-CoV-2 infection. These results identify protective epitopes on the SRBD and provide a structure-based framework for rational vaccine design and the selection of robust immunotherapeutic agents.
Similar content being viewed by others


Main
The S protein of SARS-CoV-2 is the molecular determinant of viral attachment, fusion and entry into host cells6. The S protein is composed of an N-terminal subunit (S1) that mediates receptor binding, and a C-terminal subunit (S2) that mediates fusion between the virus and the membrane of the host cell. The S1 subunit contains an N-terminal domain (NTD) and a receptor-binding domain (RBD). SARS-CoV-2 and SARS-CoV, the genomes of which share approximately 78% sequence identity1, both use human ACE2 as an entry receptor7,8,9. Human antibodies to the S glycoprotein mediate protective immunity against other zoonotic betacoronaviruses of high pathogenicity, including SARS-CoV10,11,12,13,14 and Middle East respiratory syndrome coronavirus (MERS-CoV)15,16,17,18,19,20,21,22,23,24. The most potent S-protein-specific monoclonal antibodies appear to neutralize betacoronaviruses by binding to the region on the SRBD that directly mediates receptor engagement, and thereby blocking the attachment of the virus to host cells. Human antibodies could be used for prophylaxis, post-exposure prophylaxis or treatment of SARS-CoV-2 infection25. Many studies are ongoing—including randomized controlled trials assessing plasma from convalescent individuals with prior SARS-CoV-2 infection, and one trial evaluating hyperimmune immunoglobulin—but it is not yet clear whether these treatments can reduce morbidity or mortality26.
We isolated 389 SARS-CoV-2 S-protein-reactive monoclonal antibodies from the B cells of two convalescing individuals who had been infected with SARS-CoV-2 in Wuhan, China5. A subset of those antibodies bound to a recombinant RBD construct (SRBD) and exhibited neutralizing activity in a rapid screening assay with wild-type SARS-CoV-2 virus5. In the current study, we sought to define the antigenic landscape of SARS-CoV-2 and determine which sites of the SRBD are targets of neutralizing monoclonal antibodies. We tested 40 of the anti-S human monoclonal antibodies that were previously pre-selected by rapid neutralization screening assay in a quantitative focus reduction neutralization test (FRNT) with the WA1/2020 strain of SARS-CoV-2. The antibodies in our panel of 40 exhibited half-maximum inhibitory concentration (IC50) values that ranged from 15 to over 4,000 ng ml−1 (visualized as a heat map in Fig. 1a, values shown in Supplementary Table 1 and full curves shown in Extended Data Fig. 1). We hypothesized that many of these SRBD-reactive monoclonal antibodies neutralize virus infection by blocking the binding of the SRBD to human ACE2. Indeed, most of the neutralizing monoclonal antibodies that we tested inhibited the interaction of human ACE2 with trimeric S protein directly (Fig. 1a, Extended Data Fig. 2). Consistent with these results, these monoclonal antibodies also bound strongly to a trimeric S ectodomain (S2Pecto) protein or to monomeric SRBD (Fig. 1a, Extended Data Fig. 3). We evaluated whether the potency of the antibodies at binding S2Pecto or SRBD or blocking human ACE2 predicted binding neutralization potency independently, but none of these measurements correlated with neutralization potency (Fig. 1b–d). However, the antibodies within the highest neutralizing potency tier of the panel (IC50 < 150 ng ml−1) also had the strongest blocking activity against human ACE2 (IC50 < 150 ng ml−1) and exceptional binding activity (half-maximum effective concentration (EC50) < 2 ng ml−1) to the S2Pecto trimer (Fig. 1e). Representative neutralization curves for two potently neutralizing monoclonal antibodies designated COV2-2196 and COV2-2130 are shown in Fig. 1f. Potent neutralization was confirmed using pseudovirus neutralization assays, which revealed far-more sensitive neutralization phenotypes than the wild-type virus and demonstrated a requirement for the use of live virus in assays for assessment of monoclonal antibody potency (Fig. 1g). Both of these monoclonal antibodies (COV2-2196 and COV2-2130) bound strongly to the S2Pecto trimer and fully blocked the binding of human ACE2 (Fig. 1h, i).

a, Heat map of monoclonal antibody neutralization activity, human ACE2-blocking activity, and binding to either trimeric S2Pecto protein or monomeric SRBD. Monoclonal antibodies are ordered by neutralization potency, and dashed lines indicate the 12 antibodies with a neutralization IC50 value <150 ng ml−1. IC50 values (ng ml−1) are shown for viral neutralization (neut.) and human ACE2 blocking, and EC50 values (ng ml−1) for binding. The cross-reactive SARS-CoV SRBD monoclonal antibody rCR3022 is shown as a positive control and the anti-dengue monoclonal antibody r2D22 as a negative control. Data are representative of at least two independent experiments performed in technical duplicate. No inhibition or no binding indicates an IC50 or EC50 value >10,000 ng ml−1, respectively. b–d, Correlation of human ACE2 blocking (b), S2Pecto trimer binding (c) or SRBD binding (d) of monoclonal antibodies with their neutralization activity. e, Correlation of human ACE2 blocking and S2Pecto trimer binding. R2 values are shown for linear regression analysis of log-transformed values. Purple circles indicate monoclonal antibodies with a neutralization IC50 value <150 ng ml−1. f, Neutralization curves for COV2-2196 and COV2-2130 against wild-type SARS-CoV-2 virus. Calculated IC50 values are shown on the graph. Error bars, s.d.; data are representative of at least two independent experiments performed in technical duplicate. g, Neutralization curves for COV2-2196 and COV2-2130 in a pseudovirus neutralization assay. Error bars, s.d.; values are technical duplicates from a single experiment. Calculated IC50 values from a minimum of six experiments are shown on the graph. h, Human-ACE2-blocking curves for COV2-2196, COV2-2130 and rCR3022 in a human-ACE2-blocking ELISA. Calculated IC50 values are shown on the graph. Data are mean ± s.d. of technical triplicates from a representative experiment repeated twice. i, ELISA binding of COV2-2196, COV2-2130 and rCR3022 to trimeric S2Pecto. Calculated EC50 values are shown on the graph. Data are mean ± s.d. of technical triplicates from a representative experiment repeated twice.
We next defined the major antigenic sites on the SRBD for neutralizing monoclonal antibodies by competition-binding analysis. We first used a biolayer-interferometry-based competition assay with a minimal version of the SRBD domain to screen for monoclonal antibodies that competed for binding with the potently neutralizing monoclonal antibody COV2-2196 or a recombinant version of the previously described SARS-CoV monoclonal antibody CR3022, which recognizes a conserved cryptic epitope12,27. We identified three major groups of competing monoclonal antibodies (Fig. 2a). The largest group of antibodies blocked COV2-2196 but not recombinant CR3022 (rCR3022), whereas some monoclonal antibodies were blocked by rCR3022 but not by COV2-2196. Two monoclonal antibodies, including COV2-2130, were not blocked by either reference monoclonal antibody. Most monoclonal antibodies competed with human ACE2 for binding, suggesting that they bound near the ACE2-binding site of the SRBD. We used COV2-2196, COV2-2130 and rCR3022 in an enzyme-linked immunosorbent assay (ELISA)-based competition-binding assay with the S2Pecto trimer and found that the SRBD contained three major antigenic sites, with some monoclonal antibodies probably making contacts in more than one site (Fig. 2b). Most of the potently neutralizing monoclonal antibodies directly competed with COV2-2196 for binding. Competition-binding analyses of human ACE2 and monoclonal antibodies with serum or plasma from four previously described individuals with recent laboratory-confirmed SARS-CoV-2 infection5 showed that COV2-2196- and COV2-2130-like antibody responses are subdominant in these individuals (Extended Data Fig. 4).

a, Left, monoclonal antibody binding to the SRBD in the presence of reference monoclonal antibodies COV2-2196 or rCR3022. Values in squares are the per cent binding of the monoclonal antibody in the presence of the competing monoclonal antibody relative to a mock-competition control. Black squares, full competition (<33% relative binding); white squares, no competition (>67% relative binding). Right, biolayer-interferometry-based competition binding assay measuring the ability of monoclonal antibodies to prevent the binding of human ACE2. Values are the per cent blocking of human ACE2 by the monoclonal antibody. Red indicates high blocking activity. b, Competition of the panel of neutralizing monoclonal antibodies with reference monoclonal antibodies COV2-2130, COV2-2196 or rCR3022. Binding of reference monoclonal antibodies to trimeric S2Pecto was measured in the presence of saturating competitor monoclonal antibody in a competition ELISA and normalized to binding in the presence of r2D22. Black, full competition (<25% binding of reference antibody); grey, partial competition (25–60% binding of reference antibody); white, no competition (>60% binding of reference antibody). c, Neutralization dose–response matrix of wild-type SARS-CoV-2 by COV2-2196 and COV2-2130. Axes denote the concentration of each monoclonal antibody, with the per cent neutralization shown in each square. Data are from a representative experiment performed in technical triplicate and repeated twice. The white-to-red heat map denotes 0% neutralization to 100% neutralization, respectively. d, Synergy map calculated on the basis of the SARS-CoV-2 neutralization in c. δ-score is a synergy score. Red colour indicates areas in which synergistic neutralization was observed; black box indicates the area of maximum synergy between the two monoclonal antibodies.
As COV2-2196 and COV2-2130 did not compete for binding to the SRBD, we assessed whether these monoclonal antibodies synergize for virus neutralization—a phenomenon that has been observed previously for SARS-CoV monoclonal antibodies12. We tested combination responses (Fig. 2c) in an FRNT using SARS-CoV-2, and compared the values obtained experimentally with the expected responses calculated by synergy-scoring models28. The comparison revealed that the combination of COV2-2196 and COV2-2130 antibodies was synergistic, with an overall synergy δ-score of 17.4 (where any score greater than 10 indicates synergy; Fig. 2d). In particular, a combined monoclonal antibody dose of 79 ng ml−1 (16 ng ml−1 of COV2-2196 and 63 ng ml−1 of COV2-2130) had the same activity as 250 ng ml−1 of each individual antibody (Fig. 2c). This finding shows that by using a cocktail of two antibodies, the dose of each antibody can be reduced by more than threefold to achieve the same potency of virus neutralization in vitro.
We next defined the epitopes that are recognized by representative monoclonal antibodies in the two major competition-binding groups that synergize for neutralization. We used mutagenesis to determine critical residues in the SRBD for the binding of neutralizing monoclonal antibodies (Fig. 3a, Extended Data Fig. 5). These studies showed that F486 or N487 are critical residues for the binding of COV2-2196, and N487 is a critical residue for COV2-2165—two antibodies that compete with one another for binding. Likewise, mutagenesis studies for COV2-2130 using K444A and G447R mutants suggested that these residues (K444 and G447) are critical for recognition (Fig. 3a). Previous structural studies have defined the interaction between the SRBD and human ACE229 (Fig. 3b). Most of the interacting residues in the SRBD are contained within a 60-amino-acid linear peptide that defines the human ACE2 recognition motif (Fig. 3c). We next tested the binding of human monoclonal antibodies to this minimal peptide and found that potent neutralizing members of the largest group of antibodies from the competition-binding assay—including COV2-2196, COV2-2165 and COV2-2832—recognized this peptide (Fig. 3c), suggesting that these monoclonal antibodies make critical contacts within the human ACE2 recognition motif.

a, Identification of critical contact residues by alanine and arginine mutagenesis. Top, binding of COV2-2130, COV2-2165 or COV2-2196 to wild-type (WT) or mutant SRBD constructs, normalized to the wild type. Bottom, representative binding curves for COV2-2196 to wild-type or mutant SRBD constructs. b, Co-crystal structure of the SRBD (blue) and human ACE2 (green) (Protein Data Bank (PDB): 6M0J), with the human ACE2 recognition motif shown in orange. Critical contact residues are shown for COV2-2130 (gold spheres) and COV2-2196 (purple spheres). c, Top, ELISA binding of monoclonal antibodies to the human ACE2 recognition motif. r2D22 is shown as a negative control. Data are mean ± s.d. of technical triplicates from a single experiment repeated twice. Bottom, structure of human ACE2 recognition motif (orange) with COV2-2196 critical contact residues shown (purple). d, Fab–S2Pecto trimer complexes visualized by negative-stain electron microscopy for COV2-2130, COV2-2165 and COV2-2196. The SRBD is shown in blue, the SNTD in red and electron density in grey. The trimer state (open or closed) is denoted for each complex. Representative two-dimensional (2D) class averages for each complex are shown at the bottom (box size is 128 pixels, with 3.06 Å per pixel). Data are from a single experiment; detailed collection statistics are provided in Supplementary Table 2. e, COV2-2130 and COV2-2196 Fabs in complex with the S2Pecto trimer. Colours and data collection as in d. Representative 2D class averages for the complexes are shown at the bottom; scales as in d. f, Top, the competition-binding landscape visualized on the S2Pecto trimer. The CR3022 crystal structure was docked into the double-Fab–S2Pecto trimer model. CR3022 is shown in cyan. Bottom, quantitative Venn diagram showing the number of monoclonal antibodies in each competition group.
We used negative-stain electron microscopy of the S2Pecto trimer in complex with antigen-binding fragments (Fabs) to determine the structural epitopes for several monoclonal antibodies (Fig. 3d, e, Supplementary Table 2). The potently neutralizing antibodies COV2-2196 and COV2-2165 bound to the human ACE2 recognition motif of the SRBD and recognized the ‘open’ conformational state of the S2Pecto trimer, in which the RBD rotates upward to expose the residues that mediate ACE2 interaction30,31 (Fig. 3d). COV2-2130, which represents a different competition-binding group, bound to the RBD in the S2Pecto trimer in the ‘closed’ position (Fig. 3d). Because COV2-2196 and COV2-2130 did not compete for binding, we attempted to make complexes of both Fabs bound at the same time to the S2Pecto trimer. We found that both Fabs bound simultaneously when the S2Pecto trimer was in the open position, indicating that COV2-2130 can recognize the SRBD in both conformations (Fig. 3e). Overlaying the structure of the two-Fab complex with that of the SRBD–CR3022 complex27, we observed that these antibodies bind to three distinct sites on the SRBD, as predicted by our competition-binding studies (Fig. 3f).
Next, we tested the prophylactic efficacy of COV2-2196 or COV2-2130 monotherapy or a combination of both COV2-2196 and COV2-2130 in a model of SARS-CoV-2 infection in BALB/c mice. In this model (Fig. 4a), mice are first treated with an anti-IFNAR1 antibody and then transduced with an adenovirus that expresses human ACE2 (AdV-hACE2), which results in susceptibility to infection with SARS-CoV-2, viral replication and severe bronchopneumonia32. The mice were treated with a single dose of COV2-2196 or COV2-2130, a cocktail of COV2-2196 and COV2-2130, or an isotype control monoclonal antibody one day before intranasal challenge with a 4 × 105 plaque-forming unit (PFU) dose of SARS-CoV-2. Prophylaxis with COV2-2196, COV2-2130 or their combination prevented severe SARS-CoV-2-induced weight loss in the mice during the first week of infection (Fig. 4b). Viral RNA levels were reduced significantly at 7 days post-infection (dpi) in the lung and in distant sites including the heart and spleen (Fig. 4c). The expression of cytokine and chemokine genes—indicative of inflammation—was also reduced in the lungs of each group of COV2-antibody-treated mice at 7 dpi (Fig. 4d).

a, SARS-CoV-2 challenge model. Mice were treated with anti-IFNAR1 and transduced with AdV-hACE2 followed by the passive transfer of 200 μg of COV2-2196, COV-2130, their combination (1:1 ratio) or an isotype control monoclonal antibody (i.n., intranasal; i.p., intraperitoneal). One day later, mice were inoculated intranasally with SARS-CoV-2. Tissues were collected at 7 dpi for analysis (c, d). b, Body weight change of mice in a with comparison to isotype control using a repeated measurements two-way analysis of variance (ANOVA) with Tukey’s post hoc test. Data are mean ± s.e.m. of each experimental group. The number of mice (n) for each experimental group is shown. c, d, Viral burden (measured as log10(number of genome equivalents (GEQ) per mg)) at 7 dpi in the lungs, spleen and heart (c) and the expression of cytokine and chemokine genes (d) were measured by RT–qPCR assay. Comparisons were performed using a Kruskal–Wallis ANOVA with Dunn’s post hoc test. e, f, MA-SARS-CoV-2 challenge model. Mice were treated with the indicated monoclonal antibody and then inoculated intranasally with MA-SARS-CoV-2. e, Body weight change of mice (mean ± s.e.m. of each experimental group; n = 10 mice per group). f, Viral burden at 2 dpi in the lungs, measured by RT–qPCR (left) or plaque assay (right) from e; comparisons were made using a Kruskal–Wallis ANOVA with Dunn’s post hoc test (n = 10 mice per group). g, Haematoxylin and eosin staining of lung sections from mice that were treated and challenged as in a, shown at day 7. Images are shown at low (left), medium (middle) and high (right) magnification. Each image is representative of two separate experiments (n = 3 to 5 mice per group). Scale bars, 250 μm (left); 50 μm (middle); 25 μm (right). h, i, SARS-CoV-2 NHP challenge model. Rhesus macaques received one 50 mg kg−1 dose of COV2-2196 (n = 4 macaques per group), COV2-2381 (n = 4 macaques per group) or isotype control monoclonal antibody (n = 4 macaques per group) intravenously on day −3 and were then challenged intranasally and intratracheally with SARS-CoV-2 after three days. Subgenomic viral RNA levels were assessed in nasal swabs (h) and bronchioalveolar lavage (i) at multiple time points after challenge. Each black curve shows an individual macaque, with red lines indicating the median values within each treatment group. Data represent a single experiment. Dashed lines indicate the limit of detection (LOD) of the assay.
We also tested COV2-2196, COV2-2130 and their combination for prophylactic efficacy in an immunocompetent model using a mouse-adapted SARS-CoV-2 (MA-SARS-CoV-2) virus33 (Fig. 4e, f). Each of the monoclonal antibody treatments reduced viral RNA levels by up to 105-fold at 2 dpi in the lung, compared to the isotype control group (Fig. 4f). All of the mice from the COV2-2196 and the combined COV2-2196 and COV2-2130 treatment groups, and 8 out of 10 mice from the COV2-2130 treatment group, no longer had infectious virus in the lung at 2 dpi (as measured by a plaque assay of lung tissue homogenates; Fig. 4f).
We evaluated the effect of treatment with monoclonal antibodies on SARS-CoV-2-induced lung pathology. At 7 dpi, lungs from anti-IFNAR1-treated, AdV-hACE2-transduced mice that were treated with isotype control monoclonal antibody and then inoculated with SARS-CoV-2- showed perivascular, peribronchial and alveolar inflammation, with the infiltration of immune cells and alveolar damage that are characteristic of viral pneumonia (Fig. 4g, Supplementary Table 3). By contrast, mice under the same conditions that were treated with COV2-2196, COV2-2130 or their combination developed notably less lung disease, and their lung pathology was similar to that observed in AdV-hACE2-transduced control mice that were not infected with SARS-CoV-2 (Fig. 4g, Supplementary Table 3).
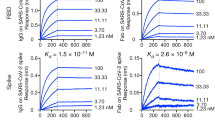
We next tested the protective efficacy of monoclonal antibodies using a recently described non-human primate (NHP) model of SARS-CoV-234,35. In this model, we tested two monoclonal antibodies as monotherapy: COV2-2196 and another of the most potent antibodies identified, COV2-2381—a neutralizing monoclonal antibody that is encoded by the same variable gene segments as COV2-2196 but which contains a number of amino acid differences in the heavy-chain complementarity-determining region 3 (HCDR3) and light-chain complementarity-determining region 3 (LCDR3) (Extended Data Fig. 6a). Notably, other groups have identified highly similar monoclonal antibodies from multiple donors, demonstrating that these monoclonal antibodies constitute a public clonotype36. Rhesus macaques received one 50 mg kg−1 dose of COV2-2196, COV2-2381 or isotype control monoclonal antibody intravenously on day −3, and were then challenged intranasally and intratracheally on day 0 with a 1.1 × 104 PFU dose of SARS-CoV-2. After the challenge, we used quantitative PCR with reverse transcription (RT–qPCR) to quantify the levels of subgenomic viral RNA generated by viral replication in the bronchoalveolar lavage and in nasal swabs. High levels of subgenomic viral RNA were observed in the macaques that were treated with isotype control monoclonal antibody, with a median peak of 7.53 (range 5.37–8.23) RNA copies per swab in nasal swabs and 4.97 (3.81–5.24) log10 RNA copies per ml in the bronchoalveolar lavage (Fig. 4h, i). Subgenomic viral RNA was not detected in samples from either of the antibody-treated groups (limit of detection = 50 (1.7 log10) RNA copies per swab or per ml), indicating that these antibodies conferred protection against SARS-CoV-2. A pharmacokinetics analysis showed that the concentrations of circulating human monoclonal antibodies were similar in macaques from each treatment group (Extended Data Fig. 6b).
We next assessed the therapeutic efficacy of treatment with COV2-2196, COV2-2130 or their combination using the MA-SARS-CoV-2 mouse model. All treatments reduced the levels of infectious virus in the lungs of mice at 2 dpi. The antibody cocktail (1:1) delivered at a dose of 400 μg per mouse (around 20 mg kg−1) was the most efficient; this treatment significantly reduced the viral burden in the lung by up to 3 × 104-fold, and four out of five mice from this treatment group did not have detectable levels of infectious virus in the lung (Fig. 5a). Similarly, treatment of AdV-hCE2-transduced mice with 400 μg per mouse of the cocktail 12 hours after challenge with wild-type SARS-CoV-2 virus revealed that infectious virus was fully neutralized in the lungs in vivo (Fig. 5b). Inflammation was also reduced in the lungs of mice that were treated with the antibody cocktail compared to the lungs of isotype-control-treated mice (Fig. 5c). Collectively, these in vivo results suggest that either of the potently neutralizing monoclonal antibodies COV2-2196 or COV2-2381 alone, and the combination of both COV2-2196 and COV2-2130, are promising candidates for the prevention or treatment of COVID-19.

a, Mice were inoculated intranasally with MA-SARS-CoV-2 and 12 hours later given the indicated monoclonal antibody treatments by intraperitoneal injection. Viral burden in the lungs at 2 dpi was measured by plaque assay. The number of mice per group (n) is indicated. Data represent one experiment. b, Mice were treated with anti-IFNAR1 and transduced with AdV-hACE2. Mice were then inoculated intranasally with wild-type SARS-CoV-2 and 12 hours later given the indicated monoclonal antibody treatments by intraperitoneal injection. Viral burden in the lungs at 2 dpi was measured by plaque assay. Two experiments were performed with n =3 to 5 mice per group. Controls for plaque neutralization assay performance were included: lung homogenates from individual mice (n = 3) that were treated with isotype control monoclonal antibody were mixed 1:1 (v:v) with lung homogenates from individual naive untreated mice or antibody-cocktail-treated mice. The latter mixture ensures that neutralization of infection did not occur ex vivo after tissue homogenization. For a, b, measurements from individual mice and median titre are shown, and each group was compared to the isotype-control-treated group using a Kruskal–Wallis ANOVA with Dunn’s post hoc test. c, Expression of cytokine and chemokine genes was measured by qPCR analysis in lungs from b. Measurements from individual mice and median values are shown. Groups were compared using the two-sided Mann–Whitney U-test. The number of mice per group (n) is indicated. Two experiments were performed with n = 3 to 5 mice per group.
Since the start of the SARS-CoV-2 pandemic, several groups have identified human monoclonal antibodies that bind to the SRBD and neutralize the virus36,37,38,39,40,41,42,43,44. Here, we have defined the antigenic landscape for a number of potently neutralizing monoclonal antibodies against SARS-CoV-2 that were derived from a larger panel of hundreds of antibodies5. These studies demonstrate that although a wide range of human neutralizing antibodies are elicited by natural infection with SARS-CoV-2, only a small subset of those monoclonal antibodies are of high potency (IC50 < 50 ng ml−1 against wild-type SARS-CoV-2 virus). Biochemical and structural analysis of these potent monoclonal antibodies defined three principal antigenic sites of vulnerability on the SRBD for SARS-CoV-2 neutralization. Representative monoclonal antibodies from two antigenic sites were shown to synergize in vitro and confer protection as an in vivo cocktail in both prophylactic and therapeutic treatment. Our findings reveal critical features of effective humoral immunity to SARS-CoV-2 and suggest that the role of synergistic neutralization activity in polyclonal responses should be investigated further. Moreover, as SARS-CoV-2 continues to circulate, population-level immunity elicited by natural infection may start to select for antigenic variants that escape the selective pressure of neutralizing antibodies. Other groups have reported the selection of SARS-CoV-2 RBD escape mutations in the presence of single monoclonal antibodies, but not in the presence of a mixture of two antibodies45, which reinforces the need to target multiple epitopes of the S protein in vaccines or immunotherapies. So far, the gene that encodes the S protein has been found to be limited in diversity—with the exception of a D614G substitution46, which is far away from the amino acid positions identified in our mutational studies for the antibodies we have considered here. Rationally selected therapeutic cocktails such as the one we describe are likely to offer greater resistance to SARS-CoV-2 escape than single antibodies. Our results provide a basis for the preclinical evaluation and development of the identified monoclonal antibodies as candidates for use as COVID-19 immunotherapeutic agents in humans.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and, with the exception of pathology scoring, the investigators were not blinded to allocation during experiments and outcome assessment.
Antibodies
The human antibodies studied in this paper were isolated from blood samples from two individuals in North America with previous laboratory-confirmed symptomatic SARS-CoV-2 infection that was acquired in China. The original clinical studies to obtain specimens after written informed consent were previously described5 and had been approved by the Institutional Review Board of Vanderbilt University Medical Center, the Institutional Review Board of the University of Washington and the Research Ethics Board of the University of Toronto. The individuals (a 56-year-old male and a 56-year-old female) are a married couple and residents of Wuhan, China who travelled to Toronto, Canada, where PBMCs were obtained by leukopheresis 50 days after symptom onset. The antibodies were isolated using diverse tools for isolation and cloning of single antigen-specific B cells and the antibody variable genes that encode monoclonal antibodies5.
Cell culture
Vero E6 (ATCC, CRL-1586), Vero (ATCC, CCL-81), HEK293 (ATCC, CRL-1573) and HEK293T (ATCC, CRL-3216) cells were maintained at 37 °C in 5% CO2 in Dulbecco’s minimal essential medium (DMEM) containing 10% (v/v) heat-inactivated fetal bovine serum (FBS), 10 mM HEPES pH 7.3, 1 mM sodium pyruvate, 1 × non-essential amino acids and 100 U ml−1 of penicillin–streptomycin. Vero-furin cells were obtained from T. Pierson and have been described previously47. FreeStyle 293F cells (Thermo Fisher Scientific, R79007) were maintained at 37 °C in 8% CO2. Expi293F cells (Thermo Fisher Scientific, A1452) were maintained at 37 °C in 8% CO2 in Expi293F Expression Medium (Thermo Fisher Scientific, A1435102). ExpiCHO cells (Thermo Fisher Scientific, A29127) were maintained at 37 °C in 8% CO2 in ExpiCHO Expression Medium (Thermo Fisher Scientific, A2910002). Authentication analysis was not performed for the cell lines used. Mycoplasma testing of Expi293F and ExpiCHO cultures was performed on a monthly basis using a PCR-based mycoplasma detection kit (ATCC, 30-1012K).
Viruses
SARS-CoV-2 strain 2019 n-CoV/USA_WA1/2020 was obtained from the Centers for Disease Control and Prevention (a gift from N. Thornburg). Virus was passaged in Vero CCL81 cells and titrated by plaque assay on Vero E6 cells. MA-SARS-CoV-2 virus was generated as described previously33. Virus was propagated in Vero E6 cells grown in DMEM supplemented with 10% Fetal Clone II and 1% penicillin–streptomycin. The virus titre was determined by plaque assay. In brief, virus was diluted serially and inoculated onto confluent monolayers of Vero E6 cells, followed by an agarose overlay. Plaques were visualized on day 2 post-infection after staining with neutral red dye. All work with infectious SARS-CoV-2 was approved by the Washington University School of Medicine or UNC Chapel Hill Institutional Biosafety Committees and conducted in approved BSL3 facilities using appropriate powered air-purifying respirators and personal protective equipment.
Recombinant antigens and proteins
A gene encoding the ectodomain of a prefusion conformation-stabilized SARS-CoV-2 spike (S2Pecto) protein was synthesized and cloned into a DNA plasmid expression vector for mammalian cells. A similarly designed S protein antigen with two prolines and removal of the furin cleavage site for stabilization of the prefusion form of S was reported previously30. In brief, this gene includes the ectodomain of SARS-CoV-2 (to residue 1,208), a T4 fibritin trimerization domain, an AviTag site-specific biotinylation sequence and a C-terminal 8×His tag. To stabilize the construct in the prefusion conformation, we included substitutions K986P and V987P and mutated the furin cleavage site at residues 682–685 from RRAR to ASVG. This recombinant spike 2P-stabilized protein (designated here as S2Pecto) was isolated by metal affinity chromatography on HisTrap Excel columns (GE Healthcare), and protein preparations were purified further by size-exclusion chromatography on a Superose 6 Increase 10/300 column (GE Healthcare). The presence of trimeric, prefusion conformation S protein was verified by negative-stain electron microscopy5. For electron microscopy with S and Fabs, we expressed a variant of S2Pecto lacking an AviTag but containing a C-terminal Twin-Strep-tag, similar to that described previously30. Expressed protein was isolated by metal affinity chromatography on HisTrap Excel columns (GE Healthcare), followed by further purification on a StrepTrap HP column (GE Healthcare) and size-exclusion chromatography on TSKgel G4000SWXL (TOSOH). To express the SRBD subdomain of the SARS-CoV-2 S protein, residues 319–541 were cloned into a mammalian expression vector downstream of an IL-2 signal peptide and upstream of a thrombin cleavage site, an AviTag and a 6×His tag. RBD protein fused to the mouse IgG1 Fc domain (designated RBD–mFc), was purchased from Sino Biological (40592-V05H). For epitope mapping by alanine scanning, wild-type SARS-CoV-2 RBD (residues 334–526) or RBD single-mutation variants were cloned with an N-terminal CD33 leader sequence and C-terminal GSSG linker, AviTag, GSSG linker and 8×His tag. Spike proteins were expressed in FreeStyle 293 cells (Thermo Fisher Scientific) or Expi293 cells (Thermo Fisher Scientific) and isolated by affinity chromatography using a HisTrap column (GE Healthcare), followed by size-exclusion chromatography with a Superdex200 column (GE Healthcare). Purified proteins were analysed by SDS–PAGE to ensure purity and appropriate molecular weights.
Electron microscopy stain grid preparation, imaging and processing of SARS-CoV-2 S2Pecto protein or S2Pecto–Fab complexes
To perform electron microscopy imaging, Fabs were produced by digesting recombinant chromatography-purified IgGs using resin-immobilized cysteine protease enzyme (FabALACTICA, Genovis). The digestion occurred in 100 mM sodium phosphate and 150 mM NaCl pH 7.2 (PBS) for around 16 h at ambient temperature. To remove cleaved Fc and intact IgG, the digestion mix was incubated with CaptureSelect Fc resin (Genovis) for 30 min at ambient temperature in PBS buffer. If needed, the Fab was buffer-exchanged into Tris buffer by centrifugation with a Zeba spin column (Thermo Fisher Scientific).
For screening and imaging of negatively stained SARS-CoV-2 S2Pecto protein in complex with human Fabs, the proteins were incubated at a molar ratio of 4 Fab:3 spike monomer for around 1 hour and approximately 3 μl of the sample at concentrations of about 10–15 μg ml−1 was applied to a glow-discharged grid with continuous carbon film on 400 square mesh copper electron microscopy grids (Electron Microscopy Sciences). The grids were stained with 0.75% uranyl formate48. Images were recorded on a Gatan US4000 4k × 4k CCD camera using an FEI TF20 (TFS) transmission electron microscope operated at 200 keV and control with SerialEM49. All images were taken at 50,000× magnification with a pixel size of 2.18 Å per pixel in low-dose mode at a defocus of 1.5–1.8 μm. The total dose for the micrographs was around 25–38 e− per Å2. Image processing was performed using the cryoSPARC software package50. Images were imported, and particles were CTF-estimated. The images were then denoised and picked with Topaz51,52. The particles were extracted with a box size of 256 pixels and binned to 128 pixels. 2D class averages were performed and good classes selected for ab initio model and refinement without symmetry. For electron microscopy model docking of SARS-CoV-2 S2Pecto protein, the closed model (PDB: 6VXX) was used in Chimera53 for docking to the electron microscopy map (see also Supplementary Table 2 for details). For the SARS-CoV-2 S2Pecto–Fab COV2-2165 and SARS-CoV-2 S2Pecto–Fab COV2-2196 complexes, the open model of SARS-CoV-2 (PDB: 6VYB) and Fab (PDB: 12E8) was used in Chimera for docking to the electron microscopy maps (see also Supplementary Table 2 for details). For the SARS-CoV-2 S2Pecto–Fab COV2-2130 complex, the closed model and Fab (PDB: 12E8) were used in Chimera for docking to the electron microscopy map (see also Supplementary Table 2 for details). All images were made with Chimera. PyMOL (Schrödinger) was used to visualize previously solved molecular structures of the SARS-CoV-2 RBD–human ACE2 complex and the 60-amino-acid human ACE2 recognition motif (PDB: 6M0J).
Monoclonal antibody production and purification
Sequences of monoclonal antibodies that had been synthesized (Twist Bioscience) and cloned into an IgG1 monocistronic expression vector (designated as pTwist-mCis_G1) were used for monoclonal antibody secretion in mammalian cell culture. This vector contains an enhanced 2A sequence and GSG linker that allows the simultaneous expression of monoclonal antibody heavy and light chain genes from a single construct upon transfection54. We previously described microscale expression of monoclonal antibodies in 1 ml ExpiCHO cultures in 96-well plates5. For larger-scale monoclonal antibody expression, we performed transfection (1–300 ml per antibody) of CHO cell cultures using the Gibco ExpiCHO Expression System and protocol for 50 ml mini bioreactor tubes (Corning) as described by the vendor. Culture supernatants were purified using HiTrap MabSelect SuRe (Cytiva, formerly GE Healthcare Life Sciences) on a 24-column parallel protein chromatography system (Protein BioSolutions). Purified monoclonal antibodies were buffer-exchanged into PBS, concentrated using Amicon Ultra-4 50-kDa centrifugal filter units (Millipore Sigma) and stored at 4 °C until use. Purified monoclonal antibodies were tested routinely for endotoxin levels (found to be less than 30 EU per mg IgG for mouse studies and less than 1 EU per mg IgG for NHP studies). Endotoxin testing was performed using the PTS201F cartridge (Charles River), with a sensitivity range from 10 to 0.1 EU per ml, and an Endosafe Nexgen-MCS instrument (Charles River).
ELISA binding assays
Wells of 96-well microtitre plates were coated with purified recombinant SARS-CoV-2 S protein or SARS-CoV-2 SRBD protein at 4 °C overnight. Plates were blocked with 2% non-fat dry milk and 2% normal goat serum in Dulbecco’s phosphate-buffered saline (DPBS) containing 0.05% Tween-20 (DPBS-T) for 1 h. The bound antibodies were detected using goat anti-human IgG conjugated with horseradish peroxidase (HRP) (Southern Biotech, cat. 2040-05, lot B3919-XD29, 1:5,000 dilution) and a 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Thermo Fisher Scientific). Colour development was monitored, 1M hydrochloric acid was added to stop the reaction, and the absorbance was measured at 450 nm using a spectrophotometer (Biotek). For dose–response assays, serial dilutions of purified monoclonal antibodies were applied to the wells in triplicate, and antibody binding was detected as detailed above. EC50 values for binding were determined using Prism v.8.0 software (GraphPad) after log transformation of the monoclonal antibody concentration using sigmoidal dose–response nonlinear regression analysis.
RBD minimal human ACE2 recognition motif peptide binding ELISA
Wells of 384-well microtitre plates were coated with 1 μg ml−1 streptavidin at 4 °C overnight. Plates were blocked with 0.5% bovine serum albumin (BSA) in DPBS-T for 1 h. Plates were washed 4 times with 1× PBST and 2 μg ml−1 biotinylated ACE2 binding motif peptide (LT5578, from LifeTein, LLC) was added to bind streptavidin for 1 h at ambient temperature. Purified monoclonal antibodies were diluted in blocking buffer, added to the wells and incubated for 1 h at ambient temperature. The bound antibodies were detected using goat anti-human IgG conjugated with HRP (2014-05, Southern Biotech) and a TMB substrate (Thermo Fisher Scientific). Colour development was monitored, 1M hydrochloric acid was added to stop the reaction, and the absorbance was measured at 450 nm using a spectrophotometer (Biotek). For dose–response assays, serial threefold dilutions starting at a 10 μg ml−1 concentration of purified monoclonal antibodies were applied to the wells in triplicate, and antibody binding was detected as detailed above.
Analysis of binding of antibodies to variant RBD proteins with alanine or arginine point mutations
Biolayer light interferometry was performed using an Octet RED96 instrument (ForteBio; Pall Life Sciences) and wild-type RBD protein or a mutant RBD protein with a single amino acid change at defined positions to alanine or arginine. Binding of the RBD proteins was confirmed by first capturing 8×His-tagged RBD wild-type or mutant protein from a 10 μg ml−1 (around 200 nM) solution onto Penta-His biosensors for 300 s. The biosensor tips then were submerged in binding buffer (PBS/0.2% Tween 20) for a 60 s wash, followed by immersion in a solution containing 150 nM of monoclonal antibody for 180 s (association), followed by a subsequent immersion in binding buffer for 180 s (dissociation). The response for each RBD mutant protein was normalized to that of the wild-type RBD protein.
FRNT
Serial dilutions of monoclonal antibodies were incubated with 102 FFU of SARS-CoV-2 for 1 h at 37 °C. The antibody–virus complexes were added to Vero E6 cell-culture monolayers in 96-well plates for 1 h at 37 °C. Cells were then overlaid with 1% (w/v) methylcellulose in minimum essential medium (MEM) supplemented to contain 2% heat-inactivated FBS. Plates were fixed 30 h later by removing overlays and fixed with 4% paraformaldehyde (PFA) in PBS for 20 min at room temperature. The plates were incubated sequentially with 1 μg ml−1 of rCR3022 anti-S antibody12 and HRP-conjugated goat anti-human IgG (Sigma-Aldrich, A6029) in PBS supplemented with 0.1% (w/v) saponin (Sigma) and 0.1% BSA. SARS-CoV-2-infected cell foci were visualized using TrueBlue peroxidase substrate (KPL) and quantitated on an ImmunoSpot 5.0.37 Macro Analyzer (Cellular Technologies). Data were processed using Prism v.8.0 (GraphPad). IC50 values were determined by nonlinear regression analysis using Prism software.
Generation of S protein pseudotyped lentivirus
Suspension cultures of 293 cells were seeded and transfected with a third-generation HIV-based lentiviral vector expressing luciferase along with packaging plasmids encoding for the following: SARS-CoV-2 spike protein with a C-terminal 19 amino acid deletion, Rev, and Gag-pol. The medium was changed 16–20 h after transfection, and the supernatant containing virus was collected 24 h later. Cell debris was removed by low-speed centrifugation, and the supernatant was passed through a 0.45-μm filter unit. The pseudovirus was pelleted by ultracentrifugation and resuspended in PBS for a 100-fold concentrated stock.
Pseudovirus neutralization assay
Serial dilutions of monoclonal antibodies were prepared in a 384-well microtitre plate and pre-incubated with pseudovirus for 30 min at 37 °C, to which 293 cells that stably express human ACE2 were added. The plate was returned to the 37 °C incubator, and then 48 h later luciferase activity was measured on an EnVision 2105 Multimode Plate Reader (Perkin Elmer) using the Bright-Glo Luciferase Assay System (Promega), according to the manufacturer’s recommendations. Per cent inhibition was calculated relative to pseudovirus-only control. IC50 values were determined by nonlinear regression using Prism v.8.1.0 (GraphPad). The average IC50 value for each antibody was determined from a minimum of three independent experiments.
Measurement of synergistic neutralization by a combination of antibodies
Synergy was defined as higher neutralizing activity mediated by a cocktail of two monoclonal antibodies when compared to that mediated by individual monoclonal antibodies at the same total concentration of antibodies in vitro. To assess whether two monoclonal antibodies synergize in a cocktail to neutralize SARS-CoV-2, we used a previously reported approach to quantify synergy11. To evaluate the significance of the beneficial effect from combining monoclonal antibodies, the observed combination responses (dose–response matrix) were compared with the expected responses calculated by means of synergy-scoring models11. Virus neutralization was measured in a conventional FRNT assay using wild-type SARS-CoV-2 and Vero E6 cell-culture monolayers. The individual monoclonal antibodies COV2-2196 and COV2-2130 were mixed at different concentrations to assess the neutralizing activity of different ratios of monoclonal antibodies in the cocktail. Specifically, each of seven two-fold dilutions of COV2-2130 (starting from 500 ng ml−1) was mixed with each of the nine two-fold dilutions of COV2-2196 (starting from 500 ng ml−1) in a total volume of 50 μl for each condition and then incubated with 50 μl of wild-type SARS-CoV-2 in cell culture medium (RPMI-1640 medium supplemented with 2% FBS) before applying to confluent Vero E6 cells grown in 96-well plates. The control values included those for determining the dose–response of the neutralizing activity measured separately for the individual monoclonal antibody COV2-2196 or COV2-2130, which were assessed at the same doses as in the cocktail. Each measurement was performed in duplicate. We next calculated the per cent virus neutralization for each condition and then calculated the synergy score value, which defines the interaction between these two monoclonal antibodies in the cocktail. A synergy score of less than −10 indicates antagonism, a score from −10 to 10 indicates an additive effect, and a score greater than 10 indicates a synergistic effect28.
Quantification of monoclonal antibodies
Quantification of purified monoclonal antibodies was performed by UV spectrophotometry using a NanoDrop spectrophotometer and accounting for the extinction coefficient of human IgG.
Competition-binding analysis through biolayer interferometry
Anti-mouse IgG Fc capture biosensors (FortéBio 18-5089) on an Octet HTX biolayer interferometry instrument (FortéBio) were soaked for 10 min in 1× kinetics buffer (Molecular Devices 18-1105), followed by a baseline signal measurement for 60 s. Recombinant SARS-CoV-2 RBD fused to mouse IgG1 (RBD–mFc, Sino Biological 40592-V05H) was immobilized onto the biosensor tips for 180 s. After a wash step in 1× kinetics buffer for 30 s, the reference antibody (5 μg ml−1) was incubated with the antigen-containing biosensor for 600 s. Reference antibodies included the SARS-CoV human monoclonal antibodies CR3022 and COV2-2196. After a wash step in 1× kinetics buffer for 30 s, the biosensor tips then were immersed into the second antibody (5 μg ml−1) for 300 s. The maximum binding of each antibody was normalized to a buffer-only control. Self-to-self blocking was subtracted. A comparison between the maximum signal of each antibody was used to determine the per cent binding of each antibody. A reduction in maximum signal to less than 33% of the un-competed signal was considered full competition of binding for the second antibody in the presence of the reference antibody. A reduction in maximum signal to between 33% and 67% of the un-competed signal was considered intermediate competition of binding for the second antibody in the presence of the reference antibody. A per cent binding of the maximum signal of more than 67% was considered absence of competition of binding for the second antibody in the presence of the reference antibody.
Human ACE2 inhibition analysis
Wells of 384-well microtitre plates were coated with 1 μg ml−1 purified recombinant SARS-CoV-2 S2Pecto protein at 4 °C overnight. Plates were blocked with 2% non-fat dry milk and 2% normal goat serum in DPBS-T for 1 h. For screening assays, purified monoclonal antibodies from microscale expression were diluted twofold in blocking buffer starting from 10 μg ml−1 in triplicate, added to the wells (20 μl per well) and incubated for 1 h at ambient temperature. Recombinant human ACE2 with a C-terminal Flag tag peptide was added to wells at 2 μg ml−1 in a 5 μl per well volume (final 0.4 μg ml−1 concentration of human ACE2) without washing of antibody and then incubated for 40 min at ambient temperature. Plates were washed and bound human ACE2 was detected using HRP-conjugated anti-Flag antibody (Sigma-Aldrich, cat. A8592, lot SLBV3799, 1:5,000 dilution) and TMB substrate. ACE2 binding without antibody served as a control. The signal obtained for binding of the human ACE2 in the presence of each dilution of tested antibody was expressed as a percentage of the human ACE2 binding without antibody after subtracting the background signal. For dose–response assays, serial dilutions of purified monoclonal antibodies were applied to the wells in triplicate, and monoclonal antibody binding was detected as detailed above. IC50 values for inhibition by monoclonal antibody of S2Pecto protein binding to human ACE2 was determined after log transformation of antibody concentration using sigmoidal dose–response nonlinear regression analysis (Prism v.8.0, GraphPad).
Human-ACE2-blocking assay using biolayer interferometry biosensor
Anti-mouse IgG biosensors on an Octet HTX biolayer interferometry instrument (FortéBio) were soaked for 10 min in 1× kinetics buffer, followed by a baseline signal measurement for 60 s. Recombinant SARS-CoV-2 RBD fused to mouse IgG1 (RBD–mFc, Sino Biological, 40592-V05H) was immobilized onto the biosensor tips for 180 s. After a wash step in 1× kinetics buffer for 30 s, the antibody (5 μg ml−1) was incubated with the antigen-coated biosensor for 600 s. After a wash step in 1× kinetics buffer for 30 s, the biosensor tips then were immersed into the human ACE2 receptor (20 μg ml−1) (Sigma-Aldrich, SAE0064) for 300 s. The maximum binding of human ACE2 was normalized to a buffer-only control. Per cent binding of human ACE2 in the presence of antibody was compared to human ACE2 maximum binding. A reduction in maximal signal to less than 30% was considered human-ACE2-blocking.
High-throughput competition-binding analysis
Wells of 384-well microtitre plates were coated with 1 μg ml−1 purified SARS-CoV-2 S2Pecto protein at 4 °C overnight. Plates were blocked with 2% BSA in DPBS-T for 1 h. Microscale purified unlabelled monoclonal antibodies were diluted tenfold in blocking buffer, added to the wells (20 μl per well) in quadruplicates and incubated for 1 h at ambient temperature. A biotinylated preparation of a recombinant monoclonal antibody based on the variable gene sequence of the previously described monoclonal antibody CR302212, as well as the newly identified monoclonal antibodies COV2-2130 and COV2-2196 that recognized distinct antigenic regions of the SARS-CoV-2 S protein, were added to each of four wells with the respective monoclonal antibody at 2.5 μg ml−1 in a volume of 5 μl per well (final concentration of biotinylated monoclonal antibody 0.5 μg ml−1) without washing of unlabelled antibody, and then incubated for 1 h at ambient temperature. Plates were washed and bound antibodies were detected using HRP-conjugated avidin (Sigma) and a TMB substrate. The signal obtained for binding of the biotin-labelled reference antibody in the presence of the unlabelled tested antibody was expressed as a percentage of the binding of the reference antibody alone after subtracting the background signal. Tested monoclonal antibodies were considered competing if their presence reduced the reference antibody binding to less than 41% of its maximal binding and non-competing if the signal was greater than 71%. A level of 40–70% was considered intermediate competition.
Plasma or serum antibody competition-binding assays
Wells of 384-well microtitre plates were coated with 1 μg ml−1 purified SARS-CoV-2 S2Pecto at 4 °C overnight. Plates were blocked with 2% BSA in DPBS-T for 1 h. Plasma or serum samples were diluted in blocking buffer twofold starting from 1:10 sample dilution, added to the wells (20 μl per well) in triplicate and incubated for 1 h at ambient temperature. For self-blocking controls, unlabelled monoclonal antibodies COV2-2196 or COV2-2130 were added at 10 μg ml−1 to separate wells coated with S2Pecto. Serum from a donor without an exposure history to SARS-CoV-2 was used as a negative control for monoclonal antibody binding inhibition. A biotinylated monoclonal antibody COV2-2196 or COV2-2130 was added to the respective wells at 2.5 μg ml−1 in a volume of 5 μl per well (final concentration of biotinylated monoclonal antibody 0.5 μg ml−1) without washing of unlabelled antibody, and then incubated for 30 min at ambient temperature. Binding of biotinylated monoclonal antibodies COV2-2196 or COV2-2130 alone to S2Pecto served as a control for maximum binding. Plates were washed and bound antibodies were detected using HRP-conjugated avidin (Sigma) and a TMB substrate. Inhibition of COV2-2196 or COV2-2130 binding in the presence of each dilution of tested plasma or serum was calculated as a percentage of the maximum COV2-2196 or COV2-2130 binding inhibition using values from COV2-2196 or COV2-2130 binding alone (maximum binding) and the corresponding self-blocking controls (maximum inhibition) after subtracting the background signal. For the human ACE2 inhibition assay by plasma or serum antibodies, plasma or serum samples were diluted and added to wells with S2Pecto as detailed above. Recombinant human ACE2 was added to wells at 2 μg ml−1in a volume of 5 μl per well (final concentration of human ACE2 0.4 μg ml−1) without washing of antibody, and then incubated for 40 min at ambient temperature. Plates were washed and bound human ACE2 was detected using HRP-conjugated anti-Flag antibody (Sigma) and a TMB substrate. Human ACE2 binding without antibody served as a control. The signal obtained for binding of the human ACE2 in the presence of each dilution of tested plasma or serum was expressed as a percentage of the ACE2 binding without antibody after subtracting the background signal.
Protection against wild-type SARS-CoV-2 in mice transduced with human ACE2
Animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (assurance number A3381–01). Viral inoculations were performed under anaesthesia, which was induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize animal suffering.
Wild-type, female BALB/c mice were purchased from The Jackson Laboratory (strain 000651). Mice were housed in groups of up to 5 mice per cage at 18–24 °C ambient temperatures and 40–60% humidity. Mice were fed a 20% protein diet (PicoLab 5053, Purina) and maintained on a 12-h light–dark cycle (06:00 to 18:00). Food and water were available ad libitum.
Mice (10–11 weeks old) were given a single intraperitoneal injection of 2 mg of anti-IFNAR1 monoclonal antibody (MAR1-5A355, Leinco) one day before intranasal administration of 2.5 × 108 PFU of AdV-hACE2. Five days after AdV transduction, mice were inoculated with 4 × 105 PFU of SARS-CoV-2 via the intranasal route. Anti-SARS-CoV-2 human monoclonal antibodies or isotype control monoclonal antibodies were administered 24 h before (prophylaxis) or 12 h after (therapy) SARS-CoV-2 inoculation. Weights were monitored on a daily basis, mice were euthanized at 2 or 7 dpi and tissues were collected.
Measurement of viral burden
For RT–qPCR, tissues were weighed and homogenized with zirconia beads in a MagNA Lyser instrument (Roche Life Science) in 1 ml of DMEM medium supplemented with 2% heat-inactivated FBS. Tissue homogenates were clarified by centrifugation at 10,000 rpm for 5 min and stored at −80 °C. RNA was extracted using a MagMax mirVana Total RNA isolation kit (Thermo Fisher Scientific) and a Kingfisher Flex 96-well extraction machine (Thermo Fisher Scientific). TaqMan primers were designed to target a conserved region of the N gene using SARS-CoV-2 (MN908947) sequence as a guide (L primer: ATGCTGCAATCGTGCTACAA; R primer: GACTGCCGCCTCTGCTC; probe: /56-FAM/TCAAGGAAC/ZEN/AACATTGCCAA/3IABkFQ/). To establish an RNA standard curve, we generated concatenated segments of the N gene in a gBlocks fragment (IDT) and cloned this into the PCR-II topo vector (Invitrogen). The vector was linearized, and in vitro T7-DNA-dependent RNA transcription was performed to generate materials for a quantitative standard curve.
For the plaque assay, homogenates were diluted serially tenfold and applied to Vero-furin cell monolayers in 12-well plates. Plates were incubated at 37 °C for 1 h with rocking every 15 min. Cells were then overlaid with 1% (w/v) methylcellulose in MEM supplemented with 2% FBS. Plates were collected 72 h later by removing overlays and fixed with 4% PFA in PBS for 20 min at ambient temperature. After removing the 4% PFA, plaques were visualized by adding 1 ml per well 0.05% crystal violet in 20% methanol for 20 min at ambient temperature. Excess crystal violet was washed away with PBS, and plaques were counted.
Cytokine and chemokine mRNA measurements
RNA was isolated from lung homogenates at 7 dpi as described above. cDNA was synthesized from DNase-treated RNA using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) with the addition of RNase inhibitor, following the manufacturer’s protocol. Cytokine and chemokine expression was determined using TaqMan Fast Universal PCR master mix (Thermo Fisher Scientific) with commercial primers and probe sets specific for Ifng (IDT: Mm.PT.58.41769240), Il6 (Mm.PT.58.10005566), Cxcl10 (Mm.PT.58.43575827) and Ccl2 (Mm.PT.58.42151692) and results were normalized to Gapdh (Mm.PT.39a.1) levels. Fold change was determined using the 2−ΔΔCt method comparing anti-SARS-CoV-2-specific or isotype-control monoclonal-antibody-treated mice to naive controls.
Histology
Mice were euthanized, and tissues were collected before lung inflation and fixation. The left lung lobe was tied off at the left main bronchus and collected for viral RNA analysis. The right lung lobe was inflated with around 1.2 ml of 10% neutral buffered formalin using a 3-ml syringe and catheter inserted into the trachea. For fixation after infection, inflated lungs were kept in a 40-ml suspension of neutral buffered formalin for 7 days before further processing. Tissues were embedded in paraffin, and sections were stained with haematoxylin and eosin. Tissue sections were visualized using a Leica DM6B microscope equipped with a Leica DFC7000T camera. The sections were scored by an immunopathology expert blinded to the compositions of the groups.
Viral challenge studies using MA-SARS-CoV-2 and wild-type mice
Animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the UNC Chapel Hill School of Medicine (NIH/PHS animal welfare assurance number D16-00256 (A3410-01)). Virus inoculations were performed under anaesthesia that was induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize animal suffering.
Protection against MA-SARS-CoV-2 in wild-type mice
BALB/c mice (12 months old) from Envigo were used in experiments. Mice were housed in groups of up to 5 mice per cage at 18–24 °C ambient temperatures and 40–60% humidity. Mice were fed a 20% protein diet (PicoLab 5053, Purina) and maintained on a 12-h light–dark cycle (08:00 to 20:00). Food and water were available ad libitum. Mice were acclimated in the BSL3 for at least 72 h before start of experiments. At 6 h before infection, mice were treated with 200 μg of human monoclonal antibodies via intraperitoneal injection. The next day, mice were anaesthetized with a mixture of ketamine and xylazine and intranasally inoculated with 105 PFU of MA-SARS-CoV-2 diluted in PBS. Daily weight loss was measured, and at 2 dpi mice were euthanized by isoflurane overdose before tissue collection. For the post-exposure therapy study, mice were inoculated intranasally with 105 PFU of MA-SARS-CoV-2 and 12 h later given the indicated antibody treatments by intraperitoneal injection. The lungs were collected at 2 dpi.
Plaque assay of lung tissue homogenates
The lower lobe of the right lung was homogenized in 1 ml PBS using a MagnaLyser (Roche). Serial dilutions of virus were titrated on Vero E6 cell-culture monolayers, and virus plaques were visualized by neutral red staining two days after inoculation. The limit of detection for the assay is 100 PFU per lung.
NHP challenge study
The NHP research studies adhered to principles stated in the eighth edition of the Guide for the Care and Use of Laboratory Animals. The facility in which this research was conducted (Bioqual, Rockville) is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) and approved by the Office of Laboratory Animal Welfare (NIH/PHS assurance number D16-00052). NHP studies were conducted in compliance with all relevant local, state and federal regulations and were approved by the Institutional Animal Care and Use Committee (IACUC) at Bioqual.
Twelve healthy adult rhesus macaques (Macaca mulatta) of Indian origin (5–15 kg body weight) were studied. Rhesus macaques were 5–7 years old and mixed male and female. Macaques were allocated randomly to two anti-SARS-CoV-2 monoclonal antibody treatment groups (n = 4 per group) and one control (isotype-treated) group (n = 4 per group). Macaques received one 50 mg kg−1 dose of COV2-2196, COV2-2381 or an isotype control monoclonal antibody intravenously on day −3 and were challenged three days later with 1.1 × 104 PFU SARS-CoV-2, administered as 1 ml via the intranasal route and 1 ml via the intratracheal route. After challenge, viral RNA was assessed by RT–qPCR in bronchoalveolar lavage and nasal swabs at multiple time points as described previously34,35. All macaques were given physical examinations. In addition, all macaques were monitored daily with an internal scoring protocol approved by the IACUC. These studies were not blinded.
Detection of circulating human monoclonal antibodies in NHP serum
ELISA plates were coated overnight at 4 °C with 1 μg ml−1 of goat anti-human IgG (H+L) secondary antibody (monkey pre-adsorbed) (Novus Biologicals, NB7487) and then blocked for 2 h. The serum samples were assayed at threefold dilutions starting at a 1:3 dilution in Blocker Casein in PBS (Thermo Fisher Scientific) diluent. Samples were incubated for 1 h at ambient temperature and then removed, and plates were washed. Wells then were incubated for 1 h with HRP-conjugated goat anti-human IgG (monkey pre-adsorbed) (Southern Biotech, 2049-05) at a 1:4,000 dilution. Wells were washed and then incubated with SureBlue Reserve TMB Microwell Peroxidase Substrate (Seracare) (100 μl per well) for 3 min followed by TMB Stop Solution (Seracare) to stop the reaction (100 μl per well). Microplates were read at 450 nm. The concentrations of the human monoclonal antibodies were interpolated from the linear range of purified human IgG (Sigma) standard curves using Prism v.8.0 (GraphPad).
Quantification and statistical analysis
Mean ± s.e.m. or mean ± s.d. were determined for continuous variables as noted. Technical and biological replicates are described in the figure legends. In the mouse studies, the comparison of weight-change curves was performed using a repeated measurements two-way ANOVA with Tukey’s post hoc test using Prism v.8.0 (GraphPad). Viral burden and gene-expression measurements were compared using a Kruskal–Wallis ANOVA with Dunn’s post hoc test or a two-sided Mann–Whitney U-test using Prism v.8.0 (GraphPad). The analyses of synergy score and the dose–response matrix were performed using a web application, SynergyFinder28.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The electron microscopy maps have been deposited at the Electron Microscopy Data Bank (EMDB) with accession codes EMD-21974, EMD-21975, EMD-21976 and EMD-21977 (Supplementary Table 2). The electron microscopy map EMD-21965 is publicly available. The accession numbers for the cryo-electron-microscopy and crystal structures used for structural analysis, including structures of the closed conformation of SARS-CoV-2 S (PDB: 6VXX), the open conformation of SARS-CoV-2 (PDB: 6VYB), the Fab used for docking (PDB: 12E8) and the SARS-CoV-2 RBD–human ACE2 complex (PDB: 6M0J) are publicly available. Sequences of the monoclonal antibodies characterized here are available from GenBank under the following accession numbers: MT665032–MT665070, MT665419–MT665457, MT763531 and MT763532. Materials used in this study will be made available but may require execution of a Materials Transfer Agreement. Source data are provided with this paper.
References
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Tse, L. V., Meganck, R. M., Graham, R. L. & Baric, R. S. The current and future state of vaccines, antivirals and gene therapies against emerging coronaviruses. Front. Microbiol. 11, 658 (2020).
Siracusano, G., Pastori, C. & Lopalco, L. Humoral immune responses in COVID-19 patients: a window on the state of the art. Front. Immunol. 11, 1049 (2020).
Zost, S. J. et al. Rapid isolation and profiling of a diverse panel of human monoclonal antibodies targeting the SARS-CoV-2 spike protein. Nat. Med. https://doi.org/10.1038/s41591-020-0998-x (2020).
Pillay, T. S. Gene of the month: the 2019-nCoV/SARS-CoV-2 novel coronavirus spike protein. J. Clin. Pathol. 73, 366–369 (2020).
Wan, Y., Shang, J., Graham, R., Baric, R. S. & Li, F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 94, e00127-20 (2020).
Hoffmann, M., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003).
Sui, J. et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl Acad. Sci. USA 101, 2536–2541 (2004).
ter Meulen, J. et al. Human monoclonal antibody as prophylaxis for SARS coronavirus infection in ferrets. Lancet 363, 2139–2141 (2004).
ter Meulen, J. et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 3, e237 (2006).
Zhu, Z. et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl Acad. Sci. USA 104, 12123–12128 (2007).
Rockx, B. et al. Structural basis for potent cross-neutralizing human monoclonal antibody protection against lethal human and zoonotic severe acute respiratory syndrome coronavirus challenge. J. Virol. 82, 3220–3235 (2008).
Chen, Z. et al. Human neutralizing monoclonal antibody inhibition of Middle East respiratory syndrome coronavirus replication in the common marmoset. J. Infect. Dis. 215, 1807–1815 (2017).
Choi, J. H. et al. Characterization of a human monoclonal antibody generated from a B-cell specific for a prefusion-stabilized spike protein of Middle East respiratory syndrome coronavirus. PLoS One 15, e0232757 (2020).
Niu, P. et al. Ultrapotent human neutralizing antibody repertoires against Middle East respiratory syndrome coronavirus from a recovered patient. J. Infect. Dis. 218, 1249–1260 (2018).
Wang, L. et al. Importance of neutralizing monoclonal antibodies targeting multiple antigenic sites on the Middle East respiratory syndrome coronavirus spike glycoprotein to avoid neutralization escape. J. Virol. 92, e02002-17 (2018).
Wang, N., et al. Structural definition of a neutralization-sensitive epitope on the MERS-CoV S1-NTD. Cell Rep. 28, 3395–3405 (2019).
Zhang, S. et al. Structural definition of a unique neutralization epitope on the receptor-binding domain of MERS-CoV spike glycoprotein. Cell Rep. 24, 441–452 (2018).
Corti, D. et al. Prophylactic and postexposure efficacy of a potent human monoclonal antibody against MERS coronavirus. Proc. Natl Acad. Sci. USA 112, 10473–10478 (2015).
Jiang, L. et al. Potent neutralization of MERS-CoV by human neutralizing monoclonal antibodies to the viral spike glycoprotein. Sci. Transl. Med. 6, 234ra59 (2014).
Tang, X. C. et al. Identification of human neutralizing antibodies against MERS-CoV and their role in virus adaptive evolution. Proc. Natl Acad. Sci. USA 111, E2018–E2026 (2014).
Ying, T. et al. Exceptionally potent neutralization of Middle East respiratory syndrome coronavirus by human monoclonal antibodies. J. Virol. 88, 7796–7805 (2014).
Jiang, S., Hillyer, C. & Du, L. Neutralizing antibodies against SARS-CoV-2 and other human coronaviruses. Trends Immunol. 41, 355–359 (2020).
Valk, S. J. et al. Convalescent plasma or hyperimmune immunoglobulin for people with COVID-19: a rapid review. Cochrane Database Syst. Rev. 5, CD013600 (2020).
Yuan, M. et al. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 368, 630–633 (2020).
Ianevski, A., Giri, A. K. & Aittokallio, T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res. 48, W488–W493 (2020).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Walls, A.C., et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292 (2020).
Hassan, A. O. et al. A SARS-CoV-2 infection model in mice demonstrates protection by neutralizing antibodies. Cell https://doi.org/10.1016/j.cell.2020.06.011 (2020).
Dinnon, K. H. et al. A mouse-adapted SARS-CoV-2 model for the evaluation of COVID-19 medical countermeasures. Preprint at bioRxiv https://doi.org/10.1101/2020.05.06.081497 (2020).
Chandrashekar, A. et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science https://doi.org/10.1126/science.abc4776 (2020).
Yu, J. et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science https://doi.org/10.1126/science.abc6284 (2020).
Robbiani, D. F. et al. Convergent antibody responses to SARS-CoV-2 infection in convalescent individuals. Nature https://doi.org/10.1038/s41586-020-2456-9 (2020).
Brouwer, P. J. M. et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science https://doi.org/10.1126/science.abc5902 (2020).
Cao, Y. et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 182, 73–84 (2020).
Ju, B. et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature https://doi.org/10.1038/s41586-020-2380-z (2020).
Rogers, T. F. et al. Rapid isolation of potent SARS-CoV-2 neutralizing antibodies and protection in a small animal model. Science https://doi.org/10.1126/science.abc7520 (2020).
Shi, R. et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 584, 120–124 (2020).
Wec, A. Z. et al. Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science https://doi.org/10.1126/science.abc7424 (2020).
Wu, Y. et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 368, 1274–1278 (2020).
Hansen, J. et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science https://doi.org/10.1126/science.abd0827 (2020).
Baum, A. et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science https://doi.org/10.1126/science.abd0831 (2020).
Laha, S. et al. Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect. Genet. Evol. 85, 104445 (2020).
Mukherjee, S. et al. Enhancing dengue virus maturation using a stable furin over-expressing cell line. Virology 497, 33–40 (2016).
Ohi, M., Li, Y., Cheng, Y. & Walz, T. Negative staining and image classification – powerful tools in modern electron microscopy. Biol. Proced. Online 6, 23–34 (2004).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Bepler, T., Noble, A. J. & Berger, B. Topaz-Denoise: general deep denoising models for cryoEM. Preprint at bioRxiv https://doi.org/10.1101/838920 (2019).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Chng, J. et al. Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. MAbs 7, 403–412 (2015).
Sheehan, K. C. et al. Blocking monoclonal antibodies specific for mouse IFN-α/β receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J. Interferon Cytokine Res. 26, 804–819 (2006).
Acknowledgements
We thank A. Jones and the staff of the Vanderbilt Technologies for Advanced Genomics (VANTAGE) core laboratory for expedited sequencing; R. Trosseth for assistance with data management and analysis; R. Bombardi and C. Soto of VUMC for technical consultation on genomics approaches; A. Kim, A. Bailey, L. VanBlargan and J. Earnest of WUSTL for experimental assistance and key reagents; K. M. Tuffy, S. Diallo, P. M. McTamney and L. Clarke of AstraZeneca for the generation of protein and pseudovirus reagents and related data; and H. Andersen, M. G. Lewis, R. Nityanandam, M. Kirilova and K. Verrington for research assistance with the NHP studies. This study was supported by Defense Advanced Research Projects Agency (DARPA) grants HR0011-18-2-0001 and HR00 11-18-3-0001; NIH contracts 75N93019C00074 and 75N93019C00062; NIH grants U01 AI150739, R01 AI130591 and R35 HL145242; the Dolly Parton COVID-19 Research Fund at Vanderbilt; and NIH grant S10 RR028106 for the Next Generation Nucleic Acid Sequencer, housed in VANTAGE and the Vanderbilt Institute for Clinical and Translational Research with grant support from UL1TR002243 from NCATS/NIH. S.J.Z. was supported by NIH T32 AI095202; J.B.C. was supported by a Helen Hay Whitney Foundation postdoctoral fellowship; B.T.M. was supported by NIH F32 AI138392; D.R.M. was supported by NIH T32 AI007151 and a Burroughs Wellcome Fund Postdoctoral Enrichment Program Award; L.E.W. was supported by NIH F31 AI145189; E.C.C. was supported by NIH T32 AI138932; and J.E.C. is the recipient of the 2019 Future Insight Prize from Merck KGaA, which supported this research with a research grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the US government or the other sponsors.
Author information
Authors and Affiliations
Contributions
S.J.Z., P.G., R.H.C., L.B.T., M.S.D. and J.E.C. conceived the project; J.E.C. and M.S.D. obtained funding; S.J.Z., P.G., J.B.C., E.B., R.E.C., J.P.N., A.S., J.X.R., A.T., R.S.N., R.E.S., N.S., D.R.M., L.E.W., A.O.H., N.M.K., E.S.W., J.M.F., S.S., B.K.M., A.C., N.B.M., J.J.S., K.R., Y.-M.L., S.P.K., M.J.H., L.E.G. and L.B.T. performed laboratory experiments; E.C.C., T.J., S.D., L.M. and B.T.M. performed computational work; J.M., N.L.K., D.H.B., R.S.B., L.B.T., M.S.D., R.H.C. and J.E.C. supervised the research; and S.J.Z., P.G., R.H.C. and J.E.C. wrote the first draft of the paper. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
R.S.B. has served as a consultant for Takeda and Sanofi Pasteur on issues related to vaccines. M.S.D. is a consultant for Inbios, Vir Biotechnology, NGM Biopharmaceuticals and Eli Lilly; is on the Scientific Advisory Board of Moderna; is a past recipient of an unrelated research grant from Moderna; and is a current recipient of an unrelated research grant from Emergent BioSolutions. J.E.C. has served as a consultant for Sanofi; is on the Scientific Advisory Boards of CompuVax and Meissa Vaccines; is a recipient of previous unrelated research grants from Moderna and Sanofi; and is a founder of IDBiologics. Vanderbilt University has applied for patents concerning SARS-CoV-2 antibodies that are related to this work. AstraZeneca has filed patents for materials and findings that are related to this work. J.J.S., K.R., Y.-M.L. and N.L.K. are employees of AstraZeneca and currently hold AstraZeneca stock or stock options. M.J.H. is a member of a data safety monitoring board for AstraZeneca and a founder of NuPeak Therapeutics. All other authors declare no competing interests.
Additional information
Peer review information Nature thanks Linda Saif and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 SARS-CoV-2 neutralization curves for monoclonal antibody panel.
Neutralization of wild-type SARS-CoV-2 by human monoclonal antibodies. Data are mean ± s.d. of technical duplicates, and represent one of two or more independent experiments.
Extended Data Fig. 2 Inhibition curves for monoclonal antibody inhibition of S2Pecto binding to human ACE2.
Blocking of human ACE2 binding to S2Pecto by anti-SARS-CoV-2 neutralizing human monoclonal antibodies. Data are mean ± s.d. of triplicates of one experiment. Antibodies rCR3022 and r2D22 served as controls.
Extended Data Fig. 3 ELISA binding of anti-SARS-CoV-2 neutralizing human monoclonal antibodies to trimeric SRBD, S2Pecto or SARS-CoV S2Pecto antigen.
Data are mean ± s.d. of triplicates, and are representative of two experiments. Antibodies rCR3022 and r2D22 served as controls.
Extended Data Fig. 4 Competition-binding analysis of serum or plasma antibodies with human ACE2 and monoclonal antibodies.
a, Inhibition of human ACE2 binding to S2Pecto by serum or plasma of four SARS-CoV-2 immune individuals or one non-immune control individual in an ELISA using SARS-CoV-2 S2Pecto. Monoclonal antibodies were isolated from individuals 3 and 4 as described previously5. Data are mean ± s.d. of triplicates of one experiment. Dotted line indicates full inhibition (100%) of human ACE2 by 500 ng ml−1 of monoclonal antibody COV2-2196 or COV2-2130 that were used as controls for full human ACE2 inhibition. b, Inhibition of monoclonal antibody COV2-2130 (left) or COV2-2196 (right) binding to S2Pecto by serum or plasma of four SARS-CoV-2 immune individual or one non-immune control individual in an ELISA using SARS-CoV-2 S2Pecto. Data are mean ± s.d. of triplicates, and are representative of two experiments. Dotted line indicates the percentage of self-competition of monoclonal antibodies COV2-2196 and-2130 on the SARS-CoV-2 S2Pecto antigen.
Extended Data Fig. 5 Mapping of critical contact residues for monoclonal antibodies by alanine and arginine mutagenesis and biolayer interferometry.
a, Bar graphs show response values for monoclonal antibody binding to wild-type or mutant SRBD constructs normalized to the wild type. Asterisks denote residues where increased dissociation of monoclonal antibody was observed, probably indicating that the residue is proximal to the monoclonal antibody epitope. Full response curves for monoclonal antibody association and dissociation with wild-type or mutant SRBD constructs are also shown. b, Structure of the RBD, highlighting the critical contact residues for several monoclonal antibodies and their location on the structure.
Extended Data Fig. 6 Sequence features of the human monoclonal antibodies used in animal studies and monoclonal antibody pharmacokinetics following their administration to NHPs.
a, Sequence features of human monoclonal antibodies tested in animal models. Inferred variable genes are indicated and CDR3 amino acids are shown for heavy and light chains. b, Macaques received one 50 mg kg−1 dose of COV2-2196, COV2-2381 or an isotype control monoclonal antibody (n = 4 macaques per group) intravenously on day −3 and then were challenged intranasally and intratracheally with SARS-CoV-2 at day 0. The concentration of human monoclonal antibodies was determined at indicated time points. Each curve shows an individual macaque. Data represent a single experiment.
Supplementary information
Rights and permissions
About this article

Cite this article
Zost, S.J., Gilchuk, P., Case, J.B. et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 584, 443–449 (2020). https://doi.org/10.1038/s41586-020-2548-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2548-6
This article is cited by
-
Single-domain antibodies against SARS-CoV-2 RBD from a two-stage phage screening of universal and focused synthetic libraries
BMC Infectious Diseases (2024)
-
The SARS-CoV-2 neutralizing antibody response to SD1 and its evasion by BA.2.86
Nature Communications (2024)
-
SARS-CoV-2 immunity in animal models
Cellular & Molecular Immunology (2024)
-
Safety, Efficacy and Pharmacokinetics of AZD7442 (Tixagevimab/Cilgavimab) for Treatment of Mild-to-Moderate COVID-19: 15-Month Final Analysis of the TACKLE Trial
Infectious Diseases and Therapy (2024)
-
Emerging variants develop total escape from potent monoclonal antibodies induced by BA.4/5 infection
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
