
Abstract
Chemical modifications of histones can mediate diverse DNA-templated processes, including gene transcription1,2,3. Here we provide evidence for a class of histone post-translational modification, serotonylation of glutamine, which occurs at position 5 (Q5ser) on histone H3 in organisms that produce serotonin (also known as 5-hydroxytryptamine (5-HT)). We demonstrate that tissue transglutaminase 2 can serotonylate histone H3 tri-methylated lysine 4 (H3K4me3)-marked nucleosomes, resulting in the presence of combinatorial H3K4me3Q5ser in vivo. H3K4me3Q5ser displays a ubiquitous pattern of tissue expression in mammals, with enrichment observed in brain and gut, two organ systems responsible for the bulk of 5-HT production. Genome-wide analyses of human serotonergic neurons, developing mouse brain and cultured serotonergic cells indicate that H3K4me3Q5ser nucleosomes are enriched in euchromatin, are sensitive to cellular differentiation and correlate with permissive gene expression, phenomena that are linked to the potentiation of TFIID4,5,6 interactions with H3K4me3. Cells that ectopically express a H3 mutant that cannot be serotonylated display significantly altered expression of H3K4me3Q5ser-target loci, which leads to deficits in differentiation. Taken together, these data identify a direct role for 5-HT, independent from its contributions to neurotransmission and cellular signalling, in the mediation of permissive gene expression.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
Data from ChIP-seq and RNA-seq experiments have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database under accession numbers GSE106495 and GSE117910. The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE48,49 partner repository with the dataset identifier PXD008106. Additional raw data files are available at https://chorusproject.org under project no. 1513. We declare that the data supporting findings for this study are available within the article and Supplementary Information (Supplementary Fig. 1). Related data are available from the corresponding author upon reasonable request. No restrictions on data availability apply.
References
Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007).
Jenuwein, T. & Allis, C. D. Translating the histone code. Science 293, 1074–1080 (2001).
Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000).
Lauberth, S. M. et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 152, 1021–1036 (2013).
Vermeulen, M. et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 (2007).
van Ingen, H. et al. Structural insight into the recognition of the H3K4me3 mark by the TFIID subunit TAF3. Structure 16, 1245–1256 (2008).
Colgan, L. A., Putzier, I. & Levitan, E. S. Activity-dependent vesicular monoamine transporter-mediated depletion of the nucleus supports somatic release by serotonin neurons. J. Neurosci. 29, 15878–15887 (2009).
Young, A. B., Pert, C. D., Brown, D. G., Taylor, K. M. & Snyder, S. H. Nuclear localization of histamine in neonatal rat brain. Science 173, 247–249 (1971).
Walther, D. J. et al. Serotonylation of small GTPases is a signal transduction pathway that triggers platelet α-granule release. Cell 115, 851–862 (2003).
Watts, S. W., Priestley, J. R. & Thompson, J. M. Serotonylation of vascular proteins important to contraction. PLoS ONE 4, e5682 (2009).
Hummerich, R., Thumfart, J. O., Findeisen, P., Bartsch, D. & Schloss, P. Transglutaminase-mediated transamidation of serotonin, dopamine and noradrenaline to fibronectin: evidence for a general mechanism of monoaminylation. FEBS Lett. 586, 3421–3428 (2012).
Ballestar, E., Abad, C. & Franco, L. Core histones are glutaminyl substrates for tissue transglutaminase. J. Biol. Chem. 271, 18817–18824 (1996).
Kim, J. H. et al. Histone cross-linking by transglutaminase. Biochem. Biophys. Res. Commun. 293, 1453–1457 (2002).
Sileno, S. et al. A possible role of transglutaminase 2 in the nucleus of INS-1E and of cells of human pancreatic islets. J. Proteomics 96, 314–327 (2014).
Lin, J. C. et al. Characterization of protein serotonylation via bioorthogonal labeling and enrichment. J. Proteome Res. 13, 3523–3529 (2014).
Nurminskaya, M. V. & Belkin, A. M. Cellular functions of tissue transglutaminase. Int. Rev. Cell Mol. Biol. 294, 1–97 (2012).
Hummerich, R., Costina, V., Findeisen, P. & Schloss, P. Monoaminylation of fibrinogen and glia-derived proteins: indication for similar mechanisms in posttranslational protein modification in blood and brain. ACS Chem. Neurosci. 6, 1130–1136 (2015).
Lu, J. et al. Generation of serotonin neurons from human pluripotent stem cells. Nat. Biotechnol. 34, 89–94 (2016).
Shen, L. et al. diffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLoS ONE 8, e65598 (2013).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), 90–97 (2016).
Daubert, E. A. & Condron, B. G. Serotonin: a regulator of neuronal morphology and circuitry. Trends Neurosci. 33, 424–434 (2010).
White, L. A. et al. Distinct regulatory pathways control neurofilament expression and neurotransmitter synthesis in immortalized serotonergic neurons. J. Neurosci. 14, 6744–6753 (1994).
Maze, I. et al. Critical role of histone turnover in neuronal transcription and plasticity. Neuron 87, 77–94 (2015).
Tessarz, P. et al. Glutamine methylation in histone H2A is an RNA-polymerase-I-dedicated modification. Nature 505, 564–568 (2014).
Eaton, M. J. & Whittemore, S. R. Autocrine BDNF secretion enhances the survival and serotonergic differentiation of raphe neuronal precursor cells grafted into the adult rat CNS. Exp. Neurol. 140, 105–114 (1996).
Ollivier, N. et al. Tidbits for the synthesis of bis(2-sulfanylethyl)amido (SEA) polystyrene resin, SEA peptides and peptide thioesters. J. Pept. Sci. 20, 92–97 (2014).
Dann, G. P. et al. ISWI chromatin remodellers sense nucleosome modifications to determine substrate preference. Nature 548, 607–611 (2017).
Nguyen, U. T. et al. Accelerated chromatin biochemistry using DNA-barcoded nucleosome libraries. Nat. Methods 11, 834–840 (2014).
Simon, M. D. & Shokat, K. M. A method to site-specifically incorporate methyl-lysine analogues into recombinant proteins. Methods Enzymol. 512, 57–69 (2012).
Debelouchina, G. T., Gerecht, K. & Muir, T. W. Ubiquitin utilizes an acidic surface patch to alter chromatin structure. Nat. Chem. Biol. 13, 105–110 (2017).
Lowary, P. T. & Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42 (1998).
Dyer, P. N. et al. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44 (2004).
Wojcik, F. et al. Functional crosstalk between histone H2B ubiquitylation and H2A modifications and variants. Nat. Commun. 9, 1394 (2018).
Peterson, A. C., Russell, J. D., Bailey, D. J., Westphall, M. S. & Coon, J. J. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475–1488 (2012).
Dignam, J. D., Lebovitz, R. M. & Roeder, R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 (1983).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007).
Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 (2014).
Käll, L., Canterbury, J. D., Weston, J., Noble, W. S. & MacCoss, M. J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 (2007).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Schwanhäusser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Sidoli, S., Bhanu, N. V., Karch, K. R., Wang, X. & Garcia, B. A. Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: from histone extraction to data analysis. J. Vis. Exp. 17, e54112 (2016).
Tsankova, N. M. et al. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 9, 519–525 (2006).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Gundemir, S. & Johnson, G. V. Intracellular localization and conformational state of transglutaminase 2: implications for cell death. PLoS ONE 4, e6123 (2009).
Vizcaíno, J. A. et al. A guide to the Proteomics Identifications Database proteomics data repository. Proteomics 9, 4276–4283 (2009).
Vizcaíno, J. A. et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44 (D1), D447–D456 (2016).
Acknowledgements
We thank R. Cagan (ISMMS), J. Coplan (SUNY Downstate) and C. Tamminga (UTSW) for providing Drosophila, macaque and human samples, respectively, for analysis of H3K4me3Q5ser (Fig. 1e), and G. Johnson (University of Rochester Medical Center) for wild-type and catalytically dead TGM2 constructs. This work was supported by grants from the National Institutes of Health: DP1 DA042078 (I.M.), R01 MH116900 (I.M.), R21 DA044767 (I.M.), P50 MH096890 (I.M.), R37 GM086868 (T.W.M.), P01 CA196539 (T.W.M. and B.A.G.), R01 GM110174 (B.A.G.), R21 DA040837 (B.A.G.), R01 CA129325 (R.G.R.), R01 CA204639 (R.G.R.), T32 DA007135 (R.M.B.), as well as awards from: MQ Mental Health Research Charity, MQ15FIP100011 (I.M.), Alfred P. Sloan Foundation, Fellowship in Neuroscience (I.M.), the JPB Foundation (F.H.G.), the Bob and Mary Jane Engman Foundation (F.H.G.), the Volkswagen Foundation (N.A.) and the National Natural Science Foundation of China (31430020 and 31621092, H.L.).
Reviewer information
Nature thanks Tatiana Kutateladze and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
I.M. conceived of the project with input from L.A.F., O.B. and T.W.M. L.A.F., F.H.G., B.A.G., H.L., T.W.M. and I.M. designed the experiments and interpreted the data. L.A.F., R.E.T., S.Z., A.E.L., Y.L., N.V.B., B.Z., K.C.V., K.J.H., R.M.B., B.J.L., H.M. and I.M. collected and analysed the data. Y.-H.E.L., A.R., G.E. and L.S. performed the sequencing-based bioinformatics with input from I.M. T.N. and R.G.R. provided the Flag–TBP purified TFIID complex. H.Z.III provided a subset of chemically modified peptides. N.A. and M.B. provided tissues from Tph1 and Tph2 knockout mice. L.A.F., H.L., T.W.M. and I.M. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Bioorthogonal labelling by 5-PT identifies H3 serotonylation in chromatin.
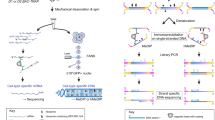
a, Structures of 5-HT and 5-PT in transamidation reactions. b, Immunofluorescence of intracellular 5-PT in HeLa cells (bottom) after exogenous application of the molecule (versus vehicle, top). Intracellular 5-PT was imaged in fixed cells after chemical labelling with Alexa Fluor 488 azide. DAPI was used as a nuclear co-stain. Results confirmed in at least two independent experiments. Scale bars, 500 μm. c, Cell versus lysate donor competition assays indicating that application of excess 5-HT to live HeLa cells, but not to processed lysates, before chemical labelling and 5-PT-based pulldowns results in loss of H3 signal after immunoprecipitation. Input and immunoprecipitate western blots are shown. d, Cellular fractionation analysis identifying H3 serotonylation post-immunoprecipitation in HeLa cell chromatin, but not in soluble nuclear or cytosolic fractions. Input and immunoprecipitate western blots are shown. H3 results confirmed in at least three independent experiments.
Extended Data Fig. 2 TGM2 is both necessary and sufficient to promote H3 serotonylation in cells.
a, Western blot analysis of TGM2 expression across mouse brain and peripheral tissues (including blood) validate TGM2 as a ubiquitously expressed enzyme in mammals. Whereas two isoforms of TGM2 exist and are expressed differentially across tissues, the functions of these two variants remain unknown (n = 1 per brain region or organ tissue). b, Bioorthogonal labelling of 5-PT-modified H3 from HeLa cell extracts in the absence or presence of the transglutaminase inhibitor cystamine (4 mM). Input and immunoprecipitate western blots are shown. Results confirmed in at least two independent experiments. c, Quantitative PCR with reverse transcription (RT–qPCR) analysis of TGM genes comparing HeLa and HEK 293T cells (n = 1 per cell type). Insert western blot validating that TGM2 is differentially expressed across cell types. d, Bioorthogonal labelling of 5-PT-modified H3 from mock (non-transfected) versus nuclear expression of TGM2 in HEK 293T cell extracts. Catalytically dead TGM2(W241A) was used as a negative control. Input and immunoprecipitate western blots are shown. Results confirmed in at least two independent experiments. e, Recombinant histones (H3 versus H4) and reconstituted nucleosomes, as well as fibrinogen (positive control) and BSA (negative control), were subjected to radioactive serotonylation (5-3HT) reactions in vitro (n = 3), in the absence or presence of cystamine (one-way ANOVA: fibrinogen, F2,6 = 33.47, ***P = 0.0006; Dunnett’s post hoc test comparing +TGM2 and –TGM2, P < 0.001; BSA, F2,6 = 1.000, P = 0.4219; H3, F2,6 = 130.0, ****P < 0.0001; Dunnett’s post hoc test comparing +TGM2 and –TGM2, P < 0.001; H4, F2,6 = 0.7345, P = 0.5184; nucleosomes, F2,6 = 88.52, ****P < 0.0001; Dunnett’s post hoc test comparing +TGM2 and –TGM2, P < 0.001, and comparing +TGM2/+cystamine and –TGM2, P < 0.05). f, LC–MS/MS analysis of a TGM2-transamidated H3 peptide (H31–15), identifying Gln5 as a substrate for serotonylation. The y and b series indicate peptide fragments at amide bonds. Results confirmed in at least two independent experiments. g, TGM2 monoaminylation assays examining MDC transfer to recombinant wild-type H3 versus H3(Q5A), indicating Q5 as a dominant reactive amino acid substrate for H3. Coomassie staining of loading is shown. H3 results confirmed in at least three independent experiments. Data are mean ± s.e.m.
Extended Data Fig. 3 Histone semisynthesis and nucleosome assembly.
a, Representative semisynthesis of serotonylated histone H3 comprising (i) native chemical ligation between the serotonylated H31–13 α-thioester and a truncated histone H314–135, followed by (ii) cysteine alkylation to give K14 thialysine. b, c, RP-HPLC and mass spectrometry characterization of semisynthetic histone proteins, including H3K4me3 (b) and H3Q5Ser (c). Purified proteins were eluted from a C18 RP-HPLC column using a gradient of 0–73% solvent B (0.1% TFA in 9:1 acetonitrile:water) in solvent A (0.1% TFA in water) detecting absorption at 214 nm. Mass spectra of purified proteins were deconvoluted (inset) and observed versus calculated masses are shown. Results confirmed in at least three independent experiments. d, e, Validation of octamer (d) and mononucleosome (e) assembly post-semisynthesis of unmodified, K4me3 and Q5ser proteins. Results confirmed in at least three independent experiments. f, TGM2 monoaminylation assays on unmodified versus H3K4me3 nucleosomes using biotin cadaverine in place of 5-HT (n = 3, two-tailed Student’s t-test; t4 = 0.500, P = 0.64). H4 is provided as a loading control. g, MLL1-complex methyltransferase assays on unmodified versus Q5ser mononucleosmes (−SAM/−MLL1, +SAM/−MLL1 and +SAM/+MLL1; n = 3, two-tailed Student’s t-test, +/+ versus +/+; t4 = 0.3100, P = 0.76). Data are mean ± s.e.m.
Extended Data Fig. 4 H3Q5ser and H3K4me3Q5ser antibody validations.
a, Synthesis of peptide antigens on 2-Cl trityl resin by (i) iterative Fmoc solid-phase peptide synthesis incorporating Fmoc–Glu(OAII)-OH at position 5 and either Fmoc–Lys(Boc)-OH or Fmoc–Lys(Me3)-OH at position 4, (ii) followed by Pd(0) deallylation, (iii) 5-HT coupling and (iv) acidolytic cleavage from the resin and global deprotection. Side-chain protecting groups are omitted for clarity. b, Peptide dot blot titrations testing the H3Q5ser antibody’s reactivity against unmodified versus Q5ser peptides; note that linear signal was observed only with the Q5ser peptide. Direct blue staining was used to control for peptide loading. c, Western blot analysis of TGM2 serotonylation assays on unmodified mononucleosomes, which reveals that the H3Q5ser antibody only detects signal when the nucleosomes have been transamidated with 5-HT. Direct blue staining was used to control for protein loading. d, Peptide competition western blot analysis of lysates from RN46A-B14 cells indicating the specificity of the H3Q5ser antibody. e, Peptide dot blot titrations testing the H3K4me3Q5ser antibody’s reactivity against various peptides; note that linear signal was observed only with the K4me3Q5ser peptide. Direct blue staining was used to control for peptide loading. f, Western blot analysis of TGM2 serotonylation assays on monomeric EPL K4me3 H3, revealing that the H3K4me3Q5ser antibody only detects signal when the K4me3 histone has been transamidated with 5-HT. Direct blue staining was used to control for protein loading. g, Peptide competition western blot analysis of lysates from RN46A-B14 cells indicating the specificity of the H3K4me3Q5ser antibody in vivo; note that a minimum of 20 μg protein was loaded for all other comparisons made throughout the study. h, Peptide competition western blot analysis of lysates from hPSC-derived 5-HT neurons indicating the specificity of the H3K4me3Q5ser antibody in human cells. For experiments in b–h, similar results were confirmed in at least two independent experiments or assays.
Extended Data Fig. 5 LC–MS/MS identification of H3 serotonylation in cells and brain.
LC–MS/MS analysis of endogenous and synthetic (N-terminally labelled with D5) H3Q5ser or H3K4me3Q5ser peptides in RN46A-B14 cells (post-differentiation, top) and brain (mouse DRN, bottom) following immunoprecipitation using our H3Q5ser and H3K4me3Q5ser antibodies, respectively. a, Liquid chromatography–mass spectrometry (LC–MS, MS1) chromatograms showing elution profiles for the samples including the endogenous peptide versus a synthetic peptide standard. Top to bottom: (i) total ion current chromatogram depicting the summed intensity of all signals; (ii) base peak chromatogram showing the intensity of the top signal; (iii) extracted ion chromatogram of the m/z of the endogenous peptide; (iv) extracted ion chromatogram of the m/z value of the synthetic, heavy-labelled peptide. b, MS/MS (MS2) spectrum (aligned by m/z, and magnified where indicated on either side of the precursor ion) of the H3Q5ser and H3K4me3Q5ser peptides obtained by HCD fragmentation. b+ and y+ fragment ions were annotated manually, and the top insets show good coverage. Results confirmed in at least two independent experiments (RN46A-B14 cells pre- versus post-differentiation and/or in mouse brain).
Extended Data Fig. 6 Euchromatic H3K4me3Q5ser distribution in DRN.
a, Immunofluorescence of H3K4me3Q5ser in brain (DRN) with or without peptide competition as indicated. DAPI was used as a nuclear co-stain and a secondary-antibody-only control is also included. Scale bars, 100 μm. b, Immunofluorescence of H3K4me3Q5ser in DRN (no peptide block) revealing a euchromatic distribution pattern in the nucleus with near-total exclusion from DAPI-rich chromocentres typical of neurons. Scale bars, 10 μm. c, Immunofluorescence images of H3K4me3Q5ser in DRN, counterstained with DAPI, NeuN (a neuronal cell marker) and TPH2 (a marker of serotonergic neurons). Merged images reveal that H3K4me3Q5ser is not only expressed in serotonergic neurons (NeuN+TPH2+, yellow circles) but also in non-serotonergic neurons (NeuN+TPH2–, white circles) and in non-neuronal cells (NeuN–TPH2–, red circles). Aq, aqueduct. Scale bars, 100 μm. d, Western blot validation of TPH2 knockout (KO) in DRN of TPH1 and TPH2 KO mice. Direct blue was used to control for loading. e, Immunofluorescence validation of 5-HT depletion in DRN of TPH1 and TPH2 knockout mice versus wild-type littermate controls. Scale bars, 100 μm. For all immunofluorescence experiments (a–e), results were confirmed in at least two independent experiments. f, Western blot validation that TPH1 and TPH2 knockout results in loss of H3K4me3Q5ser signal in DRN (n = 11 wild type versus n = 7 TPH1 and TPH2 knockout, two-tailed Student’s t-test; t16 = 3.425, **P = 0.0035); no effects on H3K4me3 or total H3 expression were observed. Data are mean ± s.e.m.
Extended Data Fig. 7 H3K4me3Q5ser positively correlates with gene expression in developing mouse brain.
a, RT–qPCR analysis of Tph2 gene expression in embryonic mouse brain (E9.5 (n = 6) versus E17.5 (n = 8), two-tailed Student’s t-test; t12 = 3.100, **P = 0.0092). Actb was used as a normalization control. b, Quantitative western blot analysis of H3K4me3 and H3K4me3Q5ser expression in embryonic mouse brain at E9.5 versus E17.5 (two-tailed Student’s t-test, two-sided, n = 3 independent biological replicates per age; K4me3: t4 = 3.168, *P = 0.0339; K4me3Q5ser: t4 = 4.277, *P = 0.0129). H3 was used as a loading control. c, MACS v.2.1.1-based peak-calling for H3K4me3 versus H3K4me3Q5ser ChIP-seq data in embryonic mouse brain at E17.5 (FDR <0.05, FC >1.2 cut-offs applied after adjusting for multiple comparisons; n = 3 independent biological replicates per age). d, Analysis of H3K4me3Q5ser differential enrichment comparing E17.5 and E9.5 embryonic mouse brain using diffReps (FDR <0.05, FC >1.2 cut-offs applied after adjusting for multiple comparisons; n = 3 independent biological replicates per age). Pie chart indicates distribution of genic differential enrichment events for the mark comparing promoter and gene body regions. e, Overlap of differential enrichment events at protein-coding genes for H3K4me3 and H3K4me3Q5ser versus differential gene expression during mouse brain development indicates significant associations between the two marks, as well as positive correlations with gene expression. f, Odds-ratio analysis (using Fisher’s exact tests) of overlapping genes displaying differential H3K4me3Q5ser enrichment versus differential gene expression (n = 3 independent biological replicates per age, FDR <0.05 cut-off applied after adjusting for multiple comparisons) indicating positive correlations between mark enrichment and permissive gene expression during development. Insert numbers indicate respective P values for associations, followed by the number of protein-coding genes overlapping per significant category. Data are mean ± s.e.m.
Extended Data Fig. 8 Functional validations of the role of H3 serotonyl in permissive gene expression.
a, Peptide dot blot titrations testing the ability of the H3K4me3 antibody to detect the mark in the absence or presence of Q5ser; note that Q5ser does not occlude antibody recognition of the mark. Results confirmed in at least two independent experiments. b, Peptide competition qChIP validation of H3K4me3Q5ser enrichment at two target loci (Actb and Vim) identified via ChIP-seq in RN46A-B14 cells (undifferentiated, n = 1 per group). c, Overlap of differential enrichment events at protein-coding genes for H3K4me3 and H3K4me3Q5ser versus differential gene expression in response to RN46A-B14 cell differentiation indicates positive correlations between H3K4me3Q5ser gene expression in the absence of significant changes in H3K4me3 enrichment (FDR <0.05, FC >1.2 cut-offs applied after adjusting for multiple comparisons. n = 3 independent biological replicates per differentiation state per antibody). d, Recombinant methylation assays (MLL1-complex-mediated) on wild-type H3 versus H3(Q5A), followed by western blot for H3K4me3, indicating that the Q5A mutation does not affect methylation capacity at K4. Direct blue was used to control for protein loading. Results confirmed in at least two independent experiments. e, Odds-ratio analysis (using Fisher’s exact tests) of overlapping genes displaying differential H3K4me3Q5ser enrichment (or not) versus differential gene expression (DEx, n = 5 (H3.3(WT)) versus n = 6 (H3.3(Q5A)) independent biological replicates per virus), FDR <0.05 cut-off applied after adjusting for multiple comparisons) comparing differentiated RN46A-B14 cells expressing either H3.3(WT) or H3.3(Q5A). Insert numbers indicate respective P values for associations, followed by the number of protein-coding genes overlapping per significant category. f, Heat map of RNA-seq data comparing undifferentiated and differentiated (n = 5 H3.3(WT) versus n = 6 H3.3(Q5A)-expressing) RN46A-B14 cells using normalized RNA expression values (averaged between replicates) to generate z-scores for each row. The genes that displayed differential enrichment during differentiation are represented, along with altered gene expression (see Fig. 3), and along with opposing regulation in the context of H3.3(Q5A). These genes were found to significantly enrich for pathways associated with axon guidance signalling via KEGG analysis (see Supplementary Table 28). g, RT–qPCR analysis of candidate gene expression in RN46A-B14 cells (n = 5 DMSO versus n = 6 LDN 27219/Tgm2i; one-tailed Student’s t-test; Iws1: t9 = 2.559, *P = 0.0154; Sema3e: t8 = 3.982, **P = 0.0020; Robo1: t9 = 3.344, **P = 0.0043; Srgap1: t8 = 3.312, **P = 0.0053; Nrp1: t9 = 2.452, *P = 0.0183; Zmat3: t9 = 3.820, **P = 0.0020; Actg1: t8 = 7.836, ****P < 0.0001; Reln: t9 = 2.209, *P = 0.0273; Qser1: t8 = 2.513, *P = 0.0181; Ppp3ca: t8 = 2.418, *P = 0.0210; Arid5b: t9 = 1.754, *P = 0.0567; Pten: t9 = 1.936, *P = 0.0424). Actb was used as a normalization control. h, Immunofluorescence of RN46A-B14 cells infected during differentiation with lentiviruses expressing either wild-type H3.3–HA or H3.3(Q5A)–HA. Results confirmed at least three independent coverslips per viral treatment. Scale bars, 20 μm. i, Neurite outgrowth analysis examining RN46A-B14 cellular length post-differentiation (n = 54 H3.3(WT) versus n = 44 H3.3(Q5A)-expressing cells; two-tailed Student’s t-test, t96 = 4.664, ****P < 0.0001). Data are mean ± s.e.m.
Extended Data Fig. 9 Genome-wide associations between H3K4me3Q5ser and TFIID.
a, Western blot quantifications related to Fig. 4a (Student’s t-tests, two-sided) of modified H31–10 peptide immunoprecipitations from HeLa nuclear extracts (TAF2: n = 3 per peptide, t4 = 2.724, *P = 0.05; TAF3: n = 3 per peptide, t4 = 2.920, *P = 0.04; TAF5: n = 3 per peptide, t4 = 3.685, *P = 0.02; TAF7: n = 3 per peptide, t4 = 8.885, ***P = 0.0009; TBP: n = 4 per peptide, t6 = 5.383, **P = 0.0017). b, Modified H31–10 peptide immunoprecipitations against purified TFIID from Flag-tagged TBP expressing soluble HeLa nuclear extracts (left, silver stain of the purified complex), followed by western blots (right). Inputs are provided (n = 1 per protein examined). c, Gene plot (ngs.plot) of TAF3–TFIID enrichment (fold change (expressed in log2) versus input) comparing signals pre- and post-differentiation (n = 3 independent biological replicates per differentiation states). d, IGV genome browser tracks of the Prrg4 locus for H3K4me3, H3K4me3Q5ser and TAF3–TFIID (versus DNA input) in RN46A-B14 cells pre- and post differentiation (example locus was chosen on the basis of MACS v.2.1.1- and diffReps-based statistical comparisons). Odds-ratio analysis (using Fisher’s exact tests) of overlapping genes displaying differential H3K4me3 or H3K4me3Q5ser enrichment (or those genes containing differential sites for both or neither of the two marks, FDR <0.05, FC >1.2) versus TAF3 peaks (e; n = 3 independent biological replicates per differentiation state, MACS v.2.1.1, FDR <0.05, FC >1.2 cut-offs applied after adjusting for multiple comparisons; normalized to respective DNA inputs) or differential TAF3–TFIID enrichment (f; n = 3 independent biological replicates per differentiation state, diffReps, FDR <0.05, FC >1.2 cut-offs applied after adjusting for multiple comparisons) post-differentiation. Insert numbers indicate respective P values for associations, followed by the number of protein-coding genes overlapping per significant category. g, Model of the effect of Q5ser on K4me3-mediated gene expression in vivo; our data suggest that the presence of combinatorial H3K4me3Q5ser alters interactions with certain K4me3 reader proteins, such as the TFIID complex, to potentiate or stabilize permissive gene expression in mammalian cells.
Supplementary information
Supplementary Figure 1
This file contains uncropped western blots and other gels, as well as images/quantification for each of the human PSC lines used to generate 5-HT neurons.
Supplementary Tables
This file contains Supplementary Tables 1-34, containing processed ChIP- and RNA-seq data from cells and brain, as well as proteomic binding data and qPCR/qChIP primer sets.
Rights and permissions
About this article

Cite this article
Farrelly, L.A., Thompson, R.E., Zhao, S. et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567, 535–539 (2019). https://doi.org/10.1038/s41586-019-1024-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1024-7
This article is cited by
-
Chromatin accessibility and H3K9me3 landscapes reveal long-term epigenetic effects of fetal-neonatal iron deficiency in rat hippocampus
BMC Genomics (2024)
-
Interrogating epigenetic mechanisms with chemically customized chromatin
Nature Reviews Genetics (2024)
-
Histone butyrylation in the mouse intestine is mediated by the microbiota and associated with regulation of gene expression
Nature Metabolism (2024)
-
Epigenomic insights into common human disease pathology
Cellular and Molecular Life Sciences (2024)
-
New Insights on NLRP3 Inflammasome: Mechanisms of Activation, Inhibition, and Epigenetic Regulation
Journal of Neuroimmune Pharmacology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
