
Abstract
Zika virus (ZIKV) is a mosquito-transmitted flavivirus that has emerged as a global health threat because of its potential to generate explosive epidemics and ability to cause congenital disease in the context of infection during pregnancy. Whereas much is known about the biology of related flaviviruses, the unique features of ZIKV pathogenesis, including infection of the fetus, persistence in immune-privileged sites and sexual transmission, have presented new challenges. The rapid development of cell culture and animal models has facilitated a new appreciation of ZIKV biology. This knowledge has created opportunities for the development of countermeasures, including multiple ZIKV vaccine candidates, which are advancing through clinical trials. Here we describe the recent advances that have led to a new understanding of the causes and consequences of the ZIKV epidemic.
Similar content being viewed by others

Main
ZIKV was first isolated from a febrile sentinel monkey in Uganda in 1947. Serological data suggest that ZIKV was distributed widely throughout Africa and subsequently in Asia despite the absence of described morbidity1. The first ZIKV outbreak to garner international attention occurred on Yap Island in the Western Pacific Ocean in 2007. Forty-nine confirmed human cases were reported2. More than half of the inhabitants of Yap were believed to have been infected, with many experiencing rash, fever and arthralgia. ZIKV activity was next detected in the islands of French Polynesia in 2013, with a larger number of infections. Some of the unique clinical features of ZIKV (for example, Guillain–Barré syndrome, congenital malformations and the presence of the virus in semen) were identified during this outbreak or later in retrospective studies3. ZIKV was introduced in Brazil in late 2013 or early 2014, spread rapidly within the northeast part of the country, and was repeatedly introduced into regions of the Americas4. The large number of infections and links to congenital neurodevelopmental defects identified this epidemic as an international public health emergency. ZIKV activity in the Americas peaked in the early spring of 2016, followed by a marked decrease in reported cases in 2017, which is probably attributable to the effect of herd immunity. Seroprevalence studies suggest that 63% of the inhabitants of Salvador, Brazil were infected during this outbreak5. By 2017, more than 220,000 confirmed and 580,000 suspected cases were reported in 52 countries or territories in the Americas (PAHO Zika Cumulative Cases; 4 January 2018).
Viral structure
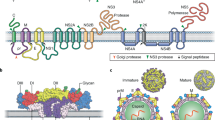
Flaviviruses encapsulate a positive-stranded RNA genome, which encodes a single open reading frame flanked by two structured untranslated regions (UTRs) (Fig. 1). The single viral polyprotein is processed by host and viral proteases into three structural proteins (capsid (C), pre-membrane (prM) and envelope (E)) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5), the latter of which mediate genome replication, viral polyprotein processing and modulation of the host response. ZIKV virions are composed of the three structural proteins, a lipid envelope and the viral RNA genome. The E protein is an elongated protein, which consists of three ectodomains connected by short flexible loops and is anchored to the viral membrane by a helical stem and two antiparallel transmembrane domains (Fig. 1). In most ZIKV strains, the E protein is modified by a single N-linked glycan at position E154 located in domain I (E-DI); some pre-epidemic ZIKV strains from Africa lack this N-linked glycan and are less neuroinvasive6. Although ZIKV is most closely related to another African flavivirus called Spondweni virus (approximately 68% E protein amino acid identity), it shares sequence similarity with other flaviviruses. Approximately 50% of the E protein is conserved among ZIKV and dengue (DENV) virus strains. Although conserved regions could be targeted by broadly reactive protective antibodies7,8, this feature complicates the development of virus-specific diagnostics, and raises the prospect of adverse immune reactions in individuals exposed sequentially to ZIKV and DENV9. Two lineages of ZIKV (African and Asian) differ from each other by approximately 10% at the nucleotide level1. ZIKV strains in the Americas descend from the Asian lineage10.

a, ZIKV encapsulates a positive-stranded genomic RNA sequence that encodes a single polyprotein, which is cleaved into three structural and seven non-structural proteins. b, E consists of three ectodomains (E-DI, E-DII and E-DIII, which are shown in red, yellow and blue, respectively) and is anchored into the viral membrane by two anti-parallel transmembrane domains (TMD; grey). A highly conserved fusion loop (DII-FL) is located at the distal end of DII (green). c, Mature virions incorporate 180 copies of the E protein arranged with icosahedral symmetry. E proteins are in three environments defined by their proximity to the two-, three- and fivefold symmetry axes (shades of purple)12,13.
The prM protein is a small glycoprotein that is connected to the viral membrane by antiparallel transmembrane helices. Immature ZIKV virions assemble on endoplasmic reticulum-derived membranes as particles on which trimers of prM–E heterodimers form spiked projections arranged with icosahedral symmetry11. These non-infectious virions transit through the secretory pathway and undergo a conformational change upon exposure to the mildly acidic environment in the trans-Golgi network that enables cleavage of prM by a host furin-like protease. Mature virions are characterized by a relatively smooth structure created by 90 E protein dimers orientated parallel to the plane of the viral membrane12,13 (Fig. 1). However, prM cleavage can be inefficient, resulting in partially mature infectious virions that retain uncleaved prM. Although the efficiency of prM cleavage on ZIKV relative to other flaviviruses is not yet known, partially mature ZIKV virions may be infectious. Although there has been rapid progress into the structural biology of ZIKV virions, these studies capture only a single state of mature and immature virions. Insights into the structural ensembles that are adopted by the virus throughout its replication cycle await further study14. Additional comparative analyses of prM–E proteins and virion structures of pre- and post-epidemic ZIKV strains could provide further insights into how ZIKV evolved to become more pathogenic and cause distinct clinical syndromes.
ZIKV infection in different clinical contexts
The clinical syndrome caused by ZIKV in humans was historically reported as a mild influenza-like illness that resolved within days and occurred in approximately 20% of infected individuals2 (Fig. 2). However, in French Polynesia, the rate of symptomatic infections was higher (approximately 50%)15. The most common signs and symptoms of ZIKV infection in the French Polynesian and American outbreaks occurred within 3–7 days of being bitten by a mosquito and included fever (72%), arthralgia and myalgia (65%), conjunctivitis (63%), headache (46%), fatigue and/or rash3. During the recent epidemics, ZIKV infection also has been associated, albeit infrequently, with severe disease in adults, including multi-organ failure16, meningitis and encephalitis17, and thrombocytopenia18. Although ZIKV generally does not cause fatal disease in adults, mortality has been described in children with sickle cell disease, adults with cancer16 and those cases who develop Guillain–Barré syndrome19, a progressive polyneuropathy linked to ZIKV infection, which occurred in 1/6,500 to 1/17,000 individuals in endemic regions3,20 (Fig. 2).

a, Transmission. ZIKV is transmitted in an epidemic cycle between Aedes mosquitoes and humans. ZIKV can also be transmitted through sexual contact or vertical transmission from an infected pregnant mother to her fetus. b, Clinical syndromes. Most ZIKV infections are asymptomatic. Among symptomatic cases, most patients develop an illness characterized by fever, rash, conjunctivitis, headaches and muscle and/or joint pain. During pregnancy, ZIKV infection can result in microcephaly, congenital ZIKV syndrome (CZS) and fetal demise. In a subset of adults, infection is linked to Guillain–Barré syndrome, which can result in muscle weakness and paralysis.
A distinguishing feature of ZIKV infection during the recent epidemic is an apparent broadening of cellular tropism and persistence in multiple organs; this has resulted in ostensibly new clinical manifestations. It remains unclear whether this reflects a fundamental change in ZIKV virulence or whether this is now appreciated owing to the greater number of diagnosed infections. ZIKV persists in whole-blood and immune-sanctuary sites. In multiple case reports, whole blood from non-pregnant adults remained positive for ZIKV RNA for 60–100 days, long after serum and other body fluids became negative21,22. In a pediatric study that evaluated the acute phase of infection, ZIKV principally infected CD14+CD16+ monocytes in the blood23. ZIKV can replicate persistently within cells of the anterior and posterior chambers of the eye24, which causes conjunctivitis, maculopathy and uveitis, the latter of which can result in blindness25,26. ZIKV persistence in the eye has been detected in mice and humans for up to 30 days24,27, and is speculated to be a means of direct transmission16,24.
Another site of ZIKV persistence is the male reproductive tract. Persistent ZIKV RNA in sperm and semen has been reported in humans for months28, although infectious virus was limited to the first few weeks after disease onset29. Experiments in monkeys also show persistence of ZIKV in male reproductive tract tissues30. Studies in mice have demonstrated that ZIKV can replicate in cells of the testis, including spermatogonia, Sertoli cells and Leydig cells, which results in destruction of testicular architecture, reduction in sperm counts, lower levels of sex hormones and reduced fertility31,32. Oligospermia and haematospermia have been observed in humans after ZIKV infection and are speculated to affect fertility33. The high viral load in seminal fluid can lead to sexual transmission from men-to-women34 and men-to-men35 (Fig. 2).
ZIKV is linked to the development of Guillain–Barré syndrome in a small percentage of adults36,37, although causality has not been proven. Guillain–Barré syndrome is an acute inflammatory immune-mediated polyneuropathy that typically presents with paresthesia, weakness and pain, but can progress to paralysis and even death. Case reports have been published of ZIKV-associated Guillain–Barré syndrome in French Polynesia in 2013 and in the Americas38,39. Although more investigations into its cause are needed, leading hypotheses include B or T cell-mediated immunopathology due to viral antigen mimicry or direct viral infection and injury of cells of the peripheral nervous system.
Considerable effort has been made to define why ZIKV has teratogenic abilities. Initial studies focused on the placenta, because it acts as a structural and immunologic barrier between the maternal uterine-derived decidua and the developing fetus during pregnancy40 (Fig. 3). The maternal–fetal interface is characterized by an apposition of maternal decidua with fetally derived proliferative cytotrophoblasts and terminally differentiated syncytiotrophoblasts, the latter of which create a multi-nucleated cell barrier. In the first trimester, the human placenta becomes haemochorial, directly contacting maternal blood, which allows for the exchange of gases, nutrients and wastes. Syncytiotrophoblasts also function as a physical and immunological barrier between the maternal and fetal circulation to prevent spread of microbial agents. The maternal–fetal interface is defined by the differentiation of floating and anchoring villous structures. The core of the villous structure beneath the syncytiotrophoblast layer consists of fetal Hofbauer macrophages, fibroblasts and endothelial cells.

Vertical transmission requires ZIKV to spread to the immunologically privileged fetus. Maternal–fetal interactions occur at placental villous structures that are in contact with maternal blood and the adjacent decidua. Placental villi are lined by syncytiotrophoblasts that form a physical barrier against infection and have active roles in antiviral immunity through production of IFNλ and microRNAs43,147. ZIKV targets several placental cells including trophoblasts, Hofbauer macrophages and endothelial cells.
The importance of trophoblasts to fetal ZIKV infection was first identified in cell culture studies. Human trophoblast cells isolated early during pregnancy propagated ZIKV to high levels41,42. However, human fetal-derived chorionic villi or trophoblast cells obtained later during gestation supported less ZIKV infection, which probably reflects pregnancy stage-dependent effects of trophoblast differentiation and immunological maturation43. The reduced susceptibility to ZIKV infection at later gestational stages results in part from distinct innate immune profiles, including production or response to type-III interferon-λ (IFNλ)43,44 or differential expression of putative entry receptors. ZIKV can also replicate in placental cytotrophoblasts or primitive trophoblasts41,42, fetal Hofbauer macrophages45 and fetal endothelial cells46. Infection of fetal Hofbauer macrophages may be particularly important in humans, as ZIKV RNA and antigen have been localized to these cells in the placentas of women who had pregnancy losses during the first or second trimester47. These data suggest that ZIKV may be transmitted to fetuses by a transplacental route. This tropism of ZIKV may not be unique among flaviviruses, as inoculation of human placental explants or pregnant mice with the related West Nile and Powassan viruses also resulted in infection of trophoblasts and injury to the placenta48.
Thousands of infants have been born with ZIKV-induced neurological sequelae (CZS) that will impair their future neurodevelopmental function49. Insight into the molecular basis of CZS has been informed by experiments with cultured neuroprogenitor stem cells, cortical organoids, brain slices and mouse brain inoculation studies50. In these models, ZIKV preferentially targets progenitor cells of the cerebral cortex and results in reduced cell proliferation and differentiation, and increased inflammation and increased cell death51. ZIKV may also infect microglia in the brain, which can produce inflammatory cytokines (for example, TNF, IL-6 and IL-1β) that inhibit proliferation and differentiation of neuronal precursor cells52. Studies have also suggested that ZIKV can infect and modulate immune responses of astrocytes, in a manner that depends on expression of the cell surface protein AXL53,54.
There is a range of penetrance of CZS in different geographical regions for reasons that remain unclear. In one case series of 182 symptomatic, ZIKV-infected pregnant women in Brazil, a remarkable 42% of fetuses had abnormal clinical or brain imaging findings, and adverse outcomes were noted regardless of trimester of infection55. Retrospective analysis in French Polynesia also found an increased risk of microcephaly associated with ZIKV infection56. In a study of ZIKV infection during pregnancy in the territories of the United States, 5% of fetuses or infants had birth defects, with deleterious outcomes occurring in all trimesters57. Neuroimaging of the brains of congenitally infected neonates have reported hypoplasia of the cerebellum and brainstem, ventriculomegaly, myelination defects, calcifications and cortical malformations. The clinical presentation of CZS is variable and includes fetal demise and microcephaly55, as well as sensorineural hearing loss, ocular abnormalities and arthrogryposis (joint contractures). Severe microcephaly is associated with mental retardation, learning disabilities, behavioural abnormalities, muscle weakness and altered muscle tone. The devastating effects of CZS are reflected by data from one longitudinal cohort of infants that was followed for eight months after birth: 85% had irritability, 56% had altered muscle tone and movement, 50% had epileptic seizures, 15% had dysphagia, and all of these infants had abnormal brain imaging studies58. In a second study of CZS-affected children from Brazil who were followed for 19–24 months, most had severe motor impairment, seizure disorders, hearing and vision abnormalities and sleep difficulties; this resulted in these children falling far behind in achieving developmental milestones, indicating the need for long-term support59.
Key questions related to CZS remain: (1) What is the risk of vertical transmission and disease in mothers who contract an asymptomatic ZIKV infection? (2) For neonates of mothers infected with ZIKV during the different stages of pregnancy who appear normal at birth, what is the risk of neurodevelopmental disorders? (3) How does pre-existing maternal flavivirus immunity impact pregnancy-associated disease?
Pathogenesis models in animals
There has been remarkable progress establishing mouse models of ZIKV infection and disease. Peripheral challenge of adult wild-type mice with ZIKV isolates results in little virus replication and no disease60. In comparison, infection of neonatal mice often causes non-fatal yet severe neurological disease characterized by tremors, ataxia, seizures and microcephaly. Because of the failure of immunocompetent adult mice to sustain a ZIKV infection, several groups evaluated the capacity of mice with IFN signalling deficiencies to support ZIKV replication, as this strategy had been used for other flaviviruses (for example, DENV and yellow fever virus). Mice that lacked the type-I Ifnar1 gene, or both Ifnar1 and the type-II Ifngr receptors, or that received a neutralizing anti-Ifnar1 antibody were susceptible to infection by most strains of ZIKV, and this resulted in central nervous system disease and death following inoculation through several different routes.
Mouse models of ZIKV infection during pregnancy have been developed to study transmission, teratogenicity and vaccine protection. Subcutaneous inoculation of pregnant Ifnar1−/− or wild-type dams treated with anti-Ifnar1 antibodies resulted in ZIKV infection of trophoblasts in the placenta, which enabled transplacental transmission, spread to the fetal brain and fetal demise41. The gestational stage of the dam affects clinical outcome: ZIKV infection during early pregnancy (embryonic day (E)6) resulted in placental insufficiency and fetal demise, infections at mid-pregnancy (E9) resulted in reduced cranial dimensions consistent with microcephaly, and infection during late pregnancy (E12) caused no apparent fetal disease44. These results correlate with human studies, which show ZIKV-associated microcephaly occurs more commonly when infections happen during the first and early second trimesters61.
Pregnant immunocompetent mice do not transmit ZIKV to the placenta or fetus when the virus is inoculated subcutaneously, presumably because of the failure to evade type-I IFN immunity41. However, intravenous inoculation of pregnant mice with high doses of ZIKV caused cortical brain malformations and fetal ocular abnormalities62,63. As an alternative model, direct ZIKV inoculation into the uterine wall of pregnant mice resulted in placental infection and inflammation, reduced neonatal brain cortical thickness and reduced fetal viability64. One study showed that direct viral infection of the fetus is not essential for demise, as placental pathology may be a stronger contributor to adverse pregnancy outcomes65. Other groups have injected ZIKV directly into the developing fetus in the cerebroventricular space; this resulted in decreased brain size, thinning of cortical layers, reduced numbers of cortical neural progenitors and neuronal cell death66. Intravaginal transmission of ZIKV during pregnancy has also been modelled in Ifnar1−/− mice during the progesterone-high, diestrous phase of the estrous cycle67. Finally, a recent study generated an immunocompetent mouse model of ZIKV infection and placental transmission by introducing the human STAT2 gene into the mouse Stat2 locus68.
In Africa, ZIKV undergoes a sylvatic phase with infection of non-human primates (NHPs) occurring in an endemic cycle. NHP models have advantages over rodent models, as ZIKV naturally targets primates and, thus, is more likely to overcome species-specific immune barriers to infection. Moreover, NHPs are evolutionary closer to humans than rodents, and the findings on pathogenesis and host restriction therefore are probably more relevant. Consequently, experimental ZIKV infection in rhesus, cynomolgus and pigtail macaques as well as marmosets has been used to evaluate ZIKV biology69,70,71,72. Because there is concern that ZIKV could establish a sylvatic cycle in the Americas, more recent studies have evaluated ZIKV infection in New World marmosets73.
Experimental inoculation of rhesus macaques with ZIKV resulted in weight loss, elevated body temperature, rash and mild hepatitis. These animals developed viraemia that peaks within the first week and then becomes undetectable by day 10, and viral RNA was detected in urine, saliva, lacrimal fluid, cerebrospinal fluid, seminal fluid and vaginal secretions69,70. ZIKV infected multiple tissues of rhesus and cynomolgus macaques including lymphoid organs, the male reproductive tract, the intestines, and the brain and spinal cord70,72,74. Because infected rhesus macaques also developed ZIKV-specific humoral and cell-mediated immune responses69,70,71,74, this model has been used to study sequential ZIKV and DENV infections and vaccine efficacy75,76.
Although challenging, ZIKV infection of pregnant NHPs is important because the haemochorial placenta and gestational development are more similar to humans than mice. Pregnant rhesus macaques infected with ZIKV developed viraemia that lasted for 30–55 days69, which is similar to viraemia observed in pregnant women77. The first NHP model of in utero transmission was established after inoculation of a pigtail macaque with an Asian ZIKV strain78. Infection resulted in reduced growth of the fetal brain, white matter gliosis and axonal damage, and ZIKV RNA was detected in the placenta, fetal brain and liver. In other studies, pregnant rhesus macaques infected with an Asian or American ZIKV strain also resulted in prolonged maternal viraemia79,80. Fetal head growth in the last month of gestation was decreased, and ZIKV RNA was detected in fetal tissues at birth. Pathological analysis showed neutrophil infiltration at the maternal–fetal interface and brain lesions in fetuses, including microcalcifications, haemorrhage, vasculitis and apoptosis of neuroprogenitor cells79,80. Because 26% of NHPs infected with ZIKV during early gestation experienced fetal demise despite showing few clinical signs, pregnancy loss due to asymptomatic infection may be underrecognized81. Vertical transmission in NHP models may provide a platform for testing vaccines and antibody-based therapeutics82,83. In other studies, postnatal ZIKV infection was associated with abnormalities in brain structure, function and behaviour in infant macaques84.
Innate immune responses to ZIKV infection
Although the initial innate immune events following ZIKV infection are beginning to be characterized, paradigms for recognition and control by the cytoplasmic (RIG-I-like receptors) and endosomal (Toll-like receptors) viral RNA sensors and signalling through downstream adaptor molecules and transcription factors have been extrapolated largely from studies with other flaviviruses.
Type-I and type-III IFNs induce antiviral states through induction of IFN-stimulated genes (ISG) that control viral replication. Type-I IFNs (for example, IFNα and IFNβ) bind to their heterodimeric receptor (IFNAR1/IFNAR2) and promote phosphorylation of JAK1 and TYK2. This activates STAT1 and STAT2 to bind IRF9 and form the IFN-stimulated gene factor 3 complex (ISGF3), which transcriptionally activates hundreds of ISGs. Type-III IFNλ binds to a selectively expressed, heterodimeric receptor (IFNLR1/IL10Rβ), and analogously promotes ISGF3 nuclear translocation and ISG induction. In addition, IFNλ has antiviral functions against ZIKV in the maternal decidua and placenta during pregnancy43,44. Constitutive secretion of IFNλ by syncytiotrophoblasts correlated with their ability to restrict ZIKV infection43. The importance of IFN signalling in mediating host restriction of ZIKV was shown by the pathogenicity of ZIKV in Ifnar1−/− and Stat2−/− but not immunocompetent mice60,85,86. Several antiviral effector genes induced by type-I and type-III IFNs reportedly have antiviral effects against ZIKV. Members of the IFITM family and their interacting proteins inhibit ZIKV infection at an entry step in the viral life cycle87,88. Expression of viperin also inhibited ZIKV replication in cells89.
ZIKV evades IFN responses by impairing induction and signalling pathways at multiple steps. Human dendritic cells can be infected productively by ZIKV90, but do not secrete pro-inflammatory cytokines or type-I IFN, probably because antiviral pathogen-recognition receptors and their downstream signalling pathways are downregulated or evaded91. ZIKV targets the IFN signalling pathways by inhibiting JAK1 and STAT activity. ZIKV NS5 targets human STAT2, but not mouse Stat2 for proteasomal degradation92,93. In addition, ZIKV infection prevents STAT1 phosphorylation90. ZIKV NS1 and NS4B also appear to inhibit IFNβ induction at the level of TBK1 activation, and the ZIKV NS2B–NS3 protease impairs IFNAR induction and signalling pathways by targeting human STING but not mouse Sting for cleavage94 and by degrading JAK195. Finally, ZIKV NS4B induces elongation of mitochondria, which physically contact the membranes associated with the endoplasmic reticulum that are sites of replication. This restructuring attenuates RIG-I-dependent activation of IFN responses. Beyond viral protein-mediated evasion mechanisms, ZIKV also generates a subgenomic viral RNA that antagonizes RIG-I-induced type-I IFN responses96.
Apart from active innate immune evasion mechanisms, ZIKV targets some cells that are inherently deficient in innate immune responses. Primary neural progenitor cells have a delayed innate response to ZIKV infection89 and glioblastoma cancer stem cells, which are highly permissive to ZIKV infection, show an absence of IFN signatures97. Analogously, in the vagina, ZIKV replication induces a weak antiviral IFN response98.
Adaptive immune responses to ZIKV infection
During primary infection, anti-ZIKV IgM becomes detectable as early as three days after onset of illness with most individuals developing responses by day 8. This early antibody response originates from extrafollicular ZIKV-specific plasmablasts, which comprise a large fraction of the circulating B cells99,100. This plasmablast response, however, is transient and lasts only a few weeks100, with germinal centre-derived plasma cells starting to produce antibody at this time. Neutralizing antibodies develop within the first week of illness, and as the IgG response matures, inhibitory antibodies in sera accumulate and neutralize virus strains from both Asian and African lineages101.
The functional quality and antigenic targets of ZIKV-induced B cell responses have been evaluated102. Prior flavivirus immunity is associated with serological cross-reactivity after ZIKV infection99,103,104. In humans with prior DENV immunity, a substantial proportion of anti-ZIKV antibodies generated during acute infection targets the highly conserved fusion loop in E-DII. Plasmablasts from acutely ZIKV-infected, DENV-immune individuals exhibited high levels of somatic hypermutation, with many derived from common memory B cell clones99. By contrast, plasmablasts from ZIKV-infected, flavivirus-naive individuals exhibited less somatic hypermutation or clonal expansion99, and antibody responses were more ZIKV-specific105. In general, cross-reactive antibodies had poorer neutralizing capacity in vitro and limited protective activity in vivo against ZIKV106,107. Prior flavivirus immunity triggers cross-reactive responses because the memory B cells formed during the first flavivirus infection encounter conserved epitopes present on ZIKV antigens102. The magnitude and durability of the cross-reactive response may depend on the duration separating the two flavivirus infections and the number of prior exposures108,109.
Functional and structural studies have revealed epitopes on all domains of the ZIKV E protein for engagement by highly neutralizing monoclonal antibodies102. One class, which consists of antibodies that are cross-reactive with DENV and recognize the quaternary envelope dimer epitope110, neutralized ZIKV infection with high potency7 and protected mice and NHPs from ZIKV infection8,83 or transplacental transmission8. A second class of highly neutralizing and protective anti-ZIKV monoclonal antibodies binds to residues within the lateral ridge epitope of E-DIII and blocks infection at a post-attachment step111. E-DIII antibodies appear important for controlling ZIKV, as their depletion from human serum resulted in reduced neutralizing activity against ZIKV112. A third class of ZIKV monoclonal antibodies also protects against vertical transmission of ZIKV in mice104. The only described monoclonal antibody of this class, ZIKV-117, recognizes an epitope across neighbouring E protein dimers and probably prevents the conformational changes required for pH-dependent fusion in the endosome. Finally, neutralizing monoclonal antibodies binding to additional sites within E-DI and E-DII have also been reported113.
We are beginning to understand T cell responses against ZIKV. In mice, polyfunctional, cytotoxic CD8+ T cells become activated114 and can reduce ZIKV burden, whereas their depletion or genetic absence resulted in greater ZIKV infection and mortality115. Consistent with this observation, adoptive transfer of ZIKV-immune CD8+ T cells can protect against ZIKV infection116. Another study identified human-relevant ZIKV CD8+ T cell epitopes in naive and DENV-experienced HLA-transgenic mice and demonstrated that both ZIKV-specific and ZIKV–DENV cross-reactive CD8+ T cells can protect against ZIKV infection117. Thus, CD8+ T cells probably have a protective activity against ZIKV. Nevertheless, CD8+ T cells could have pathological consequences in the brain118 and result in ZIKV-associated paralysis119.
Less is known about human T cell responses to ZIKV. In one study, ZIKV-infected patients developed polyfunctional CD4+ T cell responses that produced antiviral cytokines105. In another study, ZIKV infection induced CD4+ T effector cells with a low frequency of IFNγ production120. In comparison, whereas CD8+ T cells were highly activated during the viraemic phase (15% to >25% of blood CD8+ T cells), low levels of antigen-specific CD8+ T cells were identified105, although others have reported tetramer-positive ZIKV-specific CD8+ T cells in blood100. Another study found that memory T cell responses elicited by prior infection or vaccination with DENV were restimulated with ZIKV-derived peptides121; the consequence of this expanded cross-reactive response (beneficial or detrimental) remains to be determined. Almost 60% of the ZIKV-specific CD8+ T cell response was directed against the structural proteins122, which could be beneficial for vaccines that exclusively target the structural proteins (see Figs. 1, 4).

a, Flavivirus prM–E proteins form non-infectious subviral particles that share functional and antigenic features with infectious virions. Subviral particles are smaller with T = 1 icosahedral symmetry, although they may be heterogeneous in size. Multiple ZIKV vaccine platforms that encode prM–E proteins have been evaluated in humans. DNA vaccines GLS-5700 (NCT02809443), VRC-5288 (NCT02840487) and VRC 5283 (NCT02996461) differ with respect to ZIKV strain and signal sequence preceding prM. The C terminus of VRC5288 is a chimaera of JEV. Nucleoside-modified mRNAs (mRNA-1325) and a measles vector (MV-ZIKV) expressing prM–E have also been evaluated (NCT03014089 and NCT02996890, respectively). b, Vaccine candidates derived from infectious ZIKV. Four inactivated vaccine candidates are being assessed. Phase I studies of the ZPIV vaccine construct developed by WRAIR have been conducted (NCT02963909, NCT02952833 and NCT02937233). Studies of the Takeda PIZV (NCT03343626), Emergent Biosolutions VLA1601 (NCT03425149) and Bharat Biotech ZikaVac are underway. Clinical trials of a chimaeric live-attenuated vaccine derived from the NIAID DENV vaccine platform are anticipated.
Possible explanations for the emergence of ZIKV
How ZIKV has changed to cause massive epidemics, congenital defects, infection in immune sanctuary sites, sexual transmission and Guillain–Barré syndrome remains an area of intensive study. Multiple factors may be responsible for the changing epidemiology and disease pathogenesis.
The presence of NS1 in human blood facilitates flavivirus acquisition by mosquito vectors because this protein suppresses the immune functions of the mosquito midgut. A single alanine to valine (A188V) substitution in NS1 of the epidemic ZIKV strains facilitated greater infectivity in Aedes aegypti mosquitoes and enzootic transmission123. Clinical isolates from the Americas with a valine at position 188 had higher NS1 antigenemia in mice and were more infectious in mosquitoes than pre-epidemic strains. This same mutation promotes the binding of NS1 to TBK1, resulting in reduced levels of TBK1 phosphorylation and IFNβ expression in human cells95. Thus, sequence changes in NS1 during the pre-epidemic to epidemic transition appear to have facilitated immune evasion and enhanced ZIKV transmissibility from hosts to vectors, which creates conditions for epidemic transmission.
Sequence changes have also been hypothesized to affect ZIKV pathogenicity and explain its tropism for fetal neuroprogenitor cells10. A single serine to asparagine substitution (S139N) in the viral polyprotein (residue 17 of prM) increased ZIKV infectivity in neural progenitor cells that resulted in more severe microcephaly and higher mortality rates in neonatal mice124. Evolutionary analysis indicated that the S139N substitution arose just before the 2013 outbreak in French Polynesia and has been maintained during the American epidemic. The mechanistic basis for how the S139N in prM mediates this effect remains uncertain.
Noncoding sequence adaptations that affect the RNA structure may contribute to neuropathogenicity of epidemic strains. One group identified a Musashi protein binding element in the SL2 stem loop of the 3′ UTR, with sequence changes between ancestral and contemporary strains immediately upstream of this site125. Because Musashi proteins regulate mRNA translation and can modulate progenitor cell growth and differentiation, this group postulated that nucleotide polymorphisms in the 3′ UTR might be linked to alterations in Musashi protein binding activity and neurovirulence. A second group showed that Musashi-1 interacts directly with ZIKV genomic RNA and facilitates viral replication126. ZIKV infection disrupted the binding of Musashi-1 to its endogenous targets, which deregulated expression of factors that have been implicated in neural stem cell function.
A feature of DENV pathogenesis is that antibodies to one serotype can exacerbate infection with a second serotype via antibody-dependent enhancement (ADE)9. ADE occurs when cross-reactive, non-neutralizing quantities of antibodies bind to a heterologous DENV serotype and facilitate infection of myeloid cells that express Fc-γ receptors. Because of the structural similarity between ZIKV and DENV, antibodies produced against these flaviviruses can cross-react103,127. ADE between DENV and ZIKV has been proposed to contribute to the severity of ZIKV disease in the Americas, since the epidemics occurred in regions in which most people are DENV-immune. Indeed, cross-reactive anti-DENV antibodies can enhance ZIKV infection in cell culture106,127. Notwithstanding this observation, ADE in cell culture has been demonstrated for many viruses without evidence of worsened disease in humans. One recent study showed that passive transfer of immune plasma against DENV or West Nile virus can enhance ZIKV infection and pathogenesis in Stat2−/− mice128, which suggest that pre-existing anti-flavivirus immunity can promote ZIKV pathogenesis; a caveat to this model is that these mice lacked immune T cells, which can limit the effects of ADE in the context of DENV infection. However, no differences in ZIKV infection were observed after inoculation of naive and flavivirus-immune rhesus macaques75. By contrast, prior exposure to ZIKV enhanced DENV infection in rhesus macaques76, which has implications for ZIKV vaccine development and deployment. More detailed epidemiological evidence from humans is necessary to confirm whether clinically relevant ADE of ZIKV pathogenesis occurs, especially in the context of vertical transmission. To date, studies in Brazilian cohorts have not found any evidence of ADE, greater disease severity, or effects on birth outcomes in patients with acute ZIKV infection who had previously been exposed to DENV129,130.
Development of ZIKV vaccines
Efforts to develop a vaccine were initiated rapidly after the threat of ZIKV congenital disease became clear. Multiple vaccine platforms have been evaluated in preclinical and clinical studies (Fig. 4). Because ZIKV circulates as a single serotype and infection provides immunity to re-challenge by heterologous strains, a requirement for only a single vaccine antigen is anticipated71,101.
Synthetic nucleic acids provide a platform for the rapid development of vaccines. Multiple ZIKV prM–E DNA vaccine configurations have been evaluated that vary with respect to the sequence of the structural genes, method of codon optimization, signal sequence used to direct prM into the endoplasmic reticulum lumen and the plasmid backbone131,132. A construct that lacks the ‘pr’ portion of prM has also been developed133,134. Three of these DNA vaccines have been evaluated in human studies132,135. DNA vaccine GLS-5700, which encodes a consensus of prM–E sequences from divergent ZIKV strains lacking the N-linked glycosylation site at E154, was safe and immunogenic in humans; neutralizing antibodies were present in 62% of recipients132. DNA vaccine candidates VRC5283 and VRC5288 encode a codon-optimized prM–E derived from an Asian strain downstream of the signal sequence of Japanese encephalitis virus (JEV). The stem and transmembrane domains at the C terminus of these constructs differ due to the replacement of this sequence in VRC5288 with the analogous sequence of JEV. Phase I studies revealed superior immunogenicity of VRC5283135. Three doses of VRC5283 delivered at four-week intervals elicited neutralizing antibodies in all subjects. A phase II study to confirm immunogenicity and define efficacy is underway at twenty sites in the Americas.
Nucleoside-modified mRNAs drive protein expression at high levels in vivo, due in part to an ability to limit recognition by sensors of foreign nucleic acids in transduced cells. Capped mRNAs are synthesized using modified nucleosides, encode a codon-optimized open reading frame flanked by untranslated regions, and are encapsulated in lipid nanoparticles or complexed with lipids. Multiple ZIKV mRNA vaccine candidates expressing prM–E elicit neutralizing antibodies at high titre and protect against viraemia after challenge136,137,138. Similar to DNA vaccine platforms, these constructs differ with respect to the sequence of prM–E and signal sequence at the N terminus of prM; the contribution of these properties to differences in immunogenicity are not yet understood. Protection of NHPs following a single dose highlights the promise of this platform136. One of these mRNAs (mRNA-1325) has been evaluated in phase I clinical studies.
The chemical inactivation of flaviviruses is an established method for creating protective immunogens for use in humans. Inactivated vaccines are currently in use against tick-borne encephalitis virus and JEV. The Walter Reed Army Institute of Research (WRAIR) developed an inactivated ZIKV vaccine by formalin-inactivation of a Puerto Rican ZIKV isolate. Administration of this ZIKV-purified inactivated vaccine (ZPIV) into mice and NHPs elicited neutralizing antibodies and conferred protection against viraemia following challenge133,134. Subsequent studies demonstrated that two doses of ZPIV conferred protection for more than one year after vaccination139. Safety and immunogenicity of ZPIV in humans was demonstrated in three placebo-controlled clinical studies of two doses at a 28-day interval. Although the WRAIR ZPIV candidate is not being evaluated further, similar inactivated vaccines are being developed by Takeda Pharmaceuticals (PIZV), Emergent Biosolutions (VLA1601) and Bharat Biotech. In contrast to other ZIKV vaccine candidates, the Bharat inactivated virus (ZikaVac) is derived from an African lineage strain.
Live-attenuated vaccines (LAVs) have been safe and cost-effective approaches to control flaviviruses. Whereas the first LAV was created by extensive passage of a strain of yellow fever virus in animals, molecular clone technology has enabled the rational design of attenuated vaccine candidates. ZIKV LAVs that use multiple attenuation strategies including deletions of the 3′ UTR, mutations to remove N-linked glycans on NS1, and chimerization with other flaviviruses have been evaluated in preclinical studies140,141,142. In each case, vaccine-mediated protection was established in mice or NHPs. Clinical studies of a ZIKV chimeric LAV (National Institute of Allergy and Infectious Diseases) will begin enrollment in 2018.
Viral vectored vaccines engineered to express ZIKV antigens also have promise. An adenovirus vector expressing ZIKV M–E elicited neutralizing antibodies and a protective immune response in mice and NHP models133,134. Vesicular stomatitis and measles virus vectors expressing ZIKV prM–E are in preclinical development and phase I clinical trials, respectively143. Vaccinia virus vectors expressing prM–E144 or NS1145 elicit protective responses in mouse models. Because NS1 is not present on virions, the NS1 antigen elicits antibodies that probably contribute to protection by recruiting host effector functions. Finally, partially purified recombinant proteins or virus-like particles elicit protective immune responses in mouse models146.
A goal of ZIKV vaccine development is to protect against congenital disease. Most preclinical studies of ZIKV vaccine candidates evaluate protection from viraemia following peripheral challenge. The degree to which viral replication must be inhibited to prevent infection of the fetus or access to other immune privileged sites associated with persistence or transmission is unknown. The requirements for vaccine-mediated protection may also depend on the route of infection (mosquito versus sexual transmission). Several vaccines have the capacity to protect against vertical transmission of ZIKV to the fetus138,142. Vaccine-elicited maternal immunity in the context of neonatal infection has also been shown using a vesicular stomatitis virus-vectored vaccine platform against ZIKV143. Passive transfer of vaccine-elicited antibodies and the identification of neutralization titres that relate to protection suggest ZIKV-reactive antibody levels can be a functional correlate131,133,139. It remains unclear whether estimates of a protective quantity of neutralizing antibody will apply uniformly to all vaccine platforms. Preclinical approaches to identify humoral and cellular correlates of protection may have a key role in vaccine licensure.
Conclusions
The epidemic of ZIKV and its clinical consequences resulted in a rapid research response, which has begun to provide answers as to why this virus transitioned from obscurity to notoriety. The scientific community is now answering questions related to viral evolution, structure and function, virulence, tropism and immune evasion, which begin to explain how ZIKV causes congenital disease. Unanswered questions remain with regard to transmission dynamics, viral persistence, cross-immunity with related viruses, as well as the neurodevelopmental sequelae of congenital infection. Although the last year has seen a waning of ZIKV cases, our new knowledge of ZIKV biology has informed the development of candidate vaccines and therapies, which will hopefully be implemented before a new epidemic. The lessons we have learned from ZIKV may be applicable to other viruses that cause future unanticipated clinical syndromes.
References
Weaver, S. C. et al. Zika virus: history, emergence, biology, and prospects for control. Antiviral Res. 130, 69–80 (2016).
Duffy, M. R. et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 360, 2536–2543 (2009).
Musso, D. et al. Zika virus in French Polynesia 2013–14: anatomy of a completed outbreak. Lancet Infect. Dis. 18, e172–e182 (2018).
Metsky, H. C. et al. Zika virus evolution and spread in the Americas. Nature 546, 411–415 (2017).
Netto, E. M. et al. High Zika virus seroprevalence in Salvador, northeastern Brazil limits the potential for further outbreaks. MBio 8, e01390-17 (2017).
Annamalai, A. S. et al. Zika virus encoding non-glycosylated envelope protein is attenuated and defective in neuroinvasion. J. Virol. e01348-17 (2017).
Barba-Spaeth, G. et al. Structural basis of potent Zika–dengue virus antibody cross-neutralization. Nature 536, 48–53 (2016).
Fernandez, E. et al. Human antibodies to the dengue virus E-dimer epitope have therapeutic activity against Zika virus infection. Nat. Immunol. 18, 1261–1269 (2017).
Culshaw, A., Mongkolsapaya, J. & Screaton, G. R. The immunopathology of dengue and Zika virus infections. Curr. Opin. Immunol. 48, 1–6 (2017).
Faria, N. R. et al. Zika virus in the Americas: early epidemiological and genetic findings. Science 352, 345–349 (2016). Key phylogenetic analysis of ZIKV entry into the Americas.
Prasad, V. M. et al. Structure of the immature Zika virus at 9 Å resolution. Nat. Struct. Mol. Biol. 24, 184–186 (2017).
Kostyuchenko, V. A. et al. Structure of the thermally stable Zika virus. Nature 533, 425–428 (2016).
Sirohi, D. et al. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 352, 467–470 (2016). Two papers 12 , 13 provide high-resolution cryo-EM structures of ZIKV.
Rey, F. A., Stiasny, K. & Heinz, F. X. Flavivirus structural heterogeneity: implications for cell entry. Curr. Opin. Virol. 24, 132–139 (2017
Aubry, M. et al. Zika virus seroprevalence, French Polynesia, 2014–2015. Emerg. Infect. Dis. 23, 669–672 (2017).
Swaminathan, S., Schlaberg, R., Lewis, J., Hanson, K. E. & Couturier, M. R. Fatal Zika virus infection with secondary nonsexual transmission. N. Engl. J. Med. 375, 1907–1909 (2016).
Carteaux, G. et al. Zika virus associated with meningoencephalitis. N. Engl. J. Med. 374, 1595–1596 (2016).
Karimi, O. et al. Thrombocytopenia and subcutaneous bleedings in a patient with Zika virus infection. Lancet 387, 939–940 (2016).
Dirlikov, E. et al. Postmortem findings in patient with Guillain–Barré syndrome and Zika virus infection. Emerg. Infect. Dis. 24, 114–117 (2018).
Styczynski, A. R. et al. Increased rates of Guillain–Barré syndrome associated with Zika virus outbreak in the Salvador metropolitan area, Brazil. PLoS Negl. Trop. Dis. 11, e0005869 (2017).
Murray, K. O. et al. Prolonged detection of Zika virus in vaginal secretions and whole blood. Emerg. Infect. Dis. 23, 99–101 (2017).
Mansuy, J. M. et al. Zika Virus infection and prolonged viremia in whole-blood specimens. Emerg. Infect. Dis. 23, 863–865 (2017).
Michlmayr, D., Andrade, P., Gonzalez, K., Balmaseda, A. & Harris, E. CD14+CD16+ monocytes are the main target of Zika virus infection in peripheral blood mononuclear cells in a paediatric study in Nicaragua. Nat. Microbiol. 2, 1462–1470 (2017).
Miner, J. J. et al. Zika virus infection in mice causes panuveitis with shedding of virus in tears. Cell Rep. 16, 3208–3218 (2016).
Kodati, S. et al. Bilateral posterior uveitis associated with Zika virus infection. Lancet 389, 125–126 (2017).
Parke, D. W., III et al. Serologically confirmed Zika-related unilateral acute maculopathy in an adult. Ophthalmology 123, 2432–2433 (2016).
Tan, J. J. L. et al. Persistence of Zika virus in conjunctival fluid of convalescence patients. Sci. Rep. 7, 11194 (2017).
Mansuy, J. M. et al. Zika virus in semen and spermatozoa. Lancet Infect. Dis. 16, 1106–1107 (2016).
Mead, P. S. et al. Zika virus shedding in semen of symptomatic infected men. N. Engl. J. Med. 378, 1377–1385 (2018).
Hirsch, A. J. et al. Zika virus infection of rhesus macaques leads to viral persistence in multiple tissues. PLoS Pathog. 13, e1006219 (2017).
Govero, J. et al. Zika virus infection damages the testes in mice. Nature 540, 438–442 (2016).
Ma, W. et al. Zika virus causes testis damage and leads to male infertility in mice. Cell 167, 1511–1524 (2016).
Joguet, G. et al. Effect of acute Zika virus infection on sperm and virus clearance in body fluids: a prospective observational study. Lancet Infect. Dis. 17, 1200–1208 (2017).
Russell, K. et al. Male-to-female sexual transmission of Zika virus—United States, January–April 2016. Clin. Infect. Dis. 64, 211–213 (2017).
Deckard, D. T. et al. Male-to-male sexual transmission of Zika virus—Texas, January 2016. MMWR Morb. Mortal. Wkly Rep. 65, 372–374 (2016).
Oehler, E. et al. Zika virus infection complicated by Guillain–Barré syndrome—case report, French Polynesia, December 2013. Euro Surveill. 19, 20720 (2014).
Parra, B. et al. Guillain–Barré syndrome associated with Zika virus infection in Colombia. N. Engl. J. Med. 375, 1513–1523 (2016).
dos Santos, T. et al. Zika virus and the Guillain–Barré Syndrome — case series from seven countries. N. Engl. J. Med. 375, 1598–1601 (2016). Description of ZIKV-associated Guillain–Barré Syndrome in the Americas.
Dirlikov, E. et al. Acute Zika virus infection as a risk factor for Guillain–Barré syndrome in Puerto Rico. J. Am. Med. Assoc. 318, 1498–1500 (2017).
Arora, N., Sadovsky, Y., Dermody, T. S. & Coyne, C. B. Microbial vertical transmission during human pregnancy. Cell Host Microbe 21, 561–567 (2017).
Miner, J. J. et al. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165, 1081–1091 (2016). Establishment of a mouse model of the fetal injury caused by ZIKV.
Sheridan, M. A. et al. Vulnerability of primitive human placental trophoblast to Zika virus. Proc. Natl Acad. Sci. USA 114, E1587–E1596 (2017).
Bayer, A. et al. Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe 19, 705–712 (2016).
Jagger, B. W. et al. Gestational Stage and IFN-λ signaling regulate ZIKV infection in utero. Cell Host Microbe 22, 366–376 (2017).
Quicke, K. M. et al. Zika virus infects human placental macrophages. Cell Host Microbe 20, 83–90 (2016).
Richard, A. S. et al. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc. Natl Acad. Sci. USA 114, 2024–2029 (2017).
Martines, R. B. et al. Pathology of congenital Zika syndrome in Brazil: a case series. Lancet 388, 898–904 (2016).
Platt, D. J. et al. Zika virus-related neurotropic flaviviruses infect human placental explants and cause fetal demise in mice. Sci. Transl. Med. 10, eaao7090 (2018).
Delaney, A. et al. Population-Based surveillance of birth defects potentially related to Zika virus infection — 15 States and U.S. Territories, 2016. MMWR Morb. Mortal. Wkly Rep. 67, 91–96 (2018).
Li, H., Saucedo-Cuevas, L., Shresta, S. & Gleeson, J. G. The neurobiology of Zika virus. Neuron 92, 949–958 (2016).
Tang, H. et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18, 587–590 (2016). Key paper describing ZIKV infection and injury of neuroprogenitor cells.
Lum, F. M. et al. Zika virus infects human fetal brain microglia and induces inflammation. Clin. Infect. Dis. 64, 914–920 (2017).
Meertens, L. et al. Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep. 18, 324–333 (2017).
Retallack, H. et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl Acad. Sci. USA 113, 14408–14413 (2016).
Brasil, P. et al. Zika virus infection in pregnant women in Rio de Janeiro. N. Engl. J. Med. 375, 2321–2334 (2016). Study describing the effects of ZIKV during pregnancy in Brazil.
Cauchemez, S. et al. Association between Zika virus and microcephaly in French Polynesia, 2013–15: a retrospective study. Lancet 387, 2125–2132 (2016).
Shapiro-Mendoza, C. K. et al. Pregnancy outcomes after maternal Zika virus infection during pregnancy — U.S. Territories, January 1, 2016–April 25, 2017. MMWR Morb. Mortal. Wkly Rep. 66, 615–621 (2017).
Moura da Silva, A. A. et al. Early growth and neurologic outcomes of infants with probable congenital Zika virus syndrome. Emerg. Infect. Dis. 22, 1953–1956 (2016).
Satterfield-Nash, A. et al. Health and development at age 19–24 months of 19 children who were born with microcephaly and laboratory evidence of congenital Zika virus infection during the 2015 Zika virus outbreak — Brazil, 2017. MMWR Morb. Mortal. Wkly Rep. 66, 1347–1351 (2017).
Lazear, H. M. et al. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–730 (2016).
Honein, M. A. et al. Birth defects among fetuses and infants of US women with evidence of possible Zika virus infection during pregnancy. J. Am. Med. Assoc. 317, 59–68 (2017).
Cugola, F. R. et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534, 267–271 (2016). Establishment of a mouse model of fetal injury and microcephaly caused by ZIKV infection.
Xavier-Neto, J. et al. Hydrocephalus and arthrogryposis in an immunocompetent mouse model of ZIKA teratogeny: a developmental study. PLoS Negl. Trop. Dis. 11, e0005363 (2017).
Vermillion, M. S. et al. Intrauterine Zika virus infection of pregnant immunocompetent mice models transplacental transmission and adverse perinatal outcomes. Nat. Commun. 8, 14575 (2017).
Szaba, F. M. et al. Zika virus infection in immunocompetent pregnant mice causes fetal damage and placental pathology in the absence of fetal infection. PLoS Pathog. 14, e1006994 (2018).
Li, C. et al. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19, 120–126 (2016).
Yockey, L. J. et al. Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell 166, 1247–1256 (2016). Animal study showing that intravaginal transmission of ZIKV can result in fetal brain injury.
Gorman, M. J. et al. An immunocompetent mouse model of Zika virus infection. Cell Host Microbe 23, 672–685 (2018).
Dudley, D. M. et al. A rhesus macaque model of Asian-lineage Zika virus infection. Nat. Commun. 7, 12204 (2016).
Osuna, C. E. et al. Zika viral dynamics and shedding in rhesus and cynomolgus macaques. Nat. Med. 22, 1448–1455 (2016).
Aliota, M. T. et al. Heterologous protection against Asian Zika virus challenge in rhesus macaques. PLoS Negl. Trop. Dis. 10, e0005168 (2016).
Koide, F. et al. Development of a Zika virus infection model in cynomolgus macaques. Front. Microbiol. 7, 2028 (2016).
Chiu, C. Y. et al. Experimental Zika virus inoculation in a new world monkey model reproduces key features of the human infection. Sci. Rep. 7, 17126 (2017).
Li, X. F. et al. Characterization of a 2016 clinical isolate of Zika virus in non-human primates. EBioMedicine 12, 170–177 (2016).
McCracken, M. K. et al. Impact of prior flavivirus immunity on Zika virus infection in rhesus macaques. PLoS Pathog. 13, e1006487 (2017).
George, J. et al. Prior exposure to Zika virus significantly enhances peak dengue-2 viremia in rhesus macaques. Sci. Rep. 7, 10498 (2017).
Driggers, R. W. et al. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N. Engl. J. Med. 374, 2142–2151 (2016).
Adams Waldorf, K. M. et al. Fetal brain lesions after subcutaneous inoculation of Zika virus in a pregnant nonhuman primate. Nat. Med. 22, 1256–1259 (2016).
Nguyen, S. M. et al. Highly efficient maternal–fetal Zika virus transmission in pregnant rhesus macaques. PLoS Pathog. 13, e1006378 (2017).
Martinot, A. J. et al. Fetal neuropathology in Zika virus-infected pregnant female rhesus monkeys. Cell 173, 1111–1122 (2018).
Dudley, D. M. et al. Miscarriage and stillbirth following maternal Zika virus infection in nonhuman primates. Nat. Med. (2018).
Morrison, T. E. & Diamond, M. S. Animal models of Zika virus infection, pathogenesis, and immunity. J. Virol. 91, e00009-17 (2017).
Abbink, P. et al. Therapeutic and protective efficacy of a dengue antibody against Zika infection in rhesus monkeys. Nat. Med. 24, 721–723 (2018).
Mavigner, M. et al. Postnatal Zika virus infection is associated with persistent abnormalities in brain structure, function, and behavior in infant macaques. Sci. Transl. Med. 10, eaao6975 (2018).
Rossi, S. L. et al. Characterization of a novel murine model to study Zika virus. Am. J. Trop. Med. Hyg. 94, 1362–1369 (2016).
Tripathi, S. et al. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog. 13, e1006258 (2017).
Savidis, G. et al. The IFITMs inhibit Zika virus replication. Cell Rep. 15, 2323–2330 (2016).
Monel, B. et al. Zika virus induces massive cytoplasmic vacuolization and paraptosis-like death in infected cells. EMBO J. 36, 1653–1668 (2017).
Van der Hoek, K. H. et al. Viperin is an important host restriction factor in control of Zika virus infection. Sci. Rep. 7, 4475 (2017).
Bowen, J. R. et al. Zika virus antagonizes type I interferon responses during infection of human dendritic cells. PLoS Pathog. 13, e1006164 (2017).
Sun, X. et al. Transcriptional changes during naturally acquired Zika virus infection render dendritic cells highly conducive to viral replication. Cell Rep. 21, 3471–3482 (2017).
Grant, A. et al. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19, 882–890 (2016). A study that explains in part how ZIKV evades the interferon response in humans but not mice.
Kumar, A. et al. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 17, 1766–1775 (2016).
Ding, Q. et al. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl Acad. Sci. USA 115, E6310–E6318 (2018).
Xia, H. et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 9, 414 (2018).
Donald, C. L. et al. Full genome sequence and sfRNA Interferon antagonist activity of Zika virus from Recife, Brazil. PLoS Negl. Trop. Dis. 10, e0005048 (2016).
Zhu, Z. et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 214, 2843–2857 (2017).
Khan, S. et al. Dampened antiviral immunity to intravaginal exposure to RNA viral pathogens allows enhanced viral replication. J. Exp. Med. 213, 2913–2929 (2016).
Rogers, T. F. et al. Zika virus activates de novo and cross-reactive memory B cell responses in dengue-experienced donors. Sci. Immunol. 2, eaan6809 (2017).
Ricciardi, M. J. et al. Ontogeny of the B- and T-cell response in a primary Zika virus infection of a dengue-naïve individual during the 2016 outbreak in Miami, FL. PLoS Negl. Trop. Dis. 11, e0006000 (2017).
Dowd, K. A. et al. Broadly neutralizing activity of Zika virus–immune sera identifies a single viral serotype. Cell Rep. 16, 1485–1491 (2016).
Priyamvada, L., Suthar, M. S., Ahmed, R. & Wrammert, J. Humoral immune responses against Zika virus infection and the importance of preexisting flavivirus immunity. J. Infect. Dis. 216, S906–S911 (2017).
Stettler, K. et al. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353, 823–826 (2016).
Sapparapu, G. et al. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 540, 443–447 (2016). First two papers 103 , 104 describing neutralizing human monoclonal antibodies against ZIKV.
Lai, L. et al. Innate, T-, and B-cell responses in acute human Zika patients. Clin. Infect. Dis. 66, 1–10 (2018).
Priyamvada, L. et al. Human antibody responses after dengue virus infection are highly cross-reactive to Zika virus. Proc. Natl Acad. Sci. USA 113, 7852–7857 (2016).
Zhao, H. et al. Structural basis of Zika virus-specific antibody protection. Cell 166, 1016–1027 (2016).
Swanstrom, J. A. et al. Dengue Virus envelope dimer epitope monoclonal antibodies isolated from dengue patients are protective against Zika virus. MBio 7, e01123-16 (2016).
Collins, M. H. et al. Lack of durable cross-neutralizing antibodies against Zika virus from dengue virus infection. Emerg. Infect. Dis. 23, 773–781 (2017).
Rouvinski, A. et al. Recognition determinants of broadly neutralizing human antibodies against dengue viruses. Nature 520, 109–113 (2015).
Wang, J. et al. A Human bi-specific antibody against Zika virus with high therapeutic potential. Cell 171, 229–241 (2017).
Yu, L. et al. Delineating antibody recognition against Zika virus during natural infection. JCI Insight 2, 93042 (2017).
Wang, Q. et al. Molecular determinants of human neutralizing antibodies isolated from a patient infected with Zika virus. Sci. Transl. Med. 8, 369ra179 (2016).
Pardy, R. D. et al. Analysis of the T cell response to Zika virus and identification of a novel CD8+ T cell epitope in immunocompetent mice. PLoS Pathog. 13, e1006184 (2017).
Elong Ngono, A. et al. Mapping and role of the CD8+ T cell response during primary Zika virus infection in mice. Cell Host Microbe 21, 35–46 (2017).
Huang, H. et al. CD8+ T cell immune response in immunocompetent mice during Zika virus infection. J. Virol. 91, e00900-17 (2017).
Wen, J. et al. Identification of Zika virus epitopes reveals immunodominant and protective roles for dengue virus cross-reactive CD8+ T cells. Nat. Microbiol. 2, 17036 (2017).
Manangeeswaran, M., Ireland, D. D. & Verthelyi, D. Zika (PRVABC59) infection is associated with T cell infiltration and neurodegeneration in CNS of immunocompetent neonatal C57BL/6 mice. PLoS Pathog. 12, e1006004 (2016).
Jurado, K. A. et al. Antiviral CD8 T cells induce Zika-virus-associated paralysis in mice. Nat. Microbiol. 3, 141–147 (2018).
Cimini, E. et al. Human Zika infection induces a reduction of IFN-γ producing CD4 T-cells and a parallel expansion of effector Vδ2 T-cells. Sci. Rep. 7, 6313 (2017).
Grifoni, A. et al. Prior Dengue virus exposure shapes T cell immunity to Zika virus in humans. J. Virol. e01469-17 (2017).
Weiskopf, D. et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc. Natl Acad. Sci. USA 110, E2046–E2053 (2013). (2013).
Liu, Y. et al. Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 545, 482–486 (2017).
Yuan, L. et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 358, 933–936 (2017). Two papers 123 , 124 describe the genetic changes in epidemic ZIKV strains that may explain altered epidemiology and pathogenicity.
Klase, Z. A. et al. Zika fetal neuropathogenesis: etiology of a viral syndrome. PLoS Negl. Trop. Dis. 10, e0004877 (2016).
Chavali, P. L. et al. Neurodevelopmental protein Musashi-1 interacts with the Zika genome and promotes viral replication. Science 357, 83–88 (2017).
Dejnirattisai, W. et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with Zika virus. Nat. Immunol. 17, 1102–1108 (2016).
Bardina, S. V. et al. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 356, 175–180 (2017).
Terzian, A. C. B. et al. Viral load and cytokine response profile does not support antibody-dependent enhancement in dengue-primed Zika virus-infected patients. Clin. Infect. Dis. 65, 1260–1265 (2017).
Halai, U. A. et al. Maternal Zika virus disease severity, virus load, prior dengue antibodies, and their relationship to birth outcomes. Clin. Infect. Dis. 65, 877–883 (2017).
Dowd, K. A. et al. Rapid development of a DNA vaccine for Zika virus. Science 354, 237–240 (2016).
Tebas, P. et al. Safety and immunogenicity of an anti-Zika virus DNA vaccine — preliminary report. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa1708120 (2017).
Abbink, P. et al. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 353, 1129–1132 (2016).
Larocca, R. A. et al. Vaccine protection against Zika virus from Brazil. Nature 536, 474–478 (2016).
Gaudinski, M. R. et al. Safety, tolerability, and immunogenicity of two Zika virus DNA vaccine candidates in healthy adults: randomised, open-label, phase 1 clinical trials. Lancet 391, 552–562 (2018). Five papers 131,132,133,134, – 135 describe the DNA and inactivated vaccine platforms under development against ZIKV.
Pardi, N. et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543, 248–251 (2017).
Richner, J. M. et al. Modified mRNA vaccines protect against Zika virus infection. Cell 168, 1114–1125 (2017).
Richner, J. M. et al. Vaccine mediated protection against Zika virus-induced congenital disease. Cell 170, 273–283 (2017). Three papers 136,137, – 138 describe the use of mRNA-based vaccines against ZIKV.
Abbink, P. et al. Durability and correlates of vaccine protection against Zika virus in rhesus monkeys. Sci. Transl. Med. 9, eaao4163 (2017).
Xie, X. et al. Understanding Zika virus stability and developing a chimeric vaccine through functional analysis. MBio 8, e02134-16 (2017).
Shan, C. et al. A live-attenuated Zika virus vaccine candidate induces sterilizing immunity in mouse models. Nat. Med. 23, 763–767 (2017).
Shan, C. et al. A single-dose live-attenuated vaccine prevents Zika virus pregnancy transmission and testis damage. Nat. Commun. 8, 676 (2017).
Betancourt, D., de Queiroz, N. M., Xia, T., Ahn, J. & Barber, G. N. Cutting edge: innate immune augmenting vesicular stomatitis virus expressing Zika virus proteins confers protective immunity. J. Immunol. 198, 3023–3028 (2017).
Prow, N. A. et al. A vaccinia-based single vector construct multi-pathogen vaccine protects against both Zika and chikungunya viruses. Nat. Commun. 9, 1230 (2018).
Brault, A. C. et al. A Zika vaccine targeting NS1 protein protects immunocompetent adult mice in a lethal challenge model. Sci. Rep. 7, 14769 (2017).
Salvo, M. A., Kingstad-Bakke, B., Salas-Quinchucua, C., Camacho, E. & Osorio, J. E. Zika virus like particles elicit protective antibodies in mice. PLoS Negl. Trop. Dis. 12, e0006210 (2018).
Bayer, A. et al. Chromosome 19 microRNAs exert antiviral activity independent from type III interferon signaling. Placenta 61, 33–38 (2018).
Acknowledgements
This work was supported by NIH grants (R01 AI073755, R01 AI104972, U19 AI083019 and R01 HD091218) and by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH. We thank E. Tyler (NIH) for assistance with figure preparation of virion models. This publication is the responsibility of the authors and does not necessarily represent the official view of the NIH.
Reviewer information
Nature thanks J. Jung and H. Tang for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
T.C.P. and M.S.D. conceived and wrote the review.
Corresponding authors
Ethics declarations
Competing interests
M.S.D. is a consultant for Inbios and on the Scientific Advisory Board of Moderna. T.C.P. is a co-inventor of NIAID ZIKV vaccine candidates.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pierson, T.C., Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 560, 573–581 (2018). https://doi.org/10.1038/s41586-018-0446-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-018-0446-y
This article is cited by
-
Development of a quantitative NS1 antigen enzyme-linked immunosorbent assay (ELISA) for Zika virus detection using a novel virus-specific mAb
Scientific Reports (2024)
-
Defined microbial communities and their soluble products protect mice from Clostridioides difficile infection
Communications Biology (2024)
-
Laboratory diagnosis of CNS infections in children due to emerging and re-emerging neurotropic viruses
Pediatric Research (2024)
-
Nuclear membrane protein SUN2 promotes replication of flaviviruses through modulating cytoskeleton reorganization mediated by NS1
Nature Communications (2024)
-
Flavivirus prM interacts with MDA5 and MAVS to inhibit RLR antiviral signaling
Cell & Bioscience (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
