
Abstract
Mechanisms that integrate the metabolic state of a cell with regulatory pathways are necessary to maintain cellular homeostasis. Endogenous, intrinsically reactive metabolites can form functional, covalent modifications on proteins without the aid of enzymes1,2, and regulate cellular functions such as metabolism3,4,5 and transcription6. An important ‘sensor’ protein that captures specific metabolic information and transforms it into an appropriate response is KEAP1, which contains reactive cysteine residues that collectively act as an electrophile sensor tuned to respond to reactive species resulting from endogenous and xenobiotic molecules. Covalent modification of KEAP1 results in reduced ubiquitination and the accumulation of NRF27,8, which then initiates the transcription of cytoprotective genes at antioxidant-response element loci. Here we identify a small-molecule inhibitor of the glycolytic enzyme PGK1, and reveal a direct link between glycolysis and NRF2 signalling. Inhibition of PGK1 results in accumulation of the reactive metabolite methylglyoxal, which selectively modifies KEAP1 to form a methylimidazole crosslink between proximal cysteine and arginine residues (MICA). This posttranslational modification results in the dimerization of KEAP1, the accumulation of NRF2 and activation of the NRF2 transcriptional program. These results demonstrate the existence of direct inter-pathway communication between glycolysis and the KEAP1–NRF2 transcriptional axis, provide insight into the metabolic regulation of the cellular stress response, and suggest a therapeutic strategy for controlling the cytoprotective antioxidant response in several human diseases.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
RNA-seq primary data are deposited in the Gene Expression Omnibus (GEO) under accession number GSE116642. Source data for all mouse experiments have been provided. Full scans for western blots and gels are available in the Supplementary Information. All other data are available on reasonable request.
References
Lin, H., Su, X. & He, B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem. Biol. 7, 947–960 (2012).
Wagner, G.R., et al. A class of reactive acyl-CoA species reveals the non-enzymatic origins of protein acylation. Cell Metab. 25, 823–837 (2017).
Zhang, Z. et al. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63 (2011).
Moellering, R. E. & Cravatt, B. F. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 341, 549–553 (2013).
Weinert, B. T. et al. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell 51, 265–272 (2013).
Sabari, B. R., Zhang, D., Allis, C. D. & Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18, 90–101 (2016).
Kobayashi, A. et al. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 26, 221–229 (2006).
Lo, S. C., Li, X., Henzl, M. T., Beamer, L. J. & Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 25, 3605–3617 (2006).
Jaramillo, M. C. & Zhang, D. D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 27, 2179–2191 (2013).
Scannevin, R. H. et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 341, 274–284 (2012).
Khor, T. O. et al. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 66, 11580–11584 (2006).
Uruno, A. et al. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 33, 2996–3010 (2013).
Sykiotis, G. P. & Bohmann, D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev. Cell 14, 76–85 (2008).
Cleasby, A. et al. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS One 9, e98896 (2014).
Hur, W. et al. A small-molecule inducer of the antioxidant response element. Chem. Biol. 17, 537–547 (2010).
Saw, C. L. et al. Impact of Nrf2 on UVB-induced skin inflammation/photoprotection and photoprotective effect of sulforaphane. Mol. Carcinog. 50, 479–486 (2011).
Tao, S., Justiniano, R., Zhang, D. D. & Wondrak, G. T. The Nrf2-inducers tanshinone I and dihydrotanshinone protect human skin cells and reconstructed human skin against solar simulated UV. Redox Biol. 1, 532–541 (2013).
El-Abaseri, T. B., Putta, S. & Hansen, L. A. Ultraviolet irradiation induces keratinocyte proliferation and epidermal hyperplasia through the activation of the epidermal growth factor receptor. Carcinogenesis 27, 225–231 (2006).
Martinez Molina, D. et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 (2013).
Chang, J. W., Lee, G., Coukos, J. S. & Moellering, R. E. Profiling reactive metabolites via chemical trapping and targeted mass spectrometry. Anal. Chem. 88, 6658–6661 (2016).
Zhang, D. D. & Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 23, 8137–8151 (2003).
Wakabayashi, N. et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl Acad. Sci. USA 101, 2040–2045 (2004).
Ogura, T. et al. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc. Natl Acad. Sci. USA 107, 2842–2847 (2010).
Lo, T. W., Westwood, M. E., McLellan, A. C., Selwood, T. & Thornalley, P. J. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with Nα-acetylarginine, Nα-acetylcysteine, and Nα-acetyllysine, and bovine serum albumin. J. Biol. Chem. 269, 32299–32305 (1994).
Rabbani, N. & Thornalley, P. J. Dicarbonyl proteome and genome damage in metabolic and vascular disease. Biochem. Soc. Trans. 42, 425–432 (2014).
Chaplen, F. W., Fahl, W. E. & Cameron, D. C. Evidence of high levels of methylglyoxal in cultured Chinese hamster ovary cells. Proc. Natl Acad. Sci. USA 95, 5533–5538 (1998).
Dhar, A., Desai, K., Liu, J. & Wu, L. Methylglyoxal, protein binding and biological samples: are we getting the true measure? J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 877, 1093–1100 (2009).
Moraru, A. et al. Elevated levels of the reactive metabolite methylglyoxal recapitulate progression of type 2 diabetes. Cell Metab. 27, 926–934 (2018).
Ravichandran, M. et al. Impairing l-threonine catabolism promotes healthspan through methylglyoxal-mediated proteohormesis. Cell Metab. 27, 914–925 (2018).
Nishimoto, S., Koike, S., Inoue, N., Suzuki, T. & Ogasawara, Y. Activation of Nrf2 attenuates carbonyl stress induced by methylglyoxal in human neuroblastoma cells: Increase in GSH levels is a critical event for the detoxification mechanism. Biochem. Biophys. Res. Commun. 483, 874–879 (2017).
Mitsuishi, Y. et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79 (2012).
Hur, W. & Gray, N. S. Small molecule modulators of antioxidant response pathway. Curr. Opin. Chem. Biol. 15, 162–173 (2011).
Rabbani, N. & Thornalley, P. J. Measurement of methylglyoxal by stable isotopic dilution analysis LC–MS/MS with corroborative prediction in physiological samples. Nat. Protoc. 9, 1969–1979 (2014).
Bücher, T. Phosphoglycerate kinase from Brewer’s yeast. Methods Enzymol. 1, 415–422 (1955).
Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V. & Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 (2007).
Ruifrok, A. C. & Johnston, D. A. Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 23, 291–299 (2001).
Acknowledgements
We thank S. Zhu for discussions about target identification experiments. Animal experiments were approved by the Scripps Research Institute Institutional Review Board. We are grateful for financial support of this work from the following: Kwanjeong Educational Fellowship (to G.L.); NIH MSTP Training Grant (T32GM007281 to J.S.C.); NIH R00CA175399, R01CA211916 and DP2GM128199 (R.E.M.); V Foundation for Cancer Research (V2015-020 to R.E.M.); Damon Runyon Cancer Research Foundation (DFS08-14); The Skaggs Institute for Chemical Biology, and The University of Chicago.
Reviewer information
Nature thanks H. Christofk, A. Dinkova-Kostova, H. Lin and the anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
All authors reviewed the manuscript. M.J.B., G.L., J.S.C., H.Y., C.Z., J.W.C., I.A., L.L.L. and R.E.M. designed and performed biochemical and cell-based biological experiments. H.Y., J.W.C., J.S.C. and A.K.C. synthesized and characterized chemical probes and reagents. E.N.C., M.J.B., L.L.L. and P.G.S. designed, performed and analysed in vivo experiments. G.L., J.S.C., J.W.C. and R.E.M. designed, performed and analysed metabolomic, proteomic and structural characterization experiments. L.L.L., P.G.S. and R.E.M. conceived of the study and supervised research. M.J.B. and R.E.M. wrote the manuscript with considerable input from all authors.
Corresponding authors
Ethics declarations
Competing interests
Declared none.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 A high-throughput screen identifies a non-covalent NRF2 activator chemical series that activate a robust NRF2 transcriptional program in multiple cell types.
a, Plate-based Z-scores of ARE-LUC luminance measurements of all test compounds from a 30,000 compound screen in IMR32 cells. b, Structure of screening hit CBR-470-0. c, Relative ARE-LUC luminance measurements from IMR32 cells treated for 24 h with a concentration response of CBR-470-0 and reported NRF2 activators TBHQ and AI-1 (n = 3 biologically independent samples, mean and s.e.m.). d, LC–MS quantification of CBR-470-1 (50 μM) incubated in the presence or absence of GSH (1 mM) in PBS for 1 h (left) and 48 h (right). Relative ion intensities within each time point were compared with representative chromatograms shown (n = 2). e, Relative ARE-LUC luminance values from IMR32 cells transfected with wild-type (wt) or mutant (mt; two core nucleotides necessary for NRF2 binding were changed from GC to AT) ARE-LUC reporter constructs and treated with the indicated doses of CBR-470-1 for 24 h (n = 3, mean and s.e.m.). f, Relative abundance of NRF2-dependent transcripts as determined by qRT–PCR from IMR32 cells treated for 24 h with 5 μM CBR-470-1 (n = 3). g, Western blot analyses of total NRF2 protein content or NRF2-controlled genes (NQO1 and HMOX1) from IMR32 cells treated for 24 h with 5 μM CBR-470-1 (n = 5). h, Western blot analyses of total NRF2 protein content from the indicated cell types treated for 4 h with 5 μM CBR-470-1 (n = 3). i, Relative expression levels of NQO1 and HMOX1 from the indicated cell types treated for 24 h with 5 μM CBR-470-1 (n = 3, mean and s.d.). j, Relative ARE-LUC luminescence values from HEK293T cells transfected with the indicated shRNA constructs and pTI-ARE-LUC and then treated with TBHQ (10 μM) or CBR-470-1 (5 μM) for 24 h (n = 3). k, Relative viability measurements of SH-SY5Y cells treated with either CBR-470-1 (5 μM) or TBHQ (10 μM) for 48 h and then challenged with the indicated doses of TBHP for 8 h (n = 4). Data are mean and s.d. or s.e.m. of biologically independent samples. *P < 0.05, **P < 0.005, ***P < 0.001, univariate two-sided t-test.
Extended Data Fig. 2 CBR-470-2 pharmacokinetics and in vivo activity.
a, Structure of CBR-470-2. b, Relative ARE-LUC luminance values from IMR32 cells transfected with pTI-ARE-LUC and treated with the indicated doses of CBR-470-1 and CBR-470-2 for 24 h (n = 3 biologically independent samples). c, Plasma concentrations of CBR-470-2 from mice treated with a single 20 mg kg−1 dose of compound (n = 3 animals, mean and s.e.m.). d, e, Relative transcript levels of Nqo1 and Hmox1 from mouse epidermal keratinocytes (d) and mouse dermal fibroblasts (e) treated for 24 h with the indicated doses of compound (n = 3 biologically independent samples, mean and s.d.). f, Blinded erythema scores from mice treated with vehicle, CBR-470-2 or bardoxolone after exposure to UV (n = 8 animals, mean and s.e.m.). *P < 0.05, *** P< 0.005, one-way ANOVA with Dunnett’s correction. g, Representative images of UV-exposed dorsal regions of animals at day 10 of the study.
Extended Data Fig. 3 A photoactivatable affinity probe-based approach identifies PGK1 as the relevant cellular target of CBR-470-1.
a, Structure of CBR-470-PAP. b, Relative ARE-LUC luminance values from IMR32 cells transfected with pTI-ARE-LUC and then treated with the indicated doses of CBR-470-PAP for 24 h (n = 3). c, Silver staining and anti-biotin western blots of ammonium sulfate fractionated lysates from UV-irradiated IMR32 cells treated with 5 μM for 1 h with or without CBR-470-1 competition (250 μM) (n = 3). Shown on the right are initial proteomic target results from gel-band digestion and LC–MS/MS analysis. d, Anti-biotin western blots from in vitro crosslinking assays with recombinant PGK1 and EBP1 in the presence of the indicated doses of CBR-470-PAP (n = 2). e, Anti-biotin western blot analyses from an in vitro crosslinking assay with recombinant PGK1 in the presence of CBR-470-PAP (1 μM) and indicated concentration of soluble CBR-470-1 competitor (n = 2). f, Anti-biotin western blot analyses of cells treated with 5 μM CBR-470-PAP after transduction with anti-PGK1 and anti-EBP1 shRNA for 48 h. Depletion of PGK1 protein selectively reduces CBR-470-PAP-dependent labelling (n = 2). g, Dye-based thermal denaturation assay with recombinant PGK1 in the presence CBR-470-1 (20 μM) or vehicle alone (n = 3). Calculated melting temperature (Tm) values are listed. h, i, Dose-dependent thermal stability assay of recombinant PGK1 and GAPDH in the presence of increasing doses of CBR-470-1 near the Tm of both proteins (57 °C) (n = 5) (h) or room temperature (n = 3) (i). Western blot of sample supernatants after centrifugation (13,000 r.p.m.) detected total PGK1 and GAPDH protein, which were plotted in Prism (below). j, ARE-LUC reporter activity in HEK293T cells with transient shRNA knockdown of ENO1 (n = 3). Data are mean ± s.e.m. of biologically independent samples.
Extended Data Fig. 4 CBR-470-1 inhibits PGK1 in vitro and in situ.
a, Schematic of the GAPDH/PGK1 coupled assay. Pre-equilibration of the GAPDH reaction (top left) results in an NAD+/NADH equilibrium, which after addition of PGK1 and ADP pulls the reaction to the right, producing more NADH. Monitoring NADH absorbance after the addition of PGK1 (bottom right) can be used to monitor PGK1 activity in the forward direction (right). Kinetic monitoring of NADH absorbance (340 nm) after established equilibrium with GAPDH shows little change (black curve), but is significantly increased after the addition of PGK1, pulling the equilibrium to the right (red curve). b, CBR-470-1 does not affect the GAPDH equilibrium alone, but significantly inhibits PGK1-dependent activity and accumulation of NADH (n = 5). c, d, Relative level of central metabolites in IMR32 cells treated with viral knockdown of PGK1 for 72 h (c) (n = 4) and with CBR-470-1 relative DMSO alone for the indicative times (d) (n = 3). Each metabolite is normalized to the control condition at each time point. Statistical analysis was by univariate two-sided t-test (b). Data are mean ± s.e.m. of biologically independent samples.
Extended Data Fig. 5 Modulation of PGK1 induces HMM-KEAP1.
a, Anti-phosphoglyceryl-lysine (pgK) and anti-GAPDH western blots analysis of CBR-470-1 or DMSO-treated IMR32 cells at early (30 min) and late (24 h) time points (n = 6). b, Anti-Flag (left) and anti-pgK (right) western blot analysis of affinity purified Flag–KEAP1 from HEK293T cells treated with DMSO or CBR-470-1 for 30 min. Duplicate samples were run under non-reducing (left) and reducing (DTT, right) conditions (n = 6). c, Densitometry quantification of total endogenous KEAP1 levels (combined bands at approximately 70 and 140 kDa) in IMR32 cells treated with DMSO or CBR-470-1 for the indicated times (n = 6). d, Western blot detection of Flag–KEAP1 in HEK293T cells comparing non-reducing reagent to DTT (left), and the stability of CBR-470-1-dependent HMM-KEAP1 to the presence of DTT (12.5 mM final concentration, middle) or β-mercaptoethanol (βME; 5% (v/v) final concentration, right) during sample preparation. e, Time-dependent CBR-470-1 treatment of HEK293T cells expressing Flag–KEAP1. Time-dependent assays were run with 20 μM CBR-470-1 with western blot analysis at the indicated time-points (n = 8). f, g, Western blot detection (f) and quantification (g) of endogenous KEAP1 and β-actin in IMR32 cells treated with DMSO or CBR-470-1 for the indicated times (n = 6). Arrows indicate monomeric KEAP1 (70 kDa) and HMM-KEAP1 (140 kDa) bands. h, i, Western blot (h) detection and quantification (i) of Flag–KEAP1 in HEK293T cells exposed to increasing doses of CBR-470-1 (n = 3). j, Kinetic qRT–PCR measurement of NQO1 mRNA levels from IMR32 cells treated with TBHQ (10 μM) or CBR-470-1 (10 μM) for the indicated times (n = 3). k, Quantification of HMM-KEAP1 formation after treatment with CBR-470-1 or the direct KEAP1 alkylator TBHQ, in the presence or absence of GSH or N-acetylcysteine (NAC) (n = 3). All measurements were taken after 8 h of treatment in Flag–KEAP1-expressing HEK293T cells. l, Transient shRNA knockdown of PGK1 induced HMM-KEAP1 formation, which was blocked by co-treatment of cells with GSH (n = 3). m, Anti-Flag western blot analysis of Flag–KEAP1 monomer and the HMM–KEAP1 fraction, with dose-dependent incubation of distilled MGO in lysates from HEK293T cells expressing Flag–KEAP1 (n = 4). n, SDS–PAGE gel (silver stain) and anti-Flag western blot analysis of purified KEAP1 treated with MGO under the indicated reducing conditions for 2 h at 37 °C (n = 3). Purified protein reactions were quenched in 4× SDS loading buffer containing β-mercaptoethanol and processed for gel analysis as in d. Data are mean ± s.e.m. of biologically independent samples.
Extended Data Fig. 6 MGO and glyoxylase activity regulates NRF2 activation.
CBR-470-1 causes increased levels of MGO in cells. a, Schematic depicting chemical derivatization and trapping of cellular MGO for analysis by targeted metabolomics using two unique fragment ions. b, c, Daughter ion fragments (b) and resulting MS/MS quantification of MGO levels (c) in IMR32 cells treated with CBR-470-1, relative to DMSO (n = 4). d, Quantitative LC–MS/MS measurement of cellular MGO levels in IMR32 cells treated for 2 h with CBR-470-1 or co-treated for 2 h with CBR-470-1 and N-acetylcysteine (2 mM) relative to DMSO (n = 4). e, Relative ARE-LUC luminance values from IMR32 cells transfected with pTI-ARE-LUC and co-treated with the indicated doses of CBR-470-1 and GSH (n = 3). f, Relative levels of NQO1 and HMOX1 transcripts from IMR32 cells co-treated with CBR-470-1 (10 µM) and the indicated concentrations of GSH for 24 h (n = 3). g, Fractional ARE-LUC values from HEK293T cells transiently co-transfected with pTI-ARE-LUC and the indicated shRNAs and then treated for 24 h with the indicated doses of CBR-470-1 (n = 3). h, ARE-LUC reporter activity in HEK293T cells treated with CBR-470-1 alone (black) and with a cell-permeable small-molecule GLO1 inhibitor (red) (n = 3). Statistical analysis was by univariate two-sided t-test (d, h). Data are mean ± s.e.m. of biologically independent samples.
Extended Data Fig. 7 Schematic of SILAC-based proteomic mapping of KEAP1 modifications in response to CBR-470-1 and NMR characterization of CR-MGO peptide.
a, SILAC experiments in which Flag-tagged KEAP1 was treated with vehicle (‘light’) and CBR-470-1 or MGO (‘heavy’). Subsequent mixing of the cell lysates, anti-Flag enrichment, tryptic digestion and LC–MS/MS analysis permitted the detection of unmodified portions of KEAP1, which retained approximate 1:1 SILAC ratios relative to the median ratios for all detected KEAP1 peptides. By contrast, peptides that are modified under one condition will no longer match tryptic MS/MS searches, resulting in skewed SILAC ratios that ‘drop out’ (bottom). b, SILAC ratios for individual tryptic peptides from Flag–KEAP1-enriched DMSO-treated light cells and CBR-470-1-treated heavy cells, relative to the median ratio of all KEAP1 peptides. Highlighted tryptic peptides were significantly reduced by three- to fourfold relative to the KEAP1 median, indicative of structural modification (n = 8). c, Structural depiction of potentially modified stretches of human KEAP1 (red) using published X-ray crystal structure of the BTB (PDB code 4CXI) and KELCH (PDB code 1U6D) domains. Intervening protein stretches are depicted as unstructured loops in green. d, SILAC ratios for individual tryptic peptides from Flag–KEAP1-enriched MGO-treated heavy cell lysates and non-treated light cell lysates, relative to the median ratio of all KEAP1 peptides. Highlighted tryptic peptides were significantly reduced by 2- to 2.5-fold relative to the KEAP1 median, indicative of structural modification (n = 12). e, Representative western blotting analysis of Flag–KEAP1 dimerization from HEK293T cells pre-treated with BARD followed by CBR-470-1 treatment for 4 h (n = 3). f, 1H-NMR of CR-MGO peptide (isolated product of MGO incubated with Ac-NH-VVCGGGRGG-C(O)NH2 peptide). 1H-NMR (500 MHz, d6-DMSO) δ 12.17 (s, 1H), 12.02 (s, 1H), 8.44 (t, J = 5.6 Hz, 1H), 8.32–8.29 (m, 2H), 8.23 (t, J = 5.6 Hz, 1H), 8.14 (t, J = 5.9 Hz, 1H), 8.05 (t, J = 5.9 Hz, 1H), 8.01 (t, J = 5.9 Hz, 1H), 7.93 (d, J = 8.5 Hz, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.26 (s, 1H), 7.09 (s, 1H), 4.33–4.28 (m, 1H), 4.25-4.16 (m, 3H), 3.83 (dd, J = 6.9 Hz, J = 16.2 Hz, 1H), 3.79-3.67 (m, 6H), 3.63 (d, J = 5.7 Hz, 2H), 3.54 (dd, J = 4.9 Hz, J = 16.2 Hz, 1H), 3.18-3.13 (m, 2H), 3.04 (dd, J = 4.9 Hz, J = 13.9 Hz, 1H), 2.88 (dd, J = 8.6 Hz, J = 13.6 Hz, 1H), 2.04 (s, 3H), 1.96 (sep, J = 6.8 Hz, 2H), 1.87 (s, 3H), 1.80–1.75 (m, 1H), 1.56–1.47 (m, 3H), 0.87–0.82 (m, 12H). g, 1H-NMR of CR peptide (Ac-NH-VVCGGGRGG-C(O)NH2). 1H-NMR (500 MHz, d6-DMSO) δ 8.27–8.24 (m, 2H), 8.18 (t, J = 5.7 Hz, 1H), 8.13–8.08 (m, 3H), 8.04 (t, J = 5.7 Hz, 1H), 7.91 (d, J = 8.8 Hz), 7.86 (d, J = 8.8 Hz, 1H), 7.43 (t, J = 5.4 Hz, 1H), 7.28 (s, 1H), 7.10 (s, 1H), 4.39 (dt, J = 5.6 Hz, J = 7.4 Hz, 1H), 4.28 (dt, J = 5.7 Hz, J = 7.2 Hz, 1H), 4.21-4.13 (m, 2H), 3.82-3.70 (m, 8H), 3.64 (d, J = 5.8, 2H), 3.08 (dt, J = 6.5 Hz, J = 6.5 Hz, 2H), 2.80-2.67 (m, 2H), 2.43 (t, J = 8.6 Hz, 1H), 1.94 (sep, J = 6.8 Hz, 2H), 1.85 (s, 3H), 1.75–1.68 (m, 1H), 1.54–1.42 (m, 3H), 0.85–0.81 (m, 12H) h, 1H–1H total correlation spectroscopy (TOCSY) of CR-MGO peptide. i, Peak assignment for CR-MGO peptide TOCSY spectrum. Data are mean ± s.e.m. of biologically independent samples.
Extended Data Fig. 8 MS2 analysis of CR-MGO-crosslinked KEAP1 peptide.
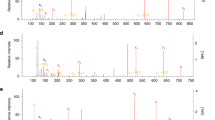
a, Targeted PRM transitions (n = 6). b, Annotated MS/MS spectrum from the crosslinked C151–R135 KEAP1 peptide.
Supplementary information
Supplementary Figure 1
The uncropped gel and Western blots.
Source data
Rights and permissions
About this article

Cite this article
Bollong, M.J., Lee, G., Coukos, J.S. et al. A metabolite-derived protein modification integrates glycolysis with KEAP1–NRF2 signalling. Nature 562, 600–604 (2018). https://doi.org/10.1038/s41586-018-0622-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-018-0622-0
Keywords
This article is cited by
-
Ferroptosis in early brain injury after subarachnoid hemorrhage: review of literature
Chinese Neurosurgical Journal (2024)
-
The pleiotropic functions of reactive oxygen species in cancer
Nature Cancer (2024)
-
Methylglyoxal, a highly reactive dicarbonyl compound, as a threat for blood brain barrier integrity
Fluids and Barriers of the CNS (2023)
-
Regulation of phosphoglycerate kinase 1 and its critical role in cancer
Cell Communication and Signaling (2023)
-
TNFAIP2 confers cisplatin resistance in head and neck squamous cell carcinoma via KEAP1/NRF2 signaling
Journal of Experimental & Clinical Cancer Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
