
Abstract
Epigenetic research has accelerated rapidly in the twenty-first century, generating justified excitement and hope, but also a degree of hype. Here we review how the field has evolved over the last few decades and reflect on some of the recent advances that are changing our understanding of biology. We discuss the interplay between epigenetics and DNA sequence variation as well as the implications of epigenetics for cellular memory and plasticity. We consider the effects of the environment and both intergenerational and transgenerational epigenetic inheritance on biology, disease and evolution. Finally, we present some new frontiers in epigenetics with implications for human health.
Similar content being viewed by others

Main
Biologists have long sought to understand how a fertilized egg can form an organism composed of hundreds of specialized cell types, each expressing a defined set of genes. Cellular identity is now accepted to be the result of the expression of specific combinations of genes (Fig. 1). This expression pattern must be established and maintained—two distinct, but connected, processes. The pluripotency of the initial cell and the establishment of cell types depend to a large extent on the coordinated deployment of hundreds of transcription factors that bind to specific DNA sequences to activate or repress the transcription of cell lineage genes1. This establishment phase corresponds most closely to what is generally cited as the first definition of epigenetics by Conrad Waddington, namely the study of the mechanisms by which the genotype produces the phenotype in the context of development2. The maintenance phase often involves a plethora of non-DNA sequence specific chromatin cofactors that set up and maintain chromatin states through cell division and for extended periods of time—sometimes in the absence of the initial transcription factors3. This phase is more akin to a definition of epigenetics put forward by Nanney4, then elaborated on by Riggs and Holliday5,6,7 and further modified by Bird8 and others9 to mean the inheritance of alternative chromatin states in the absence of changes in the DNA sequence. DNA methylation was proposed early on as a carrier of epigenetic information with subsequent work revealing that chromatin proteins and noncoding RNAs are also important for this process10,11,12,13,14. For example, histone variants and histone modifications can influence local chromatin structure, either directly or indirectly. Such modifications can be heritable but reversible and are governed by a series of writers (that deposit them), readers (to interpret them) and erasers (to remove them). Finally, higher-order 3D chromosome folding is also thought to modulate gene expression and might contribute to inheritance15.

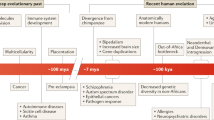
Starting from the zygotic genome, stage- and cell-type-specific transcription factors initiate regulatory cascades that induce cell differentiation. Epigenetic components (for example, Polycomb PRC1/2 and Trithorax group proteins) maintain the ‘off’ states of certain genes and the ‘on’ states of others, in a cell-type- and time-specific manner (the bottom panels show three genes, depicted schematically as chromatinized templates, in which transcription is triggered by specific transcription factors and silent or active states are maintained by PRC1/2 or Trithorax proteins, respectively). In doing so, they constitute barriers against accidental reprogramming that maintain developmental and physiological homeostasis. Altered epigenomes can lead to changes in programmed cell differentiation or, when accidental, to disease (bottom right). Germline reprogramming resets the majority (but not all) of the epigenome to achieve reproduction (top right).

Since 1942, when the word was first coined, epigenetics has been redefined multiple times16 (Table 1). In this Review, we use epigenetics to mean “the study of molecules and mechanisms that can perpetuate alternative gene activity states in the context of the same DNA sequence”. This operational definition has several implications. First, it encompasses transgenerational inheritance as well as mitotic inheritance and the persistence of gene activity or chromatin states through extended periods of time, even without cell division—for instance, in long-lived post-mitotic cells such as adult neurons. Second, the DNA sequence to be considered depends on the biological system. In mitotic inheritance, one should consider the genomic sequence of individual cells, whereas in transgenerational inheritance one should consider the DNA of the whole organism (including its microbiota, if this can contribute to inheritance). Finally, this definition explicitly extends the usage of ‘epigenetic’ to regulatory processes that involve molecules known to participate in epigenetic inheritance, even when not addressing the epigenetic memory function per se. We argue that this common practice should be accepted, as it conveys to non-specialists the broader field of epigenetic research. We also note that cases of inheritance that do not involve chromosomal components have been documented14,17,18 and it will be important to study how widespread they are and whether similar phenomena occur in humans.
Here, we review the interplay between regulatory plasticity and stable epigenetic heritability, including cell fate and reprogramming events that occur during development, in response to physiological stimuli, and in disease. We discuss how noncoding RNAs, DNA methylation, heterochromatin, Polycomb and Trithorax proteins and 3D genome architecture (Box 1) can regulate both inheritance and gene expression plasticity, and how new technologies allow these phenomena to be analysed in a spatiotemporal fashion, in small numbers of cells or even single cells, and at multiple scales from the nucleotide to the chromosome (Box 2). We discuss evidence for a hotly debated topic—epigenetic inheritance across generations—particularly focusing on mammalian examples because of the potential biomedical implications. We also consider two other important new research areas: the potential influence of the environment and the effects of epigenetic changes on genome integrity. In closing, we highlight how epigenetic research may benefit human health.
Epigenetic inheritance versus plasticity
An appreciation of the role of chromatin as a carrier of epigenetic information that can propagate active and silent activity states during cell division came from the study of different biological processes and model organisms. These include, to name but a few, heterochromatin inheritance in yeast, X-chromosome inactivation (the process by which one of the copies of the female X chromosome is silenced), or genomic imprinting (the parent-of-origin-specific repression of certain genes) in mammals; vernalization (the induction of flowering by exposure to prolonged cold during winter) in plants; position effect variegation (the silencing of a gene in some cells through its abnormal juxtaposition to heterochromatin) in Drosophila. These studies demonstrated that differentially expressed states can be transmitted across cell divisions, once they are established and in the absence of the original signal. Studies of cellular reprogramming in the germline and early embryogenesis19,20,21,22, during induced pluripotency (iPS)23,24, or upon somatic nuclear transfer25,26 have shown that chromatin and DNA methylation act as important ‘epigenetic barriers’ (Fig. 1) that prevent changes in gene expression and cell identity.
Epigenetic systems (Box 1) include heterochromatin (HP1 and H3K9me3 (trimethylation of histone 3 lysine 9)), Polycomb (PRC1 and PRC2) and Trithorax (COMPASS (complex proteins associated with SET1)) complexes. These complexes are thought to perpetuate functional responses by modifying histone proteins in chromatin and by binding their own histone marks in order to convey stable inheritance. Indeed, nucleosomes are subject to constant remodelling, histones are exchanged and all DNA and histone marks discovered so far are reversible, although the rates of exchange and the stability of the marks vary in different genomic domains27. Therefore, most regulatory signals would be rapidly lost in the absence of tight self-reinforcing loops that maintain the memory of the chromatin state28. Furthermore, the inheritance of epigenetic marks through cell division requires that they survive DNA replication and mitosis (Fig. 2). This is particularly relevant for histone modifications, because nucleosomes do not have a DNA template-based duplication system. Deposition of parental H3 and H4 histones occurs within few hundred base pairs of their pre-replication position and, upon replication, they are roughly equally distributed to the leading and the lagging strand daughter DNA molecules, through the action of dedicated molecular complexes29,30. Chromatin maturation factors, including DNMT1–UHRF1, EZH2 and HP1, use the proliferating cell nuclear antigen (PCNA; a DNA clamp that is essential for replication) or origin recognition complex (ORC) proteins as tethering components31,32,33,34 (Fig. 2a). In addition, Polycomb components utilize their DNA-anchoring factors to propagate mitotic memory. Loss of the target DNA sequence elements results in loss of PcG proteins and of gene silencing within a few cell divisions in Drosophila35,36, although sequence-independent propagation of silencing can be maintained in mammalian cell culture37. Mitotic retention of regulatory components (Fig. 2c), including transcription factors and some of the epigenetic machineries described above38,39, has been well-documented in recent years40,41. Inheritance through meiosis is also possible at least to some extent, as shown by the ability of maternally deposited H3K27me3 to control DNA methylation-independent imprinting42,43. An additional possibility is that only a fraction of the marks can be meiotically transmitted, but this might be sufficient to reconstruct chromatin organization in the subsequent generation44.

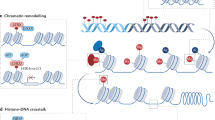
a, DNA replication during the S phase of the cell cycle is a challenge to the maintenance of nucleosome marks. Epigenetic components, such as HMTs and UHRF1, interact with components of the DNA replication machinery, such as the PCNA clamp, in order to reconstitute chromatin domains after the passage of the fork. The case of DNA methylation is depicted schematically. Newly replicated DNA is unmethylated (empty lollipops; the methylated template DNA strand is not shown here for simplicity). The UHRF1/DNMT1 complex associated with PCNA facilitates remethylation of hemimethylated DNA after DNA replication. b, Both constitutive (involving H3K9 methylases and HP1) and facultative (involving PRC1 and PRC2) heterochromatin, as well as euchromatic features (involving an interplay between PRC1, PRC2, Trithorax/COMPASS and ATP-dependent chromatin remodelling complexes), are stably maintained during interphase in order to prevent genes from inappropriately switching their functional states. SWI/SNF is a nucleosome remodelling complex. c, During mitosis, most chromosome-associated factors are evicted during chromosome condensation, but ‘mitotic bookmarking’ of genes is achieved by the maintenance of key components (such as certain transcription factors or RNA polymerase III) bound to their target loci.
Owing to the lack of a precise ‘replication’ process for parental nucleosomes and to the loss of many DNA-binding factors and chromatin-associated components during mitosis and meiosis, the inheritance of single nucleosome marks poses specific challenges28. Mathematical modelling and biological evidence suggest that chromatin heritability requires the establishment of domains of several or even hundreds of kilobases in size45,46,47. Indeed, the genome is now known to be hierarchically organized in a series of 3D structures, starting from nucleosome clutches, to chromatin loops, to chromosomal domains called topologically associating domains (TADs), and finally to active or repressive compartments and chromosome territories15,46,48,49,50. TADs and compartments might stabilize functional states and drive their own inheritance. Furthermore, multiple epigenetic machineries often act together to stabilize heritable states. For example, PRC2 collaborates with PRC1 complexes and DNA methylation is sustained by heterochromatin proteins and/or small RNA pathways51. In summary, epigenetic inheritance can involve multiple layers; and usually entails the cooperation of partially overlapping signals, initially dependent on DNA sequence (elicited by transcription factor binding or RNA-mediated mechanisms). Each of these layers adds a degree of stability, but each of them is also reversible, allowing plasticity in the presence of regulatory cues47,52. The inheritance of chromatin states in the absence of chromatin domains, or without self-reinforcing mechanisms, is more challenging28. This might require retention of transcription factors, histone variants and histone modifiers during DNA replication and mitotic bookmarking53.
Epigenetics and DNA sequence variation
DNA sequence variation and epigenetics are inextricably linked. Chromatin states can influence transcription factor binding54, and DNA sequence polymorphism influences chromatin states. Chromatin and DNA methylation display extensive variation in humans55. Furthermore they regulate genome stability and mutability. Transposable elements are frequent targets of epigenetic silencing that can sometimes be environmentally influenced and can influence gene expression as well as genome integrity.
Genetic effects on epigenetics
The genome of each individual experiences both natural and environmentally induced mutations. While most mutations are neutral, sequence polymorphisms can affect epigenomic landscapes. For example, analysis of chromatin accessibility and ‘CCCTC-binding factor’ (CTCF) DNA binding in parents and children from families with different ancestry found a substantial percentage of bound sites that were unique to each ancestry, with differential binding being explained mainly by genetic variation56. As CTCF can affect 3D genome architecture and gene expression, this finding suggests that rewiring of epigenomic landscapes might frequently occur as a consequence of mutations. On the other hand, mutations that affect histone and DNA methyltransferases, or demethylases (TET enzymes), chromatin remodellers and other chromatin factors including histones, are frequently found in disease57 and their effects may be specifically targeted by therapeutic interventions58. The often cryptic relationship between DNA sequence and epigenetic changes can mean that mutations may be overlooked or mistaken for epimutations, leading to misconceptions about the driver versus passenger role for epigenetic changes59. This issue should be partly addressed by cheaper and faster sequencing methods, which will be able to produce genomic and epigenomic information from the same sample.
Chromatin and DNA methylation in mutagenesis
Mutation rates vary in different parts of the genome, at different stages of the life cycle and in diseases such as cancer, where they depend on the cell of origin, environmental exposure and cancer type60,61,62. Mutation rates can be affected by DNA methylation63 and nucleosome positioning64. Higher-order chromosome folding also influences mutagenicity. A large-scale survey of balanced chromosomal abnormalities in patients with congenital anomalies revealed disruption of TADs encompassing known syndrome-linked loci in 7.3% of cases65, and a combination of Hi-C chromosome capture with whole-genome sequencing in multiple myeloma showed significant enrichment of copy number variation breakpoints at TAD boundaries66 that are frequently bound by CTCF. Furthermore, CTCF is frequently mutated in human cancer57. Hypermutation of the heterochromatic inactive X chromosome has also been noted in cancer and may be due to DNA replication stress in aberrantly proliferating cells67.
The role of the repetitive genome
Transposable elements are intimate components of genomes, with gene regulatory potential that may lead to phenotypic diversity. Indeed, transposable elements and their relics constitute a major fraction of most eukaryotic genomes. McClintock proposed that transposons were turned on or off by environmental changes or during development, acting as ‘control elements’. We now know that transposons can influence gene activity in multiple ways, acting as regulatory elements or interfering with transcription68. Genomes have evolved species-specific mechanisms to limit transposon activity, for example by targeting repressive heterochromatin machinery, either through specific RNAs or DNA binding factors. In Drosophila, heterochromatin-dependent mechanisms allow the expression of specific clusters of transposon relics in order to produce PIWI-interacting RNAs (piRNAs) that, in turn, inhibit transposition. piRNAs are maternally heritable and can be amplified via a ping-pong system, effectively allowing the organism to resist new invasions and adapt their genome to them69. Caenorhabditis elegans uses heterochromatin components to prevent illegitimate repetitive DNA transcription and genome instability70. Plants produce small RNAs derived from double-stranded precursors, which are synthesized by dedicated polymerases and target DNA methylation and the H3K9 methylation machinery71. Finally, numerous strategies are deployed in mammals, the most recently characterized being the repression of endogenous retroviruses (ERVs) by the KAP1 protein (also known as TRIM28), which co-recruits heterochromatin proteins such as SETDB1 by interacting with Krüppel-associated box (KRAB) domain-containing zinc-finger proteins (KZFPs). This strategy also enables the rapid evolution of gene regulation strategies via binding of KZFP to ERVs near genes72, thus influencing gene expression dynamics and levels.
Environmental epigenetics
Recently the influence of the environment in development and physiology has been underlined. Gene × environment interactions determine how individuals with the same or different genotypes will respond to environmental variation. The importance of epigenetics in environmental responses is well-established in plants, particularly in Polycomb-based vernalization73, but similar processes appear to take place in some animal species.
Environmental epigenetic regulation in animals
In Drosophila, environmentally induced phenotypes that depend on epigenetic regulation involve transmission across several generations74,75,76. C. elegans has been shown to translate several environmental stimuli, such as viral infection, starvation or elevated temperatures, into modification of epigenetic components77,78,79. Whereas starvation and viral infection induce inheritance via the production of small RNAs77,78, apparently without involvement of chromatin, temperature-dependent epigenetic inheritance involves the H3K9 methylation machinery (SET-25)79, without RNA interference (RNAi), suggesting that, depending on the type of stimulus, the RNAi machinery and chromatin regulators can act differently to drive inheritance.
Examples of environmental effects are by no means limited to model organisms. Temperature is a major sex-determining factor in many reptiles. In a turtle species in which sex is determined by temperature during egg incubation, the KDM6B H3K27me3-specific demethylase exhibits sexually dimorphic, temperature-dependent expression that regulates the sex-determining gene Dmrt180. In Australian central bearded dragons, chromosomal sex determination is overridden by high temperatures to produce sex-reversed female offspring. Temperature induces alternative splicing of KDM6B and of JARID2, a PRC2-recruiting component81. It is intriguing that temperature affects PRC2 factors in diverse animal and plant species, suggesting that temperature sensing by PRC2 might be evolutionarily conserved, although this is not the only environmental effect that can stably modify chromatin. Another form of environmentally induced chromatin regulation is found in social insects such as the carpenter ant Camponotus floridanus, in which the balance between morphologically distinct worker castes depends on the levels of histone acetylation, which may be influenced by feeding behaviour82.
Metabolism and epigenetics in mammals
DNA and chromatin modifications use metabolic products. For example, S-adenosylmethyonine (SAM) is the methyl donor in DNA and histone methylation; folate and vitamins B6 and B12 induce SAM production; α-ketoglutarate (αKG) is required for DNA and histone demethylation; succinate and fumarate inhibit DNA and histone demethylases; acetyl-coenzyme A is the acetyl donor for histone acetylation; β-hydroxybutyrate inhibits class I histone deacetylases; and the NAD+/NADH ratio regulates the sirtuins (class III histone deacetylases). Therefore, metabolic alterations can induce global perturbations of the epigenome and mutant metabolic components represent potential therapeutic targets83,84. On the other hand, metabolic changes can affect specific loci and induce long-lasting epigenetic modifications, including intergenerational epigenetic inheritance85,86,87. The effectors of these perturbations are DNA methylation, Polycomb components, and transfer RNA (tRNA) fragments, which, among other effects, repress genes associated with endogenous retroelements and might thereby help to preserve genome integrity87,88,89. A protein restriction diet in mice can also induce DNA methylation and repression of a subset of ribosomal DNA (rDNA) genes90, although the inducer and the roles of this rDNA ‘epiallele’ remain to be identified. In summary, there are compelling examples in which the environment is linked to epigenetic regulation. However, confounding effects, the impact of multifactorial exposure, access to appropriate tissues and assessment of causality for DNA sequence versus epigenetic variation remain major challenges, particularly in humans. Most importantly, there is an urgent need to identify direct links between environmental changes, metabolic changes and epigenetic components. The recent discovery that histone demethylases KDM5A and KDM6A (also known as UTX) can sense oxygen concentrations and thereby modulate H3K4me3 and H3K27me3 levels91,92 is a first step in this direction.
Transgenerational epigenetics
The modern evolutionary synthesis93 postulates that evolution acts mainly via natural selection on phenotypes, ultimately affecting DNA sequences. The discovery that non-DNA sequence information, such as parental, ecological, behavioural and cultural information, can be heritable94 has not broken the modern framework of evolutionary synthesis. Indeed, one could postulate that complex chains of DNA-driven events ultimately drive parental and ecological behaviours and, therefore, DNA sequence alone would still explain these complex forms of inheritance. A direct demonstration that other molecules, in addition to DNA, carry substantial heritable information would represent an important conceptual change in evolutionary biology.
When adults are exposed to a stimulus or an intervention, their germline, as well as the germline of the fetus in pregnant females, is exposed. We thus distinguish intergenerational inheritance, in the F1 of exposed males and up to the F2 of exposed females, from transgenerational inheritance, starting from the F2 of exposed males and the F3 of exposed females44,95. There is abundant evidence for intergenerational inheritance in plants and some animals86,95,96,97,98,99,100, suggesting that this phenomenon might be involved in establishing early developmental patterning. What about transgenerational epigenetic inheritance (TEI)100? In yeast, TEI has been well-documented101 and is known to involve RNAi-dependent heterochromatin deposition, leading to Clr4-dependent H3K9me3 marking of silent heterochromatin47,102. By contrast, in the absence of RNAi-dependent amplification, H3K9me3 alone is insufficient to drive stable epigenetic memory, unless the histone demethylase Epe1 is mutated47,102. In Tetrahymena, TEI participates in the phenomenon called ‘programmed DNA elimination’ from the transcriptionally active somatic nucleus and, again, it involves small RNA-dependent formation of heterochromatin on the DNA elements to be eliminated103. In plants, TEI has also been well-described. Plant epialleles can be stable over many generations, and TEI is generally conveyed by RNA-directed DNA methylation (Fig. 3a), a mechanism that can also promote recovery from loss of DNA methylation in a subset of epialleles in Arabidopsis95,104. Chromatin components such as the histone chaperone CAF-1 also modulate DNA methylation-dependent TEI105. Future work is needed to elucidate the link between nucleosome dynamics and inheritance of DNA methylation.

Hallmarks of TEI in plants (a), C. elegans (b) and flies (c). From top to bottom, the Figure shows the triggering mechanisms, the molecules involved in establishment and transmission of transgenerational memory (carrier molecular machinery), the phenotypic consequences of epigenetic changes and the stability of TEI phenomena in terms of the number of generations (N) in which inheritance has been reported.
Unlike plants, the germline is separated from the soma in most sexually reproducing organisms, and Weismann postulated that information can flow only from germ cells to the soma106. Furthermore, a large number of epigenome features are erased in germline cell chromatin before and during meiosis. An important open question, however, is how much of the epigenome resists erasure? Evidence for substantial epigenetic inheritance of molecules other than DNA through gametes would overturn a fundamental tenet of neo-Darwinism. C. elegans epialleles (epigenetically modified alleles that induce specific phenotypes and are heritable over many generations) involve heterochromatin components (Fig. 3b), which, depending on the induction paradigm, may or may not involve piRNAs79,107. In Drosophila (Fig. 3c), heterochromatin components can induce TEI upon heat shock or osmotic stress108, whereas piRNAs produce TEI in response to transposable element activity69. A second mechanism that can lead to TEI in Drosophila relies on Polycomb proteins109. Post-eclosion dietary manipulation with a low-protein diet that resulted in elevation of the PRC2 enzymatic subunit E(z) or inhibition of PRC2 by RNAi or by an E(z) inhibitor induced a change in H3K27me3 and in longevity that could be inherited for at least two generations110. Furthermore, perturbation of chromosome architecture and of PRC2 function was shown to induce stable but reversible TEI in Drosophila111. Exposure of C. elegans to bisphenol A also induced alterations in the levels of H3K9me3 and H3K27me3 through five generations112 and, in plants, TEI of vernalization is prevented by the function of ELF6, which is a H3K27-specific demethylase52. These data suggest that both heterochromatin and Polycomb can induce TEI. Notably, the presence of a histone binding domain that recognizes the same mark as is deposited by the enzymatic moiety in both heterochromatin and PRC2 might provide these systems with amplification potential28. Differential levels of their marks might be reconstituted at each generation through differential affinity of PRC2 or other heterochromatin complexes to chromatin regions endowed with differential initial densities of marked nucleosomes (Fig. 3c).
Transgenerational inheritance in mammals
In vertebrates, DNA methylation is globally reduced twice in each generation: immediately after fertilization and in developing primordial germ cells113. Histone marks and 3D genome organization are also reprogrammed in the germline and after fertilization19. Furthermore, it is difficult in mammals and virtually impossible in humans to exclude potential confounding elements, such as maternal contribution, components of seminal fluids, changes in utero or postnatal effects114. So, what is the evidence for mammalian TEI? A classic example of multigenerational inheritance, insertion of the IAP endogenous retrovirus at the mouse agouti coat-colour locus, depends on heritable, variable methylation of the IAP retrovirus on an alternative promoter for the agouti gene115. A recent systematic survey of murine IAP insertions has indicated that multigenerational inheritance is rare, however116. Several reports have attracted attention for their suggestion that diet or exposure to chemicals and behavioural stresses can be transmitted to the progeny for multiple generations117,118,119, but some of these results have been criticized120. In humans, epidemiological evidence from the Överkalix population cohort established links between grandpaternal food supply at the beginning of the twentieth century and the mortality rate of subsequent generations121, although molecular evidence is unavailable for this cohort. DNA methylation has been suggested as a potential mechanism for these effects, and retroelements and some genes involved in neurological and metabolic disorders remain methylated during the wave of DNA demethylation in human primordial germ cells122. However, a recent report in which a high-fat diet induced insulin resistance, obesity and addictive-like behaviours up to the third generation did not identify heritable changes in the DNA methylome123. Other chromatin components might also be involved. For example, transient overexpression of the H3K4-specific KDM1A histone demethylase in mouse spermatogenesis has been shown to induce TEI124. These studies suggest that TEI is limited but possible in humans. Future work should address the underlying mechanisms of TEI, and epigenome-wide association studies should complement genome-wide association studies in order to assess the relative contributions of DNA sequence and epigenome alterations in disease59.
Epigenetics, health and disease
Changes in the levels of DNA methylation, histone modifications and changes in non-coding RNA (ncRNA) function are common in disease, as are mutations in epigenetic components57,125. The ability to distinguish driver from passenger roles for epigenetic alterations will make it possible to identify diseases in which epigenetics might affect diagnosis, prognosis and therapy. Dissecting the interplay between epigenetic components and other disease pathways will also allow the development of combinatorial intervention approaches.
The epigenetics of ageing
The application of machine learning to high-throughput DNA methylation data has identified indicators of chronological or biological age. One study found that changes in CpG methylation at 353 genomic sites produced a score that was highly correlated with age across tissues126 (epigenetic clock; Fig. 4a). Strikingly, a comparison of different molecular predictors of age indicated that the epigenetic clock is the most highly correlated to biological age127. Furthermore, epigenetic age is adversely affected (accelerated) by a high body mass index, whereas it is reduced by high levels of education or physical activity, a low body mass index and consumption of fish, poultry, fruits and vegetables128. Many of the 353 CpGs investigated are located close to poised promoters of bivalent genes (marked by H3K4me3 and H3K27me3), or to active promoters126, suggesting that ageing may correlate with reduced plasticity in the expression of some bivalent genes, which might resolve into repressed or active states, and with active genes changing their expression levels. More recently, integration with composite clinical measures of phenotypic age identified a set of CpG genomic sites that better predicts lifespan as well as healthspan129. Establishing the mechanistic links between the ageing process and variations in CpG methylation will be critical in order to identify the causes of ageing.

a, The ‘epigenetic clock’ consists of a specific set of genomic CpG sites whose levels of DNA methylation change progressively with age, leading to an estimate of age based on DNA methylation that correlates tightly with chronological age. Rather than a global change in methylation levels, some of the age-related CpG sites show increased methylation (black lollipops, red outline), whereas others show decreased methylation (white lollipops, blue outline). The relationship between changes in DNA methylation and chromatin architecture in ageing remains to be investigated, as well as the cause–consequence relationships between ageing, DNA methylation and gene expression changes. b, The genes encoding epigenetic components such as DNA methylases and demethylases, Polycomb, Trithorax, chromatin remodellers, DNA methylation components and CTCF are frequently mutated or dysregulated in cancer, often as a result of environmental insults or physiological changes such as ageing. These mutations alter cellular properties such as cell division, cell differentiation, adhesion and proliferation, and increase the heterogeneity of gene expression, thereby promoting tumorigenesis.
Developmental epigenetics and disease
Drawing initially on epidemiological studies, Barker formulated the hypothesis of the fetal or developmental origin of health and disease (DOHaD)130, which suggests that exposure to environmental factors such as chemicals, drugs, stress or infections during specific sensitive periods of intrauterine fetal development or early childhood might predispose an organism to diseases in adult life. Later work proposed that epigenetic components might mediate some of these effects131,132. Long-lasting changes to the epigenome that affect cancer susceptibility and biology have also been documented133. Other areas of intense study include obesity and diabetes134, neurological disorders125 and age-related conditions such as Parkinson’s and Alzheimer’s diseases135,136. Embryonic development and early life are two major susceptibility windows during which epigenetic programming is sensitive to environmental influences, such as diet, temperature, environmental toxins, maternal behaviour or childhood abuse137. Behavioural molecular genetics has identified a third susceptibility window, adolescence, during which adverse life experiences affect the risk of anxiety, depression and aggressive behaviour, associated with DNA methylation of specific genes138 or with alterations in levels of HDAC1139. Furthermore, memory formation, a behavioural response to environmental stimuli, is associated with changes in histone and DNA modification at selected loci140,141. Future studies should establish whether any of these alterations are in fact causal. Interestingly, one study found that low maternal care in mice decreases DNMT3a and DNA methylation at the L1 promoter and simultaneously induces the mobilization of L1 elements in the hippocampus, suggesting that environmental variation can cause genetic and epigenetic changes simultaneously142.
Cancer epigenetics
Genome-wide association studies of specific types of cancer or from the cancer genome atlas project have identified frequent mutations in genes that encode epigenetic components57,58,143. These include mutations in DNA methylases and demethylases, histones144 and histone modifiers, and genes involved in chromatin remodelling and chromosome architecture, but also metabolic genes such as IDH1 and IDH2 that affect histone and DNA methylation57 and might perturb 3D genome architecture145 (Fig. 4b). Repetitive DNA elements can also contribute to cancer. For instance, in Hodgkin lymphoma, transcription of the IRF5 transcription factor gene is induced by DNA hypomethylation of a normally dormant endogenous retroviral long terminal repeat located upstream of the promoter, a phenomenon dubbed onco-exaptation146, whereas in other tumours, DNA demethylating agents can have the opposite effect147. Although epigenetic perturbations are generally accompanied by mutations in cancer driver genes, sporadic cases in which cancer can be induced in the absence of obvious driver DNA mutations have also been reported in mice148. Furthermore, analysis of pancreatic cancer metastases did not uncover any obvious driver mutations; instead, large-scale chromatin reprogramming was observed, with changes in the level of H3K9me3 in many chromosomal domains149. These findings suggest that epigenetic changes can be major driver of oncogenic processes in certain circumstances.
Concluding remarks
Epigenetic mechanisms buffer environmental variation while allowing plastic responses to the most extreme environmental conditions. In this sense, epigenetics is returning to and expanding the original Waddington definition. A frequently held misconception about epigenetics is that it is a carrier of freedom from a presumed DNA-encoded destiny. The great discoveries of the second part of the twentieth century have generated much excitement about the role of DNA in evolution, biology and medicine, which led to the view of DNA as the ‘book of life’. The fact that the same DNA can correspond to different heritable phenotypes has now been portrayed as proof that ‘DNA isn’t your destiny’, a statement which merely reflects the level of hype about epigenetics. Most organisms buffer environmental variation in physiology and inheritance, although buffering does not erase every bit of epigenetic information (Fig. 5). Phenotypes thus depend on specific combinations of genome composition, epigenetic components and environmental inputs. The advent of increasingly sophisticated and economically feasible approaches to genomics, biochemistry and genetics can at last clarify the extent to which epigenetic mechanisms influence life, inheritance and evolution. This will allow us to progress towards personalized precision medicine, as well as to investigate and clarify the effects that lifestyle and ‘mind–body’ interventions may have on health. It has been suggested that the extrapolation of epigenetic findings from mice before they are confirmed in humans may lead to ‘serving epigenetics before its time’—that is, to rushed, unsupported conclusions that can cause harm and unnecessary anxiety150. We suggest that we are approaching ‘the right time for serving epigenetics’, for several reasons. The molecular machineries and mechanisms that enable states to be propagated are finally becoming clear; it is possible to test whether these mechanisms matter for biological processes, ageing or disease; and epigenetic alterations are more readily reversible than DNA mutations and can be targeted with increasing specificity. This allows the biomedical community to test the relevance of epigenetic components in specific diseases functionally, to exploit them as prognostic and diagnostic markers, and to use them as actionable targets for therapy. This path will deepen our knowledge and deliver benefits for human health. Therefore, the field of epigenetics is finally coming of age.

Environmental exposure can affect both the soma and the germline. When transient mutations or perturbations in epigenetic components occur (for example, PRC2 Polycomb components in Drosophila111, as shown), the germline chromatin may acquire an alternative state that can be transmitted and produce a phenotype (here, a change in eye colour) in subsequent generations. The degree of epigenetic inheritance varies and depends on the molecular features of each system and species.
References
Srivastava, D. & DeWitt, N. In vivo cellular reprogramming: the next generation. Cell 166, 1386–1396 (2016).
Waddington, C. H. The Epigenotype. Endeavour 1, 18–20 (1942).
Schuettengruber, B., Bourbon, H. M., Di Croce, L. & Cavalli, G. Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell 171, 34–57 (2017).
Nanney, D. L. Epigenetic control systems. Proc. Natl Acad. Sci. USA 44, 712–717 (1958).
Holliday, R. Epigenetics: an overview. Dev. Genet. 15, 453–457 (1994).
Holliday, R. & Pugh, J. E. DNA modification mechanisms and gene activity during development. Science 187, 226–232 (1975).
Riggs, A. D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 14, 9–25 (1975).
Bird, A. Perceptions of epigenetics. Nature 447, 396–398 (2007).
Berger, S. L., Kouzarides, T., Shiekhattar, R. & Shilatifard, A. An operational definition of epigenetics. Genes Dev. 23, 781–783 (2009).
Jenuwein, T. & Allis, C. D. Translating the histone code. Science 293, 1074–1080 (2001).
Festuccia, N., Gonzalez, I., Owens, N. & Navarro, P. Mitotic bookmarking in development and stem cells. Development 144, 3633–3645 (2017).
Chen, Q., Yan, W. & Duan, E. Epigenetic inheritance of acquired traits through sperm RNAs and sperm RNA modifications. Nat. Rev. Genet. 17, 733–743 (2016).
Neeb, Z. T. & Nowacki, M. RNA-mediated transgenerational inheritance in ciliates and plants. Chromosoma 127, 19–27 (2018).
Sharma, U. & Rando, O. J. Metabolic inputs into the epigenome. Cell Metab. 25, 544–558 (2017).
Bonev, B. & Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 17, 661–678 (2016).
Nicoglou, A. & Merlin, F. Epigenetics: A way to bridge the gap between biological fields. Stud. Hist. Philos. Biol. Biomed. Sci. 66, 73–82 (2017).
Beisson, J. & Sonneborn, T. M. Cytoplasmic inheritance of the organization of the cell cortex in Paramecium aurelia. Proc. Natl Acad. Sci. USA 53, 275–282 (1965).
Wan, G. et al. Spatiotemporal regulation of liquid-like condensates in epigenetic inheritance. Nature 557, 679–683 (2018).
Eckersley-Maslin, M. A., Alda-Catalinas, C. & Reik, W. Dynamics of the epigenetic landscape during the maternal-to-zygotic transition. Nat. Rev. Mol. Cell Biol. 19, 436–450 (2018).
Irie, N., Sybirna, A. & Surani, M. A. What can stem cell models tell us about human germ cell biology? Curr. Top. Dev. Biol. 129, 25–65 (2018).
Kobayashi, T. & Surani, M. A. On the origin of the human germline. Development 145, dev150433 (2018).
Smith, Z. D. et al. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature 549, 543–547 (2017).
Cheloufi, S. et al. The histone chaperone CAF-1 safeguards somatic cell identity. Nature 528, 218–224 (2015).
Tischler, J. et al. Metabolic regulation of pluripotency and germ cell fate through α-ketoglutarate. EMBO J. 38, e99518 (2019).
Jullien, J. et al. Gene resistance to transcriptional reprogramming following nuclear transfer is directly mediated by multiple chromatin-repressive pathways. Mol. Cell 65, 873–884.e878 (2017).
Hormanseder, E. et al. H3K4 methylation-dependent memory of somatic cell identity inhibits reprogramming and development of nuclear transfer embryos. Cell Stem Cell 21, 135–143.e136 (2017).
Allis, C. D. & Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 (2016).
Reinberg, D. & Vales, L. D. Chromatin domains rich in inheritance. Science 361, 33–34 (2018).
Yu, C. et al. A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science 361, 1386–1389 (2018).
Petryk, N. et al. MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 361, 1389–1392 (2018).
Ferry, L. et al. Methylation of DNA ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA methylation. Mol. Cell 67, 550–565.e555 (2017).
Liu, X. et al. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 4, 1563 (2013).
A, P. et al. EZH2 promotes DNA replication by stabilizing interaction of POLδ and PCNA via methylation-mediated PCNA trimerization. Epigenetics Chromatin 11, 44 (2018).
Trembecka-Lucas, D. O., Szczurek, A. T. & Dobrucki, J. W. Dynamics of the HP1β-PCNA-containing complexes in DNA replication and repair. Nucleus 4, 74–82 (2013).
Coleman, R. T. & Struhl, G. Causal role for inheritance of H3K27me3 in maintaining the OFF state of a Drosophila HOX gene. Science 356, eaai8236 (2017).
Laprell, F., Finkl, K. & Müller, J. Propagation of Polycomb-repressed chromatin requires sequence-specific recruitment to DNA. Science 356, 85–88 (2017).
Moussa, H. F. et al. Canonical PRC1 controls sequence-independent propagation of Polycomb-mediated gene silencing. Nat. Commun. 10, 1931 (2019).
Zhen, C. Y., Duc, H. N., Kokotovic, M., Phiel, C. J. & Ren, X. Cbx2 stably associates with mitotic chromosomes via a PRC2- or PRC1-independent mechanism and is needed for recruiting PRC1 complex to mitotic chromosomes. Mol. Biol. Cell 25, 3726–3739 (2014).
Ginno, P. A., Burger, L., Seebacher, J., Iesmantavicius, V. & Schübeler, D. Cell cycle-resolved chromatin proteomics reveals the extent of mitotic preservation of the genomic regulatory landscape. Nat. Commun. 9, 4048 (2018).
Festuccia, N. et al. Mitotic binding of Esrrb marks key regulatory regions of the pluripotency network. Nat. Cell Biol. 18, 1139–1148 (2016).
Teves, S. S. et al. A stable mode of bookmarking by TBP recruits RNA polymerase II to mitotic chromosomes. eLife 7, e35621 (2018).
Inoue, A., Chen, Z., Yin, Q. & Zhang, Y. Maternal Eed knockout causes loss of H3K27me3 imprinting and random X inactivation in the extraembryonic cells. Genes Dev. 32, 1525–1536 (2018).
Inoue, A., Jiang, L., Lu, F., Suzuki, T. & Zhang, Y. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547, 419–424 (2017).
Miska, E. A. & Ferguson-Smith, A. C. Transgenerational inheritance: models and mechanisms of non-DNA sequence-based inheritance. Science 354, 59–63 (2016).
Hathaway, N. A. et al. Dynamics and memory of heterochromatin in living cells. Cell 149, 1447–1460 (2012).
Jost, D. & Vaillant, C. Epigenomics in 3D: importance of long-range spreading and specific interactions in epigenomic maintenance. Nucleic Acids Res. 46, 2252–2264 (2018).
Yu, R., Wang, X. & Moazed, D. Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation. Nature 558, 615–619 (2018). The functional study of a genomic locus of Schizosaccharomyces pombe shows that a positive feedback loop involving the collaboration of RNAi and heterochromatin can maintain stable epigenetic inheritance through mitosis and meiosis, whereas inheritance is lost in the absence of RNAi inheritance because of the action of an H3K9me3-specific demethylase.
Nora, E. P. et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). Chromosome conformation capture carbon-copy (5C) and super-resolution microscopy analysis of the X chromosome region involved in the control of X-chromosome inactivation identify the existence of TADs in the mouse genome.
Sexton, T. et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472 (2012). Genome-wide analysis of the Drosophila genome by Hi-C shows that chromosomes are hierarchically organized into TADs that strongly correlate with epigenomic marking, into active and inactive compartments, and into a series of specific long-distance contacts among TADs of the same epigenomic kind.
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). Hi-C analysis shows that the mouse genome is partitioned into TADs that are evolutionarily conserved between mouse and human and are bordered by CTCF-binding sites, housekeeping genes, tRNAs and short interspersed element (SINE) retrotransposons.
Allshire, R. C. & Madhani, H. D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 19, 229–244 (2018).
Crevillén, P. et al. Epigenetic reprogramming that prevents transgenerational inheritance of the vernalized state. Nature 515, 587–590 (2014).
Blobel, G. A. et al. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol. Cell 36, 970–983 (2009).
Deplancke, B., Alpern, D. & Gardeux, V. The genetics of transcription factor DNA binding variation. Cell 166, 538–554 (2016).
Yet, I., Tsai, P. C., Castillo-Fernandez, J. E., Carnero-Montoro, E. & Bell, J. T. Genetic and environmental impacts on DNA methylation levels in twins. Epigenomics 8, 105–117 (2016).
McDaniell, R. et al. Heritable individual-specific and allele-specific chromatin signatures in humans. Science 328, 235–239 (2010).
Bailey, M. H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 174, 1034–1035 (2018).
Mohammad, F. et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 23, 483–492 (2017).
Lappalainen, T. & Greally, J. M. Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet. 18, 441–451 (2017).
Hama, N. et al. Epigenetic landscape influences the liver cancer genome architecture. Nat. Commun. 9, 1643 (2018).
Polak, P. et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 518, 360–364 (2015).
Supek, F. & Lehner, B. Clustered mutation signatures reveal that error-prone DNA repair targets mutations to active genes. Cell 170, 534–547.e523 (2017).
Poulos, R. C., Olivier, J. & Wong, J. W. H. The interaction between cytosine methylation and processes of DNA replication and repair shape the mutational landscape of cancer genomes. Nucleic Acids Res. 45, 7786–7795 (2017).
Pich, O. et al. Somatic and germline mutation periodicity follow the orientation of the DNA minor groove around nucleosomes. Cell 175, 1074–1087.e1018 (2018).
Redin, C. et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 49, 36–45 (2017).
Wu, P. et al. 3D genome of multiple myeloma reveals spatial genome disorganization associated with copy number variations. Nat. Commun. 8, 1937 (2017).
Jäger, N. et al. Hypermutation of the inactive X chromosome is a frequent event in cancer. Cell 155, 567–581 (2013).
Shapiro, J. A. Exploring the read-write genome: mobile DNA and mammalian adaptation. Crit. Rev. Biochem. Mol. Biol. 52, 1–17 (2017).
Grentzinger, T. et al. piRNA-mediated transgenerational inheritance of an acquired trait. Genome Res. 22, 1877–1888 (2012).
Zeller, P. et al. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 48, 1385–1395 (2016).
Wendte, J. M. & Pikaard, C. S. The RNAs of RNA-directed DNA methylation. Biochim. Biophys. Acta. Gene Regul. Mech. 1860, 140–148 (2017).
Imbeault, M., Helleboid, P. Y. & Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554 (2017). Analysis of KZFPs shows that most of them bind to transposable elements and that, in addition to their silencing, they exploit some of their evolutionarily conserved DNA fragments as gene regulatory platforms even in transposable element copies that have lost all transposition potential.
Friedrich, T., Faivre, L., Bäurle, I. & Schubert, D. Chromatin-based mechanisms of temperature memory in plants. Plant Cell Environ. 42, 762–770 (2019).
Roussou, I. G., Savakis, C., Tavernarakis, N. & Metaxakis, A. Stage dependent nutritional regulation of transgenerational longevity. Nutr. Healthy Aging 4, 47–54 (2016).
Stern, S., Fridmann-Sirkis, Y., Braun, E. & Soen, Y. Epigenetically heritable alteration of fly development in response to toxic challenge. Cell Rep. 1, 528–542 (2012).
Williams, Z. M. Transgenerational influence of sensorimotor training on offspring behavior and its neural basis in Drosophila. Neurobiol. Learn. Mem. 131, 166–175 (2016).
Rechavi, O., Minevich, G. & Hobert, O. Transgenerational inheritance of an acquired small RNA-based antiviral response in C. elegans. Cell 147, 1248–1256 (2011).
Rechavi, O. et al. Starvation-induced transgenerational inheritance of small RNAs in C. elegans. Cell 158, 277–287 (2014).
Klosin, A., Casas, E., Hidalgo-Carcedo, C., Vavouri, T. & Lehner, B. Transgenerational transmission of environmental information in C. elegans. Science 356, 320–323 (2017).
Ge, C. et al. The histone demethylase KDM6B regulates temperature-dependent sex determination in a turtle species. Science 360, 645–648 (2018).
Deveson, I. W. et al. Differential intron retention in Jumonji chromatin modifier genes is implicated in reptile temperature-dependent sex determination. Sci. Adv. 3, e1700731 (2017).
Simola, D. F. et al. Epigenetic (re)programming of caste-specific behavior in the ant Camponotus floridanus. Science 351, aac6633 (2016).
Intlekofer, A. M. et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 559, 125–129 (2018).
Ferrari, A. et al. Epigenome modifiers and metabolic rewiring: new frontiers in therapeutics. Pharmacol. Ther. 193, 178–193 (2019).
Lu, T. T. et al. The Polycomb-dependent epigenome controls β cell dysfunction, dedifferentiation, and diabetes. Cell Metab. 27, 1294–1308.e7 (2018).
Öst, A. et al. Paternal diet defines offspring chromatin state and intergenerational obesity. Cell 159, 1352–1364 (2014).
Sharma, U. et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 351, 391–396 (2016).
Chen, Q. et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 351, 397–400 (2016).
Schorn, A. J., Gutbrod, M. J., LeBlanc, C. & Martienssen, R. LTR-retrotransposon control by tRNA-derived small RNAs. Cell 170, 61–71.e11, (2017). Analysis of tRNA-derived fragments identifies a class of molecules targeting the tRNA primer binding site that is essential for ERV transcription, demonstrating a novel mechanism for retrotransposon silencing in addition to heterochromatin formation.
Holland, M. L. et al. Early-life nutrition modulates the epigenetic state of specific rDNA genetic variants in mice. Science 353, 495–498 (2016).
Batie, M. et al. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 363, 1222–1226 (2019).
Chakraborty, A. A. et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363, 1217–1222 (2019).
Huxley, J. S. Evolution: the Modern Synthesis (Allen and Unwin, 1942).
Danchin, É. et al. Beyond DNA: integrating inclusive inheritance into an extended theory of evolution. Nat. Rev. Genet. 12, 475–486 (2011).
Heard, E. & Martienssen, R. A. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157, 95–109 (2014).
Huypens, P. et al. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat. Genet. 48, 497–499 (2016).
Murphy, P. J., Wu, S. F., James, C. R., Wike, C. L. & Cairns, B. R. Placeholder nucleosomes underlie germline-to-embryo DNA methylation reprogramming. Cell 172, 993–1006.e13 (2018).
Ng, S. F. et al. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 467, 963–966 (2010).
Zenk, F. et al. Germline-inherited H3K27me3 restricts enhancer function during maternal-to-zygotic transition. Science 357, 212–216 (2017).
Perez, M. F. & Lehner, B. Intergenerational and transgenerational epigenetic inheritance in animals. Nat. Cell Biol. 21, 143–151 (2019).
Grewal, S. I. S. & Klar, A. J. S. Chromosomal inheritance of epigenetic states in fission yeast during mitosis and meiosis. Cell 86, 95–101 (1996). Analysis of a strain of S. pombe in which a silenced region is replaced with a marker gene provides a classic demonstration of stable epigenetic inheritance of alternative chromatin states through mitosis and meiosis.
Audergon, P. N. et al. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science 348, 132–135 (2015).
Noto, T. & Mochizuki, K. Small RNA-mediated trans-nuclear and trans-element communications in Tetrahymena DNA elimination. Curr. Biol. 28, 1938–1949.e1935 (2018).
Teixeira, F. K. et al. A role for RNAi in the selective correction of DNA methylation defects. Science 323, 1600–1604 (2009).
Mozgova, I. et al. Transgenerational phenotype aggravation in CAF-1 mutants reveals parent-of-origin specific epigenetic inheritance. New Phytol. 220, 908–921 (2018).
Weismann, A. The Germ-Plasm. A Theory of Heredity (Charles Scribner’s Sons, New York, 1893).
Ashe, A. et al. piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans. Cell 150, 88–99 (2012).
Seong, K. H., Li, D., Shimizu, H., Nakamura, R. & Ishii, S. Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell 145, 1049–1061 (2011).
Cavalli, G. & Paro, R. The Drosophila Fab-7 chromosomal element conveys epigenetic inheritance during mitosis and meiosis. Cell 93, 505–518 (1998). Analysis of a transgenic system containing a Polycomb-binding site and a heterologous inducible promoter shows that transient transcriptional induction can flip the transgene from a Polycomb-silenced state into a derepressed state that can be transgenerationally inherited through multiple generations.
Xia, B., Gerstin, E., Schones, D. E., Huang, W. & Steven de Belle, J. Transgenerational programming of longevity through E(z)-mediated histone H3K27 trimethylation in Drosophila. Aging 8, 2988–3008 (2016).
Ciabrelli, F. et al. Stable Polycomb-dependent transgenerational inheritance of chromatin states in Drosophila. Nat. Genet. 49, 876–886 (2017).
Camacho, J. et al. The memory of environmental chemical exposure in C. elegans is dependent on the Jumonji demethylases jmjd-2 and jmjd-3/utx-1. Cell Rep. 23, 2392–2404 (2018).
Smallwood, S. A. & Kelsey, G. De novo DNA methylation: a germ cell perspective. Trends Genet. 28, 33–42 (2012).
Bohacek, J. & Mansuy, I. M. A guide to designing germline-dependent epigenetic inheritance experiments in mammals. Nat. Methods 14, 243–249 (2017).
Morgan, H. D., Sutherland, H. G., Martin, D. I. & Whitelaw, E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 23, 314–318 (1999). Analysis of an ERV of the intra-cisternal A particle (IAP) class inserted upstream of the agouti gene provides a classical demonstration of maternal transgenerational epigenetic inheritance of variable gene expression in mammals.
Kazachenka, A. et al. Identification, characterization, and heritability of murine metastable epialleles: implications for non-genetic inheritance. Cell 175, 1259–1271.e13 (2018). A systematic genome-wide screen identifies IAP retrotransposons with variable levels of DNA methylation among individuals, but in most cases they are reset at every generation, suggesting that transgenerational inheritance is rare.
Anway, M. D., Cupp, A. S., Uzumcu, M. & Skinner, M. K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308, 1466–1469 (2005).
Dias, B. G. & Ressler, K. J. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat. Neurosci. 17, 89–96 (2014).
Gapp, K. et al. Alterations in sperm long RNA contribute to the epigenetic inheritance of the effects of postnatal trauma. Mol. Psychiatry (2018).
Francis, G. Too much success for recent groundbreaking epigenetic experiments. Genetics 198, 449–451 (2014).
Pembrey, M., Saffery, R. & Bygren, L. O. Human transgenerational responses to early-life experience: potential impact on development, health and biomedical research. J. Med. Genet. 51, 563–572 (2014).
Tang, W. W. et al. A unique gene regulatory network resets the human germline epigenome for development. Cell 161, 1453–1467 (2015).
Sarker, G. et al. Transgenerational transmission of hedonic behaviors and metabolic phenotypes induced by maternal overnutrition. Transl. Psychiatry 8, 195 (2018).
Siklenka, K. et al. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science 350, aab2006 (2015).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). Analysis of exome sequencing data from a large cohort of individuals with autism and control participants identifies a set of genes likely to affect risk, including those involved in synapse formation, transcriptional regulation and chromatin remodelling pathways.
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013). Analysis of a large panel of DNA methylation samples identifies an age predictor that correlates the methylation levels of a few hundred CpG sites with chronological age and is applicable to a large panel of tissues, and shows that cancer cells have accelerated ageing based on their DNA methylation levels.
Jylhävä, J., Pedersen, N. L. & Hägg, S. Biological age predictors. EBioMedicine 21, 29–36 (2017).
Quach, A. et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 9, 419–446 (2017).
Levine, M. E. et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591 (2018).
Barker, D. J. The fetal and infant origins of adult disease. Br. Med. J. 301, 1111 (1990).
Calkins, K. & Devaskar, S. U. Fetal origins of adult disease. Curr. Probl. Pediatr. Adolesc. Health Care 41, 158–176 (2011).
Heindel, J. J. & Vandenberg, L. N. Developmental origins of health and disease: a paradigm for understanding disease cause and prevention. Curr. Opin. Pediatr. 27, 248–253 (2015).
Feinberg, A. P. The key role of epigenetics in human disease prevention and mitigation. N. Engl. J. Med. 378, 1323–1334 (2018).
Panzeri, I. & Pospisilik, J. A. Epigenetic control of variation and stochasticity in metabolic disease. Mol. Metab. 14, 26–38 (2018).
Palumbo, S., Mariotti, V., Iofrida, C. & Pellegrini, S. Genes and aggressive behavior: epigenetic mechanisms underlying individual susceptibility to aversive environments. Front. Behav. Neurosci. 12, 117 (2018).
Del Blanco, B. & Barco, A. Impact of environmental conditions and chemicals on the neuronal epigenome. Curr. Opin. Chem. Biol. 45, 157–165 (2018).
McGowan, P. O. et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12, 342–348 (2009).
Gouin, J. P. et al. Associations among oxytocin receptor gene (OXTR) DNA methylation in adulthood, exposure to early life adversity, and childhood trajectories of anxiousness. Sci. Rep. 7, 7446 (2017).
Mitjans, M. et al. Violent aggression predicted by multiple pre-adult environmental hits. Mol. Psychiatry https://doi.org/10.1038/s41380-018-0043-3 (2018).
Kerimoglu, C. et al. KMT2A and KMT2B mediate memory function by affecting distinct genomic regions. Cell Rep. 20, 538–548 (2017).
Halder, R. et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat. Neurosci. 19, 102–110 (2016).
Bedrosian, T. A., Quayle, C., Novaresi, N. & Gage, F. H. Early life experience drives structural variation of neural genomes in mice. Science 359, 1395–1399 (2018).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Mohammad, F. & Helin, K. Oncohistones: drivers of pediatric cancers. Genes Dev. 31, 2313–2324 (2017).
Flavahan, W. A. et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016). Analysis of gliomas carrying mutation in IDH genes known to increase DNA methylation levels shows hypermethylation at cohesin and CCCTC-binding factor (CTCF)-binding sites that alters TAD boundaries and induces illegitimate oncogene activation.
Babaian, A. et al. Onco-exaptation of an endogenous retroviral LTR drives IRF5 expression in Hodgkin lymphoma. Oncogene 35, 2542–2546 (2016).
Roulois, D. et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 (2015).
Green, M. R. et al. Transient expression of Bcl6 is sufficient for oncogenic function and induction of mature B-cell lymphoma. Nat. Commun. 5, 3904 (2014).
McDonald, O. G. et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 49, 367–376 (2017).
Juengst, E. T., Fishman, J. R., McGowan, M. L. & Settersten, R. A. Jr Serving epigenetics before its time. Trends Genet. 30, 427–429 (2014).
Riggs, A. D., Martienssen, R. A. & Russo, V. E. A. in Epigenetic Mechanisms of Gene Regulation (ed. Russo, V. E. A., Martienssen, R. A. & Riggs, A. D.) 1–4 (Cold Spring Harbor Laboratory Press, 1996).
Müller, M. M., Fierz, B., Bittova, L., Liszczak, G. & Muir, T. W. A two-state activation mechanism controls the histone methyltransferase Suv39h1. Nat. Chem. Biol. 12, 188–193 (2016).
Machida, S. et al. Structural basis of heterochromatin formation by human HP1. Mol. Cell 69, 385–397.e388 (2018).
da Rocha, S. T. & Heard, E. Novel players in X inactivation: insights into Xist-mediated gene silencing and chromosome conformation. Nat. Struct. Mol. Biol. 24, 197–204 (2017).
Chen, H., Du, G., Song, X. & Li, L. Non-coding transcripts from enhancers: new insights into enhancer activity and gene expression regulation. Genomics Proteomics Bioinformatics 15, 201–207 (2017).
Holoch, D. & Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 16, 71–84 (2015).
Xie, S. & Qian, C. The growing complexity of UHRF1-mediated maintenance DNA methylation. Genes 9, E600 (2018).
Clark, S. J. et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun. 9, 781 (2018).
Peterson, V. M. et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 35, 936–939 (2017).
Argelaguet, R. et al. Multi-omics factor analysis—a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 14, e8124 (2018).
Nagano, T. et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). Single-cell Hi-C analysis shows that chromosomal compartments, TADs, chromatin contact insulation and long-range loops that can be described in population Hi-C maps are governed by distinct cell-cycle dynamics.
Ramani, V. et al. Massively multiplex single-cell Hi-C. Nat. Methods 14, 263–266 (2017).
Flyamer, I. M. et al. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544, 110–114 (2017).
Nagano, T. et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67 (2017).
Bintu, B. et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018).
Wu, J. et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 534, 652–657 (2016).
Kalhor, R. et al. Developmental barcoding of whole mouse via homing CRISPR. Science 361, eaat9804 (2018).
Andersson-Rolf, A. et al. One-step generation of conditional and reversible gene knockouts. Nat. Methods 14, 287–289 (2017).
Brocken, D. J. W., Tark-Dame, M. & Dame, R. T. dCas9: a versatile tool for epigenome editing. Curr. Issues Mol. Biol. 26, 15–32 (2018).
Morgan, S. L. et al. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat. Commun. 8, 15993 (2017).
Acknowledgements
We thank G. Papadopoulos for analysis of CpG methylation and epigenomic data linked to ageing. We sincerely apologize to all authors whose work could not be cited owing to space constraints. G.C. is supported by the CNRS and the ERC (Advanced Grant ERC-2018-AdG, No. 788972 (3DEpi)). E.H. is supported by CNRS, INSERM and ERC-2015-AdG, No. 671027 (XPRESS).
Author information
Authors and Affiliations
Contributions
Both authors contributed ideas and figures. G.C. wrote the draft manuscript and both authors finalized the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks John Greally, Matthew Lawrence and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cavalli, G., Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 571, 489–499 (2019). https://doi.org/10.1038/s41586-019-1411-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1411-0
This article is cited by
-
Imprinted gene detection effectively improves the diagnostic accuracy for papillary thyroid carcinoma
BMC Cancer (2024)
-
Long non-coding RNA ACTA2-AS1 suppresses metastasis of papillary thyroid cancer via regulation of miR-4428/KLF9 axis
Clinical Epigenetics (2024)
-
Liquid–liquid phase separation of H3K27me3 reader BP1 regulates transcriptional repression
Genome Biology (2024)
-
YAP represses intestinal inflammation through epigenetic silencing of JMJD3
Clinical Epigenetics (2024)
-
Neurofibromin 1 controls metabolic balance and Notch-dependent quiescence of murine juvenile myogenic progenitors
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
