
Abstract
Infectious tropical diseases have a huge effect in terms of mortality and morbidity, and impose a heavy economic burden on affected countries. These diseases predominantly affect the world’s poorest people. Currently available drugs are inadequate for the majority of these diseases, and there is an urgent need for new treatments. This Review discusses some of the challenges involved in developing new drugs to treat these diseases and highlights recent progress. While there have been notable successes, there is still a long way to go.
Similar content being viewed by others

Main
Infectious tropical diseases mainly affect low- and middle-income countries (LMICs). Symptoms include impaired cognitive and physical development in children (Box 1), complications in pregnancy, fever, nausea, diarrhoea, dehydration, anaemia, rashes, lesions, deformities, blindness, organ failure, haemorrhage, neurological problems, seizures and coma, and, in many cases, death. The WHO (World Health Organization) estimated that the diseases listed in Fig. 1 and Table 1 were responsible for more than 4 million deaths in 20151 and the loss of more than 250 million disability-adjusted life years (DALYs) owing to ill-health, premature disability or early death2.

Infectious tropical diseases include those defined by the WHO as neglected tropical diseases3 (NTDs). In addition, diseases such as malaria, tuberculosis, HIV/AIDS, multi-drug-resistant Gram-negative bacterial infections, diarrhoea (from a variety of pathogens) and hepatitis disproportionately affect tropical countries. The most common tropical diseases, grouped by causative agent, include: (i) viruses: Ebola, HIV/AIDS, Lassa fever, Marburg virus disease, Rift Valley fever, yellow fever, Zika disease, and the NTDs chikungunya, dengue fever and rabies; (ii) bacteria: bubonic plague, shigellosis, tuberculosis, typhoid fever, typhus, disease caused by drug-resistant Gram-negative bacteria, and the NTDs buruli ulcer, leprosy, mycetoma, trachoma and yaws; (iii) protozoa: cryptosporidiosis, malaria, and the NTDs Chagas disease, human African trypanosomiasis (HAT) and leishmaniases (visceral, cutaneous, post-kala-azar dermal and mucocutaneous); (iv) fungi: cryptococcosis and the NTD mycetoma; (v) helminths or metazoan worms: the NTDs cysticercosis, dracunculiasis (Guinea worm), echinococcosis, food-bourne trematodiases (clonorchiasis, opisthorchiasis, fasciolosis), lymphatic filariasis, onchocerciasis, schistosomiasis and soil-transmitted helminthiases (ascariasis, hookworm, trichuriasis, strongyloidiasis). Co-infection with these diseases is commonplace, and can exacerbate symptoms and complicate treatments (Box 2).
Infectious tropical diseases inflict a heavy economic burden, costing LMIC economies billions of dollars every year. For example, the annual global cost of dengue in 2013 was estimated at US$8.9 billion4, and a study carried out in the Philippines indicated that schistosomiasis resulted in the loss of an average of 45.4 work days per infected person per year5.
For many of these diseases the current treatments are unsatisfactory, and for some there are few or no treatment options available. There is an urgent need for new, safe, low-cost and efficacious treatments. Tackling these diseases effectively requires an integrated multi-sectoral approach that links chemotherapy with prevention initiatives such as improved water supply, sanitation and hygiene, vaccines and vector control. Various governmental and non-governmental organizations, charities, foundations, product-development partnerships, academic groups and pharmaceutical companies are supporting drug discovery for tropical diseases. A breakdown of funding is summarized in the G-Finder6 and IFPMA 20177 reports. In 2016 about two-thirds of the total funding for research and development for neglected diseases was spent on HIV/AIDS (34%), malaria (18%) and tuberculosis (18%)6. Overall research and development funding has markedly increased in the last decade but the gains have not been distributed evenly across diseases. Greater efforts, resources and funding are urgently needed to meet the considerable unmet medical needs.
In this Review, we will consider the specific challenges associated with drug discovery for tropical diseases and examine some of the recent successes.
Discovery pathways and challenges
Figure 2 shows the pathway for a typical small-molecule drug-discovery programme for an infectious disease.

Top, the stages of the drug-discovery process; middle, generic challenges; bottom, additional key challenges at particular stages of the drug-discovery process for neglected diseases. Biological and pharmacokinetic issues are shown in lilac, chemical issues are shown in light blue and clinical issues are shown in red.
The drug-discovery pathway
‘Hit discovery’ is the first step on the drug-discovery pathway, and is where chemical starting points are identified. This usually involves screening collections of compounds against a molecular target, typically an enzyme (target-based screening), or against whole organisms (cell-based or phenotypic screening). Alternative approaches in hit discovery include structure-based drug discovery, re-purposing of drugs from other disease areas, and in silico methods. Next, ‘hit expansion’ is performed to verify if a hit is genuine and whether there is scope for further work with the chemotype. The compound is then refined through a cyclic process of ‘design–make–test’ in the ‘hits to lead’ phase, until it has significant activity, typically in an animal model of infection. Subsequently, the biological activity, pharmacokinetics and safety profile of the series are optimized and balanced during the ‘lead optimization’ phase. ‘Candidate selection’ marks an important milestone in the drug-discovery pathway, as it involves selecting a single compound for progression. In this process, regulatory toxicology and scale-up are carried out to enable initial human studies. In human ‘clinical trials’, the compound is first tested in healthy volunteers to measure pharmacokinetics and safety (phase I); this is followed by studies to establish efficacy (phase II) and then large-scale efficacy and safety studies (phase III).
Target product profiles and compound progression criteria
The drug-discovery pathway should be guided by ‘target product profiles’ (TPPs). These describe the desired features required of the final drug product, such as duration of treatment, whether the treatment is oral or parenteral, cost of treatment, acceptable safety margins, among others8. TPPs have been defined for malaria9, leishmaniasis10, HAT10, Chagas disease11, cryptosporidiosis12 and dengue fever13, but are absent for many other diseases, hampering the drug-discovery process. TPPs are used to develop ‘compound progression criteria’, which inform the required disease-specific profile of a compound at every step along the drug-discovery process. These include, for example, activities in particular assays, pharmacokinetic profiles, and selectivity parameters8. Medicines for Malaria Venture (MMV) has several examples of compound progression criteria (https://www.mmv.org/research-development/information-scientists).
Challenges
For many tropical infectious diseases, there is little or no precedent for developing small-molecule drugs. This is exacerbated by insufficient understanding of the pathogen biology. As a result, there are often no relevant cellular or predictive animal models of the human disease. There is also frequently little or no data from clinically active molecules to help researchers to define the profile that a new drug should have in terms of its activity in preclinical assays and animal models, and its pharmacokinetics. The key issues facing researchers seeking to discover new drugs to treat tropical diseases are summarized in Fig. 2 and are considered below.
Biological challenges
There are very few well-validated molecular drug targets for tropical diseases, in part owing to a lack of understanding of the detailed biology of many of the pathogens. For example, the functions of many proteins are unknown, or have only been inferred from other organisms. Therefore a key challenge is the selection of a suitable molecular target to limit the risk of subsequent failure8. Criteria used to help to select appropriate targets include essentiality, druggability, assayability and the opportunity for selectivity over host orthologues8. Even if a target complies with all these criteria, inhibitors frequently fail to demonstrate cell-based activity owing to poor permeability, inability to compete with high substrate concentrations in cells, or because the compounds cannot inhibit the target sufficiently to kill the pathogen. The deficiency of genetic tools for many disease-relevant organisms is a key reason why so little essentiality data are available. However, new technologies are emerging14,15,16, including CRISPR–Cas917,18, which offer the prospect of markedly increasing the number of validated targets available in the near future.
Large-scale cell-based (phenotypic) screening is currently a popular approach to identify new chemical start points, as it does not require prior knowledge of the molecular target19,20. Developing assays that have sufficient throughput to allow screening of compound libraries while maintaining relevance to disease pathophysiology is challenging, but helps to reduce attrition when using in vivo models and, ultimately, in clinical trials21. As assays increase in relevance, they usually become more complex and require more resources, both in terms of specialist equipment as well as staff to develop and run the assays. Other challenges may include the lack of robust in vitro culture systems (for example, for Cryptosporidium), the inherent safety challenges of handling disease-causing agents in bulk, the lack of standardization between different laboratories working on the same organisms, and the use of laboratory strains and cell lines rather than recent clinical isolates and primary cells21,22.
Existing animal models of tropical disease infections are often poorly representative of the human disease. For example in onchocerciasis (river blindness), the causative worm (Onchocerca volvulus) is only infective to primates; consequently, related, but different, worm species are used in mice or cattle models. Plasmodium falciparum, the pathogen that causes the most severe form of human malaria, does not infect rodents normally; however, a SCID mouse model has been developed that can be infected by P. falciparum23. Work is also ongoing to develop a mouse model of human malaria caused by Plasmodium vivax24. Most mouse models of tuberculosis do not replicate the human disease well, and do not form the granulomas that are typical of the human disease. Encouragingly, progress is being made on developing animal models that more closely replicate human pathology25,26,27.
Recent human clinical trials of azoles, which inhibit the enzyme CYP51, for treatment of Chagas disease have not been successful. A mouse model has now been developed that can distinguish between benznidazole, which is known to have clinical efficacy, and an azole (posaconazole), which does not28. Better differentiation at this preclinical stage should improve the success rates of clinical trials.
Quiescence and dormancy
Dormant infections further complicate the aetiology of some diseases. In malaria, P. vivax and Plasmodium ovale can remain dormant (as hypnozoites) in hepatocytes of the human host for weeks to years, potentially causing relapse29. Latent tuberculosis, in which clinical symptoms are absent but there is a risk of progression to clinical disease, is estimated to affect a third of the global population30. Dormancy has recently been identified in Trypanosoma cruzi31. Quiescent and dormant pathogens have reduced metabolism compared to actively dividing pathogens and may therefore be less susceptible to drugs. Developing assays for these forms of pathogens is problematic. To effectively treat and eliminate these diseases, new drugs are needed that effectively tackle the reservoir of dormant and quiescent infections.
Chemistry
It is essential that the compound used for treatment can be dosed appropriately, usually orally, and can reach the part of the body where the pathogen is located. The physicochemical properties of a molecule, such as molecular weight, solubility, lipophilicity, charge, and number of hydrogen bond donors and acceptors are fundamental to achieving this.
In some cases, penetration into the pathogen is problematic. For example, Gram-negative bacteria are surrounded by an outer membrane; drug molecules can penetrate this through proteins that form channels through the membrane called porins, which tend to select for hydrophilic molecules. The drug then has to traverse the cell membrane, which is a lipid bilayer; this requires compounds with a degree of lipophilicity. Therefore, to penetrate into the cytoplasm, compounds require very defined chemical properties, and are likely to be more polar and charged than typical drugs32,33. Having penetrated to the cytoplasm, the drug then risks being pumped out of the bacteria through efflux transporters. Pathogens such as T. cruzi, Chlamydia trachomatis, Mycobacterium tuberculosis and viruses spend the majority of their life cycle inside human host cells, which can impede drug access. Some pathogens localize to extreme intracellular environments such as acidic organelles (for example, Leishmania spp. or Salmonella spp.) or necrotic granulomas (for example, M. tuberculosis). In the latter case, it is a major challenge to design drugs that can penetrate through the non-vascularized lipid-rich caseum to access the pathogen34. Other infections are localized at sites that are protected by a blood–tissue barrier, such as the central nervous system (for example, HAT, Cryptococcus neoformans, Taenia solium, or encephalitis- or meningitis-causing viruses or bacteria) or the eyes (for example, C. trachomatis). To reach these sites, drugs need to pass through endothelial cells without being pumped back into the blood by P-glycoprotein35.
The nature of these diseases means that the cost of treatment must be low. For malaria, the aim is to produce a treatment that costs less than US$1. This implies a short and cheap chemical synthesis and precludes complex formulations to counter issues such as poor solubility. Owing to the lack of temperature-controlled supply chains in many regions, compounds that are stable over long periods of time at high temperatures (>40 °C) and high humidity are required. Finally, patient compliance is also challenging, and is often compounded by minimal medical support. Consequently, it is essential to minimize the number of doses and complexity of dosing regimens. For example, MMV is aiming for a single-dose treatment for bloodstream malaria9.
Resistance
Drug resistance is a considerable problem for treatment of most infectious diseases, in particular for viral, bacterial and apicomplexan infections36,37,38. Poor management of treatments can accelerate the occurrence of resistance. The most publicized current example is antibacterial drug resistance. In humans, antibiotics are sometimes taken when not required, or given at sub-therapeutic doses, potentially increasing the proportion of resistant bacteria within a population. This problem is aggravated by the large amounts of antibiotics in the environment owing to indiscriminate use in agriculture and aquaculture.
Combination therapy is being adopted for many diseases in an attempt to slow development of drug resistance. This has been particularly successful in the treatment of HIV/AIDS, in which the resistance rate for single therapy39 is exceptionally high. Combination treatment has also proved very effective against tuberculosis and malaria. Worryingly, in the case of malaria, resistance to artemisinin combination therapy, the principal form of treatment38, is now developing in southeast Asia. There is therefore a need for drugs with novel modes of action to deliver new combination therapies for which there is no clinical resistance to any of the components.
Progress
Despite all these challenges, progress is being gradually made. Some examples are highlighted below, although this is not a comprehensive list. Such successes are due to coordinated and substantial investments in research, along with concerted efforts to understand the TPPs and drug-discovery pathways.
Apicomplexans
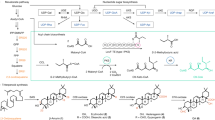
With 200 million cases a year, the human and economic cost of malaria is extremely high40. Africa bears a disproportionately high share of the burden with more than 400,000 deaths a year. Multiple new types of antimalarial drugs are needed to overcome resistance, give single-dose treatments, prevent relapse of P. vivax infections, block transmission and to act as chemopreventatives. The malaria drug-discovery portfolio has markedly improved over the past decade41. The starting points for the majority of the compounds currently in development were identified by phenotypic screening in collaborations with MMV. In many cases, target deconvolution has subsequently led to the identification of the molecular targets. There are a number of novel compounds in clinical trials supported by or in collaboration with MMV (Fig. 3). The following are derived from phenotypic screens and are in phase II trials: the spiroindolone KAE609, the imidazolopiperazine KAF15642 (a combination trial with lumefantrine), the synthetic trioxolane artefenomel (OZ439, in combination with ferroquine)43 and the 2-aminopyridine MMV048. The mode of action has been determined for some of these (P. falciparum (Pf) ATP4 for KAE60944 and PfPI4K for MMV04845), whereas the modes of action of KAF156 and OZ439 remain uncertain. KAF156 has been linked to the cyclic amine resistance locus (Pfcarl)46 and OZ439 probably acts through formation of carbon radicals, whereas ferroquine acts to prevent haem detoxification. In phase I trials are: the dihydroisoquinoline SJ733, another PfATP4 inhibitor47; and M5717, which is active against translation elongation factor 248. Two compounds derived from target-based projects are also in clinical trials: the dihydroorotate dehydrogenase inhibitor DSM26549 is in phase II trials, and the dihydrofolate reductase inhibitor P218 is in phase I trials.

Key compounds discussed in the text.
We envisage an increase in target-based drug discovery as mode-of-action studies are revealing the chemically validated targets of many of the recently identified phenotypic hits. There are systematic efforts trying to identify the mode of action of phenotypic hits50. New types of phenotypic screens have been developed that target life-cycle stages in addition to, or other than, the asexual blood stages51,52. Clinical development for malaria has accelerated thanks to the development of human challenge models in which healthy volunteers are infected with treatable malaria to obtain an early indication of drug efficacy53,54.
Cryptosporidiosis is a major cause of diarrhoea in children in tropical countries (Box 1). Nitazoxanide, the current treatment for the disease, has variable efficacy in immunocompetent patients and is not effective in immunocompromized patients55,56. There is an urgent need for new drugs to treat children under 24 months of age, especially those who are malnourished, immunocompromized or suffering from chronic diarrhoea.
There have been recent advances in the cryptosporidiosis discovery pipeline. A series of bumped kinase inhibitors of Cryptosporidium parvum calcium-dependent kinase 157 has been developed, an example of which is BKI1534. Promisingly, recent advances in C. parvum high-content imaging infection assays in human intestinal epithelial cells have made phenotypic screening of medium- and large-size compound libraries possible. A screen of compounds active against malaria has led to the identification of a preclinical candidate, KDU73158, an inhibitor of the Cryptosporidium phosphatidylinositol 4-kinase. Clofazimine, an approved drug for the treatment of leprosy, was identified from a screen of bioactive compounds59 and is entering clinical trials for cryptosporidiosis.
Kinetoplastid diseases
This area has recently been reviewed in detail60. Mortality rates for HAT (also known as sleeping sickness) have decreased substantially in recent years61; however, there is still a considerable disease burden in central Africa. The advent of nifurtimox–eflornithine (NECT) combination therapy in 2009 marked an important step in the treatment of stage two T. brucei gambiense HAT. This has reduced the duration of treatment as well as decreasing adverse effects62. Nevertheless, treatment is still problematic because the two subspecies that cause the disease (T. brucei gambiense and T. brucei rhodesiense) and the two stages (acute and central nervous system) require different drugs61. The requirement for staging patients before treatment also limits uptake, as it involves a painful and technically challenging lumbar puncture63. The ongoing development of two new drugs has the potential to transform the clinical management of HAT64. The first of these drugs is fexinidazole, a nitroaromatic compound developed in the 1970s and 1980s as a broad-spectrum antibiotic, which was more recently found to have a suitable profile for clinical development for HAT65. Drugs for Neglected Diseases initiative (DNDi) completed a phase III clinical trial in 2017 and reported non-inferiority over NECT for both stages of the disease with a ten-day oral treatment66. The second is an oxaborole compound, SCYX-7158 (acoziborole)67, which is currently undergoing a pivotal phase II/III trial10. This has the potential to provide a single-dose oral treatment for all forms of HAT.
Less progress has been made for leishmaniasis and Chagas disease. Current treatments, particularly liposomal amphotericin-B (which is given intravenously), together with other measures, such as vector control and effective surveillance, have led to progress towards elimination or control of visceral leishmaniasis (VL)68 in the Indian subcontinent. However, current drugs are less effective or unsuitable in other areas of the world. New oral drugs are urgently needed to treat VL in these regions and to provide improved treatment options in the Indian subcontinent. Furthermore, HIV–VL co-infection is also currently poorly treated. Several potential oral treatments identified through phenotypic screening are in preclinical development, including an oxaborole and a nitroimidazole69. The discovery of a pan-kinetoplastid proteasome inhibitor70 (GNF6702, Fig. 3) offers potential for development of new drugs for leishmaniasis, Chagas disease and HAT. The majority of preclinical work on leishmaniasis focuses on VL, the deadly form of the disease, rather than cutaneous leishmaniasis (CL). New oral therapies for CL are urgently needed as well. Research on Chagas disease suffered a setback recently as a consequence of the clinical failures of the CYP51 inhibitors posaconazole and fosravuconazole71,72. Currently, few treatments are under development for this challenging disease.
Viruses
Viruses such as HIV, hepatitis C virus (HCV), dengue and rabies have a large global impact (Fig. 1), resulting in over 1 million deaths in 20151. Effective treatments for HIV and HCV have been developed73,74. Other viruses, including flaviviruses, arenaviruses, coronaviruses and filoviruses still pose a considerable threat for tropical countries and others, and pose a particular risk of global pandemics, as exemplified by the recent outbreaks of Zika75, chikungunya76, dengue77, Ebola78 and Middle East respiratory syndrome (MERS)79. Since future epidemics are almost inevitable, and could be triggered by any member of these families, drug-discovery efforts rightly focus on broad-acting antivirals80. These diseases have received relatively little attention in terms of small-molecule drug discovery. However, both target-based and cell-based hit discovery approaches have yielded new compounds of interest, but to date none of these have been developed to a similar degree as the HCV and HIV drugs80. The most advanced compounds are favipiravir, GS-5734 (remdesivir) and BCX4430 (galidesivir), which target the viral polymerase (Fig. 3). Favipiravir was tested in humans during the Ebola virus epidemic of 2014. Although conditions complicated the study, it was concluded that further investigation was warranted81. GS-5734 has shown activity in rhesus monkey model of Ebola82, and was also used in humans during the 2014 Ebola outbreak. It is currently undergoing a phase II clinical trial in West Africa. Both favipiravir and GS-5734 are being considered for use in the 2018 Ebola virus outbreak in the Democratic Republic of the Congo83. BCX4430 has demonstrated complete protection against Marburg virus in non-human primates and against Ebola in rodent models when administered within 48 h of infection. This compound also shows broad activity against other viruses, including arenaviruses and flaviviruses84,85, and is currently in clinical development. Inhibitors that target the viral protease are not as far advanced (with the exception of HIV and HCV); however, they also show promise as broad-spectrum antivirals. The potential of targeting host pathways is also being explored80,86. Although new small-molecule antivirals are being discovered, there is an urgent need to translate more such treatments into the clinic, alongside ongoing vaccine development.
Bacterial diseases
Tuberculosis continues to be a major problem across the world. As with malaria, combination treatments are used against tuberculosis to combat resistance, and can circumvent issues of tuberculosis residing in multiple environments and metabolic states. The need for combination therapy complicates the progression of new compounds through clinical trials (https://www.tballiance.org/portfolio). Recently, two new drugs, bedaquiline and delamanid, have been conditionally approved for the treatment of specific levels of drug-resistant tuberculosis on the basis of phase IIb trials, and are currently undergoing phase III studies87. Bedaquiline targets ATP synthase88 and is active against the latent forms of the bacterium89. Delamanid is a nitro heterocycle; its precise mode of action has not been determined, but it is probably a prodrug that requires activation inside the bacterium. It has been implicated in inhibition of mycolic acid biosynthesis, which is critical for the structure of the cell wall of M. tuberculosis90,91.
A number of other compounds that are currently under development for treatment of tuberculosis target protein synthesis, the respiratory chain or the cell envelope (https://www.tballiance.org/portfolio and https://www.newtbdrugs.org/pipeline/clinical). Several different steps in protein synthesis are being studied. Linezolid has demonstrated activity against drug-resistant tuberculosis, and has a marked effect on patients with extensively drug-resistant tuberculosis92. It is a member of the oxazolidinone class of molecules, and targets the 50S ribosomal subunit. However adverse effects are associated with the extended treatment periods with linezolid that are required for multi-drug-resistant tuberculosis. This is linked to mitochrondrial toxicity123. Other oxazolidinones, such as sutezolid and delpazolid, are undergoing clinical trials87. Avoiding mitochondrial toxicity should allow development of oxazolidinone analogues with higher tolerability. GSK070, an oxaborole inhibitor of leucyl-tRNA synthetase, an enzyme involved in protein synthesis, is progressing through clinical trials93. GSK070 forms a covalent adduct with the 3′-adenosyl acceptor nucleotide of the tRNA, its boron binding to the 2′- and 3′-hydroxyl groups. This complex is formed in the editing site, inactivating the enzyme.
Q203 has just entered clinical trials and targets the respiratory pathway88, inhibiting cytochrome bc1. This is on the same pathway that is targeted by bedaquiline, although it acts at a different step. Hitting multiple points on this pathway appears to enhance killing of the bacterium88.
β-lactams, which target cell wall biosynthesis, are also being re-investigated for treatment of tuberculosis. Carbapenems appear to have the most potential in this class of compounds, because of the proteins that they target and because they are much weaker substrates of the M. tuberculosis β-lactamases87. Several enzymes involved in cell envelope biosynthesis are promiscuous targets, including DprE1 and MmpL3. The former is involved in the biosynthesis of an arabinogalactan polysaccharide that is critical for cell wall biosynthesis. Benzothiazones have been found to be covalent inhibitors of this enzyme; one of these compounds, BTZ043, is in preclinical development, and another, PTBZ169, is undergoing clinical evaluation (https://www.newtbdrugs.org/pipeline/clinical). Two non-covalent inhibitors of DprE1 are also undergoing clinical evaluation, both in phase I, an azaindole, TBA-7371, and OPC-167832. MmpL3 is required for the export of mycolic acid precursors, which are essential for cell wall biosynthesis. Compound SQ109, which targets this enzyme and probably others87, is in clinical trials94. Several preclinical drug-discovery programmes are targeting the enoyl reductase InhA, which is involved in the biosynthesis of mycolic acid. InhA is also the target of the prodrug isoniazid.
The nitroimidazole pretomanid (PA-824), which is active against both replicating and non-replicating mycobacteria, is in clinical trials. Like delamanid, pretomanid is likely to be a prodrug. A number of other compounds124 are undergoing clinical evaluation (https://www.newtbdrugs.org/pipeline/clinical), and several fluoroquinolines are being investigated in combination therapies, including moxifloxacin and levofloxacin.
Whereas antibacterial drug resistance is not technically a neglected or tropical disease, its impact is most clearly seen in LMICs, where there are very high levels of drug resistance. The WHO has recently published a priority list of pathogens95. The priority 1 pathogens are: Enterobacteriaceae, which are resistant to carbapenems and third generation cephalosporins, and Acinetobacter baumannii and Pseudomonas aeruginosa, which are carbapenem-resistant. New drugs are urgently needed to tackle these pathogens, but there are few drugs in clinical development that address them96. In addition, several bacterial infections, such as shigellosis and typhoid, almost exclusively affect LMICs. Drug resistance is a major issue, and worryingly, in some instances, the resistant bacteria have a higher fitness than wild-type bacteria97,98. Treatment is reliant on existing antibacterials.
Helminths
At least one billion people, and probably more, are estimated to have one or more helminth infections99. Nevertheless, the only new antihelminthic drug to emerge in the last 30 years is tribendimidine, which shows promising broad-spectrum activity. Recent clinical trials show that it is a suitable alternative for the benzimidazoles and praziquantel in various helminth infections100,101,102. Beyond this, development of new treatments is focused on repurposing drugs. This has mostly involved anthelminthics that are approved for veterinary use. Oxantel pamoate, a licensed veterinary drug since 1974, has shown promising efficacy in human clinical trials for trichuriasis (whipworm), one of the more difficult-to-treat soil-transmitted helminths103, and in combination with albendazole and pyrantel pamoate, it has recently demonstrated high clinical efficacy against hookworm infections104. Two other veterinary drugs, emodepside and moxidectin, show promising activity against several human helminths, and are currently undergoing clinical trials for onchocerciasis69,105. Some antimalarial compounds show activity against schistosomes in vitro and in vivo. Mefloquine–praziquantel and mefloquine–artesunate–praziquantel combinations were tested in clinical trials against chronic Schistosoma haematobium infection. Unfortunately, adverse event rates were high and there was no increased efficacy compared to praziquantel106. In the case of filarial worms, an alternative strategy is targeting of the Wolbachia endosymbiotic bacteria; this results in slow killing of the worms, thereby avoiding serious adverse events. For example, four weeks of treatment with doxycycline cured people suffering from onchocerciasis. The major aim of Wolbachia-directed therapy is to find shorter-course antibiotics107. Several high-throughput phenotypic screens have been carried out to discover new chemical entities that act against helminth infections108,109, but no new clinical candidates have been reported to date.
Fungal infections
Fungal infections are particularly serious in immunocompromized patients. In addition to the common fungal infections, several are found predominantly in tropical regions. One of the most important problems is meningitis due to C. neoformans, which is estimated to cause 180,000 deaths per year among people infected with HIV, the majority of whom are in Africa110. This is poorly treated at present, and there is a need for new more efficacious drugs. Mycetoma, a disfiguring infection of the skin and soft tissue, is a particularly neglected disease, and is endemic to tropical and subtropical areas. Mycetoma can be caused by bacteria (actinomycetoma) or by fungi (eumycetoma). Fungal mycetoma requires long treatment with azoles, but drug resistance, disease recurrence and side effects are common. Patients often develop deformities that require amputations. DNDi are currently running a phase II/III trial for fosravuconazole to assess whether this azole is more effective and safer than the the currently used azoles itraconazole and ketoconazole. A comprehensive review of the current antifungal pipeline has been published recently111.
Conclusion
The current treatments used against tropical diseases are suboptimal, and in some cases, there are no drugs available. However, recent progress in drug discovery in human African trypanosomiasis, tuberculosis and malaria show that headway can be made with the concerted efforts of governments, charities, foundations, product-development partnerships, academic institutions and pharmaceutical companies. There is still a long way to go, even in these disease areas. The high attrition rate in clinical trials and issues of resistance mean that there is no room for complacency. Further, multiple new agents are required for each disease area to allow combination therapies. While there has been progress in some disease areas, there is much less support and activity in others.
References
World Health Organization. Global Health Estimates 2015: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2015. (World Health Organization, Geneva, 2016).
World Health Organization. Global Health Estimates 2015: Disease burden by Cause, Age, Sex, by Country and by Region, 2000–2015. (World Health Organization, Geneva, 2016).
World Health Organization. Neglected Tropical Diseases http://www.who.int/neglected_diseases/diseases/en/ (2018).
Shepard, D. S., Undurraga, E. A., Halasa, Y. A. & Stanaway, J. D. The global economic burden of dengue: a systematic analysis. Lancet Infect. Dis. 16, 935–941 (2016).
Conteh, L., Engels, T. & Molyneux, D. H. Socioeconomic aspects of neglected tropical diseases. Lancet 375, 239–247 (2010).
G-Finder. Neglected Disease Research and Development: Reflecting on a Decade of Global Investment. (Policy Cures Research, Brisbane, 2017).
IPFMA. The Pharmaceutical Industry and Global Health. Facts and Figures 2017. (International Federation of Pharmaceutical Manufacturers & Associations, Geneva, 2017).
Wyatt, P. G., Gilbert, I. H., Read, K. D. & Fairlamb, A. H. Target validation: linking target and chemical properties to desired product profile. Curr. Top. Med. Chem. 11, 1275–1283 (2011).
Burrows, J. N. et al. New developments in anti-malarial target candidate and product profiles. Malar. J. 16, 26 (2017). A comprehensive discussion of target product and target candidate profiles for malaria.
DNDi. Diseases & Projects. https://www.dndi.org/diseases-projects (2018).
Chatelain, E. Chagas disease drug discovery: toward a new era. J. Biomol. Screen. 20, 22–35 (2015).
Huston, C. D. et al. A proposed target product profile and developmental cascade for new cryptosporidiosis treatments. PLoS Negl. Trop. Dis. 9, e0003987 (2015).
Whitehorn, J. et al. Dengue therapeutics, chemoprophylaxis, and allied tools: state of the art and future directions. PLoS Negl. Trop. Dis. 8, e3025 (2014).
Pawlowic, M. C., Vinayak, S., Sateriale, A., Brooks, C. F. & Striepen, B. Generating and maintaining transgenic Cryptosporidium parvum parasites. Curr. Protoc. Microbiol. 46, 20B.2.1–20B.2.32 (2017).
Bushell, E. et al. Functional profiling of a Plasmodium genome reveals an abundance of essential genes. Cell 170, 260–272 (2017).
de Koning-Ward, T. F., Gilson, P. R. & Crabb, B. S. Advances in molecular genetic systems in malaria. Nat. Rev. Microbiol. 13, 373–387 (2015).
Lander, N., Chiurillo, M. A. & Docampo, R. Genome editing by CRISPR/Cas9: a game change in the genetic manipulation of protists. J. Eukaryot. Microbiol. 63, 679–690 (2016).
Wang, Y. et al. A ‘suicide’ CRISPR–Cas9 system to promote gene deletion and restoration by electroporation in Cryptococcus neoformans. Sci. Rep. 6, 31145 (2016).
Zheng, W., Thorne, N. & McKew, J. C. Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today 18, 1067–1073 (2013).
Ang, M. L. & Pethe, K. Contribution of high-content imaging technologies to the development of anti-infective drugs. Cytometry A 89, 755–760 (2016).
Horvath, P. et al. Screening out irrelevant cell-based models of disease. Nat. Rev. Drug Discov. 15, 751–769 (2016).
Fux, C. A., Shirtliff, M., Stoodley, P. & Costerton, J. W. Can laboratory reference strains mirror ‘real-world’ pathogenesis? Trends Microbiol. 13, 58–63 (2005).
Angulo-Barturen, I. et al. A murine model of falciparum-malaria by in vivo selection of competent strains in non-myelodepleted mice engrafted with human erythrocytes. PLoS ONE 3, e2252 (2008). Description of a humanized mouse model for P. falciparum malaria.
Mikolajczak, S. A. et al. Plasmodium vivax liver stage development and hypnozoite persistence in human liver-chimeric mice. Cell Host Microbe 17, 526–535 (2015).
Zhan, L. J., Tang, J., Sun, M. M. & Qin, C. Animal models for tuberculosis in translational and precision medicine. Front. Microbiol. 8, 717 (2017).
Kramnik, I. & Beamer, G. Mouse models of human TB pathology: roles in the analysis of necrosis and the development of host-directed therapies. Semin. Immunopathol. 38, 221–237 (2016).
Myllymaki, H., Niskanen, M., Oksanen, K. E. & Ramet, M. Animal models in tuberculosis research—where is the beef? Expert Opin. Drug Discov. 10, 871–883 (2015).
Francisco, A. F. et al. Limited ability of posaconazole to cure both acute and chronic Trypanosoma cruzi infections revealed by highly sensitive in vivo imaging. Antimicrob. Agents Chemother. 59, 4653–4661 (2015).
White, N. J. Determinants of relapse periodicity in Plasmodium vivax malaria. Malar. J. 10, 297 (2011).
Barry, C. E. III et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7, 845 (2009).
Sanchez-Valdez, F. J., Padilla, A., Wang, W., Orr, D. & Tarleton, R. L. Spontaneous dormancy protects Trypanosoma cruzi during extended drug exposure. eLife 7, e34039 (2018).
Silver, L. L. A Gestalt approach to Gram-negative entry. Bioorg. Med. Chem. 24, 6379–6389 (2016). A description of the barriers to compound penetration in Gram-negative bacteria.
Brown, D. G., May-Dracka, T. L., Gagnon, M. M. & Tommasi, R. Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J. Med. Chem. 57, 10144–10161 (2014).
Sarathy, J. P. et al. Prediction of drug penetration in tuberculosis lesions. ACS Infect. Dis. 2, 552–563 (2016).
Fromm, M. F. Importance of P-glycoprotein at blood–tissue barriers. Trends Pharmacol. Sci. 25, 423–429 (2004).
Woodford, N. & Ellington, M. J. The emergence of antibiotic resistance by mutation. Clin. Microbiol. Infect. 13, 5–18 (2007).
Irwin, K. K., Renzette, N., Kowalik, T. F. & Jensen, J. D. Antiviral drug resistance as an adaptive process. Virus Evol. 2, vew014 (2016).
Blasco, B., Leroy, D. & Fidock, D. A. Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 23, 917–928 (2017).
Hammer, S. M. et al. Treatment for adult HIV infection: 2006 recommendations of the international AIDS society—USA panel. J. Am. Med. Assoc. 296, 827–843 (2006).
World Health Organization. World Malaria Report 2017. (World Health Organization, Geneva, 2017).
Wells, T. N., Hooft van Huijsduijnen, R. & Van Voorhis, W. C. Malaria medicines: a glass half full? Nat. Rev. Drug Discov. 14, 424–442 (2015).
White, N. J. et al. Antimalarial activity of KAF156 in falciparum and vivax malaria. N. Engl. J. Med. 375, 1152–1160 (2016).
Macintyre, F. et al. A randomised, double-blind clinical phase II trial of the efficacy, safety, tolerability and pharmacokinetics of a single dose combination treatment with artefenomel and piperaquine in adults and children with uncomplicated Plasmodium falciparum malaria. BMC Med. 15, 181 (2017).
White, N. J. et al. Spiroindolone KAE609 for falciparum and vivax malaria. N. Engl. J. Med. 371, 403–410 (2014).
Paquet, T. et al. Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci. Transl. Med. 9, eaad9735 (2017).
Lim, M. Y. et al. UDP-galactose and acetyl-CoA transporters as Plasmodium multidrug resistance genes. Nat. Microbiol, 1, 16166 (2016).
Jimenez-Diaz, M. B. et al. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc. Natl Acad. Sci. USA 111, E5455–E5462 (2014).
Baragana, B. et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 522, 315–320 (2015).
McCarthy, J. S. et al. Safety, tolerability, pharmacokinetics, and activity of the novel long-acting antimalarial DSM265: a two-part first-in-human phase 1a/1b randomised study. Lancet Infect. Dis. 17, 626–635 (2017). A description of a clinical trial for an antimalarial that includes a human challenge trial.
Cowell, A. N. et al. Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science 359, 191–199 (2018).
Plouffe, D. M. et al. High-throughput assay and discovery of small molecules that interrupt malaria transmission. Cell Host Microbe 19, 114–126 (2016).
Hovlid, M. L. & Winzeler, E. A. Phenotypic screens in antimalarial drug discovery. Trends Parasitol. 32, 697–707 (2016).
McCarthy, J. S. et al. Linking murine and human Plasmodium falciparum challenge models in a translational path for antimalarial drug development. Antimicrob. Agents Chemother. 60, 3669–3675 (2016).
Stanisic, D. I., McCarthy, J. S. & Good, M. F. Controlled human malaria infection: applications, advances, and challenges. Infect. Immun. 86, e00479-17 (2018).
Amadi, B. et al. High dose prolonged treatment with nitazoxanide is not effective for cryptosporidiosis in HIV positive Zambian children: a randomised controlled trial. BMC Infect. Dis. 9, 195 (2009).
Manjunatha, U. H., Chao, A. T., Leong, F. J. & Diagana, T. T. Cryptosporidiosis drug discovery: opportunities and challenges. ACS Infect. Dis. 2, 530–537 (2016).
Hulverson, M. A. et al. Bumped-kinase inhibitors for cryptosporidiosis therapy. J. Infect. Dis. 215, 1275–1284 (2017). Description of development of a series of compounds against cryptosporidiosis.
Manjunatha, U. H. et al. A Cryptosporidium PI(4)K inhibitor is a drug candidate for cryptosporidiosis. Nature 546, 376–380 (2017). Description of the discovery of a preclinical candidate for cryptosporidiosis by re-purposing of a series initially developed for malaria.
Love, M. S. et al. A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Negl. Trop. Dis. 11, e0005373 (2017).
Field, M. C. et al. Anti-trypanosomatid drug discovery: an ongoing challenge and a continuing need. Nat. Rev. Microbiol. 15, 217–231 (2017). A review encompassing drug discovery for the kinetoplastids.
Buscher, P., Cecchi, G., Jamonneau, V. & Priotto, G. Human African trypanosomiasis. Lancet 390, 2397–2409 (2017).
Priotto, G. et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet 374, 56–64 (2009).
Njamnshi, A. K., Gettinby, G. & Kennedy, P. G. E. The challenging problem of disease staging in human African trypanosomiasis (sleeping sickness): a new approach to a circular question. Trans. R. Soc. Trop. Med. Hyg. 111, 199–203 (2017).
Steinmann, P., Stone, C. M., Sutherland, C. S., Tanner, M. & Tediosi, F. Contemporary and emerging strategies for eliminating human African trypanosomiasis due to Trypanosoma brucei gambiense. Trop. Med. Int. Health 20, 707–718 (2015).
Torreele, E. et al. Fexinidazole—a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop. Dis. 4, e923 (2010).
Mesu, V. et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: a pivotal multicentre, randomised, non-inferiority trial. Lancet 391, 144–154 (2018). Results from a human African trypanosomiasis clinical trial demonstrating non-inferiority of fexinidazole compared to NECT treatment.
Jacobs, R. T. et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 5, e1151 (2011).
Singh, O. P., Hasker, E., Boelaert, M. & Sundar, S. Elimination of visceral leishmaniasis on the Indian subcontinent. Lancet Infect. Dis. 16, e304–e309 (2016).
DNDi. DNDi Portfolio 2017 https://www.dndi.org/diseases-projects/portfolio (2017).
Khare, S. et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 537, 229–233 (2016).
Chatelain, E. Chagas disease research and development: is there light at the end of the tunnel? Comput. Struct. Biotechnol. J. 15, 98–103 (2017).
Molina, I. et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 370, 1899–1908 (2014).
Ford, N. et al. The WHO public health approach to HIV treatment and care: looking back and looking ahead. Lancet Infect. Dis. 18, e76–e86 (2018).
Meanwell, N. A. 2015 Philip S. Portoghese Medicinal Chemistry Lectureship. Curing hepatitis C virus infection with direct-acting antiviral agents: the arc of a medicinal chemistry triumph. J. Med. Chem. 59, 7311–7351 (2016).
Basile, K., Kok, J. & Dwyer, D. E. Zika virus: what, where from and where to? Pathology 49, 698–706 (2017).
Wahid, B., Ali, A., Rafique, S. & Idrees, M. Global expansion of chikungunya virus: mapping the 64-year history. Int. J. Infect. Dis. 58, 69–76 (2017).
Gyawali, N., Bradbury, R. S. & Taylor-Robinson, A. W. The epidemiology of dengue infection: harnessing past experience and current knowledge to support implementation of future control strategies. J. Vector Borne Dis. 53, 293–304 (2016).
Coltart, C. E., Lindsey, B., Ghinai, I., Johnson, A. M. & Heymann, D. L. The Ebola outbreak, 2013–2016: old lessons for new epidemics. Phil. Trans. R. Soc. Lond. B 372, 20160297 (2017).
de Wit, E., van Doremalen, N., Falzarano, D. & Munster, V. J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 14, 523–534 (2016).
Boldescu, V., Behnam, M. A. M., Vasilakis, N. & Klein, C. D. Broad-spectrum agents for flaviviral infections: dengue, Zika and beyond. Nat. Rev. Drug Discov. 16, 565–586 (2017).
Sissoko, D. et al. Experimental treatment with favipiravir for Ebola virus disease (the JIKI Trial): a historically controlled, single-arm proof-of-concept trial in Guinea. PLoS Med. 13, e1001967 (2016).
Warren, T. K. et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 531, 381–385 (2016).
Check Hayden, E. Experimental drugs poised for use in Ebola outbreak. Nature 557, 475–476 (2018).
Taylor, R. et al. BCX4430 — a broad-spectrum antiviral adenosine nucleoside analog under development for the treatment of Ebola virus disease. J. Infect. Public Health 9, 220–226 (2016).
Warren, T. K. et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 508, 402–405 (2014).
Debing, Y., Neyts, J. & Delang, L. The future of antivirals: broad-spectrum inhibitors. Curr. Opin. Infect. Dis. 28, 596–602 (2015).
Hoagland, D. T., Liu, J., Lee, R. B. & Lee, R. E. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv. Drug Deliv. Rev. 102, 55–72 (2016).
Lamprecht, D. A. et al. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat. Commun. 7, 12393 (2016).
Kakkar, A. K. & Dahiya, N. Bedaquiline for the treatment of resistant tuberculosis: promises and pitfalls. Tuberculosis 94, 357–362 (2014).
Matsumoto, M. et al. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3, e466 (2006).
Olaru, I. D. et al. Novel drugs against tuberculosis: a clinician’s perspective. Eur. Respir. J. 45, 1119–1131 (2015).
Lee, M. et al. Linezolid for XDR-TB—final study outcomes. N. Engl. J. Med. 373, 290–291 (2015).
Li, X. et al. Discovery of a potent and specific M. tuberculosis leucyl-tRNA synthetase inhibitor: (S)-3-(aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzo[c][1,2]oxaborol-1(3H)-ol (GSK656). J. Med. Chem. 60, 8011–8026 (2017).
World Health Organization. Global Tuberculosis Report 2017. (World Health Organization, Geneva, 2017).
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327 (2018).
World Health Organization. Antibacterial Agents in Clinical Development: an Analysis of the Antibacterial Clinical Development Pipeline, Including Tuberculosis. (World Health Organization, Geneva, 2017).
Baker, S. et al. Fitness benefits in fluoroquinolone-resistant Salmonella Typhi in the absence of antimicrobial pressure. eLife 2, e01229 (2013).
Wong, V. K. et al. Phylogeographical analysis of the dominant multidrug-resistant H58 clade of Salmonella Typhi identifies inter- and intracontinental transmission events. Nat. Genet. 47, 632–639 (2015).
Hotez, P. J. et al. Helminth infections: the great neglected tropical diseases. J. Clin. Invest. 118, 1311–1321 (2018).
Moser, W. et al. Efficacy and safety of tribendimidine, tribendimidine plus ivermectin, tribendimidine plus oxantel pamoate, and albendazole plus oxantel pamoate against hookworm and concomitant soil-transmitted helminth infections in Tanzania and Cote d’Ivoire: a randomised, controlled, single-blinded, non-inferiority trial. Lancet Infect. Dis. 17, 1162–1171 (2017).
Sayasone, S. et al. Efficacy and safety of tribendimidine versus praziquantel against Opisthorchis viverrini in Laos: an open-label, randomised, non-inferiority, phase 2 trial. Lancet Infect. Dis. 18, 155–161 (2018).
Xiao, S. H., Utzinger, J., Tanner, M., Keiser, J. & Xue, J. Advances with the Chinese anthelminthic drug tribendimidine in clinical trials and laboratory investigations. Acta Trop. 126, 115–126 (2013). A comprehensive review of the discovery and development of tribendimidine, the first new anthelminthic in 30 years.
Moser, W. et al. Efficacy and safety of oxantel pamoate in school-aged children infected with Trichuris trichiura on Pemba Island, Tanzania: a parallel, randomised, controlled, dose-ranging study. Lancet Infect. Dis. 16, 53–60 (2016).
Moser, W. et al. Efficacy and tolerability of triple drug therapy with albendazole, pyrantel pamoate, and oxantel pamoate compared with albendazole plus oxantel pamoate, pyrantel pamoate plus oxantel pamoate, and mebendazole plus pyrantel pamoate and oxantel pamoate against hookworm infections in school-aged children in Laos: a randomised, single-blind trial. Lancet Infect. Dis. 18, 729–737 (2018).
Awadzi, K., Opoku, N. O., Attah, S. K., Lazdins-Helds, J. & Kuesel, A. C. A randomized, single-ascending-dose, ivermectin-controlled, double-blind study of moxidectin in Onchocerca volvulus infection. PLoS Negl. Trop. Dis. 8, e2953 (2014).
Keiser, J. et al. Praziquantel, mefloquine–praziquantel, and mefloquine–artesunate–praziquantel against Schistosoma haematobium: a randomized, exploratory, open-label trial. PLoS Negl. Trop. Dis. 8, e2975 (2014).
Klarmann-Schulz, U. et al. Comparison of doxycycline, minocycline, doxycycline plus albendazole and albendazole alone in their efficacy against onchocerciasis in a randomized, open-label, pilot trial. PLoS Negl. Trop. Dis. 11, e0005156 (2017).
Guidi, A. et al. Discovery by organism based high-throughput screening of new multi-stage compounds affecting Schistosoma mansoni viability, egg formation and production. PLoS Negl. Trop. Dis. 11, e0005994 (2017).
Mansour, N. R. et al. High throughput screening identifies novel lead compounds with activity against larval, juvenile and adult Schistosoma mansoni. PLoS Negl. Trop. Dis. 10, e0004659 (2016).
Rajasingham, R. et al. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect. Dis. 17, 873–881 (2017).
Perfect, J. R. The antifungal pipeline: a reality check. Nat. Rev. Drug Discov. 16, 603–616 (2017).
Kotloff, K. L. et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case–control study. Lancet 382, 209–222 (2013). The results of a clinical study into enteric diseases in LMICs.
Liu, J. et al. Use of quantitative molecular diagnostic methods to identify causes of diarrhoea in children: a reanalysis of the GEMS case–control study. Lancet 388, 1291–1301 (2016).
Hotez, P. J., Fenwick, A., Savioli, L. & Molyneux, D. H. Rescuing the bottom billion through control of neglected tropical diseases. Lancet 373, 1570–1575 (2009).
Checkley, W. et al. A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium. Lancet Infect. Dis. 15, 85–94 (2015).
Kublin, J. G. et al. Effect of Plasmodium falciparum malaria on concentration of HIV-1-RNA in the blood of adults in rural Malawi: a prospective cohort study. Lancet 365, 233–240 (2005).
Mayer, K. H. & Dukes Hamilton, C. Synergistic pandemics: confronting the global HIV and tuberculosis epidemics. Clin. Infect. Dis. 50, S67–S70 (2010).
Brooker, S. et al. Epidemiology of Plasmodium–helminth co-infection in Africa: populations at risk, potential impact on anemia, and prospects for combining control. Am. J. Trop. Med. Hyg. 77, 88–98 (2007).
Blanc, F. X. et al. Earlier versus later start of antiretroviral therapy in HIV-infected adults with tuberculosis. N. Engl. J. Med. 365, 1471–1481 (2011).
Josephson, F. Drug–drug interactions in the treatment of HIV infection: focus on pharmacokinetic enhancement through CYP3A inhibition. J. Intern. Med. 268, 530–539 (2010).
Pion, S. D. et al. Co-infection with Onchocerca volvulus and Loa loa microfilariae in central Cameroon: are these two species interacting? Parasitology 132, 843–854 (2006).
Hoerauf, A., Pfarr, K., Mand, S., Debrah, A. Y. & Specht, S. Filariasis in Africa—treatment challenges and prospects. Clin. Microbiol. Infect. 17, 977–985 (2011).
Song, T. et al. Linezolid trough concentrations correlate with mitochondrial toxicity-related adverse events in the treatment of chronic extensively drug-resistant tuberculosis. EBioMedicine 2, 1627–1633 (2015).
Tiberi, S. et al. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis 18, e183–e198 (2018).
Acknowledgements
We acknowledge the following organizations that have supported our drug-discovery efforts for some of these diseases: Wellcome Trust, Medicines for Malaria Venture, the Bill and Melinda Gates Foundation and Drugs for Neglected Diseases initiative. We thank members of the Drug Discovery Unit for helpful discussions and K. Read, S. Green and S. Wyllie for reading the manuscript.
Reviewer information
Nature thanks B. Melillo, E. Winzeler and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
All the authors contributed to writing the text.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
De Rycker, M., Baragaña, B., Duce, S.L. et al. Challenges and recent progress in drug discovery for tropical diseases. Nature 559, 498–506 (2018). https://doi.org/10.1038/s41586-018-0327-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-018-0327-4
This article is cited by
-
DBPP-Predictor: a novel strategy for prediction of chemical drug-likeness based on property profiles
Journal of Cheminformatics (2024)
-
Menthol- and thymol-based ciprofloxacin derivatives against Mycobacterium tuberculosis: in vitro activity, lipophilicity, and computational studies
Scientific Reports (2023)
-
Drug Repurposing for Spinal Cord Injury: Progress Towards Therapeutic Intervention for Primary Factors and Secondary Complications
Pharmaceutical Medicine (2023)
-
Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework
Nature Machine Intelligence (2022)
-
Asymmetric synthesis of bedaquiline based on bimetallic activation and non-covalent interaction promotion strategies
Science China Chemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
