
Abstract
Antibody-dependent enhancement (ADE) of disease is a general concern for the development of vaccines and antibody therapies because the mechanisms that underlie antibody protection against any virus have a theoretical potential to amplify the infection or trigger harmful immunopathology. This possibility requires careful consideration at this critical point in the pandemic of coronavirus disease 2019 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Here we review observations relevant to the risks of ADE of disease, and their potential implications for SARS-CoV-2 infection. At present, there are no known clinical findings, immunological assays or biomarkers that can differentiate any severe viral infection from immune-enhanced disease, whether by measuring antibodies, T cells or intrinsic host responses. In vitro systems and animal models do not predict the risk of ADE of disease, in part because protective and potentially detrimental antibody-mediated mechanisms are the same and designing animal models depends on understanding how antiviral host responses may become harmful in humans. The implications of our lack of knowledge are twofold. First, comprehensive studies are urgently needed to define clinical correlates of protective immunity against SARS-CoV-2. Second, because ADE of disease cannot be reliably predicted after either vaccination or treatment with antibodies—regardless of what virus is the causative agent—it will be essential to depend on careful analysis of safety in humans as immune interventions for COVID-19 move forward.
Similar content being viewed by others

Main
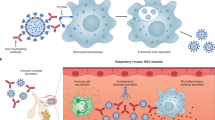
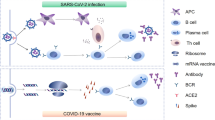
The benefit of passive antibodies in ameliorating infectious diseases was recognized during the 1918 influenza pandemic1. Since then, hyperimmune globulin has been widely used as pre- and post-exposure prophylaxis for hepatitis A, hepatitis B, chickenpox, rabies and other indications for decades without evidence of ADE of disease2 (see Box 1 for definition of terms). The detection of antibodies has also been a reliable marker of the effectiveness of the many licensed human vaccines3. The antiviral activity of antibodies is now known to be mediated by the inhibition of entry of infectious viral particles into host cells (neutralization) and by the effector functions of antibodies as they recruit other components of the immune response. Neutralizing antibodies are directed against viral entry proteins that bind to cell surface receptors, either by targeting viral proteins that are required for fusion or by inhibiting fusion after attachment4,5,6 (Fig. 1). Antibodies can cross-neutralize related viruses when the entry proteins of the viruses share epitopes—the part of a protein to which the antibody attaches. Antibodies also eliminate viruses through effector functions triggered by simultaneous binding of the antigen-binding fragment (Fab) regions of immunoglobulin G (IgG) to viral proteins on the surfaces of viruses or infected cells, and of the fragment crystallizable (Fc) portion of the antibody to Fc gamma receptors (FcγRs) that are expressed by immune cells7,8 (Fig. 2). Antibodies that mediate FcγR- and complement-dependent effector functions may or may not have neutralizing activity, can recognize other viral proteins that are not involved in host-cell entry and can be protective in vivo independent of any Fab-mediated viral inhibition9,10. Recent advances in FcR biology have identified four activating FcγRs (FcγRI, FcγRIIa, FcγRIIc and FcγRIIIa) and one inhibitory FcγR (FcγRIIb) that have various Fc ligand specificities and cell-signalling motifs10. The neonatal Fc receptor (FcRn) has been described to support antibody recycling and B and T cell immunity through dendritic cell endocytosis of immune complexes11,12. Natural killer cells recognize IgG–viral protein complexes on infected cells via FcγRs to mediate antibody-dependent cytotoxicity, and myeloid cells use these interactions to clear opsonized virions and virus-infected cells by antibody-dependent cellular phagocytosis (Fig. 2). The complement pathway is also activated by Fc binding to the complement component C1q, resulting in the opsonization of viruses or infected cells and the recruitment of myeloid cells. Antibody effector functions also contribute to antiviral T-cell-mediated immunity in vivo13. Notably, new knowledge about Fc effector functions has led to improved passive-antibody therapies through Fc modifications that reduce or enhance interactions with FcγRs, lengthen the half-life of the antibody and potentially enhance antigen presentation to T cells, providing what is termed a vaccinal effect8,11,14.

Mechanisms of antibody-mediated neutralization of viruses by functions of the IgG Fab fragment that block binding to cell surface receptors and inhibit infectivity by aggregating viral particles and inhibiting steps in the viral life cycle, such as fusion. Binding of antibodies with certain properties may enable changes in the viral entry protein that accelerate fusion.
Although their importance for protection is indisputable, the concern about ADE of disease arises from the possibility that antibodies present at the time of infection may increase the severity of an illness. The enhancement of disease by antibody-dependent mechanisms has been described clinically in children given formalin-inactivated respiratory syncytial virus (RSV) or measles vaccines in the 1960s, and in dengue haemorrhagic fever due to secondary infection with a heterologous dengue serotype15,16,17,18,19,20,21. For example, antibodies may enable viral entry into FcγR-bearing cells, bypassing specific receptor-mediated entry; this is typically followed by degradation of the virus, but could amplify infection if progeny virions can be produced. Although cytokine release triggered by interactions between the virus, antibody and FcγR is also highly beneficial—owing to direct antiviral effects and the recruitment of immune cells—tissue damage initiated by viral infection may be exacerbated22.
While recognizing that other mechanisms of immune enhancement may occur, the purpose of this Perspective is to review clinical experiences, in vitro analyses and animal models relevant to understanding the potential risks of antibody-dependent mechanisms and their implications for the development of the vaccines and antibodies that will be essential to stop the COVID-19 pandemic. Our objective is to evaluate the hypothesis that antibody-mediated enhancement is a consequence of low-affinity antibodies that bind to viral entry proteins but have limited or no neutralizing activity; antibodies that were elicited by infection with or vaccination against a closely related serotype, termed ‘cross-reactive’ antibodies; or suboptimal titres of otherwise potently neutralizing antibodies. We assess whether there are experimental approaches that are capable of reliably predicting ADE of disease in humans and conclude that this is not the case.
Principles for assessing potential ADE of disease
The use of ADE to denote enhanced severity of disease must be rigorously differentiated from ADE of infection—that is, from the binding, uptake and replication of the virus, cytokine release or other activities of antibodies detected in vitro. The first principle is that an antibody-dependent effect in vitro does not represent or predict ADE of disease without proof of a role for the antibody in the pathogenesis of a more severe clinical outcome. A second principle is that animal models for the evaluation of human polyclonal antibodies or monoclonal antibodies (mAbs) should be judged with caution because FcRs that are engaged by IgGs are species-specific23,24, as is complement activation. Antibodies can have very different properties in animals that are not predictive of those in the human host, because the effector functions of antibodies are altered by species-specific interactions between the antibody and immune cells. Animals may also develop antibodies against a therapeutic antibody that limit its effectiveness, or cause immunopathology. In addition, the pathogenesis of a model virus strain in animals does not fully reflect human infection because most viruses are highly species-specific. These differences may falsely support either protective or immunopathological effects of vaccines and antibodies. A third principle is that the nature of the antibody response depends on the form of the viral protein that is recognized by the immune system, thus determining what epitopes are presented. Protective and non-protective antibodies can be elicited to different forms of the same protein. A fourth principle is that mechanisms of pathogenesis in the human host differ substantially among viruses, or even between strains of a particular virus. Therefore, findings regarding the effects of passive antibodies or vaccine-induced immunity on outcomes cannot be extrapolated with confidence from one viral pathogen to another.
Observations about RSV, influenza and dengue
As background for considering the risks of ADE of disease caused by SARS-CoV-2, it is important to closely examine clinical circumstances relevant to the hypothesis that antibodies predispose to ADE of disease by amplifying infection or through damaging inflammatory responses. We focus on the clinical experiences with RSV, influenza and dengue to demonstrate the complexities of predicting from in vitro assays or animal models whether passively transferred or vaccine-induced antibodies will cause ADE of disease, and of differentiating ADE from a severe illness that is unrelated to pre-existing antibodies.
RSV
In a study of RSV in children under the age of 2 years, there were more cases requiring hospitalization for RSV-related bronchiolitis or pneumonia—especially in those aged between 6 and 11 months—in children who were immunized with a formalin-inactivated (FI)-RSV vaccine (10/101) than in children who were not immunized with FI-RSV (control cases; 2/173)25. This was also observed in a second study (18/23 hospitalizations of immunized children, with two deaths, compared with 1/21 control cases)16 and in two smaller studies17,26. This condition has been termed vaccine-associated enhanced respiratory disease. Later studies showed that the ratio of fusion protein (F) binding antibodies to neutralizing antibodies was higher in the sera of 36 vaccinated compared to 24 naturally infected children, suggesting that non-neutralizing antibodies to an abnormal F-protein conformation may have been a predisposing factor27. Complement activation, detected by the presence of C4d in the lungs of the two fatal cases, suggested that antibody–F protein immune complexes led to more severe disease28. However, C4d deposition can result from the lectin-binding pathway as well as from the classical pathway, and C4 can be produced by epithelial cells and activated by tissue proteases29. Whether harmful RSV-specific T cells were induced was not determined: although lymphocyte transformation frequencies were higher, this early method did not differentiate antigen-specific responses from secondary cytokine stimulation or from CD4 and CD8 T cell responses, although CD4 T cell proliferation is more likely30. Importantly, the FI-RSV clinical experience did not establish that vaccine-enhanced disease was antibody-dependent31. Subsequently, in animal studies, the production of low-avidity antibodies due to insufficient Toll-like-receptor signalling and lack of antibody maturation, and the formation of immune complexes have been implicated. However, a definitive antibody-mediated mechanism of enhancement has not been documented32, and models have also identified Th2-skewing of the T cell response and lung eosinophilia with challenge after FI-RSV, raising the possibility that T cells contribute to vaccine-induced enhancement of RSV disease31,33.
Experience with RSV also includes more than 20 years of successful prophylaxis of high-risk infants with palivizumab, a mAb directed against pre- and post-fusion F protein34. Importantly, this experience challenges a role for low neutralizing-antibody titres in the ADE of lung disease, because RSV morbidity does not increase as titres decrease. Further, if suboptimal neutralization were a factor, the failure of suptavumab—caused by F protein drift in RSV B strains—would be associated with ADE of disease; however, infections in such cases were not more severe35. Clinical trials of an RSV mAb that has an extended half-life have shown a reduction in hospitalizations of around 80%, again supporting the concept that such treatments provide protection without a secondary risk from declining titres36. mAbs against RSV have been consistently safe, even as the neutralizing capacity diminishes after administration.
Influenza
Influenza is instructive when considering the hypothesis that cross-reactive antibodies predispose to ADE of disease, because almost all humans contain antibodies that are not fully protective against antigenically drifted strains that emerge year after year. Instead, pre-existing immunity typically provides some protection against a second viral strain of the same subtype. Antibodies against neuraminidase and against the stem or head regions of haemagglutinin also correlate with protection37. When an H1N1 strain with a haemagglutinin shift emerged in the 2009 H1N1 pandemic, some epidemiological studies linked a greater incidence of medically treated illness to previous vaccination against influenza, whereas others did not38,39,40,41. One report correlated cross-reactive, low-avidity and poorly neutralizing antibodies with risk in middle-aged people—the demographic with a higher prevalence of severe 2009 H1N142. Immunopathology and C4d were reported in the lungs of six fatal cases in this age group, indicating that antibody-dependent complement activation through immune-complex formation may have been a contributing factor. However, as noted above, other mechanisms lead to C4d deposition, and lung T lymphocytosis attributed to T cell epitopes shared by 2009 H1N1 and earlier H1N1 strains was also observed, raising the possibility that T cells played a part. Another study correlated pre-existing antibodies that mediated infected cell lysis by complement activation with protection against H1N1 in children43. In a porcine model, enhanced pulmonary disease was observed after vaccination with an inactivated influenza H1N2 strain followed by heterologous H1N1 challenge44. The animals had non-neutralizing antibodies that bound haemagglutinin in the stem region, but did not block the binding of haemagglutinin to its cell receptor and accelerated fusion in vitro by a Fab-dependent mechanism (Fig. 1). Lung pathology was also observed in mice treated with a mAb that induced a conformational change in haemagglutinin that facilitated fusion45. Such a mechanism was postulated to have potential clinical relevance when the infecting influenza virus has undergone antigenic shift and the infection boosts non-neutralizing haemagglutinin-stem-binding antibodies without a neutralizing antibody response. The likelihood of these circumstances occurring is unclear. Further, human influenza vaccines are not known to elicit immunodominant antibodies with this property. Importantly, as noted above, stem antibodies correlate both with resistance to infection and to severe disease in humans, indicating that this interesting mechanism is not predictive of disease causation for stem-specific antibodies37. In addition, mAbs can be screened to avoid fusion-enhancing properties, and fusion is not intrinsically accelerated by low titres of neutralizing antibodies. Notably, infants benefit from immunization from six months of age, despite their limited capacity to produce affinity-matured, high-avidity antibodies. Overall, widespread annual surveillance of influenza does not reveal ADE of disease, even though cross-reactive strains and vaccine mismatches are common.

Antibody effector functions are mediated by binding of the IgG Fc domain to FcγRs on myeloid cells or to components of the complement system. These activities occur when the antibody binds the target virus protein either on virions or on infected cells. a, Viral particles are internalized and degraded and local cytokine release recruits immune cells. b, If cells are permissive, progeny virions could be produced. When virus–antibody complexes are taken up by the cell, a detrimental cytokine response may be generated. c, Binding of the IgG Fc fragment to C1q leads the activation of complement components C3, C3a and C5a and of the complement membrane attack complex (MAC) that disrupts membranes. C3 and C5a facilitate phagocytosis by myeloid cells. C3a and C5a are anaphylatoxins that attract inflammatory cells, which can secrete cytokines that enhance antiviral immunity but could be detrimental if produced in excess. d, e, The IgG Fc domain binds to multiple types of FcγRs on myeloid cells to trigger effector functions. The specific consequences of this interaction are dependent on the FcγR that is involved and are not detailed here. d, Antibody-dependent phagocytosis by macrophages and dendritic cells. e, Antibody-dependent cytotoxicity mediated by natural killer (NK) cells. f, Antibody-mediated antigen presentation after the uptake of virus or virus-infected cells by phagocytic cells leads to the activation of antiviral T cells.
Dengue
There are four viral serotypes of dengue that circulate in endemic areas19. Although severe dengue haemorrhagic fever and shock syndrome occurs during primary infection, possible ADE of disease has been associated with poorly neutralizing cross-reactive antibodies against a heterologous dengue serotype. Taking into account the difficulty of classification due to the overlapping signs of severe infection and ADE of disease, clinical experience indicates that ADE of disease does occur, but is rare in endemic areas (36/6,684 participants; around 0.5%) and is correlated with a narrow range of low pre-existing antibody titres (1:21–1:80)20. In the same study, high antibody titres were found to be protective. The challenge of predicting how to avoid such a rare immune-enhancing situation against the background of protection conferred by dengue neutralizing antibodies implies that it will be equally difficult for SARS-CoV-2.
When considering conditions that may result in ADE of disease, it is important to emphasize that dengue differs from other viruses because it targets monocytes, macrophages and dendritic cells and can produce progeny virus in these cells, which abundantly express both viral entry receptors and FcγRs. ADE of infection can be demonstrated in vitro with FcγR-expressing cells—typically with cross-reactive antibodies that have low or no neutralizing activity, have low affinity, or target non-protective epitopes, or if a narrow range of antibody and infectious virus concentrations is tested46,47. The mechanism of ADE of disease associated with dengue therefore depends on three factors: the circulation of multiple strains of a virus that have variable antigenicity, a virus that is capable of replication in FcγR-expressing myeloid cells and sequential infection of the same person with these different viral serotypes. Despite these pre-disposing conditions and the fact that dengue is an increasingly common infectious disease, severe dengue disease is rare.
The role of pre-existing immunity has also been a concern for the quadrivalent live attenuated dengue vaccine (Dengvaxia), because higher hospitalization rates were observed among vaccine recipients who were initially seronegative—especially children aged between two and eight years48. Other explanations for this outcome include poor efficacy against serotypes 1–3, or the failure to induce cell-mediated immunity because T cells primarily recognize non-structural proteins that are not present in the chimeric vaccine. Importantly, the cause of death in 14 fatal cases of dengue could not be determined by the WHO (World Health Organization) Global Advisory Committee on Vaccine Safety, because a failure of vaccine protection could not be distinguished from immune enhancement by clinical or laboratory criteria49. This experience underscores how difficult it is to predict the potential for vaccine-induced antibodies or a therapeutic antibody to enhance the severity of disease, because other mechanisms of pathogenesis that result in severe disease are potentially involved—even for the well-studied case of dengue.
In other assessments of the risks and benefits of cross-reactive antibodies, infection with Zika—which, as with dengue, is a flavivirus—was less common in individuals who had previously been infected with dengue50. In addition, the presence of cross-reactive antibodies has been associated with improved efficacy, as measured by the responses to a yellow fever vaccine in recipients who had received a Japanese encephalitis vaccine47, and by association of the effectiveness of Dengvaxia with seropositivity for dengue at the time of immunization51.
In summary, these clinical experiences with RSV, influenza and dengue provide strong evidence that the circumstances that are proposed to lead to ADE of disease—including low affinity or cross-reactive antibodies with limited or no neutralizing activity or suboptimal titres—are very rarely implicated as the cause of severe viral infection in the human host. Furthermore, clinical signs, immunological assays or biomarkers that can differentiate severe viral infection from a viral infection enhanced by an immune mechanism have not been established49,52.
Assessing the risk of ADE of disease with SARS-CoV-2
Given the complexities described above, it is sobering to take on the challenge of predicting ADE of disease caused by SARS-CoV-2. Here we consider whether clinical circumstances point to a role for antibodies with poor or no neutralizing activity in severe COVID-19, incorporating relevant experience from disease caused by the common human coronaviruses, as well as by severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome-related coronavirus (MERS-CoV).
Infection by SARS-CoV-2 is initiated by the binding of its fusion protein, the spike (S) protein, to the entry receptor, angiotensin-converting enzyme 2 (ACE2)53,54,55. Other receptors for SARS-CoV-2, such as CD147, have also been reported56. ACE2 is expressed on alveolar type II pneumocytes, airway epithelial cells, nasal tract goblet cells and ciliated cells, as well as on intestinal and other non-respiratory tract cells, as assessed by RNA expression57. On most such cells, ACE2 seems to be expressed at low levels; however, it can be upregulated by interferons58, which could theoretically promote infection if the virus overcomes interferon-induced barriers. FcγRIIa and FcγRIIIa were detected in alveolar, bronchial and nasal-cavity epithelial cells by single-cell RNA sequencing, but both fractions of positive cells and levels of expression per cell were considerably lower than for resident myeloid and natural killer cells59,60. The moderate prevalence of both ACE2 and FcγRs results in poor co-occurrence, although this might be underestimated because of the dropout effect in single-cell transcriptomics. Co-expression of ACE2 and FcγRs therefore seems to be limited, which would mitigate against antibody-enhanced disease caused by SARS-CoV-2 via the dual-receptor mechanism proposed in dengue infection.
When considering potential detrimental effects of antibodies, the presence or absence of cross-reactive antibodies against other human coronavirus (HCoV) strains has not been linked to whether SARS-CoV-2 infection is more severe, mild or asymptomatic, although antibodies that recognized the SARS-CoV-2 S2 subunit were detected in 12 out of 95 uninfected individuals61. In two reports, 30–50% of SARS-CoV-2 seronegative or unexposed individuals had CD4 T cells that recognized the SARS-CoV-2 S protein62,63. Previous infection with HCoV-HKU1 and HCoV-OC43 betacoronaviruses, or HCoV-NL63 and HCoV-229E alphacoronaviruses, is not known to predispose to more severe infection with the related virus from the same lineage64,65,66,67. Conversely, the endemic nature of coronavirus infections indicates that infection in the presence of low levels of antibodies is common, providing a theoretical opportunity for ADE of disease—although these illnesses are mild—and suggesting that cross-protection may be transient68. It is of interest that neither low neutralizing-antibody titres nor heterologous virus challenge were associated with enhanced disease in human studies of HCoV-229E64,65. Although HCoV-NL63 also uses the ACE2 entry receptor, the receptor-binding domain (RBD) of HCoV-NL63 is structurally very different from that of SARS-CoV-2, which would limit antibody cross-reactivity.
Antibodies to the S proteins of SARS-CoV and SARS-CoV-2—and, to a much lesser extent, MERS-CoV—can cross-react, and both high-potency neutralizing antibodies that also mediate antibody-dependent cytotoxicity and antibody-dependent cellular phagocytosis69, as well as non-neutralizing antibodies, can be elicited against conserved S epitopes70,71. However, the limited spread of SARS-CoV and MERS-CoV means that it is not feasible to assess whether there is any ADE of disease due to SARS-CoV-2 attributable to cross-reactive antibodies72. A finding that pre-existing antibodies for other coronaviruses correlate with the low incidence of symptomatic SARS-CoV-2 infection in children would support protection rather than a risk of disease enhancement73. To answer this question, the broad application of serological assays that quantify antibodies to virus-specific and cross-reactive epitopes of human coronaviruses in relation to the outcomes of natural infection and of vaccine and antibody trials is required.
The administration of passive antibodies could also reveal whether antibodies predispose to ADE of disease. In small studies, patients infected with SARS or MERS received polyclonal antibodies without apparent worsening of their illness74,75,76,77, and from a meta-analysis it was concluded that early treatment with plasma from patients that had recovered from SARS-CoV infection correlated with a better outcome76. In 10 patients with severe COVID-19 that were given plasma with neutralizing titres greater than 1:640 (200 ml) at a median of 16.5 days after disease onset, viraemia was no longer detected and clinical parameters improved within 3 days78. Similar findings were reported for 5 severely ill patients treated with plasma with neutralizing titres greater than 1:4079; however, another study found no difference in outcome between 52 treated and 51 untreated patients80. The evidence that COVID-19 does not worsen after treatment with plasma from convalescent patients has been substantially reinforced by a study of 20,000 patients who were severely ill with the disease, showing an adverse event incidence of 1–3%81. If further substantiated, these findings will markedly diminish the concern that clinically relevant amplification of infection, release of immunopathogenic cytokines or immune-complex deposition in the presence of a high viral load is mediated by SARS-CoV-2 antibody-dependent mechanisms82,83.
High-dose intravenous polyclonal IgG (IVIg)—which is used to treat systemic lupus erythematosus (SLE), idiopathic thrombocytopenia and Kawasaki syndrome84—is thought to exert its beneficial effects through the activation of FcγR inhibitory signalling. Because severe COVID-19 could reflect immune dysregulation, a benefit and/or lack of adverse effects in patients receiving plasma from convalescent individuals might reflect the suppression of inflammation induced by IgG, rather than supporting the conclusion that passive antibodies do not trigger ADE of disease through Fab- or Fc-dependent mechanisms. However, the dose of IgG administered to patients with SLE (2 g per kg over 5 days)85 is much higher than the dose received from convalescent plasma, based on the expected IgG concentrations in plasma (around 500–800 mg per 100 ml) and the amount of convalescent plasma received (200 ml)78,79. Assuming a concentration of 1,600 mg per 200 ml, the IgG levels after receiving convalescent plasma (1.6 g per 80 kg) would be approximately 100-fold less than after receiving IVIg (160 g per 80 kg). It is therefore unlikely that the immunomodulatory effects of polyclonal non-antigen-specific IgG dampened possible manifestations of enhanced illness.
Clinically, infections with SARS-CoV, MERS-CoV and SARS-CoV-2 are often biphasic, with more severe respiratory symptoms developing after a week or more and, in some patients, in association with the release of pro-inflammatory cytokines. This pattern has led to the hypothesis that an emerging immune response—including low-avidity, poorly neutralizing antibodies—could exacerbate the disease. However, reports that relate antibody titres to disease progression involve relatively few patients86,87,88, and are confounded by the higher levels of antigen seen in severe infections that are predicted to drive a stronger immune response or a heightened innate inflammatory response. One report of three cases of fatal SARS-CoV infection reported that high neutralizing anti-S antibodies and a prominent CD163+ monocyte/macrophage pulmonary infiltrate of cells were associated with reduced expression of TGF-β and CD206+, which are proposed to be markers of macrophages with beneficial functions89. However, quantitative analysis of these changes and evidence of an antibody-mediated pathology that is dependent on these cells were not reported. A recent meta-analysis found no relationship between the kinetics of antibody responses to SARS-CoV, MERS-CoV or SARS-CoV-2 and clinical outcomes90. At present, there is no evidence that ADE of disease is a factor in the severity of COVID-19. Instead, lung pathology is characterized by diffuse alveolar damage, pneumocyte desquamation, hyaline membranes, neutrophil or macrophage alveolar infiltrates and viral infection of epithelial cells and type II pneumocytes91. Further, if instances of ADE of disease occur at all, the experience with dengue suggests that this or other types of immune enhancement will be rare and will occur under highly specific conditions. The aetiology of the inflammatory, Kawasaki-like syndrome that has been associated with SARS-CoV-2 infection in children is unknown, but has not been associated with antibody responses so far92.
In summary, current clinical experience is insufficient to implicate a role for ADE of disease, or immune enhancement by any other mechanism, in the severity of COVID-19 (Table 1). Prospective studies that relate the kinetics and burden of infection and the host response—including the magnitude, antigen-specificity and molecular mechanisms of action of antibodies, antibody classes and T cell subpopulations—to clinical outcomes are needed to define the characteristics of a beneficial compared with a failed or a potentially detrimental host response to SARS-CoV-2 infection. Although it will probably continue to be difficult to prove that ADE of disease is occurring, or to predict when it might occur, it should be possible to identify correlates of protection that can inform immune-based approaches to the COVID-19 pandemic.
Effects of antibodies on SARS-CoV and MERS-CoV
In vitro studies of the effects of antibodies on viral infection have been used extensively to seek correlates or predictors of ADE of disease (Table 1). These efforts are complicated by the fact that the same antibody mechanisms that are often proposed to result in ADE of infection are responsible for protection from viral disease in vivo. Although infection was most often blocked by anti-S antibodies, several reports have shown antibody-dependent uptake of SARS-CoV or SARS-CoV S-pseudoviruses that was mediated by binding of the Fab component to the virus and the Fc component to FcγR on the target cell (Fig. 2) using in vitro methods93,94,95,96,97,98. Importantly, viral uptake did not result in productive infection. An antibody that binds the S protein and mimics receptor-mediated entry to facilitate viral uptake has been described for MERS-CoV99, but not for SARS-CoV or SARS-CoV-2. Although SARS-CoV and SARS-CoV-2 do not infect myeloid cells100,101,102,103, the productive infection of macrophages by MERS-CoV has been reported, albeit at low levels104. It is notable that higher production of immune-cell-attracting chemokines was observed in myeloid cells infected by MERS-CoV but not in cells exposed to SARS-CoV, suggesting that productive infection has a greater effect on this response104. The biology of the interactions of coronaviruses with cells expressing FcγRs is therefore very different from the targeting of FcγR-expressing myeloid cells by the dengue viruses. Conversely, in vitro methods can reliably define the properties of mAbs or of vaccine-induced antibodies—including their epitope specificity, binding affinity and avidity, and maturation as well as any potential to enhance fusion, together with their capacities for neutralization and antiviral Fc-dependent effector functions (Fig. 2).
Antibody effects in coronavirus-infected animals
Small-animal models
Several mouse, rat and other small-animal models of SARS-CoV infection have used passive-antibody administration or immunization to investigate whether pre-existing antibodies protect against or enhance disease. Although vaccine enhancement of disease in these models could occur through other mechanisms, such studies can directly assess the protective or enhancing properties of passive antibodies (Table 1).
In the ferret model of SARS-CoV infection, a human mAb was found to protect the animals from infection105; however, modified vaccinia Ankara expressing S protein (MVA-S) was not protective and liver inflammation was noted in this model106. Pre- and post-exposure administration of a mAb against MERS-CoV protected mice from challenge, as assessed by lung viral load, lung pathology and weight loss107. Three mAbs against SARS-CoV, given at a high dose before challenge, protected young and old mice against lung viral spread and inflammation, but had no effect when given after infection108. Low doses were less protective, but no ADE of disease was observed. A caveat is that human mAbs were tested in the context of mouse FcγRs; however, this can be addressed using human FcgR transgenic animals109. Both previous infection and passive transfer of mouse neutralizing antibodies partially protected 4–6-week-old mice against secondary infection with SARS-CoV110, and no ADE of disease was observed despite low neutralizing titres. In another mouse study111, passive transfer of SARS-CoV-immune serum was found to mediate protection by Fc-dependent monocyte effector function through antibody-dependent cellular phagocytosis; however, natural killer cells, antibody-dependent cytotoxicity or complement-antibody complexes did not contribute to protection. In a mouse model of vaccination, which used SARS-CoV in which the E protein had been deleted as a live attenuated vaccine, induction of antibodies and T cell immunity and protection against lethal viral challenge was observed in mice from three age groups112. By contrast, enhanced disease was observed in mice that were immunized with formalin- or ultraviolet-inactivated SARS-CoV. Whereas younger mice were protected, older mice developed pulmonary pathology with an eosinophil infiltrate; this suggests a detrimental Th2 response related to age, rather than ADE of disease113. In some models, cellular immunopathology might be linked to Th17-mediated activation of eosinophils114. In another report, mice given formalin- or ultraviolet-inactivated SARS-CoV or other vaccine formulations developed neutralizing antibodies and were protected from challenge, but also developed eosinophilic pulmonary infiltrates115. This type of immunopathology has not been reported in fatal human coronavirus infections.
Small-animal studies of SARS-CoV-2 infection are being reported rapidly. Neutralizing antibodies to SARS-CoV-2 were induced by immunizing rats with the RBD of the S protein and adjuvant94. In vitro evaluation of the potential for enhanced uptake of SARS-CoV-2 using HEK293T cells expressing rat FcγRI in the presence or absence of ACE2 expression showed neutralization but no enhancement of viral entry. Mice that were given an mRNA vaccine expressing pre-fusion SARS-CoV-2 S protein developed neutralizing antibodies and S-protein-specific CD8 T cell responses that were protective against lung infection without evidence of immunopathology116, and neutralizing mAbs against the RBD of the S protein of SARS-CoV-2 reduced lung infection and cytokine release117.
Passive transfer of a neutralizing antibody protected Syrian hamsters against high-dose SARS-CoV-2, as demonstrated by maintained weight and low lung viral titres118. Similarly, hamsters immunized with recombinant SARS-CoV S protein trimer developed neutralizing antibodies and were protected against challenge119. Whereas serum from vaccinated hamsters mediated FcγRIIb-dependent enhancement of SARS-CoV entry into B cell lines, virus replication was abortive in vitro and viral load and lung pathology were not increased in vaccinated animals98. These data underscore that enhancement of viral entry into cells in vitro does not predict negative consequences in vivo, further highlighting the important gap between in vitro findings and the causes of ADE of disease in vivo.
Unlike SARS-CoV, MERS-CoV and SARS-CoV-2, feline infectious peritonitis virus is an alphacoronavirus that, as with dengue, has tropism for macrophages. Infection with this virus has been shown to be enhanced by pre-existing antibodies, especially those against the same strain120.
Non-human primate models
In non-human primates (NHPs), infection with SARS-CoV, MERS-CoV or SARS-CoV-2 results in viral spread to multiple tissues, including lungs121,122,123. Rhesus macaques that were administered a high inoculum of SARS-CoV-2 by nasal, tracheal, ocular and oral routes had increased temperatures and respiratory rates for 1 day, and reduced appetite and dehydration for 9–16 days122. Macaques that were euthanized at 3 days and 21 days had multifocal lung lesions, with alveolar septal thickening due to oedema and fibrin, small to moderate numbers of macrophages, a few neutrophils, minimal type II pneumocyte hyperplasia and some perivascular lymphocyte cuffing. SARS-CoV-2 viral proteins were detected in a few type I and type II pneumocytes, and alveolar macrophages and virions were found in type I pneumocytes. Although these foci of lung pathology have some similarities to those observed in human infection91, NHPs develop minimal or no signs of respiratory or systemic betacoronavirus disease.
After the outbreaks of SARS-CoV and MERS-CoV disease, NHPs were used in the evaluation of several vaccine and antibody interventions (Supplementary Table 1). In one study, FI-SARS-CoV reduced viraemia and protected against lung pathology in rhesus macaques124, whereas in another study macaques given FI-SARS-CoV developed macrophage and lymphocytic infiltrates and alveolar oedema with fibrin deposition after challenge, indicating the difficulties of establishing consistent NHP models125. Synthetic peptide vaccines have also been prepared using sera from convalescent patients to define immunodominant epitopes of SARS-CoV S protein125. The vaccines were found to reduce pathology after SARS-CoV challenge unless the S protein of the vaccine included amino acids 597−603, suggesting an epitope-specific basis for the induction of lung pathology. However, these peptide constructs would not be expected to fully mimic antibody or T cell responses that would be elicited to the intact S protein.
Two studies have reported the immunization of rhesus macaques with MVA expressing SARS-CoV S protein or an MVA control. In the first report, three out of four immunized macaques had no detectable shedding or enhanced lung infection 7 days after challenge126. In the second report, immunization elicited polyclonal anti-S antibodies with neutralizing activity and reduced infection in three out of eight macaques after challenge89. However, although the challenge inoculum was the same as in the first study, areas of diffuse alveolar damage were detected in six out of eight vaccinated macaques compared with one out of eight control animals euthanized at 7 days, as well as at 35 days. Immunization with MVA-S was associated with an accumulation of monocytes and macrophages, and with the detection of activated alveolar macrophages that produced pro-inflammatory MCP-1 and IL-8, which were were not observed in control animals. In a second cohort that was given polyclonal IgG from vaccinated macaques or control animals, loss of TGF-β and increased IL-6 production by activated pulmonary macrophages was observed in macaques that were pre-treated with anti-S IgG, and lung pathology was described as skewed towards immunopathological inflammation. However, it was not stated whether the histopathology was focal or widespread in the lungs, and immunopathology was not associated with impaired respiratory function in macaques evaluated for 21 days (passive anti-S) or for 35 days (MVA-S). Although differences in macrophage markers were associated with changes in the lungs, a causal relationship between anti-S antibodies and an antibody-dependent macrophage-mediated mechanism of more severe pathological changes was not explored, and whether MVA-S might have generated non-neutralizing antibodies that enhanced lung pathology was not assessed. It will therefore be important to define the epitope specificity and serum neutralization activity in these animal models, and potential T cell mechanisms will need to be excluded before enhanced immunopathology can be attributed to antibody mechanisms.
The second study reporting immunization of rhesus macaques with MVA-S89 also described in vitro experiments using sera from patients who had recovered from SARS-CoV infection. However, only one out of eight sera samples elicited enhanced cytokine production by human macrophages in vitro. Because IL-8 production by macrophages treated with one of the serum samples was lower in the presence of FcγR-blocking antibody (no control serum), it was concluded that blocking FcγRs might be necessary to reduce lung damage caused by SARS-CoV. However, the finding was not confirmed with sera from other severe cases of SARS, and is subject to the caveat that in vitro studies cannot be taken as evidence of ADE of disease.
In contrast to the immunopathology observed after immunization with MVA-S, other studies of SARS-CoV have suggested a protective effect of vaccine-induced antibodies. Using a purified SARS-CoV-infected cell lysate as a vaccine, cynomolgus macaques were protected from challenge, and low neutralizing antibody titres were not associated with ADE of disease127. Further, African green monkeys with pre-existing antibody and/or T cells after primary SARS-CoV infection were protected from homologous re-challenge as assessed by lung virus titres, although the pulmonary inflammatory response was not different from that of primary infection128.
In additional studies, rhesus macaques immunized with a chimpanzee adenovirus (ChAdOx1 MERS) expressing MERS-CoV S protein, a recombinant S-RBD protein or a synthetic MERS-CoV S DNA vaccine, had decreased infection and no enhanced lung pathology upon challenge129,130,131.
The potential for immune enhancement of SARS-CoV-2 infection by antibody-dependent or other mechanisms has been assessed by infection and re-challenge of rhesus macaques. Out of two rhesus macaques that were re-challenged 28 days after initial infection—when neutralizing antibody titres were low (1:8–1:16)—neither exhibited viral shedding and one had no lung pathology. Immunity to SARS-CoV-2 in nine rhesus macaques—including the presence of neutralizing antibodies, antibody-mediated effector functions and antiviral CD4 and CD8 T cells—was associated with protection upon re-challenge at 35 days123. When vaccines were tested, rhesus macaques immunized with purified β-propriolactone-inactivated SARS-CoV-2 in alum showed complete or partial protection against high-inoculum SARS-CoV-2 challenge, and histopathological analyses of lungs and other organs at 29 days showed no evidence of ADE of disease compared with control macaques132. A large study involving 35 rhesus macaques, which were given prototype DNA vaccines expressing either full-length SARS-CoV-2 S protein or components of this protein, found that protection was correlated with the presence of neutralizing antibodies—and, notably, with Fc-dependent antibody effector functions—and there were no adverse outcomes after challenge133.
In studies of neutralizing mAbs (Supplementary Table 1), viral titres and lung pathology after nasal challenge were reduced in rhesus macaques that were administered a mAb directed against a proteolytic cleavage site in the SARS-CoV S protein that is required for host-cell entry134. Macaques given mAbs against MERS-CoV showed less pulmonary involvement and no worsening of disease with challenge135. The prophylactic administration of mAbs against MERS-CoV to marmosets one day before challenge was associated with reduced lung pathology compared with the administration of control mAbs136,137,138; mAbs were found to be protective when administered 2–12 h after challenge but not when given 1 day after challenge137,138. These animal studies of coronavirus infections parallel the observation that the passive transfer of mAbs against RSV that have selected properties can be protective, whereas a particular vaccine formulation (FI-RSV) that is directed to the same viral protein can enhance disease.
In summary, in most animal models—including NHPs—vaccination or the administration of passive mAbs have demonstrated protection against challenge with SARS-CoV, MERS-CoV or SARS-CoV-2, although reports on SARS-CoV-2 are limited. However, studies of an FI-SARS-CoV vaccine, one of two studies of an MVA vaccine expressing SARS-CoV S protein, and vaccination with one S-derived peptide showed enhanced lung pathology in NHPs. Thus, there are limited data to indicate that immune responses that include antibodies (and probably also T cells) induced by some vaccine formulations may be associated with more extensive lung pathology compared with infection alone, whereas the transfer of mAbs with specific properties have, so far, provided protection in animals (Supplementary Table 1).
Overall, the lack of a link between clinical measures of disease severity in NHPs and the experimental conditions associated with exacerbated lung pathology is a limitation to their utility in predicting the risks of ADE associated with passive-antibody or vaccine interventions in humans. So far, the models do not emulate the severe respiratory disease observed in COVID-19. Evaluation of T cell responses will also be needed to draw conclusions regarding mechanisms if immunopathology is observed. For example, a strong T cell response has been described as ameliorating ADE of disease in a dengue model139 and animal studies have suggested an aberrant T cell response to FI-RSV vaccination33,114. Quantitative assessments of the extent of lung involvement, and histopathological scoring of the characteristics and severity of lesions using validated markers of infected cells, patterns of cell-subtype infection and quantification of infiltrating immune cells will be also be necessary before these models can be used to better understand either protective immunity or immune enhancement—whether mediated by antibodies, T cells, intrinsic responses or a combination of factors. A critical point is that the identification of correlates of protection in humans will be necessary to understand how studies in small- and large-animal models can be designed to support or question the benefits of particular immune interventions for SARS-CoV-2 infection.
Conclusions
It is clear that after many years, and considerable attention, the understanding of ADE of disease after either vaccination or administration of antiviral antibodies is insufficient to confidently predict that a given immune intervention for a viral infection will have negative outcomes in humans. Despite the importance that such information would have in the COVID-19 pandemic, in vitro assays do not predict ADE of disease. Most animal models of vaccines and antibody interventions show protection, whereas those that suggest potential ADE of disease are not definitive and the precise mechanisms have not been defined. Although ADE is a concern, it is also clear that antibodies are a fundamentally important component of protective immunity to all of the pathogens discussed here, and that their protective effects depend both on the binding of viral proteins by their Fab fragments and on the effector functions conferred by their Fc fragments. Even when vaccine formulations such as formalin inactivation have shown disease enhancement, neutralizing antibodies with optimized properties have been protective. Further, the potential mechanisms of ADE of disease are probably virus-specific and, importantly, clinical markers do not differentiate severe infection from immune enhancement. Additional mechanism-focused studies are needed to determine whether small-animal and NHP models of virus infection, including for SARS-CoV-2, can predict the probable benefits or risks of vaccines or passive-antibody interventions in humans. Optimizing these models must be informed by understanding the correlates of protection against SARS-CoV-2 in natural human infection and as vaccines and antibodies are evaluated in humans. Such mechanistic and in vivo studies across viral pathogens are essential so that we are better prepared to face future pandemics. In the meantime, it will be necessary to directly test safety and define correlates of protection conferred by vaccines and antibodies against SARS-CoV-2 and other viral pathogens in human clinical trials.
References
Luke, T. C., Kilbane, E. M., Jackson, J. L. & Hoffman, S. L. Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann. Intern. Med. 145, 599–609 (2006).
Casadevall, A., Dadachova, E. & Pirofski, L. A. Passive antibody therapy for infectious diseases. Nat. Rev. Microbiol. 2, 695–703 (2004).
Plotkin, S. A. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 17, 1055–1065 (2010).
VanBlargan, L. A., Goo, L. & Pierson, T. C. Deconstructing the antiviral neutralizing-antibody response: implications for vaccine development and immunity. Microbiol. Mol. Biol. Rev. 80, 989–1010 (2016).
Corti, D. & Lanzavecchia, A. Broadly neutralizing antiviral antibodies. Ann. Rev. Immunol. 31, 705–742 (2013).
Walker, L. M. & Burton, D. R. Passive immunotherapy of viral infections: ‘super-antibodies’ enter the fray. Nat. Rev. Immunol. 18, 297–308 (2018).
Lu, L. L., Suscovich, T. J., Fortune, S. M. & Alter, G. Beyond binding: antibody effector functions in infectious diseases. Nat. Rev. Immunol. 18, 46–61 (2018).
Bournazos, S. & Ravetch, J. V. Fcγ receptor function and the design of vaccination strategies. Immunity 47, 224–233 (2017).
DiLillo, D. J., Tan, G. S., Palese, P. & Ravetch, J. V. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat. Med. 20, 143–151 (2014).
Bournazos, S. et al. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell 158, 1243–1253 (2014).
Pyzik, M. et al. The neonatal Fc receptor (FcRn): a misnomer? Front. Immunol. 10, 1540 (2019).
Bergtold, A., Desai, D. D., Gavhane, A. & Clynes, R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity 23, 503–514 (2005).
Nishimura, Y. et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature 543, 559–563 (2017).
Gunn, B. M. et al. A Role for Fc function in therapeutic monoclonal antibody-mediated protection against Ebola virus. Cell Host Microbe 24, 221–233.e5 (2018).
Graham, B. S. Rapid COVID-19 vaccine development. Science 368, 945–946 (2020).
Kim, H. W. et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 89, 422–434 (1969).
Kapikian, A. Z., Mitchell, R. H., Chanock, R. M., Shvedoff, R. A. & Stewart, C. E. An epidemiologic study of altered clinical reactivity to respiratory syncytial (RS) virus infection in children previously vaccinated with an inactivated RS virus vaccine. Am. J. Epidemiol. 89, 405–421 (1969).
Polack, F. P., Hoffman, S. J., Crujeiras, G. & Griffin, D. E. A role for nonprotective complement-fixing antibodies with low avidity for measles virus in atypical measles. Nat. Med. 9, 1209–1213 (2003).
Simmons, C. P., Farrar, J. J., Nguyen, V. & Wills, B. Dengue. N. Engl. J. Med. 366, 1423–1432 (2012).
Katzelnick, L. C. et al. Antibody-dependent enhancement of severe dengue disease in humans. Science 358, 929–932 (2017).
Guzman, M. G., Alvarez, M. & Halstead, S. B. Secondary infection as a risk factor for dengue hemorrhagic fever/dengue shock syndrome: an historical perspective and role of antibody-dependent enhancement of infection. Arch. Virol. 158, 1445–1459 (2013).
Iwasaki, A. & Yang, Y. The potential danger of suboptimal antibody responses in COVID-19. Nat. Rev. Immunol. 20, 339–341 (2020).
Dekkers, G. et al. Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs 9, 767–773 (2017).
Crowley, A. R. & Ackerman, M. E. Mind the gap: how interspecies variability in IgG and its receptors may complicate comparisons of human and non-human primate effector function. Front. Immunol. 10, 697 (2019).
Fulginiti, V. A. et al. Respiratory virus immunization. A field trial of two inactivated respiratory virus vaccines; an aqueous trivalent parainfluenza virus vaccine and an alum-precipitated respiratory syncytial virus vaccine. Am. J. Epidemiol. 89, 435–448 (1969).
Chin, J., Magoffin, R. L., Shearer, L. A., Schieble, J. H. & Lennette, E. H. Field evaluation of a respiratory syncytial virus vaccine and a trivalent parainfluenza virus vaccine in a pediatric population. Am. J. Epidemiol. 89, 449–463 (1969).
Murphy, B. R. et al. Dissociation between serum neutralizing and glycoprotein antibody responses of infants and children who received inactivated respiratory syncytial virus vaccine. J. Clin. Microbiol. 24, 197–202 (1986).
Polack, F. P. et al. A role for immune complexes in enhanced respiratory syncytial virus disease. J. Exp. Med. 196, 859–865 (2002).
Atkinson, J. P. et al. The human complement system: basic concepts and clinical relevance. Clin. Immunol. https://doi.org/10.1016/B978-0-7020-6896-6.00021-1 (2019).
Kim, H. W. et al. Cell-mediated immunity to respiratory syncytial virus induced by inactivated vaccine or by infection. Pediatr. Res. 10, 75–78 (1976).
van Erp, E. A., Luytjes, W., Ferwerda, G. & van Kasteren, P. B. Fc-mediated antibody effector functions during respiratory syncytial virus infection and disease. Front. Immunol. 10, 548 (2019).
Delgado, M. F. et al. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat. Med. 15, 34–41 (2009).
Ruckwardt, T. J., Morabito, K. M. & Graham, B. S. Immunological lessons from respiratory syncytial virus vaccine development. Immunity 51, 429–442 (2019).
Aranda, S. S. & Polack, F. P. Prevention of pediatric respiratory syncytial virus lower respiratory tract illness: perspectives for the next decade. Front. Immunol. 10, 1006 (2019).
Regeneron to discontinue development of Suptavumab for respiratory syncytial virus. https://investor.regeneron.com/news-releases/news-release-details/regeneron-discontinue-development-suptavumab-respiratory (2017).
Domachowske, J. B. et al. Safety, tolerability and pharmacokinetics of MEDI8897, an extended half-life single-dose respiratory syncytial virus prefusion F-targeting monoclonal antibody administered as a single dose to healthy preterm infants. Pediatr. Infect. Dis. J. 37, 886–892 (2018).
Ng, S. et al. Novel correlates of protection against pandemic H1N1 influenza A virus infection. Nat. Med. 25, 962–967 (2019).
Skowronski, D. M. et al. Association between the 2008–09 seasonal influenza vaccine and pandemic H1N1 illness during spring–summer 2009: four observational studies from Canada. PLoS Med. 7, e1000258 (2010).
Wu, J. T. et al. The infection attack rate and severity of 2009 pandemic H1N1 influenza in Hong Kong. Clin. Infect. Dis. 51, 1184–1191 (2010).
Lansbury, L. E. et al. Effectiveness of 2009 pandemic influenza A(H1N1) vaccines: a systematic review and meta-analysis. Vaccine 35, 1996–2006 (2017).
Osterholm, M. T., Kelley, N. S., Sommer, A. & Belongia, E. A. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect. Dis. 12, 36–44 (2012).
Monsalvo, A. C. et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat. Med. 17, 195–199 (2011).
Co, M. D. T. et al. Relationship of preexisting influenza hemagglutination inhibition, complement-dependent lytic, and antibody-dependent cellular cytotoxicity antibodies to the development of clinical illness in a prospective study of A(H1N1)pdm09 influenza in children. Viral Immunol. 27, 375–382 (2014).
Khurana, S. et al. Vaccine-induced anti-HA2 antibodies promote virus fusion and enhance influenza virus respiratory disease. Sci. Transl. Med. 5, 200ra114 (2013).
Winarski, K. L. et al. Antibody-dependent enhancement of influenza disease promoted by increase in hemagglutinin stem flexibility and virus fusion kinetics. Proc. Natl Acad. Sci. USA 116, 15194–15199 (2019).
Beltramello, M. et al. The human immune response to dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 8, 271–283 (2010).
de Alwis, R. et al. Dengue viruses are enhanced by distinct populations of serotype cross-reactive antibodies in human immune sera. PLoS Pathog. 10, e1004386 (2014).
Thomas, S. J. & Yoon, I.-K. A review of Dengvaxia®: development to deployment. Hum. Vaccin. Immunother. 15, 2295–2314 (2019).
WHO Report. Dengue vaccine: WHO position paper, September 2018 – Recommendations. Vaccine 37, 4848–4849 (2019).
Rodriguez-Barraquer, I. et al. Impact of preexisting dengue immunity on Zika virus emergence in a dengue endemic region. Science 363, 607–610 (2019).
Chan, K. R. et al. Cross-reactive antibodies enhance live attenuated virus infection for increased immunogenicity. Nat. Microbiol. 1, 16164 (2016).
Browne, S. K., Beeler, J. A. & Roberts, J. N. Summary of the vaccines and related biological products advisory committee meeting held to consider evaluation of vaccine candidates for the prevention of respiratory syncytial virus disease in RSV-naïve infants. Vaccine 38, 101–106 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e6 (2020).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Muus, C. et al. Integrated analyses of single-cell atlases reveal age, gender, and smoking status associations with cell type-specific expression of mediators of SARS-CoV-2 viral entry and highlights inflammatory programs in putative target cells. Preprint at https://www.biorxiv.org/content/10.1101/2020.04.19.049254v2 (2020).
Sungnak, W. et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 26, 681–687 (2020).
Ziegler, C. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is enriched in specific cell subsets across tissues. Cell 181, 1016–1035.e19 (2020).
Wellcome Sanger Institute, Human Cell Atlas & Chan Zuckerberg Initiative. COVID-19 Cell Atlas, https://www.covid19cellatlas.org/
Chan Zuckerberg Biohub & Stanford University. Lung Cell Atlas, https://hlca.ds.czbiohub.org/
Ng, K. et al. Pre-existing and de novo humoral immunity to SARS-CoV-2 in humans. Preprint at https://www.biorxiv.org/content/10.1101/2020.05.14.095414v1 (2020).
Braun, J. et al. Presence of SARS-CoV-2 reactive T cells in COVID-19 patients and healthy donors. Preprint at https://www.medrxiv.org/content/10.1101/2020.04.17.20061440v1 (2020). Detection of anti-S protein CD4 + T cells in 83% patients with COVID-19 with reactivity to epitopes in both N- and C-terminal domains, and in 34% of healthy unexposed donors, indicating cross-reactive T cell immunity against SARS-CoV-2 attributable to previous coronavirus infections, with epitopes predominantly in the C-terminal domain that has higher homology to other coronaviruses.
Grifoni, A. et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 181, 1489–1501.e15 (2020). Extensive analysis of CD4 and CD8 T cell responses to epitopes of S-, M- and N proteins as well as non-structural proteins of SARS-CoV-2 in convalescent patients with COVID-19 and detection of cross-reactive CD4 + T cells that recognized SAR-CoV-2 epitopes in 40–60% of unexposed donors.
van der Hoek, L., Pyrc, K. & Berkhout, B. Human coronavirus NL63, a new respiratory virus. FEMS Microbiol. Rev. 30, 760–773 (2006).
Callow, K. A., Parry, H. F., Sergeant, M. & Tyrrell, D. A. J. The time course of the immune response to experimental coronavirus infection of man. Epidemiol. Infect. 105, 435–446 (1990).
Reed, S. E. The behaviour of recent isolates of human respiratory coronavirus in vitro and in volunteers: evidence of heterogeneity among 229E-related strains. J. Med. Virol. 13, 179–192 (1984).
Chan, K. H. et al. Serological responses in patients with severe acute respiratory syndrome coronavirus infection and cross-reactivity with human coronaviruses 229E, OC43, and NL63. Clin. Diagn. Lab. Immunol. 12, 1317–1321 (2005).
Kissler, S. M., Tedijanto, C., Goldstein, E., Grad, Y. H. & Lipsitch, M. Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. Science 368, 860–868 (2020).
Pinto, D. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295 (2020).
Okba, N. M. A. et al. Severe acute respiratory syndrome coronavirus 2−specific antibody responses in coronavirus disease patients. Emerging Infect. Dis. 26, 1478–1488 (2020).
Lv, H. et al. Cross-reactive antibody response between SARS-CoV-2 and SARS-CoV infections. Cell Reports 31, 107725 (2020).
Guo, X. et al. Long-term persistence of IgG antibodies in SARS-CoV infected healthcare workers. Preprint at https://www.medrxiv.org/content/10.1101/2020.02.12.20021386v1 (2020).
Lavezzo, E. et al. Suppression of COVID-19 outbreak in the municipality of Vo, Italy. Nature https://doi.org/10.1038/s41586-020-2488-1 (2020).
Yeh, K.-M. et al. Experience of using convalescent plasma for severe acute respiratory syndrome among healthcare workers in a Taiwan hospital. J. Antimicrob. Chemother. 56, 919–922 (2005).
Cheng, Y. et al. Use of convalescent plasma therapy in SARS patients in Hong Kong. Eur. J. Clin. Microbiol. Infect. Dis. 24, 44–46 (2005).
Mair-Jenkins, J. et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. J. Infect. Dis. 211, 80–90 (2015).
Ko, J.-H. et al. Challenges of convalescent plasma infusion therapy in Middle East respiratory coronavirus infection: a single centre experience. Antivir. Ther. 23, 617–622 (2018).
Duan, K. et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc. Natl Acad. Sci. USA 117, 9490–9496 (2020).
Shen, C. et al. Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. J. Am. Med. Assoc. 323, 1582–1589 (2020).
Li, L. et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: a randomized clinical trial. J. Am. Med. Assoc. https://doi.org/10.1001/jama.2020.10044 (2020).
Joyner, M. J. & Wright, R. S. Safety update: COVID-19 convalescent plasma in 20,000 hospitalized patients. Mayo Clin. Proc. https://doi.org/10.1016/j.mayocp.2020.06.028 (2020). Major US-wide study of the administration of convalescent plasma to patients with COVID-19 with severe respiratory disease, followed by observation for seven days post-infusion with no evidence of disease progression associated with passive-antibody therapy.
Chen, L., Xiong, J., Bao, L. & Shi, Y. Convalescent plasma as a potential therapy for COVID-19. Lancet Infect. Dis. 20, 398–400 (2020).
de Alwis, R., Chen, S., Gan, E. S. & Ooi, E. E. Impact of immune enhancement on COVID-19 polyclonal hyperimmune globulin therapy and vaccine development. EBioMedicine 55, 102768 (2020).
Galeotti, C., Kaveri, S. V. & Bayry, J. IVIG-mediated effector functions in autoimmune and inflammatory diseases. Int. Immunol. 29, 491–498 (2017).
Zandman-Goddard, G., Levy, Y. & Shoenfeld, Y. Intravenous immunoglobulin therapy and systemic lupus erythematosus. Clin. Rev. Allergy Immunol. 29, 219–228 (2005).
Lee, N. et al. Anti-SARS-CoV IgG response in relation to disease severity of severe acute respiratory syndrome. J. Clin. Virol. 35, 179–184 (2006).
Zhang, L. et al. Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J. Med. Virol. 78, 1–8 (2006).
Ho, M.-S. et al. Neutralizing antibody response and SARS severity. Emerg. Infect. Dis. 11, 1730–1737 (2005).
Liu, L. et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 4, e123158 (2019).
Huang, A. et al. A systematic review of antibody mediated immunity to coronaviruses: antibody kinetics, correlates of protection, and association of antibody responses with severity of disease. Preprint at https://www.medrxiv.org/content/10.1101/2020.04.14.20065771v1 (2020). Meta-analysis of reports of antibody responses to SARS-CoV, MERS-CoV and initial reports of SARS-CoV-2 in infected patients, describing inconclusive evidence for a relationship between antibody titres and disease severity.
Martines, R. B. et al. Pathology and pathogenesis of SARS-CoV-2 associated with fatal coronavirus disease, United States. Emerg. Infect. Dis. https://doi.org/10.3201/eid2609.202095 (2020).
Ramcharan, T. et al. Paediatric inflammatory multisystem syndrome: temporally associated with SARS-CoV-2 (PIMS-TS): cardiac features, management and short-term outcomes at a UK tertiary paediatric hospital. Pediatr. Cardiol. https://doi.org/10.1007/s00246-020-02391-2 (2020).
Yang, Z.-Y. et al. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 78, 5642–5650 (2004).
Quinlan, B. D. et al. The SARS-CoV-2 receptor-binding domain elicits a potent neutralizing response without antibody-dependent enhancement. Preprint at https://www.biorxiv.org/content/10.1101/2020.04.10.036418v1 (2020).
Yip, M. S. et al. Antibody-dependent enhancement of SARS coronavirus infection and its role in the pathogenesis of SARS. Hong Kong Med. J. 22, 25–31 (2016).
Yip, M. S. et al. Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol. J. 11, 82 (2014).
Wang, S.-F. et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem. Biophys. Res. Commun. 451, 208–214 (2014).
Jaume, M. et al. Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH- and cysteine protease-independent FcγR pathway. J. Virol. 85, 10582–10597 (2011).
Wan, Y. et al. Molecular mechanism for antibody-dependent enhancement of coronavirus entry. J. Virol. 94, e02015–e02019 (2020).
Yilla, M. et al. SARS-coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 107, 93–101 (2005).
Lau, Y. L., Peiris, J. S. M. & Law, H. K. W. Role of dendritic cells in SARS coronavirus infection. Hong Kong Med. J. 18, 28–30 (2012).
Tynell, J. et al. Middle East respiratory syndrome coronavirus shows poor replication but significant induction of antiviral responses in human monocyte-derived macrophages and dendritic cells. J. Gen. Virol. 97, 344–355 (2016).
Hui, K. P. Y. et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: an analysis in ex-vivo and in-vitro cultures. Lancet Respir. Med. 8, 687–695 (2020).
Zhou, J. et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J. Infect. Dis. 209, 1331–1342 (2014).
ter Meulen, J. et al. Human monoclonal antibody as prophylaxis for SARS coronavirus infection in ferrets. Lancet 363, 2139–2141 (2004).
Czub, M., Weingartl, H., Czub, S., He, R. & Cao, J. Evaluation of modified vaccinia virus Ankara based recombinant SARS vaccine in ferrets. Vaccine 23, 2273–2279 (2005).
Corti, D. et al. Prophylactic and postexposure efficacy of a potent human monoclonal antibody against MERS coronavirus. Proc. Natl Acad. Sci. USA 112, 10473–10478 (2015).
Rockx, B. et al. Structural basis for potent cross-neutralizing human monoclonal antibody protection against lethal human and zoonotic severe acute respiratory syndrome coronavirus challenge. J. Virol. 82, 3220–3235 (2008).
Smith, P., DiLillo, D. J., Bournazos, S., Li, F. & Ravetch, J. V. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc. Natl Acad. Sci. USA 109, 6181–6186 (2012).
Subbarao, K. et al. Prior infection and passive transfer of neutralizing antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. J. Virol. 78, 3572–3577 (2004).
Yasui, F. et al. Phagocytic cells contribute to the antibody-mediated elimination of pulmonary-infected SARS coronavirus. Virology 454–455, 157–168 (2014).
Fett, C., DeDiego, M. L., Regla-Nava, J. A., Enjuanes, L. & Perlman, S. Complete protection against severe acute respiratory syndrome coronavirus-mediated lethal respiratory disease in aged mice by immunization with a mouse-adapted virus lacking E protein. J. Virol. 87, 6551–6559 (2013).
Bolles, M. et al. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J. Virol. 85, 12201–12215 (2011).
Hotez, P. J., Corry, D. B. & Bottazzi, M. E. COVID-19 vaccine design: the Janus face of immune enhancement. Nat. Rev. Immunol. 20, 347–348 (2020).
Tseng, C.-T. et al. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE 7, e35421 (2012).
Corbett, K. S. et al. SARS-CoV-2 mRNA vaccine development enabled by prototype pathogen preparedness. Preprint at https://www.biorxiv.org/content/10.1101/2020.06.11.145920v1 (2020).
Zost, S. J. et al. Potently neutralizing human antibodies that block SARS-CoV-2 receptor binding and protect animals. Preprint at https://www.biorxiv.org/content/10.1101/2020.05.22.111005v1 (2020). Protection of mice against SARS-CoV-2 by human mAbs targeting distinct epitopes of the S protein, some of which had synergistic effects in vitro, without evidence of ADE of disease in the animal model.
Rogers, T. F. et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science https://doi.org/10.1126/science.abc7520 (2020). Protective effects of neutralizing mAbs against RBD and non-RBD epitopes of SARS-CoV-2 S protein without evidence of ADE of disease in a Syrian hamster model.
Kam, Y. W. et al. Antibodies against trimeric S glycoprotein protect hamsters against SARS-CoV challenge despite their capacity to mediate FcγRII-dependent entry into B cells in vitro. Vaccine 25, 729–740 (2007).
Pedersen, N. C. An update on feline infectious peritonitis: virology and immunopathogenesis. Vet. J. 201, 123–132 (2014).
Rockx, B. et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 368, 1012–1015 (2020).
Munster, V. et al. Respiratory disease and virus shedding in rhesus macaques inoculated with SARS-CoV-2. Preprint at https://www.biorxiv.org/content/10.1101/2020.03.21.001628v1 (2020).
Chandrashekar, A. et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science https://doi.org/10.1126/science.abc4776 (2020). Infection of rhesus macaques with SARS-CoV-2 and a comprehensive analysis of antibody neutralizing and Fc-mediated effector function showing multi-factorial correlation with protection against re-challenge.
Zhou, J. et al. Immunogenicity, safety, and protective efficacy of an inactivated SARS-associated coronavirus vaccine in rhesus monkeys. Vaccine 23, 3202–3209 (2005).
Wang, Q. et al. Immunodominant SARS coronavirus epitopes in humans elicited both enhancing and neutralizing effects on infection in non-human primates. ACS Infect. Dis. 2, 361–376 (2016).
Chen, Z. et al. Recombinant modified vaccinia virus Ankara expressing the spike glycoprotein of severe acute respiratory syndrome coronavirus induces protective neutralizing antibodies primarily targeting the receptor binding region. J. Virol. 79, 2678–2688 (2005).
Qin, E. et al. Immunogenicity and protective efficacy in monkeys of purified inactivated Vero-cell SARS vaccine. Vaccine 24, 1028–1034 (2006).
Clay, C. et al. Primary severe acute respiratory syndrome coronavirus infection limits replication but not lung inflammation upon homologous rechallenge. J. Virol. 86, 4234–4244 (2012).
Muthumani, K. et al. A synthetic consensus anti-spike protein DNA vaccine induces protective immunity against Middle East respiratory syndrome coronavirus in nonhuman primates. Sci. Transl. Med. 7, 301ra132 (2015).
Lan, J. et al. Recombinant receptor binding domain protein induces partial protective immunity in rhesus macaques against Middle East respiratory syndrome coronavirus challenge. EBioMedicine 2, 1438–1446 (2015).
van Doremalen, N. et al. A single dose of ChAdOx1 MERS provides protective immunity in rhesus macaques. Sci. Adv. 6, eaba8399 (2020).
Gao, Q. et al. Rapid development of an inactivated vaccine candidate for SARS-CoV-2. Science 369, 77–81 (2020). Protection of rhesus macaques against SARS-CoV-2 challenge after immunization with purified inactivated SARS-CoV-2 virus without evidence of ADE of disease.
Yu, J. et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science https://doi.org/10.1126/science.abc6284 (2020). Immunization of rhesus macaques with DNA vaccines expressing forms of the SARS-CoV-2 S protein resulted in reduced infection following challenge after administration of full-length S protein without evidence of ADE of disease.
Miyoshi-Akiyama, T. et al. Fully human monoclonal antibody directed to proteolytic cleavage site in severe acute respiratory syndrome (SARS) coronavirus S protein neutralizes the virus in a rhesus macaque SARS model. J. Infect. Dis. 203, 1574–1581 (2011).
Johnson, R. F. et al. 3B11-N, a monoclonal antibody against MERS-CoV, reduces lung pathology in rhesus monkeys following intratracheal inoculation of MERS-CoV Jordan-n3/2012. Virology 490, 49–58 (2016).
de Wit, E. et al. Prophylactic and therapeutic efficacy of mAb treatment against MERS-CoV in common marmosets. Antiviral Res. 156, 64–71 (2018).
de Wit, E. et al. Prophylactic efficacy of a human monoclonal antibody against MERS-CoV in the common marmoset. Antiviral Res. 163, 70–74 (2019).
Chen, Z. et al. Human neutralizing monoclonal antibody inhibition of Middle East respiratory syndrome coronavirus replication in the common marmoset. J. Infect. Dis. 215, 1807–1815 (2017).
Lam, J. H. et al. Dengue vaccine-induced CD8+ T cell immunity confers protection in the context of enhancing, interfering maternal antibodies. JCI Insight 2, e94500 (2017).
Acknowledgements
The authors thank D. Ma for her contributions to preparing this review.
Author information
Authors and Affiliations
Contributions
A.M.A. and H.W.V. drafted the manuscript, K.F., M.A.S., A.C., R.S., C.H.-D., A.L. and D.C. edited the draft, and A.M.A. and H.W.V. prepared the final manuscript, which was reviewed and approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors of this manuscript are employees of, or have other affiliations with, Vir Biotechnology. Vir had to choose how to proceed with mAbs to treat or prevent COVID-19 disease in the light of the evidence surrounding the possibility of ADE as detailed in this review. This review reflects the result of the team of authors carefully reviewing the literature to assess these choices and is provided as a service to the community. Vir has elected to test human mAbs with Fc activity preserved or enhanced, based on the lack of consistent evidence for ADE of disease noted above and evidence that the protective activities and potency of antibodies often involves antibody effector functions. We could have elected to take forward mAbs engineered to lack effector function, and so our antibody-related clinical programmes for SARS-CoV-2 could have moved forward regardless of the outcome of our review. A.M.A.’s contributions were part of her personal outside consulting arrangement with Vir Biotechnology and were not associated with Stanford University.
Additional information
Peer review information Nature thanks James Crowe, Gary Nabel and Stanley Perlman for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Table 1
| Summary of studies of SARS-CoV, MERS-CoV and SARS-CoV-2 in non-human primates.
Rights and permissions
About this article

Cite this article
Arvin, A.M., Fink, K., Schmid, M.A. et al. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 584, 353–363 (2020). https://doi.org/10.1038/s41586-020-2538-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2538-8
This article is cited by
-
Beyond neutralization: Fc-dependent antibody effector functions in SARS-CoV-2 infection
Nature Reviews Immunology (2023)
-
Antiviral neutralizing antibodies: from in vitro to in vivo activity
Nature Reviews Immunology (2023)
-
Temporary Cross-Immunity as a Plausible Driver of Asynchronous Cycles of Dengue Serotypes
Bulletin of Mathematical Biology (2023)
-
COVID-19 vaccines adverse events: potential molecular mechanisms
Immunologic Research (2023)
-
Immunity in SARS-CoV-2 Infection: Clarity or Mystery? A Broader Perspective in the Third Year of a Worldwide Pandemic
Archivum Immunologiae et Therapiae Experimentalis (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
