
Abstract
Recent advances in machine learning and experimental biology have offered breakthrough solutions to problems such as protein structure prediction that were long thought to be intractable. However, despite the pivotal role of the T cell receptor (TCR) in orchestrating cellular immunity in health and disease, computational reconstruction of a reliable map from a TCR to its cognate antigens remains a holy grail of systems immunology. Current data sets are limited to a negligible fraction of the universe of possible TCR–ligand pairs, and performance of state-of-the-art predictive models wanes when applied beyond these known binders. In this Perspective article, we make the case for renewed and coordinated interdisciplinary effort to tackle the problem of predicting TCR–antigen specificity. We set out the general requirements of predictive models of antigen binding, highlight critical challenges and discuss how recent advances in digital biology such as single-cell technology and machine learning may provide possible solutions. Finally, we describe how predicting TCR specificity might contribute to our understanding of the broader puzzle of antigen immunogenicity.
Similar content being viewed by others
Introduction
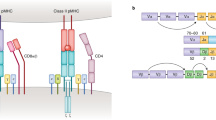
T cells typically recognize antigens presented on members of the MHC protein family via highly diverse heterodimeric T cell receptors (TCRs) expressed at their surface (Fig. 1). These antigens are commonly short peptide fragments of eight or more residues, the presentation of which is dictated in large part by the structural preferences of the MHC allele1. Lipid, metabolite and oligosaccharide T cell antigens have also been reported2,3,4. TCRs typically engage antigen–MHC complexes via one or more of their six complementarity-determining loops (CDRs), three contributed by each chain of the TCR dimer.

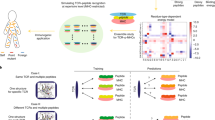
a, Cartoon illustrating cancer cell antigen presentation to a naive T cell; T cell activation and expansion and effector T cell engagement of the cancer cell. b, Antigen recognition by conventional T cells through the interaction of the αβ T cell receptor (TCR) heterodimer with peptide antigen presented by an MHC class I molecule. c, Crystal structure of the affinity-enhanced A3A TCR engaging with melanoma-associated antigen 3 (MAGE-A3)-derived peptide presented by HLA-A*01 (ref. 101) (generated with data from ref. 101 and visualized with PyMOL (see Related links)).
The pivotal role of the TCR in surveillance and response to disease, and in the development of new vaccines and therapies, has driven concerted efforts to decode the rules by which T cells recognize cognate antigen–MHC complexes. However, cost and experimental limitations have restricted the available databases to just a minute fraction of the possible sample space of TCR–antigen binding pairs (Box 1). As we discuss later, these data sets5,6,7,8 are also poorly representative of the universe of self and pathogenic epitopes and of the varied MHC contexts in which they may be presented (Fig. 2).

a, Number of T cell receptors (TCRs) containing α-chains, β-chains or paired chains, showing variation in numbers according to the data set (manually curated catalogue of pathology-associated TCR sequences (McPas-TCR), VDJ database (VDJdb), Immune Epitope Database (IEDB) and multiplex identification of TCR antigen specificity (MIRA)). b, Number of TCRs per antigen species of origin, showing that the majority of all antigens reported as binding a TCR are of viral origin. c, Cumulative frequency of antigens, showing that a group of 100 antigens makes up 70% of TCR–antigen pairs. d, Number of TCRs by HLA-A type, showing that known antigens are reported in complex with only a few common HLA alleles. e, Frequency histogram showing that most antigens have only one known cognate TCR in the combined data set5,6,7,8.
The research community has therefore turned to machine learning models as a means of predicting the antigen specificity of the so-called orphan TCRs having no known experimentally validated cognate antigen. Accurate prediction of TCR–antigen specificity can be described as deriving computational solutions to two related problems: first, given a TCR of unknown antigen specificity, which antigen–MHC complexes is it most likely to bind; and second, given an antigen–MHC complex, which are the most likely cognate TCRs?
A critical requirement of models attempting to answer these questions is that they should be able to make accurate predictions for any combination of TCR and antigen–MHC complex. These should cover both ‘seen’ pairs included in the data on which the model was trained and novel or ‘unseen’ TCR–epitope pairs to which the model has not been exposed9. Impressive advances have been made for specificity inference of seen epitopes in particular disease contexts. For example, clusters of TCRs having common antigen specificity have been identified for Mycobacterium tuberculosis10 and SARS-CoV-2 (ref. 11), providing possible avenues for new vaccine and pharmaceutical development. However, as discussed later, performance for seen epitopes wanes beyond a small number of immunodominant viral epitopes and is generally poor for unseen epitopes9,12. This matters because many epitopes encountered in nature will not have an experimentally validated cognate TCR, particularly those of human or non-viral origin (Fig. 2). In the text to follow, we refer to the case for generalizable TCR–antigen specificity inference, meaning prediction of binding for both seen and unseen antigens in any MHC context.
We must also make an important distinction between the related tasks of predicting TCR specificity and antigen immunogenicity. The former, and the focus of this article, is the prediction of binding between sets of TCRs and antigen–MHC complexes. The latter can be described as predicting whether a given antigen will induce a functional T cell immune response: a complex chain of events spanning antigen expression, processing and presentation, TCR binding, T cell activation, expansion and effector differentiation. Although great strides have been made in improving prediction of antigen processing and presentation for common HLA alleles, the nature and extent to which presented peptides trigger a T cell response are yet to be elucidated13. A significant gap also remains for the prediction of T cell activation for a given peptide14,15, and the parameters that influence pathological peptide or neoantigen immunogenicity remain under intense investigation16. We believe that only by integrating knowledge of antigen presentation, TCR recognition, context-dependent activation and effector function at the cell and tissue level will we fully realize the benefits to fundamental and translational science (Box 2).
State of the art
From deepening our mechanistic understanding of disease to providing routes for accelerated development of safer, personalized vaccines and therapies, the case for constructing a complete map of TCR–antigen interactions is compelling. We now explore some of the experimental and computational progress made to date, highlighting possible explanations for why generalizable prediction of TCR binding specificity remains a daunting task.
Experimental methods
The development of recombinant antigen–MHC multimer assays17 has proved transformative in the analysis of TCR–antigen specificity, enabling researchers to track and study T cell populations under various conditions and disease settings18,19,20. Nonetheless, critical limitations remain that hamper high-throughput determination of TCR–antigen specificity. We direct the interested reader to a recent review21 for a thorough comparison of these technologies and summarize some of the principal issues subsequently.
Antigen–MHC multimers may be used to determine TCR specificity using bulk (pooled) T cell populations, or newer single-cell methods. Bulk methods are widely used and relatively inexpensive, but do not provide information on αβ TCR chain pairing or function. As a result, single chain TCR sequences predominate in public data sets (Fig. 2). However, both α-chains and β-chains contribute to antigen recognition and specificity22,23. We shall discuss the implications of this for modelling approaches later. Multimodal single-cell technologies provide insight into chain pairing and transcriptomic and phenotypic profiles at cellular resolution, but remain prohibitively expensive, return fewer TCR sequences per run than bulk experiments and show significant bias towards TCRs with high specificity24,25,26. The appropriate experimental protocol for the reduction of nonspecific multimer binding, validation of correct folding and computational improvement of signal-to-noise ratios remain active fields of debate25,26. Indeed, concerns over nonspecific binding have led recent computational studies to exclude data derived from a 10× study of four healthy donors27.
Although bulk and single-cell methods are limited to a modest number of antigen–MHC complexes per run, the advent of technologies such as lentiviral transfection assays28,29 provides scalability to up to 96 antigen–MHC complexes through library-on-library screens. However, previous knowledge of the antigen–MHC complexes of interest is still required. This precludes epitope discovery in unknown, rare, sequestered, non-canonical and/or non-protein antigens30.
The advent of synthetic peptide display libraries (Fig. 3a) permits the extension of binding analysis to hundreds of thousands of peptides per TCR30,31,32,33. Using transgenic yeast expressing synthetic peptide–MHC constructs from a library of 2 × 108 peptides, Birnbaum et al.31 dissected the binding preferences of autoreactive mouse and human TCRs, providing clues as to the mechanisms underlying autoimmune targeting in multiple sclerosis. High-throughput library screens such as these provide opportunities for improved screening of the antigen–MHC space, but limit analysis to individual TCRs and rely on TCR–MHC binding instead of function. There remains a need for high-throughput linkage of antigen specificity and T cell function, for example, through mammalian or bead display34,35,36,37.

a, Multiplex analysis of T cell receptor (TCR)–peptide–MHC (pMHC) antigen specificity and synthetic peptide library screens for interrogation of peptide specificity of a single TCR. b, Representation of a deep neural network for supervised prediction of TCR–antigen specificity. c, Unsupervised clustering analysis of TCR–antigen specificity showing (centre) an example clustering visualization and (right) complementarity-determining loop 3 sequence logos.
As a result of these barriers to scalability, only a minuscule fraction of the total possible sample space of TCR–antigen pairs (Box 1) has been validated experimentally. At the time of writing, fewer than 1 million unique TCR–epitope pairs are available from VDJdb, McPas-TCR, the Immune Epitope Database and the MIRA data set5,6,7,8 (Fig. 2). Just 4% of these instances contain complete chain pairing information (Fig. 2a). About 97% of all antigens reported as binding a TCR are of viral origin, and a group of just 100 antigens makes up 70% of TCR–antigen pairs (Fig. 2b,c). Where the HLA context of a given antigen is known, the training data are dominated by antigens presented by a handful of common alleles (Fig. 2d). Many antigens have only one known cognate TCR (Fig. 2e). These limitations have simultaneously provided the motivation for and the greatest barrier to computational methods for the prediction of TCR–antigen specificity.
Computational methods
A comprehensive survey of computational models for TCR specificity inference is beyond the scope intended here but can be found in the following helpful reviews15,38,39,40,41,42. Broadly speaking, current models can be divided into two categories, which we dub supervised predictive models (SPMs) (Fig. 3b) and unsupervised clustering models (UCMs) (Fig. 3c) on account of their respective use of supervised learning and unsupervised learning. A non-exhaustive summary of recent open-source SPMs and UCMs can be found in Table 1.
Supervised predictive models
SPMs are those which attempt to learn a function that will correctly predict the cognate epitope for a given input TCR of unknown specificity, given some training data set of known TCR–peptide pairs. The past 2 years have seen an acceleration of publications aiming to address this challenge with deep neural networks (DNNs). Although there are many possible approaches to comparing SPM performance, among the most consistently used is the area under the receiver-operating characteristic curve (ROC-AUC). One would expect to observe 50% ROC-AUC from a random guess in a binary (binding or non-binding) task, assuming a balanced proportion of negative and positive pairs.
Performance by this measure surpasses 80% ROC-AUC for a handful of ‘seen’ immunodominant viral epitopes presented by MHC class I9,43. However, representation is not a guarantee of performance: 60% ROC-AUC has been reported for HLA-A2*01–CMV-NLVPMVATV44, possibly owing to the recognition of this immunodominant antigen by diverse TCRs. Critically, few models explicitly evaluate the performance of trained predictors on unseen epitopes using comparable data sets. Weber et al.12 achieved an average of 62 ± 6% ROC-AUC for TITAN, compared with 50% for ImRex on a reference data set of unseen epitopes from VDJdb and COVID-19 data sets. Values of 56 ± 5% and 55 ± 3% were reported for TITAN and ImRex, respectively, in a subsequent paper from the Meysman group45. Other groups have published unseen epitope ROC-AUC values ranging from 47% to 97%; however, many of these values are reported on different data sets (Table 1), lack confidence estimates following validation46,47,48,49 and have not been consistently reproducible in independent evaluations50.
Together, these results highlight a critical need for a thorough, independent benchmarking study conducted across models on data sets prepared and analysed in a consistent manner27,50. Until then, newer models may be applied with reasonable confidence to the prediction of binding to immunodominant viral epitopes by common HLA alleles. However, SPMs should be used with caution when generalizing to prediction of any epitope, as performance is likely to drop the further the epitope is in sequence from those in the training set9.
Unsupervised clustering models
Unlike SPMs, UCMs do not depend on the availability of labelled data, learning instead to produce groupings of the TCR, antigen or HLA input that reflect the underlying statistical variations of the data19,51 (Fig. 3c). Applied to TCR repertoires, UCMs take as their input single or paired TCR CDR3 amino acid sequences, with or without gene usage information, and return a mapping of sequences to unique clusters. Clustering is achieved by determining the similarity between input sequences, using either ‘hand-crafted’ features such as sequence distance or enrichment of short sub-sequences, or by comparing abstract features learnt by DNNs (Table 1).
Clustering provides multiple paths to specificity inference for orphan TCRs39,40,41. Epitope specificity can be predicted by assuming that if an unlabelled TCR is similar to a receptor of known specificity, it will bind the same epitope52. One may also co-cluster unlabelled and labelled TCRs and assign the modal or most enriched epitope to all sequences that cluster together51. Finally, DNNs can be used to generate ‘protein fingerprints’, simple fixed-length numerical representations of complex variable input sequences that may serve as a direct input for a second supervised model25,53.
As for SPMs, quantitative assessment of the relative merits of hand-crafted and neural network-based UCMs for TCR specificity inference remains limited to the proponents of each new model. Although some DNN-UCMs allow for the integration of paired chain sequences and even transcriptomic profiles48, they are susceptible to the same training biases as SPMs and are notably less easy to implement than established clustering models such as GLIPH and TCRdist19,54. However, these established clustering models scale relatively poorly to large data sets compared with newer releases51,55. Recent analyses27,53 suggest that there is little to differentiate commonly used UCMs from simple sequence distance measures. Here again, independent benchmarking analyses would be valuable, work towards which our group is dedicating significant time and effort.
Key challenges
Despite the exponential growth of unlabelled immune repertoire data and the recent unprecedented breakthroughs in the fields of data science and artificial intelligence, quantitative immunology still lacks a framework for the systematic and generalizable inference of T cell antigen specificity of orphan TCRs. Among the most plausible explanations for these failures are limitations in the data, methodological gaps and incomplete modelling of the underlying immunology.
Data
As we have set out earlier, the single most significant limitation to model development is the availability of high-quality TCR and antigen–MHC pairs. The need is most acute for under-represented antigens, for those presented by less frequent HLA alleles, and for linkage of epitope specificity and T cell function. Meanwhile, single-cell multimodal technologies have given rise to hundreds of millions of unlabelled TCR sequences8,56, linked to transcriptomics, phenotypic and functional information. However, these unlabelled data are not without significant limitations. Notably, biological factors such as age, sex, ethnicity and disease setting vary between studies and are likely to influence immune repertoires. Differences in experimental protocol, sequence pre-processing, total variation filtering (denoising) and normalization between laboratory groups are also likely to have an impact: batch correction may well need to be applied57. Therefore, thoughtful approaches to data consolidation, noise correction, processing and annotation are likely to be crucial in advancing state-of-the-art predictive models.
Modelling
The exponential growth of orphan TCR data from single-cell technologies, and cutting-edge advances in artificial intelligence and machine learning, has firmly placed TCR–antigen specificity inference in the spotlight. However, we believe that several critical gaps must be addressed before a solution to generalized epitope specificity inference can be realized.
First, models whose TCR sequence input is limited to the use of β-chain CDR3 loops and VDJ gene codes are only ever likely to tell part of the story of antigen recognition, and the extent to which single chain pairing is sufficient to describe TCR–antigen specificity remains an open question. Structural58 and statistical59 analyses suggest that α-chains and β-chains contribute equally to specificity, and incorporating both chains has improved predictive performance44. However, chain pairing information is largely absent (Fig. 2a), and many state-of-the-art SPMs and UCMs rely on single chain information alone (Table 1). Although CDR3 loops may be primarily responsible for antigen recognition, residues from CDR1, CDR2 and even the framework region of both α-chains and β-chains may be involved58. Subtle compensatory changes in interaction networks between peptide–MHC and TCR, altered binding modes and conformational flexibility in both TCR and MHC may underpin TCR cross-reactivity60,61. Explicit encoding of structural information for specificity inference has until recently been limited to studies of a limited set of crystal structures19,62. However, the advent of automated protein structure prediction with software programs such as RoseTTaFold, ESMFold and AlphaFold-Multimer provide potential opportunities for large-scale sequence and structure interpretations of TCR epitope specificity63,64,65. This has been illustrated in a recent preprint in which a modified version of AlphaFold-Multimer has been used to identify the most likely binder to a given TCR, achieving a mean ROC-AUC of 82% on a small pool of eight seen epitopes66.
To train models, balanced sets of negative and positive samples are required. In the absence of experimental negatives, negative instances may be produced by shuffling or drawing randomly from healthy donor repertoires9. However, these approaches assume, on the one hand, that TCRs do not cross-react and, on the other hand, that the healthy donor repertoires do not include sequences reactive to the epitopes of interest. A recent study from Jiang et al.67 provides interesting strategies to address this challenge.
Finally, developers should use the increasing volume of functionally annotated orphan TCR data to boost performance through transfer learning: a technique in which models are trained on a large volume of unlabelled or partially labelled data, and the patterns learnt from those data sets are used to inform a second predictive task. This technique has been widely adopted in computational biology, including in predictive tasks for T and B cell receptors49,66,68. Indeed, the best-performing configuration of TITAN made used a TCR module that had been pretrained on a BindingDB database (see Related links) of 471,017 protein–ligand pairs12. Incorporating evolutionary and structural information through sequence and structure-aware representations of the TCR and of the antigen–MHC complex69,70 may yield further benefits.
Immunology
It is now evident that the underlying immunological correlates of T cell interaction with their cognate ligands are highly variable and only partially understood, with critical consequences for model design. Importantly, TCR–antigen specificity inference is just one part of the larger puzzle of antigen immunogenicity prediction16,18, which we condense into three phases: antigen processing and presentation by MHC, TCR recognition and T cell response.
Antigen processing and presentation pathways have been extensively studied, and computational models for predicting peptide binding affinity to some MHC alleles, especially class I HLAs, have achieved near perfect ROC-AUC15,71 for common alleles. However, this problem is far from solved, particularly for less-frequent MHC class I alleles and for MHC class II alleles7.
A key challenge to generalizable TCR specificity inference is that TCRs are at once specific for antigens bearing particular motifs and capable of considerable promiscuity72,73. This contradiction might be explained through specific interaction of conserved ‘hotspot’ residues in the TCR CDR loops with corresponding two to three residue clusters in the antigen, balanced by a greater tolerance of variations in amino acids at other positions60. TCRs may also bind different antigen–MHC complexes using alternative docking topologies58. Despite the known potential for promiscuity in the TCR, the pre-processing stages of many models assume that a given TCR has only one cognate epitope. Another under-explored yet highly relevant factor of T cell recognition is the impact of positive and negative thymic selection and more specifically the effect of self-peptide presentation in formation of the naive immune repertoire74.
Many groups have attempted to bypass this complexity by predicting antigen immunogenicity independent of the TCR14, as a direct mapping from peptide sequence to T cell activation. However, similar limitations have been encountered for those models as we have described for specificity inference. Many predictors are trained using epitopes from the Immune Epitope Database labelled with readouts from single time points7. However, Achar et al.75 illustrated that integrating cytokine responses over time improved prediction of quality. Antigen load and affinity can also play important roles74,76. Thus, models capable of predicting functional T cell responses will likely need to bridge from antigen presentation to TCR–antigen recognition, T cell activation and effector differentiation and to integrate complex tissue-specific cytokine, cell phenotype and spatiotemporal data sets. Our view is that, although T cell-independent predictors of immunogenicity have clear translational benefits, only after we can dissect the relative contribution of the three stages described earlier will we understand what determines antigen immunogenicity.
New experimental and computational techniques that permit the integration of sequence, phenotypic, spatial and functional information and the multimodal analyses described earlier provide promising opportunities in this direction75,77. Integrating TCR sequence and cell-specific covariates from single-cell data has been shown to improve performance in the inference of T cell antigen specificity48. By taking a graph theoretical approach, Schattgen et al.78 reported an association between clonotype clustering with the cellular phenotypes derived from gene expression and surface marker expression. We believe that such integrative approaches will be instrumental in unlocking the secrets of T cell antigen recognition.
Conclusions and call to action
Together, the limitations of data availability, methodology and immunological context leave a significant gap in the field of T cell immunology in the era of machine learning and digital biology. We believe that by harnessing the massive volume of unlabelled TCR sequences emerging from single-cell data, applying data augmentation techniques to counteract epitope and HLA imbalances in labelled data, incorporating sequence and structure-aware features and applying cutting-edge computational techniques based on rich functional and binding data, improvements in generalizable TCR–antigen specificity inference are within our collective grasp. To aid in this effort, we encourage the following efforts from the community.
First, a consolidated and validated library of labelled and unlabelled TCR data should be made available to facilitate model pretraining and systematic comparisons. Second, a coordinated effort should be made to improve the coverage of TCR–antigen pairs presented by less common HLA alleles and non-viral epitopes. We encourage the continued publication of negative and positive TCR–epitope binding data to produce balanced data sets. Third, an independent, unbiased and systematic evaluation of model performance across SPMs, UCMs and combinations of the two (Table 1) would be of great use to the community. Such a comparison should account for performance on common and infrequent HLA subtypes, seen and unseen TCRs and epitopes, using consistent evaluation metrics including but not limited to ROC-AUC and area under the precision–recall curve. We encourage validation strategies such as those used in the assessment of ImRex and TITAN9,12 to substantiate model performance comparisons. In the future, TCR specificity inference data should be extended to include multimodal contextual information as a means of bridging from TCR binding to immunogenicity prediction.
The scale and complexity of this task imply a need for an interdisciplinary consortium approach for systematic incorporation of the latest immunological understandings of cellular immunity at the tissue level and cutting-edge developments in the field of artificial intelligence and data science. This should include experimental and computational immunologists, machine-learning experts and translational and industrial partners. Considering the success of the critical assessment of protein structure prediction series79, we encourage a similar approach to address the grand challenge of TCR specificity inference in the short term and ultimately to the prediction of integrated T and B cell immunogenicity. Competing models should be made freely available for research use, following the commendable example set in protein structure prediction65,70.
References
Nguyen, A. T., Szeto, C. & Gras, S. The pockets guide to HLA class I molecules. Biochem. Soc. Trans. 49, 2319–2331 (2021).
de Jong, A. & Ogg, G. CD1a function in human skin disease. Mol. Immunol. 130, 14–19 (2021).
de Libero, G., Chancellor, A. & Mori, L. Antigen specificities and functional properties of MR1-restricted T cells. Mol. Immunol. 130, 148–153 (2021).
Sun, L., Middleton, D. R., Wantuch, P. L., Ozdilek, A. & Avci, F. Y. Carbohydrates as T-cell antigens with implications in health and disease. Glycobiology 26, 1029–1040 (2016).
Bagaev, D. V. et al. VDJdb in 2019: database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Res. 48, D1057–D1062 (2020).
Tickotsky, N., Sagiv, T., Prilusky, J., Shifrut, E. & Friedman, N. McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 33, 2924–2929 (2017).
Vita, R. et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019).
Nolan, S. et al. A large-scale database of T-cell receptor beta (TCRβ) sequences and binding associations from natural and synthetic exposure to SARS-CoV-2. Preprint at Res. Sq. https://www.researchsquare.com/article/rs-51964/v1 (2020).
Moris, P. et al. Current challenges for unseen-epitope TCR interaction prediction and a new perspective derived from image classification. Brief. Bioinform. 22, bbaa318 (2021).
Huang, H., Wang, C., Rubelt, F., Scriba, T. J. & Davis, M. M. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat. Biotechnol. 38, 1194–1202 (2020).
Mayer-Blackwell, K. et al. TCR meta-clonotypes for biomarker discovery with tcrdist3 enabled identification of public, HLA-restricted clusters of SARS-CoV-2 TCRs. eLife 10, e68605 (2021).
Weber, A., Born, J. & Rodriguez Martínez, M. TITAN: T cell receptor specificity prediction with bimodal attention networks. Bioinformatics 37, I237–I244 (2021).
Lee, C. H., Antanaviciute, A., Buckley, P. R., Simmons, A. & Koohy, H. To what extent does MHC binding translate to immunogenicity in humans? Immunoinformatics 3–4, 100006 (2021).
Buckley, P. R. et al. Evaluating performance of existing computational models in predicting CD8+ T cell pathogenic epitopes and cancer neoantigens. Brief. Bioinform. 23, bbac141 (2022).
Mösch, A., Raffegerst, S., Weis, M., Schendel, D. J. & Frishman, D. Machine learning for cancer immunotherapies based on epitope recognition by T cell receptors. Front. Genet. 10, 1141 (2019).
Wells, D. K. et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell 183, 818–834.e13 (2020).
Altman, J. D. et al. Phenotypic analysis of antigen-specific T lymphocytes. Science 274, 94–96 (1996).
Yao, Y., Wyrozżemski, Ł., Lundin, K. E. A., Kjetil Sandve, G. & Qiao, S.-W. Differential expression profile of gluten-specific T cells identified by single-cell RNA-seq. PLoS ONE 16, e0258029 (2021).
Glanville, J. et al. Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017).
Kurtulus, S. & Hildeman, D. Assessment of CD4+ and CD8+ T cell responses using MHC class I and II tetramers. Methods Mol. Biol. 979, 71–79 (2013).
Joglekar, A. V. & Li, G. T cell antigen discovery. Nat. Methods 18, 873–880 (2021).
Bosselut, R. et al. Single T cell sequencing demonstrates the functional role of αβ TCR pairing in cell lineage and antigen specificity. Front. Immunol. 1, 1516 (2019).
Emerson, R. O. et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat. Genet. 49, 659–665 (2017).
Wang, X., He, Y., Zhang, Q., Ren, X. & Zhang, Z. Direct comparative analyses of 10× genomics chromium and Smart-Seq2. Genomics Proteomics Bioinformatics 19, 253–266 (2021).
Zhang, W. et al. A framework for highly multiplexed dextramer mapping and prediction of T cell receptor sequences to antigen specificity. Sci. Adv. 7, eabf5835 (2021).
Gascoigne, N. et al. Optimized peptide-MHC multimer protocols for detection and isolation of autoimmune T-cells. Front. Immunol. 9, 1378 (2018).
Meysman, P. et al. Benchmarking solutions to the T-cell receptor epitope prediction problem: IMMREP22 workshop report. Preprint at bioRxiv https://doi.org/10.1101/2022.10.27.514020 (2022).
Dobson, C. S. et al. Antigen identification and high-throughput interaction mapping by reprogramming viral entry. Nat. Methods 19, 449–460 (2022).
Guo, X. Z. J. & Elledge, S. J. V-CARMA: a tool for the detection and modification of antigen-specific T cells. Proc. Natl Acad. Sci. USA 119, e2116277119 (2022).
Brophy, S. E., Holler, P. D. & Kranz, D. M. A yeast display system for engineering functional peptide-MHC complexes. J. Immunol. Methods 272, 235–246 (2003).
Birnbaum, M. E. et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157, 1073–1087 (2014).
Crawford, F. et al. Use of baculovirus MHC/peptide display libraries to characterize T-cell receptor ligands. Immunol. Rev. 210, 156–170 (2006).
Coles, C. H. et al. TCRs with distinct specificity profiles use different binding modes to engage an identical peptide–HLA complex. J. Immunol. 204, 1943–1953 (2020).
Kula, T. et al. T-Scan: a genome-wide method for the systematic discovery of T cell epitopes. Cell 178, 1016 (2019).
Pan, X. et al. Combinatorial HLA-peptide bead libraries for high throughput identification of CD8+ T cell specificity. J. Immunol. Methods 403, 72–78 (2014).
Li, G. et al. T cell antigen discovery via trogocytosis. Nat. Methods 16, 183–190 (2019).
Joglekar, A. V. et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat. Methods 16, 191–198 (2019).
Schaap-Johansen, A.-L., Vujovic, M., Borch, A., Hadrup, S. R. & Marcatili, P. T cell epitope prediction and its application to immunotherapy. Front. Immunol. 12, 712488 (2021).
Valkiers, S. et al. Recent advances in T-cell receptor repertoire analysis: bridging the gap with multimodal single-cell RNA sequencing. Immunoinformatics 5, 100009 (2022).
Lee, C. H. et al. Predicting cross-reactivity and antigen specificity of T cell receptors. Front. Immunol. 11, 2498 (2020).
Vujovic, M. et al. T cell receptor sequence clustering and antigen specificity. Comput. Struct. Biotechnol. J. 18, 2166–2173 (2020).
Katayama, Y., Yokota, R., Akiyama, T. & Kobayashi, T. J. Machine learning approaches to TCR repertoire analysis. Front. Immunol. 13, 858057 (2022).
Luu, A. M., Leistico, J. R., Miller, T., Kim, S. & Song, J. S. Predicting TCR-epitope binding specificity using deep metric learning and multimodal learning. Genes 12, 572 (2021).
Montemurro, A. et al. NetTCR-2.0 enables accurate prediction of TCR-peptide binding by using paired TCRα and β sequence data. Commun. Biol. 4, 1060 (2021).
Dens, C., Bittremieux, W., Affaticati, F., Laukens, K. & Meysman, P. Interpretable deep learning to uncover the molecular binding patterns determining TCR–epitope interactions. Preprint at bioRxiv https://doi.org/10.1101/2022.05.02.490264 (2022).
Springer, I., Tickotsky, N. & Louzoun, Y. Contribution of T cell receptor alpha and beta CDR3, MHC typing, V and J genes to peptide binding prediction. Front. Immunol. 12, 1436 (2021).
Lu, T. et al. Deep learning-based prediction of the T cell receptor–antigen binding specificity. Nat. Mach. Intell. 3, 864–875 (2021).
Fischer, D. S., Wu, Y., Schubert, B. & Theis, F. J. Predicting antigen specificity of single T cells based on TCR CDR3 regions. Mol. Syst. Biol. 16, 9416 (2020).
Wu, K. et al. TCR-BERT: learning the grammar of T-cell receptors for flexible antigen-binding analyses. Preprint at bioRxiv https://doi.org/10.1101/2021.11.18.469186 (2021).
Grazioli, F. et al. On TCR binding predictors failing to generalize to unseen peptides. Front. Immunol. 13, 1014256 (2022).
Zhang, H., Zhan, X. & Li, B. GIANA allows computationally-efficient TCR clustering and multi-disease repertoire classification by isometric transformation. Nat. Commun. 12, 4699 (2021).
Chronister, W. D. et al. TCRMatch: predicting T-cell receptor specificity based on sequence similarity to previously characterized receptors. Front. Immunol. 12, 640725 (2021).
Sidhom, J. W., Larman, H. B., Pardoll, D. M. & Baras, A. S. DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires. Nat. Commun. 12, 1605 (2021).
Dash, P. et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93 (2017).
Valkiers, S., van Houcke, M., Laukens, K. & Meysman, P. ClusTCR: a python interface for rapid clustering of large sets of CDR3 sequences with unknown antigen specificity. Bioinformatics 37, 4865–4867 (2021).
Corrie, B. D. et al. iReceptor: a platform for querying and analyzing antibody/B-cell and T-cell receptor repertoire data across federated repositories. Immunol. Rev. 284, 24–41 (2018).
Andreatta, M. et al. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat. Commun. 12, 2965 (2021).
Leem, J., de Oliveira, S. H. P., Krawczyk, K. & Deane, C. M. STCRDab: the structural T-cell receptor database. Nucleic Acids Res. 46, D406–D412 (2018).
Mayer, A. & Callan Jr, C. G. Measures of epitope binding degeneracy from T cell receptor repertoires. Preprint at bioRxiv https://doi.org/10.1101/2022.07.25.501373 (2022).
Singh, N. K. et al. Emerging concepts in TCR specificity: rationalizing and (maybe) predicting outcomes. J. Immunol. 199, 2203–2213 (2017).
Quaratino, S., Thorpe, C. J., Travers, P. J. & Londei, M. Similar antigenic surfaces, rather than sequence homology, dictate T-cell epitope molecular mimicry. Proc. Natl Acad. Sci. USA 92, 10398–10402 (1995).
Lanzarotti, E., Marcatili, P. & Nielsen, M. T-cell receptor cognate target prediction based on paired α and β chain sequence and structural CDR loop similarities. Front. Immunol. 10, 2080 (2019).
Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. Preprint at bioRxiv https://doi.org/10.1101/2021.10.04.463034 (2022).
Koehler Leman, J. et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17, 665–680 (2020).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Bradley, P. Structure-based prediction of T cell receptor: peptide–MHC interactions. Preprint at bioRxiv https://doi.org/10.1101/2022.08.05.503004 (2022).
Jiang, Y., Huo, M. & Li, S. C. TEINet: a deep learning framework for prediction of TCR-epitope binding specificity. Preprint at bioRxiv https://doi.org/10.1101/2022.10.20.513029 (2022).
Chinery, L., Wahome, N., Moal, I. & Deane, C. M. Paragraph — antibody paratope prediction using Graph Neural Networks with minimal feature vectors. Bioinformatics 39, btac732 (2022).
Alley, E. C., Khimulya, G. & Biswas, S. Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 16, 1312–1322 (2019).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, 449–454 (2020).
Mason, D. A very high level of cross-reactivity is an essential feature of the T-cell receptor. Immunol. Today 19, 395–404 (1998).
Sewell, A. K. Why must T cells be cross-reactive? Nat. Rev. Immunol. 12, 669–677 (2012).
Keck, S. et al. Antigen affinity and antigen dose exert distinct influences on CD4 T-cell differentiation. Proc. Natl Acad. Sci. USA 111, 14852–14857 (2014).
Achar, S. R. et al. Universal antigen encoding of T cell activation from high-dimensional cytokine dynamics. Science 376, 880–884 (2022).
van Panhuys, N., Klauschen, F. & Germain, R. N. T cell receptor-dependent signal intensity dominantly controls CD4+ T cell polarization in vivo. Immunity 41, 63–74 (2014).
Liu, S. et al. Spatial maps of T cell receptors and transcriptomes reveal distinct immune niches and interactions in the adaptive immune response. Immunity 55, 1940–1952.e5 (2022).
Schattgen, S. A. et al. Integrating T cell receptor sequences and transcriptional profiles by clonotype neighbor graph analysis (CoNGA). Nat. Biotechnol. 40, 54–63 (2021).
Kryshtafovych, A., Schwede, T., Topf, M., Fidelis, K. & Moult, J. Critical assessment of methods of protein structure prediction (CASP) — round XIV. Proteins 89, 1607–1617 (2021).
Pearson, K. On lines and planes of closest fit to systems of points in space. Philos. Mag. 2, 559–570 (1901).
Cai, M., Bang, S., Zhang, P. & Lee, H. ATM-TCR: TCR–epitope binding affinity prediction using a multi-head self-attention model. Front. Immunol. 13, 893247 (2022).
Pavlović, M. et al. The immuneML ecosystem for machine learning analysis of adaptive immune receptor repertoires. Nat. Mach. Intell. 3, 936–944 (2021).
Heikkilä, N. et al. Human thymic T cell repertoire is imprinted with strong convergence to shared sequences. Mol. Immunol. 127, 112–123 (2020).
10× Genomics. A new way of exploring immunity: linking highly multiplexed antigen recognition to immune repertoire and phenotype. 10× Genomics https://pages.10xgenomics.com/rs/446-PBO-704/images/10x_AN047_IP_A_New_Way_of_Exploring_Immunity_Digital.pdf (2020).
Ehrlich, R. et al. SwarmTCR: a computational approach to predict the specificity of T cell receptors. BMC Bioinformatics 22, 422 (2021).
Dean, J. et al. Annotation of pseudogenic gene segments by massively parallel sequencing of rearranged lymphocyte receptor loci. Genome Med. 7, 123 (2015).
Zhang, W. et al. PIRD: pan immune repertoire database. Bioinformatics 36, 897–903 (2020).
Jokinen, E., Huuhtanen, J., Mustjoki, S., Heinonen, M. & Lähdesmäki, H. Predicting recognition between T cell receptors and epitopes with TCRGP. PLoS Comput. Biol. 17, e1008814 (2021).
Tong, Y. et al. SETE: sequence-based ensemble learning approach for TCR epitope binding prediction. Comput. Biol. Chem. 87, 107281 (2020).
Snyder, T. M. et al. Magnitude and dynamics of the T-cell response to SARS-CoV-2 infection at both individual and population levels. Preprint at medRxiv https://doi.org/10.1101/2020.07.31.20165647 (2020).
Zhang, S. Q. et al. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat. Biotechnol. 36, 1156–1159 (2018).
Zhang, H. et al. Investigation of antigen-specific T-cell receptor clusters in human cancers. Clin. Cancer Res. 26, 1359–1371 (2020).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
Chen, S. Y., Yue, T., Lei, Q. & Guo, A. Y. TCRdb: a comprehensive database for T-cell receptor sequences with powerful search function. Nucleic Acids Res. 49, D468 (2021).
Gilson, M. K. et al. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 44, 1045–1053 (2015).
Dines, J. N. et al. The ImmuneRACE Study: a prospective multicohort study of immune response action to COVID-19 events with the ImmuneCODETM Open Access Database. Preprint at medRxiv https://doi.org/10.1101/2020.08.17.20175158 (2020).
Chen, G. et al. Sequence and structural analyses reveal distinct and highly diverse human CD8+ TCR repertoires to immunodominant viral antigens. Cell Rep. 19, 569 (2017).
Huth, A., Liang, X., Krebs, S., Blum, H. & Moosmann, A. Antigen-specific TCR signatures of cytomegalovirus infection. J. Immunol. 202, 979–990 (2019).
Springer, I., Besser, H., Tickotsky-Moskovitz, N., Dvorkin, S. & Louzoun, Y. Prediction of specific TCR-peptide binding from large dictionaries of TCR–peptide pairs. Front. Immunol. 11, 1803 (2020).
Kanakry, C. G. et al. Origin and evolution of the T cell repertoire after posttransplantation cyclophosphamide. JCI Insight 1, 86252 (2016).
Raman, M. C. C. et al. Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci. Rep. 6, 18851 (2016).
Soto, C. et al. High frequency of shared clonotypes in human T cell receptor repertoires. Cell. Rep. 32, 107882 (2020).
Woolhouse, M. E. J. & Gowtage-Sequeria, S. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 11, 1842–1847 (2005).
Robinson, J., Waller, M. J., Parham, P., Bodmer, J. G. & Marsh, S. G. E. IMGT/HLA Database — a sequence database for the human major histocompatibility complex. Nucleic Acids Res. 29, 210–213 (2001).
Waldman, A. D., Fritz, J. M. & Lenardo, M. J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020).
Linette, G. P. et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871 (2013).
Arellano, B., Graber, D. J. & Sentman, C. L. Regulatory T cell-based therapies for autoimmunity. Discov. Med. 22, 73–80 (2016).
Raffin, C., Vo, L. T. & Bluestone, J. A. Treg cell-based therapies: challenges and perspectives. Nat. Rev. Immunol. 20, 158–172 (2020).
Hernando, B. et al. The effect of age on the acquisition and selection of cancer driver mutations in sun-exposed normal skin. Ann. Oncol. 32, 412–421 (2021).
Sesma, A. et al. From tumor mutational burden to blood T cell receptor: looking for the best predictive biomarker in lung cancer treated with immunotherapy. Cancers 12, 1–19 (2020).
Scott, A. C. et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270 (2019).
Yost, K. E. et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25, 1251–1259 (2019).
Wherry, E. J. & Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015).
Daniel, B. et al. Divergent clonal differentiation trajectories of T cell exhaustion. Nat. Immunol. 23, 1614–1627 (2022).
Shakiba, M. et al. TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion. J. Exp. Med. 219, e20201966 (2022).
Dan, J. M. et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 371, eabf4063 (2021).
Swanson, P. A. et al. AZD1222/ChAdOx1 nCoV-19 vaccination induces a polyfunctional spike protein-specific TH1 response with a diverse TCR repertoire. Sci. Transl Med. 13, 7211 (2021).
Bjornevik, K. et al. Longitudinal analysis reveals high prevalence of Epstein–Barr virus associated with multiple sclerosis. Science 375, 296–301 (2022).
Acknowledgements
H.K. is supported by funding from the UK Medical Research Council grant number MC_UU_12010/3. D.H. receives support from the Biotechnology and Biological Sciences Research Council (BBSRC) (grant number BB/T008784/1) and is funded by the Rosalind Franklin Institute. The authors thank A. Simmons, B. McMaster and C. Lee for critical review. H.K. acknowledges A. Antanaviciute, A. Simmons, T. Elliott and P. Klenerman for their encouragement, support and fruitful conversations.
Author information
Authors and Affiliations
Contributions
H.K. and D.H. researched and wrote the article. R.A.F., M.B. and G.O. reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
G.O. is a co-founder of T-Cypher Bio. D.H. and R.A.F provide consultancy services to companies active in T cell antigen discovery and vaccine development. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Immunology thanks M. Birnbaum, P. Holec, E. Newell and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links:
BindingDB: https://www.bindingdb.org/rwd/bind/index.jsp
Immune Epitope Database: https://www.iedb.org/
McPas-TCR: http://friedmanlab.weizmann.ac.il/McPAS-TCR
MIRA: https://clients.adaptivebiotech.com/pub/covid-2020
PyMOL: https://www.schrodinger.com/products/pymol
VDJdb: https://vdjdb.cdr3.net/
Glossary
- Area under the receiver-operating characteristic curve
-
(ROC-AUC). ROC-AUC and the area under the precision–recall curve (PR-AUC) are measures of model tendency to different classes of error. These plots are produced for classification tasks by changing the threshold at which a model prediction falling between zero and one is assigned to the positive label class, for example, predicted binding of a given T cell receptor–antigen pair. ROC-AUC is the area under the line described by a plot of the true positive rate and false positive rate. ROC-AUC is typically more appropriate for problems where positive and negative labels are proportionally represented in the input data. PR-AUC is the area under the line described by a plot of model precision against model recall. PR-AUC is typically more appropriate for problems in which the positive label is less frequently observed than the negative label.
- Library-on-library screens
-
Experimental screens that permit analysis of the binding between large libraries of (for example) peptide–MHC complexes and various T cell receptors.
- Machine learning models
-
A broad family of computational and statistical methods that aim to identify statistically conserved patterns within a data set without being explicitly programmed to do so. Machine learning models may broadly be described as supervised or unsupervised based on the manner in which the model is trained. Many recent models make use of both approaches.
- Neural networks
-
A family of machine learning models inspired by the synaptic connections of the brain that are made up of stacked layers of simple interconnected models. Although each component of the network may learn a relatively simple predictive function, the combination of many predictors allows neural networks to perform arbitrarily complex tasks from millions or billions of instances. Neural networks may be trained using supervised or unsupervised learning and may deploy a wide variety of different model architectures. Deep neural networks refer to those with more than one intermediate layer.
- Shuffling
-
In the absence of experimental negative (non-binding) data, shuffling is the act of assigning a given T cell receptor drawn from the set of known T cell receptor–antigen pairs to an epitope other than its cognate ligand, and labelling the randomly generated pair as a negative instance.
- Supervised learning
-
Models that learn a mathematical function mapping from an input to a predicted label, given some data set containing both input data and associated labels. Common supervised tasks include regression, where the label is a continuous variable, and classification, where the label is a discrete variable.
- Synthetic peptide display libraries
-
Experimental systems that make use of large libraries of recombinant synthetic peptide–MHC complexes displayed by yeast30, baculovirus32 or bacteriophage33 or beads35 for profiling the sequence determinants of immune receptor binding. Peptide diversity can reach 109 unique peptides for yeast-based libraries.
- Training data
-
The training data set serves as an input to the model from which it learns some predictive or analytical function.
- Unsupervised learning
-
Models that learn to assign input data to clusters having similar features, or otherwise to learn the underlying statistical patterns of the data. Unlike supervised models, unsupervised models do not require labels. Common unsupervised techniques include clustering algorithms such as K-means; anomaly detection models and dimensionality reduction techniques such as principal component analysis80 and uniform manifold approximation and projection.
- Validation
-
Analysis done using a validation data set to evaluate model performance during and after training. A given set of training data is typically subdivided into training and validation data, for example, in an 80%:20% ratio. Models may then be trained on the training data, and their performance evaluated on the validation data set.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hudson, D., Fernandes, R.A., Basham, M. et al. Can we predict T cell specificity with digital biology and machine learning?. Nat Rev Immunol 23, 511–521 (2023). https://doi.org/10.1038/s41577-023-00835-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-023-00835-3
This article is cited by
-
Can AlphaFold’s breakthrough in protein structure help decode the fundamental principles of adaptive cellular immunity?
Nature Methods (2024)
-
Development and use of machine learning algorithms in vaccine target selection
npj Vaccines (2024)
-
Adaptive immune receptor repertoire analysis
Nature Reviews Methods Primers (2024)
-
Deep learning predictions of TCR-epitope interactions reveal epitope-specific chains in dual alpha T cells
Nature Communications (2024)
-
Bioengineering translational models of lymphoid tissues
Nature Reviews Bioengineering (2023)
