
Abstract
Satellite glial cells (SGCs) closely envelop cell bodies of neurons in sensory, sympathetic and parasympathetic ganglia. This unique organization is not found elsewhere in the nervous system. SGCs in sensory ganglia are activated by numerous types of nerve injury and inflammation. The activation includes upregulation of glial fibrillary acidic protein, stronger gap junction-mediated SGC–SGC and neuron–SGC coupling, increased sensitivity to ATP, downregulation of Kir4.1 potassium channels and increased cytokine synthesis and release. There is evidence that these changes in SGCs contribute to chronic pain by augmenting neuronal activity and that these changes are consistent in various rodent pain models and likely also in human pain. Therefore, understanding these changes and the resulting abnormal interactions of SGCs with sensory neurons could provide a mechanistic approach that might be exploited therapeutically in alleviation and prevention of pain. We describe how SGCs are altered in rodent models of four common types of pain: systemic inflammation (sickness behaviour), post-surgical pain, diabetic neuropathic pain and post-herpetic pain.
Similar content being viewed by others


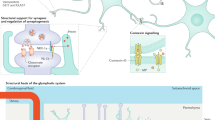
Introduction
Satellite glial cells (SGCs) are unique cells whose most distinctive morphological feature is that they wrap around neuronal cell bodies, in most cases forming a complete envelope. SGCs are found exclusively in peripheral ganglia — sensory, parasympathetic and sympathetic ganglia (Fig. 1a,b). The latter two are part of the autonomic nervous system. The gap between SGCs and the neuronal surface is about 20 nm, which is similar to that of the synaptic cleft. This organization allows for close mutual neuron–SGC interactions1,2,3. The neuron and its attendant SGCs have been termed ‘neuron–glial units’ (Fig. 1c). In most cases, neurons are wrapped individually with several SGCs, but there is evidence that a small proportion (4–9% depending on the species) of dorsal root ganglia (DRG) neurons share a common glial envelope, forming a ‘cluster’ with one or two other neurons4 (Fig. 1d). In some cases, the neurons in a cluster are separated only by a thin layer of extracellular space, whereas in other cases they are separated by a thin glial sheet, which allows neuron–SGC–neuron chemical interaction5.

a | Position of the dorsal root ganglia (DRG) in the sensory pathways leading from the skin to the brain. A paravertebral sympathetic ganglion (SG) is also indicated. These ganglia innervate most organs, including blood vessels. b | Location of the trigeminal ganglion (asterisk) which innervates the face and teeth. The three divisions of the trigeminal ganglion are indicated as V1–V3. c | Low-power electron micrograph showing the neuron–satellite glial cell (SGC) units in a DRG. Neurons are labelled N1–N6, SGCs are coloured blue. The widened area in the SGC surrounding N3 contains the cell’s nucleus. ct, connective tissue space; v, blood vessels. Scale bar, 10 µm. d | Schematic of three patterns of grouping of sensory neurons. Top: neurons are separated by a connective tissue space (indicated by arrow), and each has its own SGC sheath. Middle: a cluster of two neurons that share a common SGC sheath and are separated by a SGC process. Bottom: a cluster where the neurons share a common SGC sheath, but without an intervening SGC process. e | Schematic of a sympathetic neuron covered with an SGC envelope. The SGCs (arrows) cover the synapses. SGC processes extend beyond the neuronal soma and ensheath an axon and a dendrite. Part c adapted with permission from ref.86, Elsevier. Part e adapted with permission from ref.127, Elsevier.
Much more is known about astrocytes than SGCs, so it is instructive to compare SGCs with these as well as with other types of glia (Box 1). Two main hallmarks of astrocyte–neuron relations are that each astrocyte contacts numerous neurons, and that individual astrocytes form non-overlapping domains6. In contrast, SGCs around a given neuron are in close contact with each other, and are almost always separated from SGCs surrounding other neurons.
The close contact between SGCs and neurons is a major key to understanding their functions; for example, this contact enables them to control neuronal homeostasis, but very little is known on this topic. More is known with regard to changes that SGCs in sensory ganglia undergo following nerve damage and how they contribute to pain. Little is known about the functions of SGGs in sympathetic ganglia, but there is emerging information on their role in the control of synaptic transmission in these ganglia. Knowledge of SGCs in parasympathetic ganglia is even scarcer.

SGCs and pain
Glial cells play major roles in the function of the CNS under health and disease states, and maintain bidirectional communication with the neurons7,8,9. Glial cells express receptors for substances released from neurons, most notably ATP, acting on P2 purinergic receptors (P2Rs)10,11. Glial cells also release bioactive substances, including ATP, glutamate and cytokines such as tumour necrosis factor (TNF), IL-1β and fractalkine7,12, which enable them to communicate among themselves and with adjacent neurons. Most studies on pain and most treatments for it have focused on neurons, but Watkins and her co-workers13,14 proposed that glial cells in the spinal cord participate in pain generation and maintenance, and may serve as more suitable targets for pain therapy. This view was supported by studies of pain models in animals, showing that spinal microglia and astrocytes are essential for hyperalgesia7,15,16.
Sensory ganglia contain the somata of neurons that innervate most body parts. The main sensory ganglia are the DRG, trigeminal ganglia (TG) and nodose ganglia (Fig. 1). There is evidence that abnormal electrical activity in sensory neurons is associated with pathological pain, such as allodynia17,18,19,20. For example, it has been shown that phantom limb pain in humans is driven primarily by abnormal intrinsic DRG activity21. The ideas described above concerning CNS glia and pain prompted research into the possible role of SGCs in sensory ganglia in pain. It was found that nerve damage or inflammation activates SGCs in sensory ganglia. This included upregulation of the astrocyte marker glial fibrillary acidic protein (GFAP)22,23,24, increased coupling strength among SGCs19,25,26,27, downregulated Kir4.1 channels in SGCs28,29,30 and increased sensitivity of SGCs to the pain mediator ATP27,31 (see Box 2 for details). Activated SGCs release pro-inflammatory cytokines IL-1β, IL-6, TNF and fractalkine2,32,33,34,35,36, which can act on the neurons and increase their excitability and firing.
In this Review, we describe the normal properties of SGCs and how they change in animal pain models. We discuss the possible roles of SGCs in several models of pain in rodents — systemic inflammation, post-surgical pain (PSP), diabetic neuropathic pain (DNP) and post-herpetic pain. These studies indicate that these different pain syndromes share a rather similar chain of events that connects changes in SGCs to pain. Essentially, nerve injury leads to nitric oxide release, which induces SGC activation. SGCs in turn release compounds that lead to neuronal hyperexcitability and pain. Intercellular calcium waves play an important role in this scheme. We conclude with a brief account of SGCs in sympathetic ganglia and their possible roles under pathological conditions.

Characteristics of SGCs
Ion channels
SGCs do not have voltage-dependent Na+ or Ca2+ channels, and therefore cannot conduct action potentials37. The main K+ channel in SGCs is Kir4.1 (ref.28). Injury suppresses Kir4.1 function in SGCs, which may contribute to pain29,30. Reduced Kir4.1 permeability likely depolarizes SGCs, inducing them to release excitatory mediators such as ATP that can activate the neurons; indeed, silencing Kir4.1 in the TG leads to pain-like behaviour28. Similarly, gain or loss of Kir4.1 in astrocytes that contact the somata of CNS neurons can regulate neuronal activity38.
Gap junctions in SGCs
Glial cells in both the CNS and the periphery express gap junction channels, which provide a pathway for diffusion of ions and small molecules between cells. The most abundant connexins (Cx) in mouse DRG and TG are Cx43 and Cx32, followed by Cx30.2, Cx37, Cx26, Cx30, Cx45 and Cx36 (ref.39). Under normal conditions, SGCs contain Cx43 (refs40,41). Cx43 is upregulated in the TG and DRG following nerve injury or inflammation26,41,42,43,44,45. Cx26 expression in SGCs and neurons was increased in the TG in the setting of neuropathic pain46. These findings correlate with functional evidence for increased coupling by gap junctions among SGCs in a wide range of pain models, which include the sciatic nerve section25,47, the infraorbital nerve section37, colonic obstruction and inflammation19,48,49, injection of complete Freund’s adjuvant in mouse hindpaw50, the sciatic nerve51, the temporomandibular joint52 and chemotherapy-induced neuropathy23. Dye coupling among the SGCs not only increased within the sheaths surrounding individual neurons but extended to sheaths surrounding adjacent neurons via newly formed processes19,23,25,27,47,51,52. Moreover, injury generally results in dye spread between SGCs and neurons and from neuron to neuron, and electrophysiological recordings from the dissociated DRG and TG demonstrate electrical coupling through SGC‒SGC, neuron‒SGC and neuron–neuron gap junctions44,53.
As SGC activation is accompanied with increased coupling, it was proposed that blocking gap junctions could have an analgesic action, and this was confirmed in numerous chronic pain models19,23,26,42,43,44,45,50,54,55. Reduced Cx43 in rat TG using double-stranded RNA was analgesic in a model of neuropathic pain induced by infraorbital nerve injury26, although a potential concern was that in naive rats this treatment itself induced pain-like behaviours. A recent study has shown that increased SGC coupling contributes also to acute pain induced by capsaicin application56. Blocking gap junctions with carbenoxolone (CBX) or blocking P2X7Rs (which are expressed exclusively by SGCs) reduced pain reactions in this model, supporting a role for SGCs in acute pain.
In summary, increased coupling between SGCs and also SGC‒neuron and neuron‒neuron coupling is a common feature in pain models, and appears to contribute to chronic pain. We propose that inhibiting gap junctions might be an effective novel target for analgesic drugs.
Pannexins in SGCs
Pannexins (Panx) are homologues of gap junction proteins of multicellular invertebrates, the innexins. Although Panx1 does not form gap junctions, it does form membrane channels that allow release of ATP57,58. Panx1 expression is upregulated in the DRG following sciatic nerve ligation59, in TG SGCs in a chronic orofacial pain model60 and in nodose ganglia SGCs in a systemic inflammation model61. In the orofacial pain study60, Panx1-deleted mice did not develop tactile hypersensitivity, suggesting a role for Panx1-mediated ATP release in nociception. This analgesic effect was obtained in mice with glia-targeted but not with neuron-specific Panx1 deletion60, indicating the dominant role of SGCs in ATP release.
Receptors
Knowledge regarding the pharmacology of SGCs is still limited, and much of the information on this topic relates to purinergic transmission. ATP is a messenger in neuron–glia interactions11 and in the pain pathways15,62. Cells release ATP by vesicular or channel-mediated mechanisms, such as P2X7R or Panx1 channels. Ecto-ATPases break down ATP to ADP and other purines. In the DRG, ecto-ATPases are found in Schwann cells and SGCs, but not in neurons63. This enables these glial cells to regulate the ATP level in the ganglia.
Using calcium imaging, SGCs in the TG were found to express functional P2YRs (ref.64). Further work showed that P2Rs in the TG consist of P2Y1, P2Y2, P2Y4, P2Y6, P2Y12 and P2Y13Rs (refs62,65). In vitro incubation with the pro-inflammatory peptide bradykinin increased the SGC response to P2YR stimulation65, indicating P2YR sensitization. Release of ATP by sensory neurons can activate P2Rs in SGCs, providing one mechanism for neuron–SGC communication2,5,36,66,67,68,69 (Fig. 2a,b). In two orofacial pain models, the sensitivity of SGCs to ATP in the TG increased 100-fold31, apparently due to a switch of the P2R population from the metabotropic P2YR to the ionotropic P2XR. The exact P2XR subtype that was increased was not determined but the candidates are P2X2, P2X4 and P2X5Rs, but not P2X7Rs, which were reported to be equally present in SGCs in ganglia from both controls and treated animals31. Increased responses to ATP were also observed in a mouse model of systemic inflammatory pain in SGCs of the DRG27 and nodose ganglia61. It thus appears that P2R upregulation is part of the SGC activation after injury. There is little information on the pharmacology of SGCs in non-rodent mammals, but P2X7R expression is reportedly upregulated in DRG SGCs of patients with neuropathic pain70. These changes, combined with the increased sensitivity of sensory neurons to ATP under pathological conditions67,71, indicate that increased intercellular communication via P2Rs is likely to contribute to neuronal hyperexcitability in pain states (Fig. 2a,b).

a | Spread of calcium waves in sensory ganglia. The axon of neuron 1 (N1) is injured, which induces the firing of action potentials in the cell body. This causes release of ATP from the neuron, which acts on satellite glial cells (SGCs) by elevating intracellular Ca2+ in them, which in turn induces them to release ATP (and other factors, including cytokines). The increased gap junctions between SGCs facilitate the spread of IP3 within the coupled SGCs, which increases the intracellular calcium in these cells. The combination of the increased sensitivity to ATP and the increased coupling by gap junctions will promote the propagation of calcium waves, resulting in the excitation of N2. This excitation may relay nociceptive signals to the spinal cord. b | Example of calcium wave propagation based on experiments on cultured neurons and SGCs from mouse trigeminal ganglion. Electrical or mechanical stimulation of the neuron at time 0 resulted in higher intracellular Ca2+ in it. After a delay, the adjacent SGCs also responded with elevated intracellular Ca2+, with the amplitude and delay determined by proximity to the neuron. Similar results were obtained when an SGC was stimulated. Inset: schematic showing a neuron and three SGCs (labelled 1–3). c,d | Cross depolarization in the DRG. The method for demonstrating cross depolarization: stimulating the axon of a neuron (coloured red) evokes firing in it, and intracellular recording (blue electrode) from an unstimulated neuron reveals slow depolarization due to this firing (part c). Brief subthreshold depolarizing currents (indicated by downward-pointing arrows) were applied to the recorded cell before (1), during (2) and after (3) the stimulation of an axon of a nearby neuron. When applied while the neuron was depolarized by the excitation of the nearby neuron (marked by the bar below the lower trace), the brief electrical stimulus summed with the cross depolarization to reach the threshold and fire an action potential (red trace) (part d). The conclusion from this experiment is that electrical activity in one neuron releases a messenger (likely ATP), causing cross depolarization, which increases the excitability of other neurons. Part a adapted with permission from ref.80, Elsevier. Part b adapted with permission from ref.66, Cambridge University Press. Part d adapted from ref.78 copyright 1996, Society for Neuroscience.
Sensory neurons contain several neuroactive peptides, among which calcitonin gene-related peptide (CGRP) is one of the best studied, as it appears to be a major factor in migraine. In the TG of humans, rats and monkeys, the neurons express CGRP and SGCs express components of the CGRP receptor complex72,73, which suggests that this peptide mediates neuron–SGC communication. Recently, antibodies against CGRP and its receptors have been approved for migraine therapy, and it was found that they enter sensory ganglia but not the CNS74. This raises the possibility that the therapeutic action of these antibodies may involve disruption of abnormal neuron–SGC interactions.
Further study of SGC pharmacology is crucial for understanding the function of SGCs because chemical signalling is the main mode of glia–neuron communication. Recent technical developments allow recording of the physiological activity of sensory neurons in intact mice44,75,76, and this method can be utilized to explore the pharmacology of SGCs in vivo.
SGCs and intercellular communication
Neurons in sensory ganglia receive no synapses and are separated from each other by SGCs and the connective tissue space4 (Fig. 1c). It would therefore appear that no interactions among neurons in these ganglia are present; however, Devor and Wall77 showed that electrical activity in DRG neurons evokes depolarization in adjacent neurons, and named this ‘cross depolarization’ (Fig. 2c,d). Later work indicated that the effect is due to the release of unidentified chemical mediators78,79. Currently, there is evidence that SGCs contribute to cross depolarization, and that they interact chemically among themselves and with neurons via P2Rs (refs2,5,68,80,81) and by gap junctions45. A calcium imaging study on the DRG in intact mice found that electrical activity in one neuron can activate neighbouring ones (‘coupled activation’)44. This effect was greatly increased after nerve injury or peripheral inflammation, and was attributed to Cx43 upregulation in SGCs. Coupled activation was reduced by local or systemic application of the gap junction blocker CBX and also in Cx43 knockout mice. Pain hypersensitivity behaviour was reduced by CBX. These observations indicate that gap junctions in SGCs are a major contributor to coupled activation and pain. The underlying mechanism of coupled activation is not fully clarified, but it was suggested that neuron–SGC–SGC–neuron electrical coupling is involved44 (see Fig. 3a,b). This idea is consistent with the enhanced cell coupling observed in rodent pain models3,80, but the contribution of chemical interactions was not excluded.

a,b | Proposed mechanism of coupled activation between neurons. Synchronous activity of adjacent neurons could arise by the spread of depolarization from neuron 1 (N1) to its surrounding satellite glial cells (SGCs), then through gap junctions to SGCs of a nearby neuron and then through gap junctions from these SGCs to N2. Under control conditions, SGCs are coupled mostly to other SGCs around a given neuron (part a). After peripheral injury or inflammation to a neuron (coloured red), SGCs become more strongly coupled to other SGCs around the same neuron (and also to neurons) by newly formed gap junctions. This enables increased transfer of electrical current and small molecules among SGCs and between SGCs and neurons. Such a mechanism can account for coupled neuronal activation (part b). c,d | The chain of events connecting neuronal excitation and glial activation. Resting conditions are shown in part c. Following neuronal damage, the neuronal cell body fires a high rate of action potentials, which increases intracellular calcium that in turn activates nitric oxide synthase to produce nitric oxide. Nitric oxide diffuses from the neuron and reaches the surrounding SGCs, where it induces cyclic guanosine 5'-monophosphate (cGMP) synthesis. This second messenger can have various actions on SGCs, and may be a key factor in SGC activation. SGC activation includes the release of ATP and cytokines, gap junction formation and increased sensitivity to ATP (part d). NO, nitric oxide; P2R, P2 purinergic receptor; Panx1, pannexin 1; TNF, tumour necrosis factor; TRPV1, transient receptor potential vanilloid type 1 channel.
Studies on short-term TG cultures have shown that SGCs can transmit Ca2+ waves, which enable SGCs (together with neurons) to propagate signals over long distances66,67 (Fig. 2a,b). These waves are mediated by gap junctions and chemical messengers (ATP and glutamate). After nerve injury, gap junctions are upregulated and the sensitivity of both neurons and SGCs to ATP increases. It is thus expected that neuron–neuron interactions will be increased after injury, as indeed was observed in mouse pain models44.
Nerve damage induces prominent changes in SGCs, and as the injury site can be a large distance from the ganglion, determining which neuronal signals induce these changes has been a puzzle. A likely explanation is that the high firing rate in injured neurons causes the release of chemical mediators that act on SGCs and activate them82. Such mediators may include cytokines, growth factors and nitric oxide. Active neurons release nitric oxide, which diffuses rapidly over the narrow gap between them and the SGCs. Nitric oxide mimics the observed changes in SGCs that occur after nerve insult83, and thus appears to be a key element in SGC activation.
We have proposed a scheme to explain how increased gap junctional communication and sensitization to ATP can explain the role of SGCs in neuronal hyperexcitability in pain models45, based on the ‘ignition theory’ formulated to account for trigeminal neuralgia84. In this model, hyperexcitability of sensory neurons is evoked by nerve injury. Activation of one neuron then spreads to others, achieving synchronized and sustained activity within neuronal ensembles. In the original framework, the synchronized discharge was hypothesized to result from chemical cross-excitation. It is conceivable that the increased neuronal activity leads to ATP release through upregulated Panx1 channels and that responses are amplified as a consequence of P2R activation in SGCs, and additional opening of Panx1 channels. Release of other molecules, including nitric oxide, cytokines and growth factors, likely also contributes to SGC activation. A modified ignition model based on the ability of glial cells to sustain intercellular calcium waves would include the spread of signals among SGCs through Ca2+ wave propagation66,67 (Fig. 2a,b). Enhanced gap junction-mediated coupling among SGCs, combined with enhanced P2R transmission, would spread the activation to other sensory neurons, leading to sustained reverberating activity and recruitment of non-nociceptors into the pain response.
The ideas discussed above are summarized in Fig. 3c,d and Fig. 4, which emphasize abnormal bidirectional SGC–neuron interactions that lead to enhanced neuronal activity.

This sequence is an attempt to generalize the main events, but does not exclude additional or alternative mechanisms. The initial event in this cascade is injury to sensory neurons, such as from surgery, inflammation, diabetes or herpesvirus, which increases their firing rate. This leads to production of nitric oxide in the neurons. Nitric oxide diffuses from the injured neurons and activates satellite glial cells (SGCs) that surround them and likely also SGCs around adjacent neurons. SGC activation involves numerous processes, including greater coupling by gap junctions, increased sensitivity to ATP, upregulation of ERK and increased release of pro-inflammatory cytokines (IL-1β, tumour necrosis factor, IL-6, fractalkine and others). The increased gap junctions and sensitivity to ATP will enhance calcium waves, which lead to neuronal excitation. Cytokines released from SGCs will also contribute to neuronal sensitization and excitation. The overall effect will be greater firing (spontaneous and/or evoked) of sensory neurons and pain. TLR4, Toll-like receptor 4.
SGCs in selected pain conditions
Numerous models have been used to study pain in animals85. Most models are based on local injury to peripheral nerves, and include nerve section (axotomy) and tissue incision. Local inflammation models include chronic constriction injury or the application of substances such as carrageenan or complete Freund’s adjuvant to the paw50, face46 and teeth43. Models of systemic pain include inflammation induced by lipopolysaccharide (LPS)27,86, neuropathy following systemic administration of anticancer drugs23 and type 1 diabetes mellitus induced by streptozotocin (STZ)87,88,89. A role for SGCs in chronic pain has been documented in models of both localized and systemic types of chronic pain (somatic, visceral and orofacial). Several studies have demonstrated that reversing injury-induced changes in SGCs reduced or abolished pain behaviour in rodent models. Below, we describe the possible contributions of SGCs to several pain syndromes that are commonly encountered in clinical practice.
Systemic inflammation
Systemic inflammation is a common human disease. The ensemble of symptoms associated with this disorder is called ‘sickness behaviour’, and includes depression and pain90. As for most other pain syndromes, many investigators have emphasized the role of central mechanisms in sickness behaviour, with increased cytokine levels being a major factor. However, recent work suggests that activity initiated by LPS injection and sustained within the sensory ganglia contributes to the generation and maintenance of pain in systemic inflammation27. Moreover, a single intraperitoneal LPS injection induced changes in the DRG, which were associated with mechanical hypersensitivity that lasted for at least a month86. Apparently, the direct effects of LPS are short-lived and depend on its binding to Toll-like receptor 4 (TLR4) in sensory neurons, but the downstream sequelae can last for weeks. LPS-induced changes in SGCs were similar to those observed in local inflammatory pain models, which include SGC activation and increased SGC–SGC, neuron–SGC and neuron–neuron dye coupling by gap junctions19,50,51. Increased numbers of gap junctions in SGCs were detected by electron microscopy at both 7 and 30 days post LPS administration27,86. Neuronal gap junctions were not detected either before or after LPS injection, which is consistent with the very weak coupling of neurons to each other and to SGCs53. In addition, LPS administration increased the responses of SGCs to ATP and reduced the withdrawal threshold in response to mechanical stimulation of the paw or abdomen. Intraperitoneal injection of the gap junction blocker CBX or palmitoleic acid raised the pain threshold back to the control level, suggesting a role for gap junctions in pain. These (and other) gap junction blockers are not highly specific and may act by other mechanisms and locations (for example, those that cross the blood–brain barrier may act centrally). However, CBX does not cross the blood–brain barrier91 and is therefore likely to act largely in the periphery, and gap junction blockers did not affect the withdrawal threshold in control mice. These findings support the idea that increased gap junction expression in sensory ganglia contributes to chronic pain, and indicate that gap junction blockade in these ganglia has therapeutic potential for alleviating chronic pain.
Chronic post-surgical pain (PSP)
PSP can be acute (resolving within several days after the operation) or chronic (lasting months and even years). Chronic PSP is observed in 10–50% of patients undergoing common operations, such as hip and knee operations and mastectomy92,93. Current therapy for chronic PSP is inadequate, and preventive treatments have been largely ineffective94.
It is thought that chronic PSP is caused by nerve injury — that is, it is neuropathic — but this topic is controversial92. For example, skin and muscle incision and retraction in rats without visible nerve injury induced chronic PSP that persisted for more than 3 weeks95, suggesting that mechanisms other than nerve injury are involved in chronic PSP. It is assumed that chronic PSP is associated with central sensitization96, but peripheral mechanisms are also important97,98,99, as peripheral activity drives the central sensitization in PSP100 and in other pain states101.
Little is known regarding peripheral mechanisms in chronic PSP. Activation of P2X7R on SGCs was found to elevate phosphorylated ERK (pERK1/2) in SGCs, which led to TNF release98. TNF in turn acts on neurons, which increases neuronal excitability. These results indicate that SGCs are a key element in chronic PSP generation and may be a highly suitable therapeutic target for it. Sensory ganglia are not protected by a vascular barrier, and therefore SGCs, which surround the neurons, are ideal targets for pain therapy.
In a PSP model based on paw incision, pERK1/2 expression was found to be increased 2-fold in A-fibre neurons and SGCs in the DRG. This increase was observed as early as 2 min after the incision, and returned to baseline at 2 h99. This effect is extremely fast, especially considering that SGCs are not influenced directly by the injury and must receive signals from the neurons. The local anaesthetic levobupivacaine inhibited pERK1/2 induction. CBX, which blocks both gap junctions and Panx1 channels, inhibited the early glial pERK1/2 increase and also inhibited pain hypersensitivity99. This study indicates that early events induced in SGCs by nerve injury participate in the generation of hypersensitivity in PSP. It is thus likely that preventing these events will have therapeutic value in PSP treatment.
Diabetic neuropathic pain (DNP)
Nerve damage (neuropathy) is a common complication in diabetes mellitus type 1 and 2; it affects about 50% of the patients102 and is difficult to treat103. A frequent consequence of this neuropathy is DNP. Little is known about the mechanisms responsible for DNP, but it is clear that both central and peripheral mechanisms contribute102,104.
The most widely used diabetes mellitus type 1 model is obtained by injecting rodents with the toxin STZ, which destroys pancreatic beta cells. The key element in diabetes mellitus models is a several-fold increase in the blood glucose level, which induces numerous types of cellular damage.
Injury to the peripheral nervous system in diabetes mellitus is well recognized102. Research has mostly focused on axons, but there is evidence that functional changes in sensory neurons can contribute to DNP105,106,107,108. Ionic currents in sensory neurons are abnormal in diabetes mellitus105,106,107, indicating that sensory ganglia are highly suitable targets for research and therapy of DNP. SGCs can release the cytokines TNF and IL-1β, particularly following insults2,36,98, so neuronal excitation by SGC-derived cytokines may be one way in which SGCs contribute to DNP.
Altered enzymatic activity in SGCs might be relevant to cellular damage in diabetes mellitus. One such enzyme is aldose reductase109, which catalyses the conversion of glucose into sorbitol. Increased sorbitol production in cells leads to its accumulation, causing osmotic swelling and cell damage109,110. Aldose reductase activity is present in SGCs (but not in neurons) of rat DRG111. Targeting aldose reductase in SGCs seems to have promising potential for treating DNP, and efforts are underway to develop aldose reductase inhibitors as therapy for diabetes mellitus complications112.
There is evidence for SGC activation in the STZ model89. The underlying mechanism for this activation appears to be related to the hyperglycaemia-induced upregulation of pyruvate dehydrogenase kinases (PDK2 and PDK4) in DRG cells (SGCs, neurons and infiltrating macrophages). These enzymes play key roles in glucose metabolism105, and their enhanced activity causes lactic acidosis, which activates SGCs and macrophages105. Reactive SGCs and macrophages release pro-inflammatory cytokines (TNF, IL-1β and IL-6), which increase neuronal excitability and thus lead to pain. It can be concluded from this work105 that PDKs play a pivotal role in inducing SGC activation and creating a pro-inflammatory microenvironment in diabetic DRG, the prerequisites for pain pathogenesis.
The pharmacology of SGCs in diabetes mellitus models has received only little attention. It was found that in parallel with the activation of SGCs in the STZ model, the sensitivity of these cells to the P2X7R agonist BzATP was increased113. However, BzATP is a potent agonist of other P2XRs (ref.114), particularly P2X4R, and indeed there is evidence that P2X4R is upregulated in SGCs in the STZ model and that it is essential for the mechanical hypersensitivity observed in this model115. This topic needs to be further explored, particularly from the perspective of the degree to which SGC–neuron interaction is compromised and whether Cx43 or Panx1 is involved in the heightened P2R sensitivity in SGCs.
Post-herpetic neuralgia
Alphaherpesviruses, varicella zoster virus (VZV), herpes simplex virus type 1 (HSV-1) and HSV-2 infect the peripheral nervous system. After the primary infection, these viruses can become latent within sensory ganglia for the lifetime of the host, but retain the ability to reactivate and cause disease episodes. Reactivation of VZV results in herpes zoster, which is characterized by painful skin eruptions (shingles) and often leads to post-herpetic neuralgia116,117. Reactivation of HSV causes mucocutaneous lesions (for example, cold sores) and may also result in neuralgia118. Post-herpetic neuralgia can be severe, persistent and refractory to treatment116,119. The mechanisms underlying this condition are not known. VZV is difficult to study because it does not infect rodents, and instead HSV-1 is used in rodent studies.
The role of SGCs in herpetic infection and pain has been controversial1. Major questions are whether SGCs are actually infected and whether they contain the virus during latency. Another open question is whether abnormal SGC–neuronal interactions are present in herpes-infected ganglia, which could potentially contribute to post-herpetic pain.
To overcome the lack of a rodent model for VZV, Zerboni et al.120 grafted fetal human DRG under the kidney capsule of severe combined immunodeficiency mice. The grafted ganglia were infected with VZV, and examined histologically after various periods (up to 20 weeks). This innovative approach clearly showed that the virus was present in both DRG neurons and SGCs121, see Box 3.
Cutaneous HSV-1 infection in mice was reported to promote infiltration of leukocytes into the DRG, where they release cytokines that downregulate the expression of Kir4.1 channels in SGCs122. Although it was assumed that the inflammatory mediators were present in the skin, the authors did not evaluate whether release could be from DRG-resident cells (SGCs, for example). It should also be added that infection of DRG cells by the virus was not verified, and it could be argued that the changes in the DRG could be due to the skin lesion caused by the virus and subsequent local release of cytokines. It is notable that there is no conclusive demonstration of GFAP upregulation in SGCs in herpes, although there is evidence from western blots for GFAP elevation in the DRG after infection with HSV-1 (ref.122).
Few studies have examined the functional consequences of HSV infection on cells within sensory ganglia. In one such study, freshly dissociated TG neurons and SGCs were infected with HSV-1 and studied after 48 h in vitro67, at which time both the neurons and SGCs were infected. Calcium imaging, dye injections and intracellular electrical recordings from the neurons revealed several notable changes in these cultures. First, intercellular Ca2+ waves, which involved both neurons and SGCs, were more extensive in the infected cultures than in controls, which is consistent with greater spread of neural activity80. These waves were largely mediated by ATP acting on P2Rs in neurons and SGCs. Neuronal, but not glial, sensitivity to ATP was increased by the viral infection. Second, dye coupling among cells (neuron–neuron, neuron–SGC and SGC–SGC) was greatly increased. This coupling was not mediated by gap junctions but by cell fusion, which is known to occur following infection with alphaherpesviruses123,124, see Box 3. Apparently, the ATP-mediated Ca2+ wave propagation is facilitated by the direct connection of the cytoplasm of cells. Third, electrical recordings from the neurons showed a widening of the action potentials in neurons from infected cultures compared with controls, consistent with greater influx of Ca2+. This would raise intracellular Ca2+ levels, leading to increased release of mediators, including ATP. Thus, the enhanced Ca2+ waves are explained by greater sensitivity of neurons to ATP, by cell fusion and by greater Ca2+ influx into the neurons. The increased Ca2+ waves and neuronal hyperexcitability may lead to increased neuronal firing, and thus to pain. These results help in understanding the peripheral mechanisms of pain in herpes, but need to be validated in animal models where HSV-1 infection is made in vivo. A limitation of the in vitro work is that it does not provide information on long-term events and reactivation.

SGCs in other types of viral infections
Infections by viruses other than herpesvirus may also involve SGCs. Of special interest is HIV-1, the virus that causes AIDS, in which peripheral neuropathy and pain are common. Infection with simian immunodeficiency virus is a useful model to study HIV. In macaques infected with simian immunodeficiency virus there was an upregulation of GFAP in DRG SGCs125, but its contribution to the disease is not known. Another virus family is coronavirus that includes MERS-CoV, SARS-CoV and SARS-CoV-2 (which causes COVID-19), which are highly infective and can be fatal in humans. Information on the involvement of SGCs in coronavirus infections is minimal, but there are some clues from a study on another member of this family — swine haemagglutinating encephalomyelitis virus, which is highly lethal in pigs. An electron-microscopic study of haemagglutinating encephalomyelitis virus-infected rats found that the virus replicated in DRG neurons126. Viral particles were secreted from neurons, and were taken up by SGCs and located in lysosome-like structures, indicating that SGCs can restrict the virus spread. This protective function is consistent with the ability of SGCs to phagocytose pathogens, as described in Box 1.
SGCs in sympathetic ganglia
Most of the available knowledge on SGCs in autonomic ganglia is on sympathetic ganglia, and therefore only these ganglia are discussed here. As in sensory ganglia, SGCs in sympathetic ganglia wrap around the neurons, but gaps in the envelope may occur127. A major difference between SGCs in sympathetic and sensory ganglia is the presence of synapses in sympathetic ganglia, where SGCs form a layer over the synapses, enabling them to control synaptic transmission127 (Fig. 1e). This arrangement is similar to the ‘synaptic cradle’ proposed for astrocyte–neuron arrangement in the CNS128. Acetylcholine (ACh) is the major neurotransmitter in sympathetic ganglia, and indeed it was found that SGCs in the mouse superior cervical ganglion (sympathetic) are sensitive to ACh129. This raises the possibility that SGCs influence synaptic transmission in sympathetic ganglia. SGCs in sensory ganglia are not sensitive to ACh129, which is consistent with the idea that there is a match between chemical messengers in neurons and the receptors in adjacent glia. A later work showed that selective injury to sympathetic nerve terminals activated SGCs in superior cervical ganglia, but not in sensory ones130. Conversely, LPS-induced inflammation, which activated SGCs in sensory ganglia, had no effect on SGCs in the superior cervical ganglia130. This selective activation correlates with the presence of TLR4 in sensory neurons, and their absence in sympathetic ones. The results above are supported by a recent study131, which showed that SGCs in sympathetic ganglia release factors that augment cholinergic synaptic transmission. SGCs also promote synapse formation and contribute to neuronal survival131. Thus, SGCs play crucial roles in both development and maintenance of sympathetic function.
There is evidence that the sympathetic nervous system is involved in pain mechanisms, but this topic is controversial. Most notable is the possible role of sympathetic nerves in complex regional pain syndrome132. Animal studies have shown that nerve damage augments interactions between sympathetic and sensory ganglia. For example, damage causes sympathetic fibres to sprout in the DRG133, and in two models of DRG nerve injury there was an inflammatory response in sympathetic ganglia (infiltration of immune cells) and SGC activation134. These observations suggest that interactions between sensory and sympathetic ganglia may underlie sympathetic contribution to pain.
Sympathetic ganglia play a major role in controlling cardiovascular functions, and in a study where SGCs in mouse superior cervical ganglion were stimulated selectively using DREADD, activation of SGCs increased the heart rate and contractility135. It appears that SGC stimulation activated sympathetic fibres that innervate cardiac muscle. This is the first demonstration that SGCs can activate sympathetic nerves, and raises the possibility that sympathetic overactivity, which is a contributing factor in heart failure136, is related to abnormal SGC function. These results present a novel approach to better understanding of heart disease and may offer completely novel strategies for therapy.
Future perspectives
We expect that the increasing interest in SGCs and the application of newly developed research tools will enable progress in novel areas of this field. Future directions in SGC research that we believe may be particularly fruitful are the following. First, injury to sensory neurons causes the attraction of macrophages into the ganglia, and they even penetrate into the space between SGCs and neurons137. Macrophages release various pro-inflammatory substances, which can increase neuronal firing138,139. Studying macrophage–SGC interactions seems a promising direction. It was found that SGCs and macrophages promote regeneration of DRG axons140, and this topic needs to be probed in depth. Second, methods are now available to isolate SGCs, which allows the analysis of their transcriptomes under normal and pathological conditions141. For example, single-cell RNA sequencing will enable a search for SGC subtypes and a comparison between SGCs from different types of ganglia, topics on which we currently know nothing. Third, most available information on SGCs has been obtained in rodents, which limits its applicability to clinical situations. Human sensory ganglia are becoming available for laboratory research142, which opens exciting possibilities for validating in human material the concepts that have been developed in rodent studies. Using ganglia from patients will help in learning more about the role of SGCs in human disorders, such as migraine and post-herpetic pain. Extending the study on SGCs in viral diseases should receive high priority, as these cells may be a novel therapeutic target. Fourth, use of genetically encoded activity indicators in intact animals potentially allows the observation of interactions between neurons and SGCs in sensory ganglia44,75,76 and could provide novel information regarding such interactions in autonomic ganglia as well. These techniques are certain to provide insight into pathological activity patterns and to enable testing of pharmacological strategies to reverse the pathological changes. Finally, current knowledge of SGCs in autonomic ganglia is very limited, but in view of the recent work, which revealed novel and important roles of these cells in sympathetic ganglia, we expect that this area will attract growing attention.
Concluding remarks
We describe here possible roles of SGCs in four important pain syndromes. These syndromes have different clinical manifestations, and each may be mediated by different biochemical and physiological mechanisms. Still, the observations that SGCs in sensory ganglia are altered in a similar way in these models (as well as in other models that were not mentioned) suggest that changes in these cells are a general feature of chronic pain. The location of sensory ganglia outside the blood–brain barrier, and the arrangement of SGCs around the neurons, make SGCs an ideal therapeutic target for chronic pain. Similarly, autonomic ganglia are more accessible to intravascular agents than is the brain parenchyma74, and SGC modulation of sympathetic output might be targeted for disturbances of heart rhythm, blood pressure and other disorders.
Change history
22 October 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Hanani, M. Satellite glial cells in sensory ganglia: from form to function. Brain Res. Brain Res. Rev. 48, 457–476 (2005).
Huang, L. Y., Gu, Y. & Chen, Y. Communication between neuronal somata and satellite glial cells in sensory ganglia. Glia 61, 1571–1581 (2013).
Jasmin, L., Vit, J. P., Bhargava, A. & Ohara, P. T. Can satellite glial cells be therapeutic targets for pain control? Neuron Glia Biol. 6, 63–71 (2010).
Pannese, E. Biology and pathology of perineuronal satellite cells in sensory ganglia. Adv. Anat. Embryol. Cell Biol. 226, 1–63 (2018).
Rozanski, G. M., Li, Q. & Stanley, E. F. Transglial transmission at the dorsal root ganglion sandwich synapse: glial cell to postsynaptic neuron communication. Eur. J. Neurosci. 237, 1221–1228 (2013).
Bushong, E. A., Martone, M. E., Jones, Y. Z. & Ellisman, M. H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. Neuroscience 22, 183–192 (2002).
Ji, R. R., Donnelly, C. R. & Nedergaard, M. Astrocytes in chronic pain and itch. Nat. Rev. Neurosci. 20, 667–685 (2019).
Liddelow, S. A. & Barres, B. A. Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967 (2017).
Verkhratsky, A. & Nedergaard, M. Physiology of astroglia. Physiol. Rev. 98, 239–389 (2018).
Burnstock, G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 87, 659–797 (2007).
Fields, R. D. & Burnstock, G. Purinergic signalling in neuron–glia interactions. Nat. Rev. Neurosci. 7, 423–436 (2006).
Kettenmann, H. & Zorec, R. in Neuroglia 4th edn. (eds Kettenmann, H. & Ransom, B. R.) 197–211 (Oxford Univ. Press, 2013).
Watkins, L. R. & Maier, S. F. GLIA: a novel drug discovery target for clinical pain. Nat. Rev. Drug. Discov. 2, 973–985 (2003).
Grace, P. M., Hutchinson, M. R., Maier, S. F. & Watkins, L. R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 14, 217–231 (2014).
Inoue, K. & Tsuda, M. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 19, 138–152 (2018).
Chen, G., Zhang, Y. Q., Qadri, Y. J., Serhan, C. N. & Ji, R. R. Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron 100, 1292–1311 (2018).
Scadding, J. W. & Koltzenburg, M. in Wall and Melzack’s Textbook of Pain 6th edn (eds McMahon, S. B., Koltzenburg, M. I., Tracey, I. D. & Turk, D.) 926–951 (Elsevier Churchill Livingstone, 2013).
Devor, M. in Wall and Melzack’s Textbook of Pain 6th edn (eds McMahon, S. B., Koltzenburg, M. I., Tracey, I. D. & Turk, D.) 867–888 (Elsevier Churchill Livingstone, 2013).
Huang, T. Y., Belzer, V. & Hanani, M. Gap junctions in dorsal root ganglia: possible contribution to visceral pain. Eur. J. Pain. 14, 49.e1–49.e11 (2010).
Guha, D. & Shamji, M. F. The dorsal root ganglion in the pathogenesis of chronic neuropathic pain. Neurosurgery 63, 118–126 (2016).
Vaso, A. et al. Peripheral nervous system origin of phantom limb pain. Pain 155, 1384–1391 (2014).
Stephenson, J. L. & Byers, M. R. GFAP immunoreactivity in trigeminal ganglion satellite cells after tooth injury in rats. Exp. Neurol. 131, 11–22 (1995).
Warwick, R. A. & Hanani, M. The contribution of satellite glial cells to chemotherapy-induced neuropathic pain. Eur. J. Pain. 17, 571–580 (2013).
Woodham, P., Anderson, P. N., Nadim, W. & Turmaine, M. Satellite cells surrounding axotomized rat dorsal root ganglion cells increase expression of GFAP-like protein. Neurosci. Lett. 98, 8–12 (1989).
Hanani, M., Huang, T. Y., Cherkas, P. S., Ledda, M. & Pannese, E. Glial cell plasticity in sensory ganglia induced by nerve damage. Neuroscience 114, 279–283 (2002).
Ohara, P. T., Vit, J. P., Bhargava, A. & Jasmin, L. Evidence for a role of connexin 43 in trigeminal pain using RNA interference in vivo. J. Neurophysiol. 100, 3064–3073 (2008). This paper shows that Cx43 expression increases in SGCs in a pain model, and that blocking this expression reduces pain behaviour.
Blum, E., Procacci, P., Conte, V. & Hanani, M. Systemic inflammation alters satellite glial cell function and structure. A possible contribution to pain. Neuroscience 274, 209–217 (2014).
Vit, J. P., Ohara, P. T., Bhargava, A., Kelley, K. & Jasmin, L. Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in pain-like behavior in the absence of nerve injury. J. Neurosci. 28, 4161–4171 (2008).
Takeda, M., Takahashi, M., Nasu, M. & Matsumoto, S. Peripheral inflammation suppresses inward rectifying potassium currents of satellite glial cells in the trigeminal ganglia. Pain 152, 2147–2156 (2011).
Tang, X., Schmidt, T. M., Perez-Leighton, C. E. & Kofuji, P. Inwardly rectifying potassium channel Kir4.1 is responsible for the native inward potassium conductance of satellite glial cells in sensory ganglia. Neuroscience 166, 397–407 (2010). Together with Vit et al. (2008) and Takeda et al. (2011), this study reports that K + channel Kir4.1 expression increases in SGCs following nerve damage, which can lead to neuronal hyperexcitability.
Kushnir, R., Cherkas, P. S. & Hanani, M. Peripheral inflammation upregulates P2X receptor expression in satellite glial cells of mouse trigeminal ganglia: a calcium imaging study. Neuropharmacology 61, 739–746 (2011). This paper describes an increased sensitivity of SGCs to ATP in two pain models in mice.
Dubový, P., Klusáková, I., Svízenská, I. & Brázda, V. Satellite glial cells express IL-6 and corresponding signal-transducing receptors in the dorsal root ganglia of rat neuropathic pain model. Neuron Glia Biol. 6, 73–83 (2010).
Souza, G. R. et al. Fractalkine mediates inflammatory pain through activation of satellite glial cells. Proc. Natl Acad. Sci. USA 110, 11193–11198 (2013).
Afroz, S. et al. CGRP induces differential regulation of cytokines from satellite glial cells in trigeminal ganglia and orofacial nociception. Int. J. Mol. Sci. 20, 711 (2019).
Mitterreiter, J. G. et al. Satellite glial cells in human trigeminal ganglia have a broad expression of functional Toll-like receptors. Eur. J. Immunol. 47, 1181–1187 (2017).
Takeda, M., Takahashi, M. & Matsumoto, S. Contribution of the activation of satellite glia in sensory ganglia to pathological pain. Neurosci. Biobehav. Rev. 33, 784–792 (2009).
Cherkas, P. S. et al. The effects of axotomy on neurons and satellite glial cells in mouse trigeminal ganglion. Pain 110, 290–298 (2004).
Cui, Y. et al. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 554, 323–327 (2018).
Manteniotis, S. et al. Comprehensive RNA-Seq expression analysis of sensory ganglia with a focus on ion channels and GPCRs in trigeminal ganglia. PLoS ONE 8, e79523 (2013).
Procacci, P., Magnaghi, V. & Pannese, E. Perineuronal satellite cells in mouse spinal ganglia express the gap junction protein connexin 43 throughout life with decline in old age. Brain Res. Bull. 75, 562–569 (2008).
Lee-Kubli, C. A. et al. Analysis of the behavioral, cellular and molecular characteristics of pain in severe rodent spinal cord injury. Exp. Neurol. 278, 91–104 (2016).
Kaji, K. et al. Connexin 43 contributes to ectopic orofacial pain following inferior alveolar nerve injury. Mol. Pain. 12, 1–12 (2016).
Komiya, H. et al. Connexin 43 expression in satellite glial cells contributes to ectopic tooth-pulp pain. J. Oral. Sci. 60, 493–499 (2018).
Kim, Y. S. et al. Coupled activation of primary sensory neurons contributes to chronic pain. Neuron 91, 1085–1096 (2016). This first study of calcium imaging in sensory neurons in live mice describes the role of the SGC gap junctions in increasing neuronal interactions and pain.
Spray, D. C. & Hanani, M. Gap junctions, pannexins and pain. Neurosci. Lett. 695, 46–52 (2019).
Garrett, F. G. & Durham, P. L. Differential expression of connexins in trigeminal ganglion neurons and satellite glial cells in response to chronic or acute joint inflammation. Neuron Glia Biol. 4, 295–306 (2008).
Pannese, E., Ledda, M., Cherkas, P. S., Huang, T. Y. & Hanani, M. Satellite cell reactions to axon injury of sensory ganglion neurons: increase in number of gap junctions and formation of bridges connecting previously separate perineuronal sheaths. Anat. Embryol. 206, 337–347 (2003).
Huang, T. Y. & Hanani, M. Morphological and electrophysiological changes in mouse dorsal root ganglia after partial colonic obstruction. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G670–G678 (2005).
Song, D. D., Li, Y., Tang, D., Huang, L. Y. & Yuan, Y. Z. Neuron–glial communication mediated by TNF-α and glial activation in dorsal root ganglia in visceral inflammatory hypersensitivity. Am. J. Physiol. Gastrointest. Liver Physiol. 306, G788–G795 (2014).
Dublin, P. & Hanani, M. Satellite glial cells in sensory ganglia: their possible contribution to inflammatory pain. Brain Behav. Immun. 21, 592–598 (2007). This paper provides evidence that blocking the increased SGC coupling can reduce pain behaviour in mice.
Ledda, M., Blum, E., De Palo, S. & Hanani, M. Augmentation in gap junction-mediated cell coupling in dorsal root ganglia following sciatic nerve neuritis in the mouse. Neuroscience 164, 1538–1545 (2009).
Jin, Y. Z. et al. Connexin 43 contributes to temporomandibular joint inflammation induced-hypernociception via sodium channel 1.7 in trigeminal ganglion. Neurosci. Lett. 707, 134301 (2019).
Spray, D. C. et al. Gap junction mediated signaling between satellite glia and neurons in trigeminal ganglia. Glia 67, 791–801 (2019). This study uses dual whole-cell voltage clamp methodology to quantify gap junction-mediated coupling between SGCs and neurons in dissociated trigeminal ganglion cultures.
Durham, P. L. & Garrett, F. G. Neurological mechanisms of migraine: potential of the gap-junction modulator tonabersat in prevention of migraine. Cephalalgia 29, 1–6 (2009).
Hanstein, R. et al. Focal inflammation causes carbenoxolone-sensitive tactile hypersensitivity in mice. Open. Pain. J. 3, 123–133 (2010).
Lemes, J. B. P. et al. Participation of satellite glial cells of the dorsal root ganglia in acute nociception. Neurosci. Lett. 676, 8–12 (2018).
Sosinsky, G. et al. Pannexin channels are not gap junction hemichannels. Channels 5, 193–197 (2011).
Dahl, G., Qiu, F. & Wang, J. The bizarre pharmacology of the ATP release channel pannexin1. Neuropharmacology 75, 583–593 (2013).
Zhang, Y., Laumet, G., Chen, S. R., Hittelman, W. N. & Pan, H. L. Pannexin-1 up-regulation in the dorsal root ganglion contributes to neuropathic pain development. J. Biol. Chem. 290, 14647–14655 (2015).
Hanstein, R., Hanani, M., Scemes, E. & Spray, D. C. Glial pannexin1 contributes to tactile hypersensitivity in a mouse model of orofacial pain. Sci. Rep. 6, 38266 (2016).
Feldman-Goriachnik, R., Belzer, V. & Hanani, M. Systemic inflammation activates satellite glial cells in the mouse nodose ganglion and alters their functions. Glia 63, 2121–2132 (2015).
Magni, G., Riccio, D. & Ceruti, S. Tackling chronic pain and inflammation through the purinergic system. Curr. Med. Chem. 25, 3830–3865 (2018). This paper summarizes purinergic interactions between neurons and glia, with the emphasis on sensory ganglia.
Braun, N. et al. Association of the ecto-ATPase NTPDase2 with glial cells of the peripheral nervous system. Glia 45, 124–132 (2004).
Weick, M. et al. P2 receptors in satellite glial cells in trigeminal ganglia of mice. Neuroscience 120, 969–977 (2003).
Ceruti, S., Fumagalli, M., Villa, G., Verderio, C. & Abbracchio, M. P. Purinoceptor-mediated calcium signaling in primary neuron-glia trigeminal cultures. Cell Calcium 43, 576–590 (2008).
Suadicani, S. O. et al. Bidirectional calcium signaling between satellite glial cells and neurons in cultured mouse trigeminal ganglia. Neuron Glia Biol. 6, 43–51 (2010).
Warwick, R. A. & Hanani, M. Involvement of aberrant calcium signalling in herpetic neuralgia. Exp. Neurol. 277, 10–18 (2016).
Zhang, X., Chen, Y., Wang, C. & Huang, L. Y. Neuronal somatic ATP release triggers neuron–satellite glial cell communication in dorsal root ganglia. Proc. Natl Acad. Sci. USA 104, 9864–9869 (2007). This is the first report on the release of ATP from sensory neurons, which acts on P2X7 receptors in SGCs and induces them to release TNF, which in turn increases neuronal excitability.
Xu, G. Y., Shenoy, M., Winston, J. H., Mittal, S. & Pasricha, P. J. P2X receptor-mediated visceral hyperalgesia in a rat model of chronic visceral hypersensitivity. Gut 57, 1230–1237 (2008).
Chessell, I. P. et al. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain 114, 386–396 (2005).
Zhou, J., Chung, K. & Chung, J. M. Development of purinergic sensitivity in sensory neurons after peripheral nerve injury in the rat. Brain Res. 915, 161–169 (2001).
Eftekhari, S. et al. Differential distribution of calcitonin gene-related peptide and its receptor components in the human trigeminal ganglion. Neuroscience 169, 683–696 (2010).
Eftekhari, S. et al. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood–brain barrier. Brain Res. 1600, 93–109 (2015).
Noseda, R. et al. Fluorescently-labeled fremanezumab is distributed to sensory and autonomic ganglia and the dura but not to the brain of rats with uncompromised blood brain barrier. Cephalalgia 40, 229–240 (2020).
Chen, C. et al. Long-term imaging of dorsal root ganglia in awake behaving mice. Nat. Commun. 10, 3087 (2019).
Emery, E. C. et al. In vivo characterization of distinct modality-specific subsets of somatosensory neurons using GCaMP. Sci. Adv. 2, 11 (2016).
Devor, M. & Wall, P. D. Cross-excitation in dorsal root ganglia of nerve-injured and intact rats. J. Neurophysiol. 64, 1733–1746 (1990).
Amir, R. & Devor, M. Chemically mediated cross-excitation in rat dorsal root ganglia. J. Neurosci. 16, 4733–4741 (1996).
Oh, E. J. & Weinreich, D. Chemical communication between vagal afferent somata in nodose ganglia of the rat and the guinea pig in vitro. J. Neurophysiol. 87, 2801–2807 (2002). Together with Amir and Devor (1996), this study presents electrophysiological evidence that cross depolarization is chemically mediated.
Hanani, M. Intercellular communication in sensory ganglia by purinergic receptors and gap junctions: implications for chronic pain. Brain Res. 1487, 183–191 (2012).
Carvalho, G. B., Mulpuri, Y., Damasio, A. & Spigelman, I. A role for the P2Y1 receptor in nonsynaptic cross-depolarization in the rat dorsal root ganglia. Neuroscience 423, 98–108 (2019).
Xie, W., Strong, J. A. & Zhang, J. M. Early blockade of injured primary sensory afferents reduces glial cell activation in two rat neuropathic pain models. Neuroscience 160, 847–857 (2009).
Belzer, V. & Hanani, M. Nitric oxide as a messenger between neurons and satellite glial cells in dorsal root ganglia. Glia 67, 1296–1307 (2019).
Devor, M., Amir, R. & Rappaport, Z. H. Pathophysiology of trigeminal neuralgia: the ignition hypothesis. Clin. J. Pain. 18, 4–13 (2002).
Barrot, M. Tests and models of nociception and pain in rodents. Neuroscience 211, 39–50 (2012).
Blum, E., Procacci, P., Conte, V., Sartori, P. & Hanani, M. Long term effects of lipopolysaccharide on satellite glial cells in mouse dorsal root ganglia. Exp. Cell Res. 350, 236–241 (2017).
Peters, C. M. et al. Intravenous paclitaxel administration in the rat induces a peripheral sensory neuropathy characterized by macrophage infiltration and injury to sensory neurons and their supporting cells. Exp. Neurol. 203, 42–54 (2007).
Wang, S. et al. P2Y12 shRNA treatment decreases SGC activation to relieve diabetic neuropathic pain in type 2 diabetes mellitus rats. J. Cell Physiol. 233, 9620–9628 (2018).
Hanani, M., Blum, E., Liu, S., Peng, L. & Liang, S. Satellite glial cells in dorsal root ganglia are activated in streptozotocin-treated rodents. J. Cell Mol. Med. 18, 2367–2371 (2014).
Dantzer, R., O’Connor, J. C., Freund, G. G., Johnson, R. W. & Kelley, K. W. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56 (2008).
Leshchenko, Y. et al. Carbenoxolone does not cross the blood brain barrier: an HPLC study. BMC Neurosci. 7, 3 (2006).
Haroutiunian, S., Nikolajsen, L., Finnerup, N. B. & Jensen, T. S. The neuropathic component in persistent postsurgical pain: a systematic literature review. Pain 154, 95–102 (2013).
Steyaert, A. & De Kock, M. Chronic postsurgical pain. Curr. Opin. Anaesthesiol. 25, 584–588 (2012).
Wylde, V. et al. Systematic review of management of chronic pain after surgery. Brit. J. Surg. 104, 1293–1306 (2017).
Flatters, S. J. Characterization of a model of persistent postoperative pain evoked by skin/muscle incision and retraction (SMIR). Pain 135, 119–130 (2008).
Woolf, C. J. Central sensitization: implications for the diagnosis and treatment of pain. Pain 152, S2–S15 (2011).
Romero, A., Romero-Alejo, E., Vasconcelos, N. & Puig, M. M. Glial cell activation in the spinal cord and dorsal root ganglia induced by surgery in mice. Eur. J. Pharmacol. 702, 126–134 (2013).
Song, J. et al. The role of P2X7R/ERK signaling in dorsal root ganglia satellite glial cells in the development of chronic postsurgical pain induced by skin/muscle incision and retraction (SMIR). Brain Behav. Immun. 69, 180–189 (2018).
Yamakita, S. et al. Synergistic activation of ERK1/2 between A-fiber neurons and glial cells in the DRG contributes to pain hypersensitivity after tissue injury. Mol. Pain. 14, 1744806918767508 (2018).
Pogatzki, E. M., Vandermeulen, E. P. & Brennan, T. J. Effect of plantar local anesthetic injection on dorsal horn neuron activity and pain behaviors caused by incision. Pain 97, 151–156 (2002).
Yatziv, S. L. & Devor, M. Suppression of neuropathic pain by selective silencing of dorsal root ganglion ectopia using nonblocking concentrations of lidocaine. Pain 160, 2105–2114 (2019).
Feldman, E. L., Nave, K. A., Jensen, T. S. & Bennett, D. L. H. New horizons in diabetic neuropathy: mechanisms, bioenergetics, and pain. Neuron 93, 1296–1313 (2017).
Tesfaye, S. & Selvarajah, D. Advances in the epidemiology, pathogenesis and management of diabetic peripheral neuropathy. Diabetes Metab. Res. Rev. 28, 8–14 (2012).
Gonçalves, N. P., Vægter, C. B. & Pallesen, L. T. Peripheral glial cells in the development of diabetic neuropathy. Front. Neurol. 9, 268 (2018).
Rahman, M. H. et al. Pyruvate dehydrogenase kinase-mediated glycolytic metabolic shift in the dorsal root ganglion drives painful diabetic neuropathy. J. Biol. Chem. 291, 6011–6025 (2016).
Verkhratsky, A. & Fernyhough, P. Calcium signalling in sensory neurones and peripheral glia in the context of diabetic neuropathies. Cell Calcium 56, 362–371 (2014).
Hong, S., Morrow, T. J., Paulson, P. E., Isom, L. L. & Wiley, J. W. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J. Biol. Chem. 279, 29341–29350 (2004).
Zochodne, D. W. Diabetic polyneuropathy: an update. Curr. Opin. Neurol. 21, 527–533 (2008).
Brownlee, M. & Cerami, A. The biochemistry of the complications of diabetes mellitus. Annu. Rev. Biochem. 50, 385–432 (1981).
Schemmel, K. E., Padiyara, R. S. & D’Souza, J. J. Aldose reductase inhibitors in the treatment of diabetic peripheral neuropathy: a review. J. Diabetes Complicat. 24, 354–360 (2010).
Jiang, Y., Calcutt, N. A., Ramos, K. M. & Mizisin, A. P. Novel sites of aldose reductase immunolocalization in normal and streptozotocin-diabetic rats. J. Peripher. Nerv. Syst. 11, 274–285 (2006).
Huang, Q., Liu, Q. & Ouyang, D. Sorbinil, an aldose reductase inhibitor, in fighting against diabetic complications. Med. Chem. 15, 3–7 (2019).
Liu, S. et al. lncRNA NONRATT021972 siRNA regulates neuropathic pain behaviors in type 2 diabetic rats through the P2X7 receptor in dorsal root ganglia. Mol. Brain 9, 44 (2016).
Coddou, C., Yan, Z., Obsil, T., Huidobro-Toro, J. P. & Stojilkovic, S. S. Activation and regulation of purinergic P2X receptor channels. Pharmacol. Rev. 63, 641–683 (2011).
Teixeira, J. M. et al. Diabetes-induced neuropathic mechanical hyperalgesia depends on P2X4 receptor activation in dorsal root ganglia. Neuroscience 398, 158–170 (2019).
Johnson, R. W. & Rice, A. S. Clinical practice. Postherpetic neuralgia. N. Engl. J. Med. 371, 1526–1533 (2014).
Steiner, I. & Benninger, F. Manifestations of herpes virus infections in the nervous system. Neurol. Clin. 36, 725–738 (2018).
Fatahzadeh, M. & Schwartz, R. A. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J. Am. Acad. Dermatol. 57, 737–763 (2007).
Feller, L., Khammissa, R. A. G., Fourie, J., Bouckaert, M. & Lemmer, J. Postherpetic neuralgia and trigeminal neuralgia. Pain. Res. Treat. 2017, 1681765 (2017).
Zerboni, L., Ku, C. C., Jones, C. D., Zehnder, J. L. & Arvin, A. M. Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc. Natl Acad. Sci. USA 102, 6490–6495 (2005).
Zerboni, L. & Arvin, A. Neuronal subtype and satellite cell tropism are determinants of varicella-zoster virus virulence in human dorsal root ganglia xenografts in vivo. PLoS Pathog. 11, e1004989 (2015). This paper shows that both neurons and SGCs are infected by varicella zoster herpesvirus in human DRG grafted into mice.
Silva, J. R. et al. Neuroimmune–glia interactions in the sensory ganglia account for the development of acute herpetic neuralgia. J. Neurosci. 237, 6408–6422 (2017).
Zerboni, L., Sen, N., Oliver, S. L. & Arvin, A. M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 12, 197–210 (2014).
Weed, D. J. & Nicola, A. V. Herpes simplex virus membrane fusion. Adv. Anat. Embryol. Cell Biol. 223, 29–47 (2017).
Mangus, L. M. et al. SIV-induced immune activation and metabolic alterations in the dorsal root ganglia during acute infection. J. Neuropathol. Exp. Neurol. 78, 78–87 (2019).
Li, Y. C., Bai, W. Z., Hirano, N., Hayashida, T. & Hashikawa, T. Coronavirus infection of rat dorsal root ganglia: ultrastructural characterization of viral replication, transfer, and the early response of satellite cells. Virus Res. 163, 628–635 (2012).
Hanani, M. Satellite glial cells in sympathetic and parasympathetic ganglia: in search of function. Brain Res. Brain Res. Rev. 64, 304–327 (2010).
Verkhratsky, A. & Nedergaard, M. Astroglial cradle in the life of the synapse. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 369, 20130595 (2014).
Feldman-Goriachnik, R., Wu, B. & Hanani, M. Cholinergic responses of satellite glial cells in the superior cervical ganglia. Neurosci. Lett. 671, 19–24 (2018).
Feldman-Goriachnik, R. & Hanani, M. The effects of sympathetic nerve damage on satellite glial cells in the mouse superior cervical ganglion. Auton. Neurosci. 221, 102584 (2019).
Enes, J. et al. Satellite glial cells modulate cholinergic transmission between sympathetic neurons. PLoS ONE 15, e0218643 (2020). This paper shows that SGCs in sympathetic ganglia modulate neuron-to-neuron cholinergic neurotransmission, promote synapse formation and contribute to neuronal survival.
Shim, H., Rose, J., Halle, S. & Shekane, P. Complex regional pain syndrome: a narrative review for the practising clinician. Br. J. Anaesth. 123, e424–e433 (2109).
McLachlan, E. M., Jänig, W., Devor, M. & Michaelis, M. Peripheral nerve injury triggers noradrenergic sprouting within dorsal root ganglia. Nature 363, 543–546 (1993).
Li, A. L., Zhang, J. D., Xie, W., Strong, J. A. & Zhang, J. M. Inflammatory changes in paravertebral sympathetic ganglia in two rat pain models. Neurosci. Bull. 34, 85–97 (2018).
Xie, A. X., Lee, J. J. & McCarthy, K. D. Ganglionic GFAP+ glial Gq-GPCR signaling enhances heart functions in vivo. J. C. I. Insight 2, e90565 (2017). This study shows that stimulation of SGCs in sympathetic ganglia influences cardiac functions via actions of SGCs on sympathetic neurons.
Fukuda, K., Kanazawa, H., Aizawa, Y., Ardell, J. L. & Shivkumar, K. Cardiac innervation and sudden cardiac death. Circ. Res. 116, 2005–2019 (2015).
Hu, P. & McLachlan, E. M. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience 112, 23–38 (2002).
Ji, R. R., Chamessian, A. & Zhang, Y. Q. Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577 (2016).
Shinoda, M., Kubo, A., Hayashi, Y. & Iwata, K. Peripheral and central mechanisms of persistent orofacial pain. Front. Neurosci. 13, 1227 (2019).
Carlin, D., Halevi, A. E., Ewan, E. E., Moore, A. M. & Cavalli, V. Nociceptor deletion of Tsc2 enhances axon regeneration by inducing a conditioning injury response in dorsal root ganglia. eNeuro 6, ENEURO.0168-19.2019 (2019).
Jager, S. E. et al. Changes in the transcriptional fingerprint of satellite glial cells following peripheral nerve injury. Glia 68, 1375–1395 (2020).
Haberberger, R. V., Barry, C., Dominguez, N. & Matusica, D. Human dorsal root ganglia. Front. Cell. Neurosci. 13, 271 (2019).
Tongtako, W. Canine dorsal root ganglia satellite glial cells represent an exceptional cell population with astrocytic and oligodendrocytic properties. Sci. Rep. 7, 13915 (2017).
Takahashi, M. & Osumi, N. Identification of a novel type II classical cadherin: rat cadherin19 is expressed in the cranial ganglia and Schwann cell precursors during development. Dev. Dyn. 232, 200–208 (2005).
George, D., Ahrens, P. & Lambert, S. Satellite glial cells represent a population of developmentally arrested Schwann cells. Glia 66, 1496–1506 (2018).
Koike, T., Wakabayashi, T., Mori, T., Hirahara, Y. & Yamada, H. Sox2 promotes survival of satellite glial cells in vitro. Biochem. Biophys. Res. Commun. 464, 269–274 (2015).
Arora, D. K. et al. Evidence of postnatal neurogenesis in dorsal root ganglion: role of nitric oxide and neuronal restrictive silencer transcription factor. J. Molec. Neurosci. 32, 97–107 (2007).
Li, H. Y., Say, E. H. & Zhou, X. F. Isolation and characterization of neural crest progenitors from adult dorsal root ganglia. Stem Cell 25, 2053–2065 (2007).
Fex Svenningsen, A., Colman, D. R. & Pedraza, L. Satellite cells of dorsal root ganglia are multipotential glial precursors. Neuron Glia Biol. 1, 85–93 (2004).
Belzer, V., Shraer, N. & Hanani, M. Phenotypic changes in satellite glial cells in cultured trigeminal ganglia. Neuron Glia Biol. 6, 237–243 (2010).
Weider, M. et al. Elevated in vivo levels of a single transcription factor directly convert satellite glia into oligodendrocyte-like cells. PLoS Genet. 11, e1005008 (2015).
van Velzen, M. et al. Neuron-interacting satellite glial cells in human trigeminal ganglia have an APC phenotype. J. Immunol. 183, 2456–2461 (2009). This paper shows that SGCs in human trigeminal ganglia display some properties of immune cells and have a unique leukocyte phenotype.
Wu, H. H. et al. Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat. Neurosci. 12, 1534–1541 (2009).
Nadeau, J. R., Wilson-Gerwing, T. D. & Verge, V. M. Induction of a reactive state in perineuronal satellite glial cells akin to that produced by nerve injury is linked to the level of p75NTR expression in adult sensory neurons. Glia 62, 763–777 (2014).
Shinder, V. et al. Structural basis of sympathetic-sensory coupling in rat and human dorsal root ganglia following peripheral nerve injury. J. Neurocytol. 28, 743–761 (1999).
Yoshioka, M. et al. Expression of HIV-1 and interleukin-6 in lumbosacral dorsal root ganglia of patients with AIDS. Neurology 44, 1120–1130 (1994).
Koeppen, A. H., Becker, A. B., Qian, J. & Feustel, P. J. Friedreich ataxia: hypoplasia of spinal cord and dorsal root ganglia. J. Neuropathol. Exp. Neurol. 76, 101–108 (2017).
Acknowledgements
The authors were supported by the Israel Science Foundation (ISF 508/13 and ISF 1297/18 to M.H.), US–Israel Binational Science Foundation (BSF-2011044 to M.H. and D.C.S.) and NIH (R01NS092786, R01NS092466 and R21NS116892 to D.C.S.).
Author information
Authors and Affiliations
Contributions
Both authors wrote the article and reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Neuroscience thanks the other anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Satellite glial cells
-
(SGCs). Glial cells that surround neurons in sensory, sympathetic and parasympathetic ganglia (they should not be confused with satellite cells, which are the progenitor cells in striated muscles).
- Sympathetic ganglia
-
Clusters of neuron cell bodies that innervate smooth muscles, heart and glands; paravertebral ganglia are arranged along the spinal column, and prevertebral ones are located in the abdomen.
- Dorsal root ganglia
-
(DRG). Clusters of cells located near the spinal cord containing the cell bodies of peripheral neurons that innervate most body parts, including internal organs.
- Sensory ganglia
-
Clusters of neuron cell bodies that have a single axon that bifurcates to two branches; one branch runs to the periphery and can detect various stimuli, and the other projects into the central nervous system.
- P2 purinergic receptors
-
(P2Rs). Receptors for the neurotransmitter adenosine (P1) and ATP (P2). There are seven ionotropic receptors (P2X1–P2X7) and eight G protein-coupled receptors (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11–P2Y14).
- Trigeminal ganglia
-
(TG). Clusters of cells located at the base of the skull (but outside the brain) that contain the cell bodies of neurons that innervate the face, teeth and scalp.
- Nodose ganglia
-
Clusters of neuron cell bodies that innervate many visceral organs, such as the intestine and heart.
- Allodynia
-
Pain resulting from a non-noxious stimulus to normal skin.
- Kir4.1 channels
-
Inward rectifier channels that tend to favour the influx of potassium ions into cells over their efflux.
- Gap junctions
-
Intercellular channels that provide a pathway for diffusion of ions and small molecules between cells; they are made of connexin (Cx) proteins.
- Dye coupling
-
A method for studying gap junction-mediated coupling between cells, based on injecting a cell with a dye that passes these junctions and examining whether the dye passed to nearby cells.
- Neuralgia
-
Pain extending along the course of nerves; for example, trigeminal neuralgia.
- Lipopolysaccharide
-
(LPS). A component of the wall of Gram-negative bacteria; LPS acts on Toll-like receptor 4 (TLR4), which, in sensory ganglia, is located on the surface of the sensory neurons.
- Central sensitization
-
A state when the central nervous system becomes highly reactive, causing even mild stimuli to be sensed as painful.
- Extracellular-signal regulated kinase
-
(ERK). A member of the MAP kinase family that is involved in multiple cellular processes.
- DREADD
-
(Designer receptors exclusively activated by designer drugs). A method that utilizes G protein-coupled receptors engineered to respond exclusively to synthetic ligands.
Rights and permissions
About this article

Cite this article
Hanani, M., Spray, D.C. Emerging importance of satellite glia in nervous system function and dysfunction. Nat Rev Neurosci 21, 485–498 (2020). https://doi.org/10.1038/s41583-020-0333-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41583-020-0333-z
This article is cited by
-
Glial cell alterations in diabetes-induced neurodegeneration
Cellular and Molecular Life Sciences (2024)
-
Targeting Pannexin-1 Channels: Addressing the ‘Gap’ in Chronic Pain
CNS Drugs (2024)
-
Whole genome sequencing across clinical trials identifies rare coding variants in GPR68 associated with chemotherapy-induced peripheral neuropathy
Genome Medicine (2023)
-
Current aspects of small extracellular vesicles in pain process and relief
Biomaterials Research (2023)
-
P2X7 receptors and pannexin1 hemichannels shape presynaptic transmission
Purinergic Signalling (2023)
