
Abstract
Increasing concerns about the rising rates of antibiotic therapy failure and advances in single-cell analyses have inspired a surge of research into antibiotic persistence. Bacterial persister cells represent a subpopulation of cells that can survive intensive antibiotic treatment without being resistant. Several approaches have emerged to define and measure persistence, and it is now time to agree on the basic definition of persistence and its relation to the other mechanisms by which bacteria survive exposure to bactericidal antibiotic treatments, such as antibiotic resistance, heteroresistance or tolerance. In this Consensus Statement, we provide definitions of persistence phenomena, distinguish between triggered and spontaneous persistence and provide a guide to measuring persistence. Antibiotic persistence is not only an interesting example of non-genetic single-cell heterogeneity, it may also have a role in the failure of antibiotic treatments. Therefore, it is our hope that the guidelines outlined in this article will pave the way for better characterization of antibiotic persistence and for understanding its relevance to clinical outcomes.
Similar content being viewed by others

Introduction
More than 70 years ago, Hobby1 and Bigger2 observed that antibiotics that are considered bactericidal and kill bacteria in fact fail to sterilize cultures. Bigger realized that the small number of bacteria that manage to survive intensive antibiotic treatments are a distinct subpopulation of bacteria that he named ‘persisters’.
Fuelled in part by increasing concerns about antibiotic resistance but also by technological advances in single-cell analyses, the past 15 years have witnessed a great deal of research on antibiotic persistence by investigators with different backgrounds and perspectives. As the number of scientists that tackle the puzzles and challenges of antibiotic persistence from many different angles has profoundly increased, it is now time to agree on the basic definition of persistence and its distinction from the other mechanisms by which bacteria survive exposure to bactericidal antibiotic treatments3. Several approaches have independently emerged to define and measure persistence. Research groups following seemingly similar procedures may reach different results, and careful examination of the experimental procedures often reveals that results of different groups cannot be compared. During the European Molecular Biology Organization (EMBO) Workshop ‘Bacterial Persistence and Antimicrobial Therapy’ (10–14 June 2018) in Ascona, Switzerland, which brought together 121 investigators involved in antibiotic persistence research from 21 countries, a discussion panel laid the main themes for a Consensus Statement on the definition and detection procedure of antibiotic persistence detailed below. In light of the potential role that antibiotic persistence can have in antibiotic treatment regimens, it is our hope that clarification and standardization of experimental procedures will facilitate the translation of basic science research into practical guidelines.
Defining the persistence phenomena
We adopt here a phenomenological definition of antibiotic persistence that is based on a small set of observations that can be made from experiments performed in vitro and that does not assume a specific mechanism. We focus on the differences and similarities between antibiotic persistence and other processes enabling bacteria to survive exposure to antibiotic treatments that could kill them, such as resistance, tolerance and heteroresistance. We identify different types of persistence that should be measured differently to obtain meaningful results; therefore, the definition of these types goes beyond semantics. For the more mathematically oriented readers, we provide a mathematical definition of the various terms based on a widely used phenomenological model for survival under drug exposure in Box 1.
Persistent infection versus antibiotic persistence
First, we would like to distinguish ‘antibiotic persistence’ from ‘persistent infection’4,5 (Fig. 1). The latter is generally used to describe infections in the host that are not cleared by the host immune system, whereas antibiotic persistence describes a bacterial population that is refractory to antibiotic treatments, whether in vitro or possibly in the host. Persistent infections are typically multifactorial and involve mechanisms evolved by different pathogens to evade the immune system, such as antigenic mimicry in Helicobacter pylori, antigenic variation in Neisseria gonorrhoeae and inhibition of phagocytosis4 and immune evasion in Mycobacterium tuberculosis6. As antibiotic persistence specifically addresses the ability of bacteria to survive antibiotic treatments, it may be an additional factor for the prolongation of persistent infections despite antibiotic treatment, for example, in recurrent urinary tract infections7,8. Moreover, the same mechanisms may be involved in both immune evasion and antibiotic persistence, for example, biofilm formation9.

Persistent infection is a general term to describe infections that are not efficiently cleared by the host, in contrast to the characteristic acute response that leads to clearance of many pathogens. Antibiotic persistence specifically describes the heterogeneous response of bacterial populations, in vitro or in the host, that results in a delayed clearance of the bacterial load by antibiotics.
Historically, the term ‘persistent infection’ was used before antibiotics were available to treat infections. To avoid ambiguity, we suggest using the term antibiotic persistence to distinguish between these phenomena when first mentioned in a publication. The focus in this article is on antibiotic persistence, although for simplicity, tradition and brevity, below, we use the word persistence.
Persistence versus resistance
‘Resistance’ is the ability of bacteria to replicate and not just survive in the presence of a drug (Box 2). The most common measure of the level of resistance is the minimum inhibitory concentration (MIC), which is the lowest concentration of the antibiotic required to prevent the replication of the bacteria. A higher MIC corresponds with a higher level of resistance (Fig. 2a). Resistance is inherited and may be acquired by horizontal gene transfer of resistance-encoding genes (for example, encoding antibiotic inactivating enzymes10 or efflux pumps11) or mutations (for example, leading to modification of the antibiotic target) that confer the resistance phenotype to the bacterial population12.

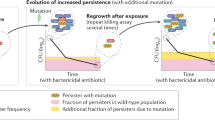
Resistance, tolerance and persistence are distinct responses to antibiotic treatment that lead to increased survival compared with susceptible cells. a | To inhibit the growth of resistant bacteria, a substantially higher minimum inhibitory concentration (MIC) of the antibiotic is needed than for susceptible bacteria. Notably, persistence and tolerance do not lead to an increase in the MIC compared with susceptible bacteria. b | By contrast, tolerance increases the minimum duration for killing (MDK; for example, for 99% of bacterial cells in the population (MDK99)) compared with susceptible bacteria. c | Persistence leads to a similar MIC and a similar initial killing of the bacterial population compared with susceptible bacteria; however, the MDK for 99.99% of bacterial cells in the population (MDK99.99) can be substantially higher owing to the survival of the persister cells. Note that pure exponential killing of the susceptible strain is rarely observed because most bacterial cultures have some level of persistence. The data shown are only illustrations and not actual measurements. Parts b and c are adapted with permission from ref.3, Springer Nature Limited (this material is excluded from the CC-BY-4.0 license).
‘Persistence’ is the ability of a subset of the population to survive exposure to a bactericidal drug concentration (Fig. 2). Therefore, persistence is defined only for bactericidal antibiotics. Several features distinguish persistence from resistance. First, the hallmark of antibiotic persistence is the biphasic killing curve (Fig. 2c); that is, the observation that not all bacteria in a clonal culture are killed at the same rate. Second, when persister cells regrow without antibiotics (see below), their progeny give rise to a population that is as susceptible to drugs as the parental population it was isolated from. Third, the level of persistence, namely, the size of the persister subpopulation, will only weakly depend on the concentration of the drug as long as it is far above the MIC. In addition, the survival advantage of persister bacteria is often observed for antibiotic treatments belonging to different classes of antibiotics, for example, β-lactams and fluoroquinolones13. Fourth, in contrast to resistant cells, persister bacteria cannot replicate in the presence of the drug any better than the non-persister cells but are killed at a lower rate than the susceptible population from which they arose. This property also distinguishes persistence from heteroresistance, a phenomenon in which a small subpopulation transiently displays a substantially (more than eightfold) higher MIC14 (see Box 1).
Persistence and tolerance
‘Tolerance’ and persistence are similar phenomena of increased survival in the presence of an antibiotic without an increase in the MIC. In studies that focus on only a qualitative understanding of the molecular mechanisms, the two terms are often interchangeable15. However, persistence has the added attribute of affecting only a subpopulation of cells, whereas tolerance is the general ability of a population to survive longer treatments, for example, by having a lower killing rate (see Fig. 2b), but without a change in the MIC16. Persister cells are simply a subpopulation of tolerant bacteria, and persistence could also be called ‘heterotolerance’. Tolerant populations survive the period of antibiotic treatment better, with, typically, a weak dependence on the antibiotic concentration. Therefore, the MIC of tolerant cells is unchanged compared with non-tolerant strains. What characterizes their slower killing, even at high concentrations of the drug, is the time required to kill a large fraction of the population, for example, the MDK99, which is the minimum duration of treatment that kills 99% of the bacterial population. Persistence is a special case of tolerance in which a subpopulation of persister cells can survive the antibiotic treatment much better than the majority of the population, as reflected in the biphasic killing curve. Not surprisingly, mechanisms linked to tolerance, such as dormancy (see definitions in Box 3), reduced metabolism and ATP levels, have also been identified in persistence9. Therefore, when studying persistence, two mechanisms are of interest, and the first one overlaps with tolerance research whereas the second is specific to persistence: (1) the molecular mechanism of tolerance that enables the persister bacteria to survive, for example, a reduction in their metabolism, and (2) the mechanism that generates heterogeneity in the population17, for example, nonlinear mechanisms leading to bimodality by amplifying stochasticity18,19. Finally, several persister subpopulations may coexist; therefore, a multimodal killing curve may occur.
Single-cell versus population phenotype
Because the definition of antibiotic persistence is anchored in the heterogeneity of the response to antibiotics in the population, it is a population-level phenotype. However, tolerance can be the attribute of a whole population that is killed at a slow rate as well as of a single cell that manages to survive an extensive treatment (see definitions in Box 3).
Genetic mutations can increase the tolerance of a strain if they result in slower killing. Similarly, genetic mutations can increase the persistence of a strain either by reducing the killing rate of the persistent subpopulation even more or by increasing the fraction of that subpopulation, as, for example, in the hipA7 high persistence mutant20. The population level of high persistence is then genetically inherited.
Types of persister bacteria
Whether a single general or multiple specific molecular mechanisms underlie persistence is still under debate21,22,23 and therefore will not be discussed in this article. However, distinct ways for generating persister bacteria in a culture have been identified. Distinguishing between the types of persistence identified thus far is crucial because each type requires a different procedure to measure the persistence level.
Triggered persistence
In most observations of persistence described to date, the fraction of persister bacteria is generated upon a stress signal, the most common one being starvation (Fig. 3). This type of persistence, here termed triggered persistence, was previously called type I persistence24. Even when the signal is removed, for example, by diluting a starved overnight culture in fresh medium, persister cells may still linger for extensive periods and be the ones found in the surviving fraction. Even when the culture is allowed to resume growth for a few hours and to reach what seems to be ‘exponential growth’, a fraction of the persisters triggered by the previous starvation may remain in a lag phase. Therefore, the lag time distribution of single cells after starvation or exposure to a stress is an important factor to take into account as it may determine the persistence level25,26.

Triggered persistence requires a trigger for bacteria to become persisters (left). The persistence level will then depend on the intensity and duration of the trigger. For example, a common trigger for persistence is starvation. Even when the trigger is removed, persister bacteria may retain their phenotype for an extended duration. Spontaneous persistence occurs when the bacteria are in steady-state exponential growth (right). A fraction of the population switches stochastically to the persister phenotype at a rate that is constant during growth. Such steady-state conditions can be found in chemostats or serially diluted cultures, and care must be taken to ensure that the persisters do not originate from the inoculum or from the culture being too close to entry into the stationary phase.
Many stress conditions have been shown to generate triggered persistence, including limitation of different nutrients27, high cell number28, acid stress, immune factors29 and exposure to immune cells30.
Confounding results can occur when the antibiotic itself serves as a trigger for growth arrest, causing drug-induced persistence31 and sometimes paradoxical lower killing at high drug concentration32. In this case, instead of killing the cells, a bactericidal antibiotic becomes bacteriostatic for a subpopulation of cells that respond to the antibiotic signal itself, for example, by activating a stress response that enables them to survive31,33. This type of persistence does not depend on the history of the culture before exposure to the drug34 and therefore may be attributed to spontaneous persistence. However, because in this case persistence relies on the response of the cells to the antibiotic, it may be more specific to the class of antibiotic used and its concentration than other forms of persistence.
Spontaneous persistence
Persistence may be observed without any trigger and when the culture is in steady-state exponential growth and all parameters are kept constant, that is, during balanced growth (Fig. 3). In this case, persisters may occur spontaneously, and their fraction remains constant as long as the steady-state growth is maintained. Spontaneous persistence was previously called type II persistence24. This form of persistence seems to be much less common than triggered persistence.
Effect of mutations
Importantly, apart from the environmental triggers mentioned above, all types of persistence may be increased (or reduced) by mutations. Although within a clonal population, persister and non-persister bacteria are typically isogenic, mutations have been identified that are able to increase the level of persistence or tolerance. For example, one of the first identified high persistence mutations, the hipA7 mutation20, increases the level of triggered persistence in Escherichia coli by orders of magnitude, reaching persistence levels of about 20% of the population, whereas it is typically below 0.1% for wild-type strains. A high persistence mutation can be viewed as a tolerance mutation with partial penetrance in the whole population; only the subpopulation of persister bacteria will exhibit a phenotype owing to the mutation and will die slower. A high tolerance mutation reduces the killing rate of the whole population. In other words, 100% persistence is equivalent to tolerance.
We stress the fact that the definition of persistence presented here is not directly linked to a specific mechanism or to a physiological state of the bacteria. Rather, it is defined by the time-kill assay; therefore, we outline below some important considerations for increasing the reproducibility and reliability of persistence detection. Typical microbiology procedures developed for measuring uniform bulk phenomena need to be carefully re-evaluated when measuring survival that is dominated by a small subpopulation of cells.
Although we focus here on antibiotic persistence of bacteria, we believe that the definitions and guidelines should be relevant for heterogeneous responses to other drugs such as antifungals35 and anticancer treatments36.
A guide to measuring persistence
The starting point for identifying persistence is the time-kill assay, which measures survival of bacteria at different time points during exposure to the antibiotic. Survival is defined as the ability to regrow when the antibiotic is removed. The hallmark of persistence is the bimodal (or multimodal) killing curve (Fig. 1). However, observing bimodal killing alone is not enough, and several additional steps are required to evaluate whether the bimodality results from persistence or from resistance and to differentiate between the different types of persistence mentioned above. A standardization of the assays and a clear description of the conditions used are required to enable comparing different strains or conditions and results from different laboratories.
Does the bimodal killing curve really reflect persistence?
First, a bimodal killing curve may be due to resistant mutants. To rule out this effect, surviving bacteria that are clearly in the tail of the survival curve should be regrown in the same conditions and exposed again to the same antibiotic treatment. Persistence requires that the killing curve remains the same as in the initial inoculation37. If resistant mutants were responsible for the slower killing rate in the first killing curve, the second assay will show reduced killing of a much higher proportion of the population than in the first assay.
Second, in order to distinguish persistence from transient modes of resistance such as heteroresistance14, the killing curve should be performed at high antibiotic concentration, at least several times the MIC, to efficiently kill bacteria that have a higher MIC than the rest of the population. The killing rate should only weakly depend on the antibiotic concentration. Strong dependence on the antibiotic concentration (scaled with MIC) reflects phenomena linked to resistance.
Third, experimental pitfalls that may result in bimodal killing should be ruled out. One of the most common reasons for a decrease in the killing rate is degradation of the drug with time. Therefore, it is important to test that the killing efficacy of the drug itself does not decrease with time because of natural degradation of the drug, uptake by bacteria or changes in the medium. Another reason for survival of some bacteria may be their adhesion to the walls of the culture vessel38, where the antibiotic may not efficiently kill them.
Finally, in addition to clearly stating in which conditions the time-kill assay is performed, care should be given to the recovery conditions, when bacteria are allowed to grow after removal of the antibiotic. The precise conditions for the evaluation of survival after treatment should be described, such as the washing out of the antibiotics, the medium in which the bacteria are recovered and the time that has passed from the exposure to the antibiotics until the exposure to the recovery conditions. For example, it has been shown that keeping the bacteria in non-growing conditions after treatment may increase their survival39. In addition, bacteria recovering from an antibiotic treatment may have a delayed regrowth either because they are in the tail of the lag time distribution26,40 or because of the post-antibiotic effect41,42, which results in the delayed growth of bacteria after treatment. Therefore, evaluating the survival by counting colonies should be done not only after the typical appearance time of colonies but also several days later.
Measuring triggered persistence
As the trigger is an integral part of triggered persistence, the trigger duration, intensity and exact conditions should be clearly mentioned and kept the same between experiments. For example, one of the most common triggers of persistence is starvation. Many reports used an ‘overnight culture’ as inoculum. This overnight culture has been exposed to several stress signals during starvation, such as high cell density, stringent response and altered pH, that may trigger persistence, and therefore inevitably still contains bacteria that experienced a trigger. In this example of triggered persistence, the persistence level will depend strongly on several parameters, including the size of the inoculum, the time that has elapsed since the inoculum was regrown and the duration of starvation during the previous overnight culture26,40. Typically, to obtain reproducible results, the time between the trigger for persistence and the exposure to antibiotics should be minimized to avoid the uncontrolled loss of persister bacteria that switch back to normal cells.
Measuring drug-induced persistence
The conditions for measuring drug-induced persistence are the same as for measuring spontaneous persistence, namely, steady-state growth, as the trigger is the drug itself and should be applied in steady-state conditions to avoid stationary-phase-induced persistence. Without further characterizations, the spontaneous and drug-induced persistence are difficult to distinguish. In this case, direct observation of single cells as they respond to the antibiotics is needed43,44, or a dissection of the molecular mechanism that allows the bacteria to respond to the drug by activating a stress response31. Earlier attempts to characterize drug-induced tolerance or drug-induced persistence made use of the minimum bactericidal concentration (MBC)38. The MBC is the concentration required to kill bacteria. Some drugs may arrest the growth at the MIC but require a higher concentration to kill. If the drug itself induces persistence at the MIC, a higher concentration may be required to reach killing. Drug-induced persister bacteria have a higher MBC than the rest of the population, but their MIC is unchanged.
Measuring spontaneous persistence
In contrast to triggered persistence, which is determined by the history of the culture, the rare spontaneous persistence should be measured in conditions of steady-state (also called balanced) growth so as to avoid the effect of the past growth conditions. This measurement can be achieved in a chemostat or by subdiluting the culture several times37 before performing the time-kill assay, making sure to dilute the inoculum to below the persistence level24. As spontaneous persistence is a steady-state phenomenon, care should be taken to evaluate whether the culture remains in steady-state growth, also after the inoculum influence has been ruled out. For example, a common pitfall is to perform the time-kill assay without diluting and subculturing the bacteria for enough time to eliminate the persister bacteria triggered by past stationary phase growth. Another common pitfall is to perform the time-kill assay when the culture is too close to the next stationary phase, which again may trigger the formation of persister cells. Even if the culture seems to be growing exponentially, it may no longer be in balanced growth and persister formation may be already triggered at a cell density that is ten times lower than the maximal density45. The spontaneous persistence fraction should remain constant with time in steady-state growth conditions. A simple way to test that the results do not depend on the cell density is to perform the same experiment at a twofold lower density and verify that the persistence fraction remains the same.
Regrowth of persister bacteria
An inherent part of the persistence phenomenon is the ability of persisters to eventually resume growth. As evidenced by the low killing rate displayed in the second phase of biphasic killing curves (Fig. 2c), persisters may resume growth at a low and constant rate, independently of the presence of the drug. Only persisters resuming growth after cessation of the antibiotic treatment will give rise to a new population of susceptible bacteria.
Single-cell observation often shows non-growing cells that remain intact during exposure to the drug. However, regrowth must be documented30 to illustrate that bacteria have survived exposure to the drug before those can be dubbed persisters.
Conclusion
There has been a sharp increase in the interest for antibiotic persistence in the past years in the background of growing concerns about antimicrobial resistance. The observation that triggered persistence evolves fast in vitro46,47 and can be followed by the evolution of resistance48 suggests that persistence may be evolving quickly in the host as well. It has been suggested that the presence of antibiotic persister cells is responsible, at least partly, for lack of clearance of pathogenic bacteria by antibiotic treatment. Indeed, even in the absence of any antibiotic resistance, many bacterial infections are hard to treat and tend to relapse (such as tuberculosis, lung infections in people with cystic fibrosis, systemic infections with Salmonella, tonsillitis and urinary tract infections). The underlying reasons are most likely multifactorial, with suboptimal pharmacodynamics in the host probably playing a major role in some instances. However, it is also clear that non-growing bacteria29 and high-persister-forming mutants are selected over time in patients exposed to repeated doses of antibiotics49,50. Further work is needed to evaluate the possible impact of persister cells on the treatment outcome of bacterial infections and to find ways to fight them15. As seen above, many pitfalls exist even for in vitro analysis of persistence, and controlling the experimental conditions is crucial. The understanding of persistence in the host, in which our knowledge of the conditions is scarce, is orders of magnitude more challenging51,52. It is our hope that the guidelines outlined in this article will enable a consensus on in vitro measurements and pave the way for designing protocols adapted to the clinical evaluation of antibiotic persistence.
Change history
29 April 2019
In Figure 2b, the minimal duration for killing (MDK) 99% of tolerant cells was erroneously labelled as MDK99.99 instead of MDK99. This has now been corrected in all versions of the Review. The publisher apologizes to the authors and to readers for this error.
References
Hobby, G. L., Meyer, K. & Chaffee, E. Observations on the mechanism of action of penicillin. Proc. Soc. Exp. Biol. (NY) 50, 281–285 (1942).
Bigger, J. W. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244, 497–500 (1944).
Brauner, A., Fridman, O., Gefen, O. & Balaban, N. Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14, 320–330 (2016).
Nataro, J. P., Blaser, M. J. & Cunningham-Rundles, S. Persistent Bacterial Infections (ASM Press, 2000).
Fisher, R. A., Gollan, B. & Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 15, 453–464 (2017).
Ernst, J. D. Mechanisms of M. tuberculosis immune evasion as challenges to TB vaccine design. Cell Host Microbe 24, 34–42 (2018).
Blango, M. G. & Mulvey, M. A. Persistence of uropathogenic Escherichia coli in the face of multiple antibiotics. Antimicrob. Agents Chemother. 54, 1855–1863 (2010).
Mulvey, M. A., Schilling, J. D. & Hultgren, S. J. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect. Immun. 69, 4572–4579 (2001).
Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5, 48–56 (2007).
Jacoby, G. A. AmpC β-lactamases. Clin. Microbiol. Rev. 22, 161–182 (2009).
Du, D. J. et al. Multidrug efflux pumps: structure, function and regulation. Nat. Rev. Microbiol. 16, 523–539 (2018).
Blair, J. M., Webber, M. A., Baylay, A. J., Ogbolu, D. O. & Piddock, L. J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51 (2015).
Wolfson, J., Hooper, D., McHugh, G., Bozza, M. & Swartz, M. Mutants of Escherichia coli K-12 exhibiting reduced killing by both quinolone and beta-lactam antimicrobial agents. Antimicrob. Agents Chemother. 34, 1938–1943 (1990).
El-Halfawy, O. M. & Valvano, M. A. Antimicrobial heteroresistance: an emerging field in need of clarity. Clin. Microbiol. Rev. 28, 191–207 (2015).
Meylan, S., Andrews, I. W. & Collins, J. J. Targeting antibiotic tolerance, pathogen by pathogen. Cell 172, 1228–1238 (2018).
Handwerger, S. & Tomasz, A. Antibiotic tolerance among clinical isolates of bacteria. Annu. Rev. Pharmacol. Toxicol. 25, 349–380 (1985).
Ackermann, M. A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Microbiol. 13, 497–508 (2015).
Rotem, E. et al. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc. Natl Acad. Sci. USA 107, 12541–12546 (2010).
Huang, G. R., Saakian, D. B. & Hu, C. K. Accurate analytic solution of chemical master equations for gene regulation networks in a single cell. Phys. Rev. E 97, 012412 (2018).
Moyed, H. S. & Bertrand, K. P. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 155, 768–775 (1983).
Levin, B. R., Concepcion-Acevedo, J. & Udekwu, K. I. Persistence: a copacetic and parsimonious hypothesis for the existence of non-inherited resistance to antibiotics. Curr. Opin. Microbiol. 21, 18–21 (2014).
Michiels, J. E., Van den Bergh, B., Verstraeten, N. & Michiels, J. Molecular mechanisms and clinical implications of bacterial persistence. Drug Resist. Updat. 29, 76–89 (2016).
Radzikowski, J. L., Schramke, H. & Heinemann, M. Bacterial persistence from a system-level perspective. Curr. Opin. Biotechnol. 46, 98–105 (2017).
Balaban, N. Q., Merrin, J., Chait, R., Kowalik, L. & Leibler, S. Bacterial persistence as a phenotypic switch. Science 305, 1622–1625 (2004).
Joers, A., Kaldalu, N. & Tenson, T. The frequency of persisters in Escherichia coli reflects the kinetics of awakening from dormancy. J. Bacteriol. 192, 3379–3384 (2010).
Levin-Reisman, I. et al. Automated imaging with ScanLag reveals previously undetectable bacterial growth phenotypes. Nat. Methods 7, 737–U100 (2010).
Gutierrez, A. et al. Understanding and sensitizing density-dependent persistence to quinolone antibiotics. Mol. Cell 68, 1147–1154 (2017).
Vega, N. M., Allison, K. R., Khalil, A. S. & Collins, J. J. Signaling-mediated bacterial persister formation. Nat. Chem. Biol. 8, 431–433 (2012).
Manina, G., Dhar, N. & McKinney, J. D. Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17, 32–46 (2015).
Helaine, S. et al. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343, 204–208 (2014).
Dorr, T., Vulic, M. & Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLOS Biol. 8, e1000317 (2010).
Eagle, H. & Musselman, A. The rate of bactericidal action of penicillin in vitro as a function of its concentration, and its paradoxically reduced activity at high concentrations against certain organisms. J. Exp. Med. 88, 99–131 (1948).
Audrain, B. et al. Induction of the Cpx envelope stress pathway contributes to Escherichia coli tolerance to antimicrobial peptides. Appl. Environ. Microbiol. 79, 7770–7779 (2013).
Johnson, P. J. T. & Levin, B. R. Pharmacodynamics, population dynamics, and the evolution of persistence in Staphylococcus aureus. PLOS Genet. 9, e1003123 (2013).
Rosenberg, A. et al. Antifungal tolerance is a subpopulation effect distinct from resistance and is associated with persistent candidemia. Nat. Commun. 9, 2470 (2018).
Kochanowski, K., Morinishi, L., Altschuler, S. & Wu, L. Drug persistence — from antibiotics to cancer therapies. Curr. Opin. Syst. Biol. 10, 1–8 (2018).
Keren, I., Kaldalu, N., Spoering, A., Wang, Y. & Lewis, K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230, 13–18 (2004).
Taylor, P. C., Schoenknecht, F. D., Sherris, J. C. & Linner, E. C. Determination of minimum bactericidal concentrations of oxacillin for Staphylococcus-aureus - influence and significance of technical factors. Antimicrob. Agents Chemother. 23, 142–150 (1983).
Mok, W. W. K. & Brynildsen, M. P. Timing of DNA damage responses impacts persistence to fluoroquinolones. Proc. Natl Acad. Sci. USA 115, E6301–E6309 (2018).
Luidalepp, H., Joers, A., Kaldalu, N. & Tenson, T. Age of inoculum strongly influences persister frequency and can mask effects of mutations implicated in altered persistence. J. Bacteriol. 193, 3598–3605 (2011).
Eagle, H. & Musselman, A. D. The slow recovery of bacteria from the toxic effects of penicillin. J. Bacteriol. 58, 475–490 (1949).
zur Wiesch, P. A. et al. Classic reaction kinetics can explain complex patterns of antibiotic action. Sci. Transl Med. 7, 287ra73 (2015).
Wakamoto, Y. et al. Dynamic persistence of antibiotic-stressed mycobacteria. Science 339, 91–95 (2013).
Maglica, Z., Ozdemir, E. & McKinney, J. D. Single-cell tracking reveals antibiotic-induced changes in mycobacterial energy metabolism. mBio 6, e02236-14 (2015).
Akerlund, T., Nordstrom, K. & Bernander, R. Analysis of cell-size and DNA content in exponentially growing and stationary-phase batch cultures of Escherichia-coli. J. Bacteriol. 177, 6791–6797 (1995).
Fridman, O., Goldberg, A., Ronin, I., Shoresh, N. & Balaban, N. Q. Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 513, 418–421 (2014).
Van den Bergh, B. et al. Frequency of antibiotic application drives rapid evolutionary adaptation of Escherichia coli persistence. Nat. Microbiol. 1, 16020 (2016).
Levin-Reisman, I. et al. Antibiotic tolerance facilitates the evolution of resistance. Science 355, 826–830 (2017).
Lafleur, M. D., Qi, Q. & Lewis, K. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrob. Agents Chemother. 54, 39–44 (2010).
Vulin, C., Leimer, N., Huemer, M., Ackermann, M. & Zinkernagel, A. S. Prolonged bacterial lag time results in small colony variants that represent a sub-population of persisters. Nat. Commun. 9, 4074 (2018).
Claudi, B. et al. Phenotypic variation of salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell 158, 722–733 (2014).
Kaiser, P. et al. Cecum lymph node dendritic cells harbor slow-growing bacteria phenotypically tolerant to antibiotic treatment. PLOS Biol. 12, e1001793 (2014).
European Committee on Antimicrobial Susceptibility Testing. Reading guide: EUCAST disk diffusion method for antimicrobial susceptibility testing. EUCAST http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Disk_test_documents/2019_manuals/Reading_guide_v_6.0_EUCAST_Disk_Test_2019.pdf (2019).
Barry, L. A. et al. Methods for Determining Bactericidal Activity of Antimicrobial Agents; Approved Guideline Vol. 19 1–3 (Clinical and Laboratory Standards Institute,1999).
Zhi, J. G., Nightingale, C. H. & Quintiliani, R. A. Pharmacodynamic model for the activity of antibiotics against microorganisms under nonsaturable conditions. J. Pharm. Sci. 75, 1063–1067 (1986).
Levin, B. R. & Udekwu, K. I. Population dynamics of antibiotic treatment: a mathematical model and hypotheses for time-kill and continuous-culture experiments. Antimicrob. Agents Chemother. 54, 3414–3426 (2010).
Nicoloff, H., Hjort, K., Levin, B. R. & Andersson, D. I. The high prevalence of antibiotic heteroresistance in pathogenic bacteria is mainly caused by gene amplification. Nat. Microbiol. 4, 504–514 (2019).
Lewis, K. Persister cells and the riddle of biofilm survival. Biochemistry Mosc. 70, 267–274 (2005).
Yang, J. H., Bening, S. C. & Collins, J. J. Antibiotic efficacy — context matters. Curr. Opin. Microbiol. 39, 73–80 (2017).
Amato, S. M., Orman, M. A. & Brynildsen, M. P. Metabolic control of persister formation in Escherichia coli. Mol. Cell 50, 475–487 (2013).
Acknowledgements
The authors thank the Congressi Stefano Franscini, the European Molecular Biology Organization (EMBO), the Federation of European Microbiological Societies (FEMS) and the University of Basel for supporting the EMBO Workshop ‘Bacterial Persistence and Antimicrobial Therapy’ and A.-C. Hiebel for taking a major role in its organization. The authors represent the groups attending the workshop but acknowledge the contributions of many other groups in the antibiotic persistence field. B.R.L. is funded by the US National Institutes of Health (NIH; R01GM 091875). N.Q.B. is funded by the European Research Council (ERC; #681619).
Author information
Authors and Affiliations
Contributions
N.Q.B., S.H. and K.L. wrote the article. All authors contributed to discussion of the content and reviewed or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Balaban, N.Q., Helaine, S., Lewis, K. et al. Definitions and guidelines for research on antibiotic persistence. Nat Rev Microbiol 17, 441–448 (2019). https://doi.org/10.1038/s41579-019-0196-3
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41579-019-0196-3
This article is cited by
-
RpoS role in antibiotic resistance, tolerance and persistence in E. coli natural isolates
BMC Microbiology (2024)
-
Phage Paride can kill dormant, antibiotic-tolerant cells of Pseudomonas aeruginosa by direct lytic replication
Nature Communications (2024)
-
Bacteria can compensate the fitness costs of amplified resistance genes via a bypass mechanism
Nature Communications (2024)
-
Cephalosporin resistance, tolerance, and approaches to improve their activities
The Journal of Antibiotics (2024)
-
Plasmid-mediated phenotypic noise leads to transient antibiotic resistance in bacteria
Nature Communications (2024)
