
Abstract
Consumption of fructose has risen markedly in recent decades owing to the use of sucrose and high-fructose corn syrup in beverages and processed foods1, and this has contributed to increasing rates of obesity and non-alcoholic fatty liver disease2,3,4. Fructose intake triggers de novo lipogenesis in the liver4,5,6, in which carbon precursors of acetyl-CoA are converted into fatty acids. The ATP citrate lyase (ACLY) enzyme cleaves cytosolic citrate to generate acetyl-CoA, and is upregulated after consumption of carbohydrates7. Clinical trials are currently pursuing the inhibition of ACLY as a treatment for metabolic diseases8. However, the route from dietary fructose to hepatic acetyl-CoA and lipids remains unknown. Here, using in vivo isotope tracing, we show that liver-specific deletion of Acly in mice is unable to suppress fructose-induced lipogenesis. Dietary fructose is converted to acetate by the gut microbiota9, and this supplies lipogenic acetyl-CoA independently of ACLY10. Depletion of the microbiota or silencing of hepatic ACSS2, which generates acetyl-CoA from acetate, potently suppresses the conversion of bolus fructose into hepatic acetyl-CoA and fatty acids. When fructose is consumed more gradually to facilitate its absorption in the small intestine, both citrate cleavage in hepatocytes and microorganism-derived acetate contribute to lipogenesis. By contrast, the lipogenic transcriptional program is activated in response to fructose in a manner that is independent of acetyl-CoA metabolism. These data reveal a two-pronged mechanism that regulates hepatic lipogenesis, in which fructolysis within hepatocytes provides a signal to promote the expression of lipogenic genes, and the generation of microbial acetate feeds lipogenic pools of acetyl-CoA.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Bray, G. A., Nielsen, S. J. & Popkin, B. M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 79, 537–543 (2004).
Jensen, T. et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 68, 1063–1075 (2018).
Hannou, S. A., Haslam, D. E., McKeown, N. M. & Herman, M. A. Fructose metabolism and metabolic disease. J. Clin. Invest. 128, 545–555 (2018).
Softic, S., Cohen, D. E. & Kahn, C. R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 61, 1282–1293 (2016).
Lambert, J. E., Ramos-Roman, M. A., Browning, J. D. & Parks, E. J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146, 726–735 (2014).
Donnelly, K. L. et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 (2005).
Fukuda, H., Katsurada, A. & Iritani, N. Effects of nutrients and hormones on gene expression of ATP citrate-lyase in rat liver. Eur. J. Biochem. 209, 217–222 (1992).
Pinkosky, S. L., Groot, P. H. E., Lalwani, N. D. & Steinberg, G. R. Targeting ATP-Citrate Lyase in Hyperlipidemia and Metabolic Disorders. Trends Mol. Med. 23, 1047–1063 (2017).
Jang, C. et al. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. 27, 351–361.e3 (2018).
Zhao, S. et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 17, 1037–1052 (2016).
Beigneux, A. P. et al. ATP-citrate lyase deficiency in the mouse. J. Biol. Chem. 279, 9557–9564 (2004).
Herman, M. A. & Samuel, V. T. The sweet path to metabolic demise: fructose and lipid synthesis. Trends Endocrinol. Metab. 27, 719–730 (2016).
Uyeda, K. & Repa, J. J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 4, 107–110 (2006).
Herman, M. A. et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484, 333–338 (2012).
Iizuka, K. The role of carbohydrate response element binding protein in intestinal and hepatic fructose metabolism. Nutrients 9, 181 (2017).
Poungvarin, N. et al. Genome-wide analysis of ChREBP binding sites on male mouse liver and white adipose chromatin. Endocrinology 156, 1982–1994 (2015).
Luong, A., Hannah, V. C., Brown, M. S. & Goldstein, J. L. Molecular characterization of human acetyl-CoA synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 275, 26458–26466 (2000).
Ikeda, Y. et al. Transcriptional regulation of the murine acetyl-CoA synthetase 1 gene through multiple clustered binding sites for sterol regulatory element-binding proteins and a single neighboring site for Sp1. J. Biol. Chem. 276, 34259–34269 (2001).
Softic, S. et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Invest. 127, 4059–4074 (2017).
Liu, X. et al. Acetate production from glucose and coupling to mitochondrial metabolism in mammals. Cell 175, 502–513.e13 (2018).
Bulusu, V. et al. Acetate recapturing by nuclear acetyl-coa synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 18, 647–658 (2017).
Lu, M. et al. ACOT12-dependent alteration of acetyl-coa drives hepatocellular carcinoma metastasis by epigenetic induction of epithelial-mesenchymal transition. Cell Metab. 29, 886–900.e5 (2019).
Ter Horst, K. W. & Serlie, M. J. Fructose consumption, lipogenesis, and non-alcoholic fatty liver disease. Nutrients 9, 981 (2017).
Bergheim, I. et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J. Hepatol. 48, 983–992 (2008).
Kaden-Volynets, V. et al. Lack of liver steatosis in germ-free mice following hypercaloric diets. Eur. J. Nutr. 58, 1933–1945 (2019).
Mews, P. et al. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386 (2017).
Li, M. V. et al. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 395, 395–400 (2010).
Lanaspa, M. A. et al. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J. Clin. Invest. 128, 2226–2238 (2018).
Ishimoto, T. et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl Acad. Sci. USA 109, 4320–4325 (2012).
Parks, E. J., Skokan, L. E., Timlin, M. T. & Dingfelder, C. S. Dietary sugars stimulate fatty acid synthesis in adults. J. Nutr. 138, 1039–1046 (2008).
den Besten, G. et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G900–G910 (2013).
Donohoe, D. R. et al. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 48, 612–626 (2012).
Zagelbaum, N. K., Yandrapalli, S., Nabors, C. & Frishman, W. H. Bempedoic acid (ETC-1002): ATP citrate lyase inhibitor: review of a first-in-class medication with potential benefit in statin-refractory cases. Cardiol. Rev. 27, 49–56 (2019).
Guo, L. et al. Enhanced acetylation of ATP-citrate lyase promotes the progression of nonalcoholic fatty liver disease. J. Biol. Chem. 294, 11805–11816 (2019).
Wang, Q. et al. Abrogation of hepatic ATP-citrate lyase protects against fatty liver and ameliorates hyperglycemia in leptin receptor-deficient mice. Hepatology 49, 1166–1175 (2009).
Wang, Q. et al. Deficiency in hepatic ATP-citrate lyase affects VLDL-triglyceride mobilization and liver fatty acid composition in mice. J. Lipid Res. 51, 2516–2526 (2010).
Postic, C. et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274, 305–315 (1999).
Nadkarni, M. A., Martin, F. E., Jacques, N. A. & Hunter, N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148, 257–266 (2002).
Guan, D. et al. Diet-induced circadian enhancer remodeling synchronizes opposing hepatic lipid metabolic processes. Cell 174, 831–842.e12 (2018).
Su, X., Lu, W. & Rabinowitz, J. D. Metabolite Spectral Accuracy on Orbitraps. Anal. Chem. 89, 5940–5948 (2017).
Titchenell, P. M., Chu, Q., Monks, B. R. & Birnbaum, M. J. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat. Commun. 6, 7078 (2015).
Iroz, A. et al. A specific ChREBP and PPARα cross-talk is required for the glucose-mediated FGF21 response. Cell Rep. 21, 403–416 (2017).
Frey, A. J. et al. LC-quadrupole/Orbitrap high-resolution mass spectrometry enables stable isotope-resolved simultaneous quantification and 13C-isotopic labeling of acyl-coenzyme A thioesters. Anal. Bioanal. Chem. 408, 3651–3658 (2016).
Trefely, S., Ashwell, P. & Snyder, N. W. FluxFix: automatic isotopologue normalization for metabolic tracer analysis. BMC Bioinformatics 17, 485 (2016).
Lee, J. V. et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca2+-NFAT signaling. Genes Dev. 32, 497–511 (2018).
Lu, W. et al. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal. Chem. 82, 3212–3221 (2018).
Chong, J. et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46 (W1), W486–W494 (2018).
Neinast, M. D. et al. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab. 29, 417–429.e4 (2018).
Acknowledgements
This work was supported by grants R01CA174761, R01CA228339 and R01DK116005 to K.E.W. S.Z. is supported by pre-doctoral fellowship F99CA222741. C.J. is supported by the American Diabetes Association through post-doctoral fellowship 1-17-PDF-076. K.U. is supported by NIAMS training grant T32AR053461. L.I. is supported by NIGMS training grant T32GM07229. S.T. is supported by the American Diabetes Association through post-doctoral fellowship 1-18-PDF-144. S.F. is supported through the Penn-PORT IRACDA grant K12 GM081259. P.M.T. is supported by K01DK111715. N.W.S. is supported by the NIH grant R03HD092630 and R01GM132261. J.D.R. is supported by the NIH Pioneer Award 1DP1DK113643 and Diabetes Research Center P30 DK019525. We thank S. Berger and P. Mews for providing the AAV.U6.shAcss2.CMV.eGFP.SV40 vector. We thank G. Wu and Y. Saimon for discussions on the microbiome.
Author information
Authors and Affiliations
Contributions
The project was conceptualized and designed by S.Z., C.J. and K.E.W. K.E.W. and J.D.R. guided the study. S.Z. generated LAKO mice and performed most of the mouse experiments, with help from J.L., S.F., A.C. and K.D.M. C.J. performed mouse experiments and most of the LC–MS analyses with help from X.Z. for LC–MS analysis of short-chain fatty acid species. S.T. and N.W.S. performed LC–MS analysis of acyl-CoA species. K.U. isolated and performed experiments on primary hepatocytes. J.L., M.G. and L.I. performed experiments. P.M.T., Z.T.S. and T.P.G. provided guidance on study design. S.Z. prepared figures with input from C.J., J.D.R. and K.E.W. S.Z., C.J. and K.E.W. wrote and edited the manuscript, with input from J.D.R. All authors read and provided feedback on manuscript and figures.
Corresponding author
Ethics declarations
Competing interests
J.D.R. is a consultant to Pfizer and to Colorado Research Partners. All other authors declare no conflicts of interest.
Additional information
Peer review information Nature thanks D. Wade Abbott, Navdeep Chandel, Catherine Postic and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Hepatic ACLY deficiency minimally affects the response to dietary fructose.
a, Schematic of fructolysis and glycolysis feeding into de novo lipogenesis. b, Body weights of wild-type and LAKO mice fed a chow diet (CD) or high-fructose diet (HFrD) for 18 weeks (n = 13 WT-CD; n = 5 LAKO-CD; n = 14 WT-HFrD; and n = 5 LAKO-HFrD mice). Data are mean ± s.d. c, Weights of liver, posterior subcutaneous white adipose tissue (sWAT) and perigonadal adipose tissue (pgWAT) in wild-type and LAKO mice on a chow diet or HFrD for 18 weeks (liver/sWAT/pgWAT: n = 7/7/7 WT-CD; n = 2/5/5 LAKO-CD; n = 6/12/12 WT-HFrD; and n = 3/5/5 LAKO-HFrD). d, Representative histology images of Periodic acid–Schiff (PAS) stain for glycogen and trichrome (TC) for fibrosis in livers from wild-type or LAKO mice on a HFrD. Scale bars, 100 μm. Images are representative of two mice per group in one experiment. e, Triglyceride content in wild-type or LAKO mice on a chow diet or HFrD for 18 weeks (n = 4 WT-CD; n = 3 LAKO-CD; n = 3 WT-HFrD; n = 4 LAKO-HFrD). P values determined by Welch’s t-test. Data in c and e denote mean values.
Extended Data Fig. 2 Hepatic ACLY deficiency results in modest metabolic alterations on a high-fructose diet.
a, Volcano plot of hepatic metabolites in wild-type or LAKO mice on a chow (CD) or high-fructose diet (HFrD) for 4 weeks. Pink dots indicate significant hits as determined by a fold-change (FC) threshold of 2.0, and P-value threshold of 0.1, assuming equal variance. b, Principle component (PC) analysis of log-transformed data in Supplementary Table 1. Each dot represents a unique sample; coloured shading denotes 95% confidence intervals. c, Relative abundance of metabolites, normalized to the wild-type chow-diet-fed (WT-CD) group. P values determined by Welch’s t-test (n = 5 WT-CD; n = 3 LAKO-CD; n = 5 WT-HFrD; and n = 4 LAKO-HFrD mice). Data are mean values.
Extended Data Fig. 3 A high-fructose diet alters hepatic lipid metabolism.
a, Hierarchical clustering of relative hepatic triglyceride abundance in wild-type or LAKO mice on a chow diet (CD) or high fructose diet (HFrD) for 4 weeks. Clustering performed using 1 − Pearson’s correlation and average linkage. b, Relative abundance of hepatic triglycerides composed of 16:0 to 18:1 fatty acids; a subset of the data in a. c, Principle component analysis of log-transformed data in Supplementary Table 2. Each dot represents a unique sample; coloured shading denotes 95% confidence intervals.
Extended Data Fig. 4 Fructose induces steatosis and contributes substantially to newly synthesized fatty acids in the liver independently of ACLY.
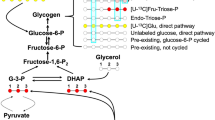
a, Schematic of experimental design of the drinking water study. b, Daily consumption of unsweetened (H2O) or 15% fructose and 15% glucose sweetened (Fruc:Gluc) water per mouse. Each dot represents a repeat measurement, and mean values are shown (n = 6 H2O, n = 7 Fruc:Gluc). P values determined by Welch’s t-test. c, Weight gain of wild-type or LAKO mice given water or fructose:glucose for 4 weeks (n = 4 WT-H2O, LAKO-H2O; n = 8 WT-Fruc:Gluc; and n = 6 LAKO-Fruc:Gluc mice). P values comparing all H2O versus fructose:glucose mice determined by Welch’s t-test. d, Representative H&E and Oil Red O histological stains of livers from mice in c. Scale bars, 100 μm. e, Experimental design for data in Fig. 1c. [U-13C] denotes uniformly labelled 13C. f, Isotopologue distribution of labelled saponified fatty acids in serum shown in Fig. 1c. Data are mean ± s.d.
Extended Data Fig. 5 Fructose signals the use of acetate for de novo lipogenesis.
a, mRNA expression of ChREBP and its target genes in livers of wild-type or LAKO mice fed a chow or high-fructose diet (n = 4 mice per group). P values for WT-CD versus WT-HFrD (blue text) and for LAKO-CD versus LAKO-HFrD (purple text) determined by two-sided t-tests with Holm–Sidak method for multiple comparisons. b, mRNA expression of lipogenic genes in livers of wild-type or LAKO mice given H2O or fructose:glucose water for 4 weeks (n = 4 mice per group). P values for WT-H2O versus WT-Fruc:Gluc, WT-H2O versus WT-Fruc:Gluc (blue font) and LAKO-H2O versus LAKO-Fruc:Gluc (purple font) were determined by two-sided t-test with Holm–Sidak method for multiple comparisons. c, Western blots of lipogenic enzymes in liver lysates of wild-type or LAKO mice given H2O or fructose:glucose water for 4 weeks. Each lane represents an individual mouse. d, Immunohistochemistry staining analysis of ACLY in livers from wild-type or LAKO mice given H2O or fructose:glucose water for 4 weeks. Yellow boxes mark the approximate location of the ×20 panels. Scale bars, 100 μm and 50 μm (for ×20). e, H3K27ac ChIP–quantitative PCR (qPCR) analysis of livers from wild-type mice provided either water for 24 h followed by an oral gavage of saline, or fructose:glucose water for 24 h followed by an oral gavage of 2.0 g kg−1 glucose and 2.0 g kg−1 fructose (n = 3 Mlxipl, Acss2; n = 4 Pklr). Livers were obtained 90 min after gavage. ‘p1’ and ‘p2’ are two different primer sets. f, ChIP–seq tracks of Mlxipl, Pklr and Acss2 genomic loci16. Red bars indicate genomic regions used to design ChIP–qPCR primers. Data in a, b, and e denote mean values.
Extended Data Fig. 6 Depletion of microbiota blocks substrate contribution, but not signalling component, of de novo lipogenesis after fructose consumption.
a, Experimental set-up for antibiotic depletion of the microbiome followed by [13C]fructose tracing into DNL. [U-13C] denotes uniformly labelled 13C. b, Representative images of caecums from a mouse treated with saline or antibiotics. c, Relative abundance of bacteria in caecal contents from mice treated with saline (n = 9) or antibiotics (n = 9), as determined by 16S RT–qPCR to a reference standard of Escherichia coli DNA. P value determined using Welch’s t-test. d, Heat map of microbial metabolite abundance in the portal blood, collected 1 h after gavage. e, f, Relative abundance of [13C]fructose (e) and percentage of total labelled carbons in glucose (f) in portal blood from wild-type or LAKO mice treated with saline or antibiotics, collected 1 h after gavage (n = 7 WT-saline, WT-antibiotics; and n = 4 LAKO-saline, LAKO-antibiotics). P values determined by Welch’s t-test. g, mRNA expression of ChREBPb, Acss2 and Fasn in liver collected 1 h after gavage (n = 4 mice per group). P values determined by two-sided t-tests with Holm–Sidak method for multiple comparisons. Data in c and e–g denote mean values.
Extended Data Fig. 7 Bolus fructose is converted into acetate in a microbiota-dependent manner.
a, TIC of labelled F1P, pyruvate, citrate and acetyl-CoA in liver, concentration of labelled acetate in portal blood, and percentage of labelled carbons in hepatic saponified fatty acids from wild-type mice treated with saline or antibiotics, and gavaged with 2.0 g kg−1 [13C]fructose plus 2.0 g kg−1 unlabelled glucose (n = 3 mice per time point). Data for saline-treated mice are also shown in Fig. 2d. b, Isotopologue distribution of saponified fatty acids in serum from wild-type or LAKO mice fed and treated as in Fig. 3b, and collected 6 h after gavage (n = 8 WT-saline, WT-antibiotics; n = 4 LAKO-saline, LAKO-antibiotics). Data are mean ± s.d.
Extended Data Fig. 8 Bolus fructose-dependent DNL requires microbial acetate and hepatic ACSS2.
a–d, Mice were gavaged with 2.0 g kg−1 [13C]fructose and 2.0 g kg−1 unlabelled glucose. a, Concentrations of labelled acetate, propionate and butyrate in caecal contents from wild-type mice treated with saline or antibiotics (n = 3 mice per time point, except for saline-180 n = 2 mice). b, Concentrations of labelled acetate, propionate and butyrate in portal blood from wild-type mice treated with saline or antibiotics (n = 8 WT-saline, WT-antibiotics; and n = 4 LAKO-saline, LAKO-antibiotics), collected 1 h after gavage. c, Heat map of hepatic triglyceride abundance in livers of mice treated with saline or antibiotics. d, Concentrations of acetate in portal and systemic blood after gavage. Each data point represents an individual mouse. P value determined by two-sided t-tests with Holm–Sidak method for multiple comparisons. e, Weight gain in wild-type and LAKO mice 1 week after tail-vein injection of AAV8-GFP or AAV8-shAcss2. P value determined by Welch’s t-test. f, Liver weight of wild-type and LAKO mice as a percentage of body weight 1 week after tail-vein injection of AAV8-GFP or AAV8-shAcss2. g, Western blots of lipogenic enzymes in liver lysates from wild-type and LAKO mice 1 week after tail-vein injection of AAV8-GFP or AAV8-shAcss2. S6 was used as a loading control.
Extended Data Fig. 9 Gradual fructose consumption promotes greater acetate usage in LAKO mice.
a, Experimental set-up for tracing of [1,2-13C]acetate into DNL before and after gradual administration of fructose. [1,2-13C] denotes 13C labelling of 1 and 2 position carbons in acetate. b, Western blots of lipogenic enzymes in liver lysates from wild-type and LAKO mice after being given fructose:glucose water for 1 or 14 days. c, Representative H&E stains of livers from wild-type and LAKO mice provided fructose:glucose water for 2 weeks. Scale bars, 100 μm. d, Relative abundance of acetate, propionate and butyrate in the caecal contents of wild-type and LAKO mice treated with saline or antibiotics for 1 week (n = 4 mice per group). P values determined by Welch’s t-test.
Extended Data Fig. 10 Fructose provides signal and substrate to promote hepatic DNL.
a, Proposed model of bolus fructose-induced hepatic DNL. Fructose catabolism in hepatocytes acts as a signal to induce DNL genes including Acss2, whereas fructose metabolism by the gut microbiota provides acetate as a substrate to feed DNL, which is mediated by ACSS2. b, Proposed model of gradual fructose-induced hepatic DNL. Similar to the bolus model, fructose catabolism in hepatocytes acts as a signal to induce DNL genes. Catabolism of hepatic fructose and glucose (made from fructose by the small intestine) provides citrate as a substrate to feed DNL, which is mediated by ACLY. Metabolism of fibres and other dietary components by the gut microbiota provides acetate as a substrate to feed DNL, after its conversion to acetyl-CoA by hepatic ACSS2. Image created with BioRender.com.
Supplementary information
Supplementary Table
Supplementary Table 1: Data presented are pool sizes of polar metabolites in livers from chow or high-fructose diet fed mice. Each row is an individual metabolite, and each column is a unique mouse liver sample. Samples from the same treatment group are indicated by name and colour.
Supplementary Table
Supplementary Table 2: Data presented are pool sizes of triglycerides in livers from chow or high-fructose diet fed mice. Each row is an individual triglyceride species, and each column is a unique mouse liver sample. Samples from the same treatment group are indicated by name and colour.
Source data
Rights and permissions
About this article

Cite this article
Zhao, S., Jang, C., Liu, J. et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579, 586–591 (2020). https://doi.org/10.1038/s41586-020-2101-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2101-7
This article is cited by
-
The gut-liver axis in hepatobiliary diseases
Inflammation and Regeneration (2024)
-
Deciphering the gut microbiome of grass carp through multi-omics approach
Microbiome (2024)
-
Interindividual differences in aronia juice tolerability linked to gut microbiome and metabolome changes—secondary analysis of a randomized placebo-controlled parallel intervention trial
Microbiome (2024)
-
Targeted metabolomics reveals plasma short-chain fatty acids are associated with metabolic dysfunction-associated steatotic liver disease
BMC Gastroenterology (2024)
-
Hyperacetylated histone H4 is a source of carbon contributing to lipid synthesis
The EMBO Journal (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
