
Abstract
Despite major progress in defining the functional roles of genes, a complete understanding of their influences is far from being realized, even in relatively simple organisms. A major milestone in this direction arose via the completion of the yeast Saccharomyces cerevisiae gene-knockout collection (YKOC), which has enabled high-throughput reverse genetics, phenotypic screenings and analyses of synthetic-genetic interactions1,2,3. Ensuing experimental work has also highlighted some inconsistencies and mistakes in the YKOC, or genome instability events that rebalance the effects of specific knockouts4,5,6, but a complete overview of these is lacking. The identification and analysis of genes that are required for maintaining genomic stability have traditionally relied on reporter assays and on the study of deletions of individual genes, but whole-genome-sequencing technologies now enable—in principle—the direct observation of genome instability globally and at scale. To exploit this opportunity, we sequenced the whole genomes of nearly all of the 4,732 strains comprising the homozygous diploid YKOC. Here, by extracting information on copy-number variation of tandem and interspersed repetitive DNA elements, we describe—for almost every single non-essential gene—the genomic alterations that are induced by its loss. Analysis of this dataset reveals genes that affect the maintenance of various genomic elements, highlights cross-talks between nuclear and mitochondrial genome stability, and shows how strains have genetically adapted to life in the absence of individual non-essential genes.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
The primary sequencing data have been deposited in the European Nucleotide Archive in the study ERP109205, under the accession numbers detailed in Supplementary Table 1. Gene-specific information is available at http://sgv.gurdon.cam.ac.uk.
Code availability
The custom code used for the analysis of primary sequencing data is available at https://github.com/fabiopuddu/augur-fermentorum (v.0.5) and it relies on Slurm workload manager (v.15.08.13); Samtools v.1.3.1 (using htslib 1.3.1); VCFtools (v.0.1.13); Bcftools v.1.3.1 (using htslib 1.3.1); BWA v.0.7.12-r1039; Python 2.7.12; Perl (v.5.22.1); Gnuplot (v.5.0 patchlevel 3). Code for secondary analyses is available on request.
References
Tong, A. H. et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 (2001).
Giaever, G. et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 (2002).
Giaever, G. & Nislow, C. The yeast deletion collection: a decade of functional genomics. Genetics 197, 451–465 (2014).
Hughes, T. R. et al. Widespread aneuploidy revealed by DNA microarray expression profiling. Nat. Genet. 25, 333–337 (2000).
Lehner, K. R., Stone, M. M., Farber, R. A. & Petes, T. D. Ninety-six haploid yeast strains with individual disruptions of open reading frames between YOR097C and YOR192C, constructed for the Saccharomyces genome deletion project, have an additional mutation in the mismatch repair gene MSH3. Genetics 177, 1951–1953 (2007).
van Leeuwen, J. et al. Exploring genetic suppression interactions on a global scale. Science 354, aag0839 (2016).
Ide, S., Saka, K. & Kobayashi, T. Rtt109 prevents hyper-amplification of ribosomal RNA genes through histone modification in budding yeast. PLoS Genet. 9, e1003410 (2013).
Saka, K., Takahashi, A., Sasaki, M. & Kobayashi, T. More than 10% of yeast genes are related to genome stability and influence cellular senescence via rDNA maintenance. Nucleic Acids Res. 44, 4211–4221 (2016).
Claypool, J. A. et al. Tor pathway regulates Rrn3p-dependent recruitment of yeast RNA polymerase I to the promoter but does not participate in alteration of the number of active genes. Mol. Biol. Cell 15, 946–956 (2004).
Mansisidor, A. et al. Genomic copy-number loss is rescued by self-limiting production of DNA circles. Mol. Cell 72, 583–593.e4 (2018).
Carlsson, M., Gustavsson, M., Hu, G.-Z., Murén, E. & Ronne, H. A Ham1p-dependent mechanism and modulation of the pyrimidine biosynthetic pathway can both confer resistance to 5-fluorouracil in yeast. PLoS ONE 8, e52094 (2013).
Askree, S. H. et al. A genome-wide screen for Saccharomyces cerevisiae deletion mutants that affect telomere length. Proc. Natl Acad. Sci. USA 101, 8658–8663 (2004).
Gatbonton, T. et al. Telomere length as a quantitative trait: genome-wide survey and genetic mapping of telomere length-control genes in yeast. PLoS Genet. 2, e35 (2006).
Rog, O., Smolikov, S., Krauskopf, A. & Kupiec, M. The yeast VPS genes affect telomere length regulation. Curr. Genet. 47, 18–28 (2005).
Murray, A. W., Schultes, N. P. & Szostak, J. W. Chromosome length controls mitotic chromosome segregation in yeast. Cell 45, 529–536 (1986).
Sullivan, M., Higuchi, T., Katis, V. L. & Uhlmann, F. Cdc14 phosphatase induces rDNA condensation and resolves cohesin-independent cohesion during budding yeast anaphase. Cell 117, 471–482 (2004).
Indjeian, V. B., Stern, B. M. & Murray, A. W. The centromeric protein Sgo1 is required to sense lack of tension on mitotic chromosomes. Science 307, 130–133 (2005).
Daniel, J. A., Keyes, B. E., Ng, Y. P. Y., Freeman, C. O. & Burke, D. J. Diverse functions of spindle assembly checkpoint genes in Saccharomyces cerevisiae. Genetics 172, 53–65 (2006).
Ben-Shitrit, T. et al. Systematic identification of gene annotation errors in the widely used yeast mutation collections. Nat. Methods 9, 373–378 (2012).
Zhu, J. et al. Single-cell based quantitative assay of chromosome transmission fidelity. G3 (Bethesda) 5, 1043–1056 (2015).
Argueso, J. L. et al. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc. Natl Acad. Sci. USA 105, 11845–11850 (2008).
Vernon, M., Lobachev, K. & Petes, T. D. High rates of “unselected” aneuploidy and chromosome rearrangements in tel1 mec1 haploid yeast strains. Genetics 179, 237–247 (2008).
Lemoine, F. J., Degtyareva, N. P., Lobachev, K. & Petes, T. D. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120, 587–598 (2005).
Yona, A. H. et al. Chromosomal duplication is a transient evolutionary solution to stress. Proc. Natl Acad. Sci. USA 109, 21010–21015 (2012).
Rancati, G. et al. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135, 879–893 (2008).
Selmecki, A. M. et al. Polyploidy can drive rapid adaptation in yeast. Nature 519, 349–352 (2015).
Veatch, J. R., McMurray, M. A., Nelson, Z. W. & Gottschling, D. E. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 137, 1247–1258 (2009).
Taylor, S. D. et al. The conserved Mec1/Rad53 nuclear checkpoint pathway regulates mitochondrial DNA copy number in Saccharomyces cerevisiae. Mol. Biol. Cell 16, 3010–3018 (2005).
Huang, M., Zhou, Z. & Elledge, S. J. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 94, 595–605 (1998).
Parsons, A. B. et al. Integration of chemical–genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat. Biotechnol. 22, 62–69 (2004).
Woolstencroft, R. N. et al. Ccr4 contributes to tolerance of replication stress through control of CRT1 mRNA poly(A) tail length. J. Cell Sci. 119, 5178–5192 (2006).
Hendry, J. A., Tan, G., Ou, J., Boone, C. & Brown, G. W. Leveraging DNA damage response signaling to identify yeast genes controlling genome stability. G3 (Bethesda) 5, 997–1006 (2015).
Sabouri, N., Viberg, J., Goyal, D. K., Johansson, E. & Chabes, A. Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res. 36, 5660–5667 (2008).
Huang, M. & Elledge, S. J. Identification of RNR4, encoding a second essential small subunit of ribonucleotide reductase in Saccharomyces cerevisiae. Mol. Cell. Biol. 17, 6105–6113 (1997).
Pereira, C., Pereira, A. T., Osório, H., Moradas-Ferreira, P. & Costa, V. Sit4p-mediated dephosphorylation of Atp2p regulates ATP synthase activity and mitochondrial function. Biochim. Biophys. Acta Bioenerg. 1859, 591–601 (2018).
Endo, K., Tago, Y., Daigaku, Y. & Yamamoto, K. Error-free RAD52 pathway and error-prone REV3 pathway determines spontaneous mutagenesis in Saccharomyces cerevisiae. Genes Genet. Syst. 82, 35–42 (2007).
Liefshitz, B., Parket, A., Maya, R. & Kupiec, M. The role of DNA repair genes in recombination between repeated sequences in yeast. Genetics 140, 1199–1211 (1995).
Zou, X. et al. Validating the concept of mutational signatures with isogenic cell models. Nat. Commun. 9, 1744 (2018).
Nik-Zainal, S. et al. Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 (2012).
Costanzo, M. et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420 (2016).
Herzog, M. et al. Detection of functional protein domains by unbiased genome-wide forward genetic screening. Sci. Rep. 8, 6161 (2018).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Seibt, K. M., Schmidt, T. & Heitkam, T. FlexiDot: highly customizable, ambiguity-aware dotplots for visual sequence analyses. Bioinformatics 34, 3575–3577 (2018).
Shampay, J., Szostak, J. W. & Blackburn, E. H. DNA sequences of telomeres maintained in yeast. Nature 310, 154–157 (1984).
Rubinstein, L. et al. Telomere length kinetics assay (TELKA) sorts the telomere length maintenance (tlm) mutants into functional groups. Nucleic Acids Res. 42, 6314–6325 (2014).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
McLaren, W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics 26, 2069–2070 (2010).
Huh, W.-K. et al. Global analysis of protein localization in budding yeast. Nature 425, 686–691 (2003).
Acknowledgements
We thank J. Warringer for providing the YKOC and discussing results; E. Alonso-Perez for help with YKOC management; the Cancer Genome Project and DNA sequencing pipelines at the Wellcome Sanger Institute for help with sample submission, tracking, library preparation and sequencing; C. Bradshaw for help with HPC cluster and nanopore sequencing; R. Wellinger for providing mutant strains; V. Mustonen, I. Vazquez-Garcia, M. Stratton, D. Adams, S. Nik-Zainal and members of the laboratory of S.P.J. for advice. This research was supported by: Wellcome Strategic Award 101126/Z/13/Z (COMSIG); Wellcome Investigator Award 206388/Z/17/Z; Wellcome PhD Fellowship 098051 to M.H.; Cancer Research UK Programme Grant C6/A18796; Cancer Research UK C6946/A24843 and Wellcome WT203144 Institute Core Funding; and the National Institutes of Health R35 GM118172 to R.L.
Author information
Authors and Affiliations
Contributions
The project was conceived by F.P. and S.P.J. YKOC duplication was carried out by I.S. and F.P. Single-colony isolation and DNA extractions were carried out by A.S. and S.W. Data analyses were carried out by F.P., M.H. and I.A. Logistic regression and telomere length analysis were carried out by R.S., R.M., S.K.-L. and M.K. Chromosome instability measurements were performed by J.Z. and M.G. under the supervision of R.L. Validation experiments were designed by F.P., and carried out by F.P. and G.M.-Z. The manuscript was written by F.P. and S.P.J., with contributions from G.M.-Z., M.H., M.K., I.S., J.Z., I.A. and R.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Peer review information Nature thanks Grant Brown, Sergei Mirkin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
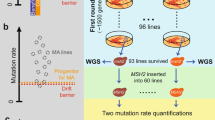
Extended Data Fig. 1 Statistics of YKOC analyses, and distribution of estimates of repetitive DNA across the YKOC.
a, Colonies that did not carry the expected deletion (red) were re-assigned by reading the barcode inserted with the deletion marker; deletion of an alternative gene was then confirmed by loss of sequencing coverage. b, Number of strains proceeding through the steps of the pipeline for data generation, and analysis used to create the dataset on which this work is based. c, Distribution of estimates of copy numbers for the indicated repeats across the YKOC. Strains are sorted by the average across the colonies sequenced, and the estimate of each colony is shown. Red zones represent values >3 s.d. of the wild-type distribution. n = 8 biologically independent samples for wild-type strains; n = 1 sample for 258 of the knockout strains; n = 2 biologically independent samples for 4,097 of the knockout strains; n = 3 biologically independent samples for 30 of the knockout strains; n = 4 biologically independent samples for 72 of the knockout strains; and n = 5 biologically independent samples for 1 of the knockout strains. d, Correlations between changes in relative copy numbers at rDNA and CUP1 tandem-repeat loci. Average correlations in all colonies of each knockout strain are shown. n = 8 biologically independent samples for wild-type strains; n = 1 sample for 258 of the knockout strains; n = 2 biologically independent samples for 4,097 of the knockout strains; n = 3 biologically independent samples for 30 of the knockout strains; n = 4 biologically independent samples for 72 of the knockout strains; and n = 5 biologically independent samples for 1 of the knockout strains. e, No overall correlation for estimates of rDNA and CUP1 copy numbers across all colonies sequenced. f, Distribution of estimates of copy number for the 2μ plasmid across the YKOC, and Gene Ontology analysis of the hits. The 2μ copy numbers did not follow a normal distribution. Instead, the maximum s.d. increases linearly with the mean copy number; this is consistent with the mode of 2μ amplification, which is activated by expression of the in cis gene FLP1 when the copy number crosses a lower threshold (we estimate this to be at 20–25 copies). Different durations of FLP1 expression will result in different increases in copy number. We detected an enrichment in gene knockouts connected to gene silencing in strains with high 2μ copy numbers, which is also consistent with this 2μ amplification mechanism.
Extended Data Fig. 2 Development of tools to study the instability of repetitive DNA.
a, Left, schematic of yeast chromosome XII. The apparent increase in sequencing coverage maps to rDNA repeats. Right, similar apparent coverage increases mapped to CUP1 and Ty transposon loci. b, WGS estimates of copy numbers of rDNA repeats linearly correlate with estimates obtained by pulsed-field gel electrophoresis. n = 3 or 4 biologically independent samples per strain. c, Per cent deviation of two sequencing technical replicates from their average. n = 2 technical replicates derived from n = 89 biologically independent samples. Median, yellow line; quartiles, blue line. Measurements were within 5% of their average. Notable exceptions were Ty5 and CUP1, probably owing to their relatively low repeat numbers. d, Relative estimated content of telomeric repeats in indicated strains (normalized to the estimated content of one wild-type colony) is plotted as a function of the minimum number of telomeric repeats in a sequencing read required to classify that read as telomeric. n = 2 biologically independent samples per strain. e, Estimates of telomere length for wild-type, tel1∆ and rif1∆ strains obtained by calculating the relative abundance of telomeric reads. Mean from n = 4–8 biologically independent samples per strain. f, Estimates of rDNA, CUP1, Ty1, Ty2 and mtDNA copy numbers, and telomeric DNA content for MATa, MATα and diploid strains in W303 and BY4743 backgrounds. Median from n = 8 biologically independent samples per strain. g, A long read spanning the CUP1 locus, derived from Oxford Nanopore Technology sequencing of a W303 (K699) genomic library. h, Comparison of CUP1 copy number estimated by qPCR or by WGS. The same DNA samples (as indicated by the labels) were analysed. Two estimates were extracted from WGS data: ‘from CUP1’ indicates estimation using a large region of the CUP1 locus and the genome-wide median for reference (the same method used for the entire YKOC); ‘from qPCR amplicon’ indicates a small region of CUP1 and a small region of GAL1 for reference (the same regions used for qPCR).
Extended Data Fig. 3 Southern blot analysis and functional connections between TLM genes.
a, Gel electrophoresis and Southern blot analysis of telomeres for 14 newly identified predicted TLM strains (‘hits’). Hits denote strains with two or more colonies with measures >3× the s.d. of the wild-type distribution. Twenty-one strains that failed these stringent hit-selection criteria (non-hits), but which displayed relatively high or low estimates of telomere length, are also shown. Representative images from two independent experiments. Purple lines, location of molecular mass markers; orange line, average telomere length for wild-type samples; green dashes, average telomere lengths for strains predicted to have longer telomeres; white dashes, average telomere lengths for strains predicted to have shorter telomeres. b, Validation of gene knockouts that failed the TLM selection criteria but that displayed relatively high or low telomere counts. c, Network-graph analysis of gene knockouts that affect telomere length, highlighting the newly identified genes that were validated by Southern blotting.
Extended Data Fig. 4 Examples of aneuploidies and chromosomal rearrangements in the YKOC.
a, Example of a strain with fractional aneuploidy of chromosome XII, which probably reflects clonal heterogeneity. b, Distribution of fractional and non-fractional aneuploidies per chromosome. n = 8,843 biologically independent samples. c, Knockout of genes that encode subunits of ribosomal proteins frequently leads to gain of the chromosome that carries the paralogue gene. d, Ploidy plots of chromosome II for two different colonies of the hta1∆, swi4∆ and spt10∆ knockout strains. hta1∆ cells (in which one of the two genes that encodes histone H2A is deleted) accumulate a specific amplification of a genome region that contains the paralogue HTA2, a centromere and two origins of replication. This is most probably transmitted as a circular genetic element formed by recombination between two adjacent transposon sequences. Only two other YKOC strains were found to carry the same genetic element: these were spt10∆ and swi4∆, which encode factors that control the transcription of cell-cycle-regulated genes (including histones).
Extended Data Fig. 5 Calculation of mtDNA copy number.
a, Sequencing coverage across the mitochondrial genome of a wild-type haploid strain (BY4741; accession ERS616991). Shaded areas indicate regions (that loosely correspond to COX1 and COX3 genes) that were used to estimate total mtDNA content. b, mtDNA regions of low sequence coverage correspond to regions with strongly reduced GC content. c, Comparison of mtDNA content estimated by qPCR and by WGS. The same DNA samples (as indicated by the labels) were analysed by qPCR and WGS. Two estimates were extracted from WGS data: ‘from COX1’ indicates estimates using a large region of the COX1 gene and the genome-wide median for reference (the same method used for the entire YKOC); ‘from qPCR amplicon’ indicates a small region of COX1 and a small region of GAL1 for reference (the same regions used for qPCR). d, Correlation between estimates of mtDNA content using COX1 or COX3 region on all sequenced strains belonging to the YKOC. Pearson R2 = 0.7596.
Extended Data Fig. 6 Connections between mtDNA and nuclear genome alterations.
a, Venn diagram showing the overlap between genes identified as rho0 by our sequencing, genes that encode mitochondria proteins (ref. 48) and gene knockouts for which respiratory growth was annotated as ‘absent’ (in the Saccharomyces Genome Database (SGD), http://www.yeastgenome.org). b, Gene Ontology of rho0 strains (estimated mtDNA copy number < 1) and rho++ strains (estimated mtDNA copy number > 20.3). Bonferroni-corrected P values. c, Sixteen gene knockouts from the top end of the mtDNA distribution were assessed for spontaneous activation of the DNA-damage response by Rad53 and histone H2A phosphorylation (representative images from two technical replicates; source data in Supplementary Fig. 1) and RNR expression (average from three technical replicates, one biological sample per strain). Strains with increased RNR expression (violet) or increased RNR expression and Rad53 hyperphosphorylation (yellow) are highlighted. Serial dilutions of the same cultures were also tested for HU sensitivity. d, Comparisons of mtDNA estimates with systematic analysis of HU sensitivity. HU-sensitive strains are highlighted in different colours depending on the study. ‘Parsons, 2004’ denotes ref. 30, n = 62 biologically independent samples; ‘Woolstencroft, 2006’ denotes ref. 31, n = 33 biologically independent samples. e, Comparison of predicted mtDNA copy number and RNR3 expression levels. Gene knockouts with increased levels of Rnr3 protein (blue, Z-score > 2); gene knockouts with increased mtDNA (yellow, mtDNA > 22.2); gene knockouts with both measures increased (green). n = 4,436 by knockout averages, of n = 8,843 biologically independent samples.
Extended Data Fig. 7 mtDNA content in knockout strains for genes that encode enzymes involved in tryptophan metabolism.
Pathway for tryptophan biosynthesis from phosphoenolpyruvate, tryptophan import and NAD biosynthesis from tryptophan are depicted along with estimates of mtDNA copy number for strains that lack each of the enzymes in the pathways. Mean from n = 8 (wild type) or 2 (knockout) biologically independent samples.
Extended Data Fig. 8 Genes that frequently carry mutations contain repetitive regions.
Self dot plots that highlight degenerate repetitive regions in the DNA sequence of genes that are found to be frequently mutated in the YKOC. Plots were obtained using FlexiDot.
Extended Data Fig. 9 The most-frequent YKOC mutations, and their distributions between different source laboratories.
The most-frequent mutations, with predicted effects on genes, detected in the YKOC (top 200) and their distribution among different source laboratories. a, Left, the mutation is indicated by its predicted effect, and the background indicates whether it is a mutation in a gene with degenerate repeats (grey), a mutation that comes from a founder effect (yellow) or a frequently mutated site (green). Genes in bold indicate a homozygous mutation. Centre, heat map of the distribution of the most-frequent mutations by the laboratory in which the strain that carries that mutation was produced; 100% indicates that all the strains with a particular mutation were generated in the same laboratory. Right, number of strains that carry the mutation. b, Not all strains derived from each laboratory share founder mutations. As in a, but a value of 100% in the heat map indicates that all the strains generated by a particular laboratory have the mutation.
Extended Data Fig. 10 Overview of genomic instability caused by knockouts of non-essential genes.
a, Overview of results from our genome instability screens. Strains with an abnormal copy number for different genomic features, aneuploidies and chromosomal rearrangements (CR) are represented by coloured boxes. b, Gene Ontology analysis for 151 genome instability genes, defined as knockouts that show 3 or more abnormal features. The numbers of genes in each Gene Ontology category, as well as Holm–Bonferroni-corrected P values, are reported. c, Genome instability genes were manually sorted into classes on the basis of their function, inferred from annotations in the SGD.
Supplementary information
Supplementary Figure 1
Spontaneous checkpoint activation in different KO strains. Source data for western blot in Extended Data Figure 6C.
Rights and permissions
About this article

Cite this article
Puddu, F., Herzog, M., Selivanova, A. et al. Genome architecture and stability in the Saccharomyces cerevisiae knockout collection. Nature 573, 416–420 (2019). https://doi.org/10.1038/s41586-019-1549-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1549-9
This article is cited by
-
Insufficient evidence for non-neutrality of synonymous mutations
Nature (2023)
-
Effects of aneuploidy on cell behaviour and function
Nature Reviews Molecular Cell Biology (2022)
-
Directed yeast genome evolution by controlled introduction of trans-chromosomic structural variations
Science China Life Sciences (2022)
-
Adaptive laboratory evolution in S. cerevisiae highlights role of transcription factors in fungal xenobiotic resistance
Communications Biology (2022)
-
The metabolic growth limitations of petite cells lacking the mitochondrial genome
Nature Metabolism (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
