
Abstract
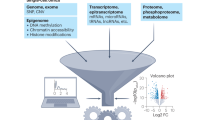
The joint analysis of the genome, epigenome, transcriptome, proteome and/or metabolome from single cells is transforming our understanding of cell biology in health and disease. In less than a decade, the field has seen tremendous technological revolutions that enable crucial new insights into the interplay between intracellular and intercellular molecular mechanisms that govern development, physiology and pathogenesis. In this Review, we highlight advances in the fast-developing field of single-cell and spatial multi-omics technologies (also known as multimodal omics approaches), and the computational strategies needed to integrate information across these molecular layers. We demonstrate their impact on fundamental cell biology and translational research, discuss current challenges and provide an outlook to the future.
Similar content being viewed by others

Introduction
Humans and many other Eukaryota are composed of billions of cells, belonging to vastly heterogeneous cell types and functional cell states determined by both cell-intrinsic and cell-extrinsic factors. Intrinsically, there is a complex interactive molecular hierarchy of the different ‘omics’ layers within a cell: from genome and epigenome to transcriptome, proteome and metabolome, and back. Extrinsically, the functional state of a cell can be modulated by its neighbouring cells through direct physical interaction (such as receptor–ligand interactions), through signalling molecules secreted by one cell that can act through receptors on remote cells (such as morphogen signalling pathways), or by other microenvironmental factors (such as chemical compound gradients)1. Consequently, investigating how multicellular organisms develop from a totipotent single cell and subsequently function, age and develop disease, necessitates single-cell and spatial multi-omics approaches (also known as multimodal omics approaches).
Robust technologies for unimodal (mono-omics) measurements of individual cells, such as single-cell RNA sequencing (scRNA-seq) methods2, have already evolved to revolutionize the discovery and understanding of cell types as well as their different functional cell states, cell plasticity upon exposure to external stimuli and drugs, and cell differentiation or reprogramming trajectories3. The power of these technologies is underscored by the instigation of the Human Cell Atlas (HCA)1,4 and other consortium-based resources afterwards, which are primarily aimed at creating cellular reference maps of organisms, including the position, function and characteristics of every cell type. However, to develop fundamental understanding of the molecular hierarchy from genome to phenome in individual cells, multi-omics methodologies at single-cell and spatial resolution are necessary. They enable investigation of the intermolecular dynamics between gene regulation on the epigenome level and gene expression on the transcriptome and/or proteome levels unambiguously in the same single cells across development, ageing and disease. Additionally, these technologies enable investigating the impact of acquired genetic variation in the genome of single cells on their own function and phenotypic features, as well as (surrounding) tissue function and more, as exemplified throughout this Review. In recent years, this field has advanced phenomenally and is rapidly maturing both technologically and computationally, enabling broad applications to understand cell biology (Fig. 1).

In addition to the year of publication, other key features of the methods are indicated. For single-cell multi-omics assays, this includes the nature of molecular analytes they analyse as well as the method used for cell barcoding. For spatial multi-omics assays, this includes the resolution, sample type, order and number of analytes that can be profiled simultaneously. FFPE, formalin-fixed paraffin-embedded; IVT, in vitro transcription; MATQ, multiple annealing and dC-tailing-based quantitative single-cell RNA-seq; STRT: single-cell tagged reverse transcription sequencing; RT, reverse transcription; TdT, terminal deoxynucleotidyl transferase; TELP, tailing extension ligation and PCR.
In this Review, we discuss the fundamental technological and computational principles, state of the art and applicative value of modern single-cell and spatial multi-omics. We focus on methods that provide a comprehensive ‘omics’ view of at least one molecular analyte, and for single-cell multi-omics are based on next-generation sequencing (NGS), whereas for spatial multi-omics we highlight both NGS-based and imaging-based methods. Although many algorithms have been tailored to analyse the individual molecular layers, here we primarily focus on computational techniques for the integration of information across the different data modalities to maximally leverage the potential of these multi-omics technologies. We end with an outlook to the future.
Single-cell multi-omics methods
Multi-ome measurements from single cells are enabled by different methodological approaches, which can be categorized according to whether the distinct molecular analytes are uncoupled before, during or after sequencing library preparation. As described in Fig. 2 and further below for specific methods, each of these different principles comes with specific advantages and limitations.

All principles are visualized with RNA and DNA as example analytes. Principle 1 is based on physical separation of the distinct molecular analytes (parts a,b). a, Following complete lysis of the isolated cell or nucleus, poly(A) RNA hybridizes to oligo-dT-coated paramagnetic beads, and following magnetic pulldown, the supernatant that contains the genomic DNA is transferred to a new reaction vessel7,9. Alternatively, biotinylated nucleotides are incorporated into RNA-derived cDNA, allowing their capture with streptavidin-coated paramagnetic beads (not shown)42. Advantages include flexibility in downstream processing of DNA and/or RNA and compatibility with intact cells and nuclei from fresh and frozen tissue. Disadvantages include potential loss of RNA and/or DNA molecules during physical separation. b, In an alternative approach, lysis conditions that rupture the cell but not the nuclear envelope allow separation of nuclear from cytoplasmic molecular analytes, either by precipitating the nucleus with centrifugation or magnetic pulldown followed by aspiration of the cytosolic supernatant13,14 or by microfluidic-controlled nucleus-from-cytoplasm separation16,17. Advantages include flexibility in downstream processing of DNA and RNA and availability of non-poly(A) RNA. Disadvantages include loss of nuclear RNA plus some cytoplasmic RNA during nuclear–cytoplasmic separation, loss of mitochondrial analytes, need for intact single cells, incompatibility with frozen tissue and likely incompatibility with mitotic cells (in which the nuclear envelope disaggregates). c, In principle 2, termed preamplification and split, distinct analytes are differentially tagged and jointly preamplified, followed by splitting the preamplification reaction for analyte-specific sequencing library preparations18. Advantages include minimal risk of analyte loss and compatibility with intact cells and nuclei from fresh and frozen tissue. Disadvantages include limited flexibility, as the preamplification protocol needs to be suitable for all analytes of interest, and risk of cross-contaminating molecular analytes. d, Principle 3, termed seq-split, involves analyte-specific barcoding and sequencing library preparation in a single-pot reaction20. Multi-omic information is uncoupled computationally following sequencing. Advantages include minimal risk of analyte loss and compatibility with intact cells and nuclei from fresh and frozen tissue. Disadvantages include that libraries cannot be sequenced separately to optimal depth for each modality, and potential risk of cross-contaminating molecular analytes. e, In principle 4, termed combinatorial indexing, molecular analytes of single cells are tagged without isolating single cells21. Multiple cells or nuclei are deposited per well of a multi-well plate, whereby each cell or nucleus serves as a reaction container. Each receives an analyte-specific tag and a well-specific barcode. By pooling, mixing and randomly re-distributing the cells or nuclei in subsequent rounds of well-specific barcoding, molecular analytes uniquely barcoded per cell are obtained. Combination with concepts of principle 2 and/or 3 achieves single-cell or single-nucleus multi-omics. Advantages include that there is no need for isolating single cells, ability to achieve extremely high throughput, and compatibility with intact cells and nuclei from fresh and frozen tissue. Disadvantages include typically lower sensitivity, risk of analyte loss and limited flexibility in whole-genome and whole-transcriptome amplification protocols. dsDNA, double-stranded DNA; NGS, next generation sequencing; poly(A), polyadenylated.
Single-cell genomics-plus-transcriptomics
Soon after the establishment of single-cell DNA sequencing (scDNA-seq) and single-cell RNA sequencing (scRNA-seq) protocols2,5, methods for genome-plus-transcriptome sequencing of individual cells were developed (Fig. 1) that rely on one of the four basic principles for multi-ome analysis (Fig. 2).
A first set of methods applies physical separation of DNA and RNA before sequencing library preparation (Fig. 2a). In G&T-seq6,7 (Supplementary Fig. 1a), oligo-dT bead-mediated precipitation of polyadenylated (poly(A)) RNA molecules enables their physical separation from the remaining molecules in the cell’s lysate, including the nuclear and mitochondrial DNA, either manually or robotically. Captured transcripts are then on-bead primed for full-length cDNA amplification through reverse transcription (RT), template switching and PCR using a Smart-seq2-like reaction8, allowing for both short-read and long-read sequencing7. Long-read sequencing is preferred for transcript isoform detection. The genomic DNA (gDNA) in the collected supernatant is subjected to whole-genome amplification (WGA) using a method of choice: multiple displacement amplification (MDA), PCR or displacement preamplification followed by PCR (DA-PCR). Single-cell transcriptogenomics applies a similar principle as G&T-seq and is compatible with targeted DNA exome sequencing following MDA9. The freedom of choice for downstream processing of the separated poly(A) RNA and gDNA presents a major advantage. Indeed, WGA is not error-free, and different WGA methods present different biases, making some more suitable for the detection of specific classes of genetic variants, as reviewed previously10,11. Similarly, different scRNA-seq methodologies present different performances in sensitivity, transcript coverage and throughput12.
As alternatives to oligo-dT bead-based separation of poly(A) RNA and gDNA, multiple methods partition cytosolic RNA from nuclear DNA using a two-step cell lysis (Fig. 2b). In SIDR-seq13 (Supplementary Fig. 1b), single cells are first subjected to hypotonic lysis, enabling the nucleus to be isolated from the supernatant containing RNA using antibody-conjugated magnetic microbeads. Then, the nucleus is lysed and subjected to MDA-based scDNA-seq, and the supernatant RNA is subjected to Smart-seq2-based scRNA-seq. In DNTR-seq14 (Supplementary Fig. 1c), the nuclei of single cells are precipitated by centrifugation after cell membrane lysis. While the supernatant cytosol is isolated for Smart-seq2-like scRNA-seq, the nuclear DNA is subjected to direct tagmentation. The latter enables direct PCR-based library preparation for scDNA-seq, thereby circumventing the classic approach of WGA before scDNA-seq library preparation and in part the resulting artefacts associated with it14,15. However, these nuclear–cytosolic partitioning methods are less amenable to comprehensive characterization of mitochondrial DNA and nuclear RNA, and they are confined to the use of intact cells as input. Furthermore, plate-based assays such as G&T-seq, transcriptogenomics, SIDR-seq and DNTR-seq are inherently low throughput and, despite (partial) automation on liquid-handling robotics, still require up to a few days’ time6. To circumvent this, integrated on-chip microfluidics approaches have been devised that enable separation and parallel processing of cytosolic RNA and nuclear DNA16,17. Such systems largely avoid the introduction of operator bias, can miniaturize the reactions and hence reduce reagent costs, and have the capacity to increase throughput, although this remains to be shown.
Another set of methods relies on a preamplification-and-split approach for uncoupling DNA and RNA molecular analytes (Fig. 2c). In DR-seq18 (Supplementary Fig. 1d), poly(A) RNA-derived first-strand cDNA is subjected to quasilinear amplification together with the gDNA in a single tube. This reaction is then split, with both aliquots containing preamplified cDNA and gDNA. In one, the preamplified gDNA is further PCR-amplified and converted to a sequencing library, with the caveat that contaminating cDNA will be co-amplified and sequenced into reads indistinguishable from the gDNA sequences. In the sister reaction, the cDNA is further amplified using in vitro transcription (IVT), followed by RT and PCR. Because only the cDNA is tagged with T7 promoter sequence for IVT, gDNA will not be co-amplified, resulting in a 3′-end scRNA-seq library. In the more recently developed TARGET-seq19, a method for targeted mutation detection and parallel transcriptome characterization of the same single cells, poly(A) cDNA is co-amplified with a mutation of interest targeted by cDNA primers and gDNA primers in a single reaction. This pot is then split to prepare the genotyping and transcriptome sequencing libraries separately. Although DR-seq and TARGET-seq minimize the risk of losing (deoxy)ribonucleic acids in comparison to above-described methods relying on physical separation of DNA and RNA, they can suffer from RNA-derived reads contaminating the gDNA analysis, and are also plate-based and low throughput.
Instead, scONE-seq20 (Supplementary Fig. 1e) follows the principle of seq-split (Fig. 2d) and differentially barcodes gDNA and RNA through, respectively, tagmentation with a 6-nucleotide DNA-barcode-containing adaptor and reverse transcription with a 6-nucleotide RNA-barcode-containing RT-primer. Differentially labelled gDNA and cDNA is then co-amplified and converted to a sequencing library in a single-tube reaction. Following NGS, gDNA-derived and RNA-derived reads are distinguished by their barcode sequence. This methodology hampers sequencing RNA-seq and DNA-seq libraries separately to optimal depths and is plate-based and low throughput, but indicates that relatively simple one-tube reactions are possible for multi-omics measurements of single cells.
Recently, a highly scalable plate-based technology was established based on the principle of single-cell combinatorial indexing (Fig. 2e) with a three-level indexing scheme and combined with linear IVT-based amplification of the cells’ genome and transcriptome (Supplementary Fig. 1f). This sci-L3-RNA/DNA co-assay enables analysis of at least tens of thousands of single nuclei, with the possibility to increase the throughput to more than 1 million cells profiled per experiment21. Proof of concept of sci-L3-RNA/DNA was presented on mixtures of male mouse and female human cell lines, proving that the single-cell transcriptomes were organized into the two expected cell clusters and that the matching single-cell genomes were of the correct sex, although high-sensitivity and high-resolution profiling per cell was not demonstrated.
The study of both the genome and the transcriptome of the same cell enables one to unambiguously investigate the impact of acquired DNA mutations, such as DNA copy number aberrations, on gene expression in the same cell. This has important applications for understanding intratumoural heterogeneity, enabling the investigation of the development of different phenotypic cancer cell states among the different genetic subclones that arise, or even within a single genetic subclone. For instance, DNTR-seq identified minor subclones having genetic copy number alterations with associated transcriptional perturbations in paediatric acute lymphoblastic leukaemia14. Additionally, transcriptional signatures of the WNT pathway activation learned from scRNA-seq could be explained by mutations detected in the scDNA-seq data from the same cells17. Furthermore, using tumour model systems exposed to treatment, or direct longitudinal sampling of patient tumour specimens before and during treatment, and analysing them by single-cell genome-plus-transcriptome sequencing will allow investigation of which genetic subclones are more fit to tolerate the drug selection. Additionally, it will allow the study of how cells within these genetic subclones putatively apply cell plasticity to change their gene expression repertoire and accommodate different phenotypic cancer cell states able to withstand drug treatment and, eventually, acquire resistance22. In turn, these approaches might enable the identification of potential cancer cell vulnerabilities, such as druggable molecular players involved in the acquisition of drug tolerance.
Beyond the field of oncology, these multi-omics methods are important for understanding the pathogenesis of other disorders in which somatic genetic variation plays a putative role, including in neurological disorders such as Alzheimer disease, Parkinson disease and others23,24,25. With the recent discovery that normal tissues are also subjected to an extraordinary amount of mutation, technologies enabling the analysis of the genome and transcriptome of the same single cells will be important to study the role or impact of acquired mutations on phenotypic and functional cellular states, and how these in turn impact development26, cellular competition between normal and genetically aberrant cells27, tissue homeostasis, normal phenotypic variation and ageing28. The power of multi-omics methods in this respect is illustrated by the detection of different transcriptional responses to acquired DNA copy number aberrations6,7,14,18 and other forms of mutations9. For instance, it was shown using DNTR-seq that structural DNA imbalances lead to both linear and nonlinear transcriptional dosage effects, whereby several genes important for cancer cell growth, such as MYC and TCF7L2, demonstrated strong dosage compensation and were shown to be mostly unaffected by copy number alterations14.
Single-cell genome-plus-transcriptome sequencing is also a valuable tool to study the efficacy and safety of genome editing in germline therapy. CRISPR–Cas9 genome editing has potential as a therapeutic tool for the correction of disease-causing mutations. Genome editing of human embryos or germ cells provides the means for introducing heritable genetic alterations, which may reduce the burden of genetic disease in specific familial situations29. Its use is currently a hot topic of international debate around ethics, safety and efficacy. Single-cell genome-plus-transcriptome sequencing approaches will be pivotal to assess on-target and off-target genome edits plus cell phenotypic consequences, as only a few cells are available for analysis from the treated and subsequently in vitro-cultured human embryo. Recently, OCT4 (also known as POU5F1) CRISPR–Cas9-targeted and control human preimplantation embryos were investigated using single-cell G&T-seq30,31 as well as single-cell or low-input mono-omics DNA-seq. Regions of loss of heterozygosity in genome-edited cells that spanned beyond the OCT4 on-target locus, as well as segmental loss and gain of the OCT4-containing chromosome 6, were detected in the genomic data, which collectively resulted in unintended genome edits being identified in ∼16% of the human embryo cells analysed. The transcriptome data suggested that the loss of heterozygosity does not lead to the misexpression of other genes adjacent to the OCT4 locus.
Furthermore, genome-plus-transcriptome sequencing approaches allow genetic variation detected in DNA sequences to be confirmed in the RNA sequences of the same cell, increasing the reliability of the genotyping call7,9,13. This principle has been shown for the detection of single nucleotide variants contained within expressed genes, forms of structural variation resulting in the expression of fusion genes, and copy number variants resulting in gene expression dosage effects. The genomic and mitochondrial DNA variants detected by genome-plus-transcriptome sequencing approaches, considering potential imperfections15, can furthermore be leveraged for the construction of a genetic lineage tree of the cells, which can be annotated with cell type and functional phenotypic states of cells using the RNA sequencing data of the same cells. As an alternative to analysing naturally occurring somatic mutations, high-throughput methods relying on CRISPR-scarring are available in model systems32, which through recent improvements may enable accurate lineage recording as well as the capturing of ancestral transcriptional states33,34. In combination with scRNA-seq readouts, these promise to revolutionize our understanding of cellular differentiation trajectories.
Single-cell epigenomics-plus-transcriptomics
Advances in the past few years have also moved the mark for methods that can profile a cell’s epigenome and transcriptome in parallel (Fig. 1). As with methods analysing the genome and transcriptome, their original designs relied on the physical separation of RNA from DNA or the nucleus, and the subsequent profiling of these separated fractions. More recently, methods for the differential marking of RNA and epigenetic information encoded in DNA have been described, relying on the separation of reads originating from both through restriction digestion, PCR or molecular barcoding. These later methods do not require upfront separation and are therefore more readily parallelized in higher throughput, via preamplification and split, seq-split or combinatorial indexing principles (Figs. 2 and 3).

Summaries of experimental workflows highlighting how and in what order different modalities are probed and separated for analysis, while retaining single-cell information. Shown are SHARE-seq38 (part a), Paired-seq240 (part b, left) and Paired-Tag48 (part b, right), SNARE-seq39 (part c) and TEA-seq82 (part d). In all methods shown, an assay for transposase-accessible chromatin (ATAC) precedes reverse transcription (RT) of polyadenylated RNA. Cell barcoding can occur through successive rounds of combinatorial indexing (parts a,b) or by compartmentalizing cells and barcoded oligonucleotides in microdroplets (parts c,d). DNA fragments originating from mRNA and DNA can be separated by binding to paramagnetic beads (part a), differential restriction digestion (part b) or using specific PCR primers (parts c,d). Part b illustrates how similar workflows can either map accessible chromatin (left) and chromatin-associated proteins or their post-translational modifications (right). TEA-seq illustrates that barcoded oligonucleotides conjugated to antibodies can be detected using approaches similar to those developed for measuring gene expression. cDNA, complementary DNA; ChIP–seq, chromatin immunoprecipitation followed by sequencing; RNA-seq, RNA sequencing; TdT, terminal deoxynucleotidyl transferase; TSO, template-switching oligonucleotide; UMI, unique molecular identifier.
Most known layers of epigenetic information, including DNA methylation, chromatin accessibility, histone modifications, and binding of transcription factors (TFs) and chromatin remodelling complexes, can be recovered from single cells in parallel to the transcriptome. Such methods abound but range in sensitivity, specificity and ease of use (Fig. 1). The presence of epigenetic modifications can be read either directly from the DNA sequence, as is the case for DNA methylation, or indirectly by first encoding them in the sequence through DNA methylation and/or tagmentation. We discuss the latter approach first, as this strategy is most widely used.
Tagmentation-based methods come in a wide range of types. Most common are methods that jointly profile the transcriptome and chromatin accessibility (Fig. 3). Here, accessible DNA is recovered as transposon-insertion-flanked regions using an assay for transposase-accessible chromatin (ATAC). Examples include scCAT-seq35 and Smart3-ATAC36, which are plate-based; sci-CAR-seq37, SHARE-seq38, SNARE-seq239 and Paired-seq40 (Fig. 3a,b), which rely on combinatorial indexing; SNARE-seq39,41, ASTAR-seq42 and the commercially available 10x Genomics Multiome technology, which rely on microfluidics for cell barcoding (Fig. 3c); and ISSAAC-seq, which is amenable to both plate-based and microfluidics-based cell barcoding43,44. scGET-seq represents an unusual type of such methods, profiling both accessible and inaccessible chromatin but not the transcriptome45. Important features to be considered in tagmentation-based method selection include the ease of use of commercially available methods, the higher throughput of combinatorial indexing-based methods, the typical lower cost (but difficulty of establishment) of non-commercial, laboratory-developed methods, and the coverage, sensitivity and specificity obtained across cellular modalities. A systematic benchmark of these methods is unfortunately currently lacking. In most methods, transcriptome and accessible chromatin libraries are prepared in a common reaction and separated after indexing using magnetic beads, restriction enzymes or specific PCR primers (Fig. 3). A key advantage of the joint profiling of transcriptome and chromatin accessibility is that the link between gene expression and TF binding is more readily evaluable. In hair follicle cells, for example, SHARE-seq analyses of differentiation trajectories revealed TFs becoming expressed, with their activity being revealed in ATAC profiles later in pseudotime as binding sites became accessible, before expression of the associated target genes38. As such, these multi-omic analyses in dynamic systems enable TF activity to be readily assigned to target genes. The ease of use of some of these methods has spearheaded their application in biomedicine and other domains.
However, the aforementioned methods quantify open chromatin without addressing the causes of accessibility changes, such as shifts in histone post-translational modifications (PTMs) or TF binding. To tackle this limitation, other methods have been developed in which tagmentation is not randomly targeting accessible regions but directed towards specific histone PTMs or TFs. Conjugating the transposase to specific antibodies enables joint profiling of transcriptome and epitopes. Cell and modality barcoding can also occur through DNA–RNA separation, preamplification and split, seq-split or combinatorial indexing principles (Fig. 2), with transcriptome and DNA libraries being separated by restriction enzymes, PCR or beads. Examples of such methods include scPCOR-seq46, coTECH47, Paired-Tag (Fig. 3b)48 and scSET-seq49. scCUT&Tag2for150 and scMulti-CUT&Tag51 are distinct variations on these methods, involving two epitopes being targeted using different antibody-conjugated transposases. The distribution of both epitopes in a single cell can be learned from their different genomic distributions (for example, broad or narrow peaks) in scCUT&Tag2for150, or from epitope-specific barcoding enabled by transposases loaded with different oligonucleotides in scMulti-CUT&Tag51. A potent illustration of these methods was the profiling of mouse brain cells for both transcripts and different histone modifications using Paired-Tag, which identified distinct categories of genes each regulated by different epigenetic mechanisms48.
Epigenetic information can also be read through DNA methylation profiling, relying either on 5-methylcytosine (5mC) or N6-methyladenine (6mA) (Fig. 4). 5mC is a prevalent endogenous epigenetic modification of DNA, found almost exclusively in a CpG context, that represses ectopic and heterochronic gene transcription initiation. In the most basic approach, endogenous 5mC DNA methylation is quantified in single cells by bisulfite sequencing. Key issues precluding more widespread adoption of single-cell 5mC analyses are the costs associated with library preparation and whole-genome sequencing, as well as the technical complexity. Indeed, bisulfite treatment of DNA causes its denaturation and fragmentation, and comes with a need for purification, leading to DNA losses. Library preparation can occur either through random-priming-based methods, such as post-bisulfite adapter tagging (PBAT), which is costly but yields genome-wide profiles covering 5–50% of the genome, or through reduced-representation bisulfite sequencing (RRBS), which is more cost-effective but only covers 1–3% of the genome. Recent evolutions in less destructive DNA methylome analytics and target capture may serve to develop more attractive workarounds52,53. DNA methylation profiled on its own through single-cell DNA sequencing enables concomitant chromosomal copy number profiling. More often, DNA methylomes are profiled together with a cell’s transcriptome, chromatin structure and/or chromatin accessibility.

Summaries of experimental workflows highlighting how and in what order different modalities are probed and separated for analysis, while retaining single-cell information. Shown are scNMT-seq59 (part a), snmCAT-seq65 (part b), scMethyl-HiC67 (part c) and EpiDamID with scDam&T-seq73 (part d). In all methods shown, several types of epigenetic information can be discerned from a single sequencing library: DNA methylation and accessibility (part a), DNA methylation, accessibility and gene expression (part b), DNA methylation and chromatin conformation (part c), and histone modifications and transcription (part d). These layers of information can be discriminated by analysing DNA methylation patterns (parts a,b), DNA methylation and read-pair mapping (part c) or read-associated barcode tags (part d). Only scNMT-seq (part a) involves physical separation of modalities to be probed (mRNA from intact cells). Each of these methods is plate-based and therefore restricted in its throughput. DNA methylation patterns can be read using bisulfite conversion of unmethylated cytosines to uracil (U) (parts a–c) or methylation-sensitive restriction digestion (part d), and reflect endogenous methylation alone (part c), endogenous methylation as well as exogenously added methylation reflecting chromatin accessibility (parts a,b), or endogenous adenine methylation added after genetic transformation (part d). cDNA, complementary DNA; Dam, DNA adenine methyltransferase; FACS, fluorescence-activated cell sorting; gDNA, genomic DNA; 5mCTP: 5-methyl-deoxycytidine triphosphate; NGS, next-generation sequencing; RT, reverse transcription; T7, T7 promoter; TdT, terminal deoxynucleotidyl transferase; TSO, template-switching oligonucleotide.
Methods that produce transcriptome profiles alongside targeted DNA methylome profiles — as in scMT-seq, scTrio-seq and Smart-RRBS — or genome-wide DNA methylome profiles — as in scM&T-seq and scTrio-seq2 — have been established54,55,56,57,58. These typically involve physical separation of DNA and RNA (Fig. 2a,b).
Endogenous CpG DNA methylation is often assessed in conjunction with chromatin accessibility. For this, a GpC methyltransferase is added to isolated nuclei where it can methylate open-chromatin-associated accessible DNA (Fig. 4a,b). Subsequent bisulfite sequencing can disclose the methylated GpC dinucleotides that mark these accessible regions, in addition to the endogenous methylation that in most cell types is nearly exclusive to CpG dinucleotides. GpC methylase-based methods show higher coverage per promoter than ATAC-based methods59, albeit at substantially higher sequencing cost per cell, and make it easier to distinguish open from truly closed regions, which are not directly detected in the sparse scATAC-seq signals. A limitation relative to regular DNA methylome analyses is that at cytosines flanked on both sides by a guanine, methylation can be attributed to endogenous as well as exogenous processes. These are discarded from analyses. Relevant methods include scCOOL-seq60, iscCOOL-seq61 and scNOME-seq62, which jointly profile accessibility and DNA methylation, and trimodal methods such as scNMT-seq59, scNOMeRe-seq63, scChaRM-seq64 and snmCAT-seq65, which in addition uncover gene expression profiles from the same cell (Fig. 4a,b). All are plate-based assays and based on DNA–RNA separation, apart from snmCAT-seq, which is seq-split based (Fig. 2d; Fig. 4b).
Noteworthy alternatives that do not profile transcriptomes are snm3C-seq66 and scMethyl-HiC67, as they profile the DNA methylome in parallel to higher-order chromatin structure (Fig. 4c). Here, a 3C or HiC-like single-cell method to capture nuclear organization is combined with bisulfite conversion, to enable joint profiling of DNA methylation and chromatin structure66,67. Methods that jointly also capture the transcriptome have yet to be described.
Despite being challenging techniques, the multi-omics nature of the resultant data enables a very in-depth analysis of cells, revealing hierarchies of changes to the epigenome and transcriptome during development, differentiation or pathogenesis. For example, stages of mouse gastrulation were profiled using scNMT-seq, which demonstrated that DNA methylation and chromatin accessibility patterns at ectodermal enhancers are pre-established in epiblast cells and stable during ectoderm differentiation, whereas mesodermal and endodermal enhancers are inactive in epiblast cells but actively remodelled following differentiation to mesoderm or endoderm68. Accessibility and DNA methylation changes seemed to be tightly coordinated. These studies illustrate that multi-omics profiling of single cells for transcriptomic and epigenetic layers is feasible and provides fundamental insights unattainable using mono-omics methods.
Apart from methylated cytosines, methylation of adenines to 6mA can also be profiled in DNA (Fig. 4d). In contrast to 5mC, 6mA is an ultra-rare base in mammalian DNA69 and can thus provide nearly unambiguous DNA marking when artificially introduced. Typically, a prokaryotic DNA adenine methyltransferase (Dam) is used to methylate adenines in GATC context for DNA-sequence encoding of epigenetic information. Dam can either be expressed to mark accessible chromatin70,71,72 or be tethered to endogenously expressed proteins using insertion mutagenesis to mark their binding sites in live cells. Two DNA–RNA separation-based methods applying adenine methylation to 6mA have been described: scDam&T-seq, with Dam tethered to chromatin-associated proteins to mark their nuclear location (DamID)70, and scDam&T-seq with EpiDamID, in which Dam is tethered to protein domains or nanobodies that recognize PTMs73 (Fig. 4d). As 6mA cannot be detected directly through short-read sequencing, quantification is relative, relying on Dam activity and a 6mA-specific restriction digest. In contrast to the other chromatin profiling methods described above, the resulting profiles typically reflect the aggregate of residence times of the proteins marked, leading to better signal-to-noise ratios but poorer temporal resolution. The reliance on endogenous tethering limits the scope of these methods to systems amenable to transgenesis. Interestingly, it extends their scope beyond only those proteins for which a specific antibody is available, and at least scDam&T-seq enables a method of analysing chromatin occupancy that is orthogonal to antibody-based methods. Additionally, these methods may suffer less from the bias towards accessible chromatin that characterizes transposase-based tagging approaches. Interestingly, these methods share several features with Fibre-seq-based approaches74, in which chromatin patterns are read through 6mA marking using single-molecule long-read sequencing. Combining both methods may thus facilitate high-throughput, haplotype-resolved and cell-type-specific characterization of chromatin structures in bulk.
Single-cell omics plus low-plex profiling of another analyte
Aside from profiling multiple comprehensive omics layers from the same cell, substantial progress was recently made in profiling single cells comprehensively for a single analyte and in a less comprehensive (low-plex) manner for another analyte. Most common among these methods are those quantifying the cell’s transcriptome and/or accessible genome, alongside a limited set of cell surface or intranuclear proteins. These methods typically rely on antibodies tagged with a specific barcoded oligonucleotide, which can be captured and amplified alongside the transcriptome. Using mixes of dozens to hundreds of such barcoded antibodies that each recognize specific epitopes, subsequent barcode counting thus enables quantification of multiple proteins of interest in single cells alongside true omics analysis. Such methods enable the profiling of protein abundances in addition to gene expression or other modalities, but can also capture other information about proteins such as protein stability, PTMs and protein isoform expression. Omics layers shown to be amenable to this include the transcriptome (REAP-seq75, CITE-seq76, inCITE-seq77, SPARC78, ECCITE-seq79 and RAID-seq80), open chromatin (ASAP-seq81 and ICICLE-seq82) or both (DOGMA-seq81 and TEA-seq82), as well as chromatin modifications (scCUT&Tag-pro83) (Fig. 3d). Epitopes available for profiling are mostly limited to the cell surface, although methods for intracellular (SPARC78 and RAID-seq80) and intranuclear (inCITE-seq77 and NEAT-seq84) epitopes have also been developed. Note that any plate-based method can theoretically also leverage antibody marking and cytometry to quantify a limited set of proteins per cell. A notable alternative approach (PHAGE-ATAC) was described recently in which barcoded phages that display a nanobody serve to bind cell surface epitopes85. The barcoded phage genomes can be quantified in conjunction with genome-wide chromatin accessibility profiles. Each of these methods is currently limited by the availability of specific antibodies or nanobodies, and although mass spectrometry-based methods for proteome-wide analysis of single cells have been developed86, such analyses alongside other omics layers are currently lacking. A related method recently described is single-cell transcriptome and translatome sequencing (T&T-seq), in which cells are sorted into plates and lysates are distributed for total RNA-seq and for affinity purification of actively translating ribosomes. T&T-seq thus enables joint profiling of all transcripts and those transcripts that are being translated into proteins87.
Finally, other molecular features have also been shown to accommodate barcoding, with guide RNAs as a key example. In cells expressing CRISPR-based gene editing, activation or inactivation systems, feature barcoding methods enable high-throughput profiling of the transcriptome or accessible chromatin in pools of single cells subjected to high-throughput genetic perturbation screens. Examples include CROP-seq, in which the guide RNA sequence is directly determined alongside the cell’s transcriptome88, and CRISP-seq89 and Perturb-seq90,91, in which each guide RNA has a unique barcode that is sequenced alongside the transcriptome. The latter two methods have lower specificity, as recombination can blur the link between barcode and guide RNA, and more recent implementations of Perturb-seq therefore apply direct guide-RNA sequencing. Methods to profile the impact of such perturbations on chromatin accessibility include CRISPR-sciATAC92, Perturb-ATAC91 and Spear-ATAC93. Recently, this approach was applied at scale when millions of single-cell transcriptomes were analysed, with each expressed gene inactivated in a subset of these cells. This strategy revealed the effect of inactivation of each expressed gene on the expression of all other genes, and thus represents a valuable resource for the in silico modelling of genetic perturbations94. Notably, a similar approach can be deployed to characterize gene interactions95. Here, cells are transfected with, on average, two different guide RNAs from a pool, generating pools of double-knockout cells. The combined impact of both perturbations can then be compared with the impact of single perturbations, providing direct quantification of a matrix of genetic interactions.
Spatial multi-omics methods
Methodologies for spatial multi-omics are developing rapidly to allow the study of different molecular analytes at up to subcellular resolution within their native tissue context (Fig. 1). Spatial multi-omics technologies were listed by Nature as one of the seven technologies to watch in 202296, with the basis for their development and ongoing innovations being a range of established spatial mono-omics methods (Box 1). Spatial multi-ome characterization of a sample, usually a fixed fresh-frozen or formalin-fixed paraffin-embedded (FFPE) tissue section, is often achieved by combining these spatial mono-omics methods. They can be applied separately on adjacent tissue sections, serially on the same tissue section if the quality of the different analytes can be maintained, or in parallel on the same tissue section if joint targeting and reading out of the different analytes is possible. The number of target analytes that can be analysed simultaneously at the different molecular levels varies between methodologies (Fig. 1). Often spatial omics measurements are also supplemented with histological stains, such as H&E (haematoxylin and eosin) staining, of the same or adjacent tissue sections, allowing integration with additional morphological annotations.
Spatial multi-omics via adjacent-section strategies
The application of spatial assays for different mono-omics layers of interest (Box 1) on adjacent or serial sections from the same tissue sample enables these techniques to be assayed in their most optimal setting and their data to be integrated computationally (Fig. 5a). This requires sample collection and preparation procedures compatible with all assays of interest, well-thought-out experimental design and good assay coordination. This strategy is mostly required when multi-omics readouts of interest are not possible yet or suboptimal on the same section. Mass spectrometry imaging (MSI) methods97, for example, can be used for spatial metabolome characterization of small biomolecules such as lipids (Box 1), but cannot easily be combined with other spatial genome, transcriptome or proteome readouts on the same section owing to specific sample preparations and limitations (for example, the need for matrix deposition in MSI97, OCT compound embedding complicating mass spectrometry98 and inherent breakdown of lipids during potential upfront assays). Conversely, MSI is compatible with H&E staining on the same section, which can also be leveraged for computational integration with other spatial assays. Additional serial sections can also be dissociated into single cells or nuclei for generating matched single-cell sequencing data that can be used for optimal deconvolution of the spatial data and additional data integration (Fig. 5a). As such, a combination of different single-cell sequencing and spatial transcriptomics and proteomics approaches was applied and integrated to assemble an atlas of healthy and obese murine and human livers and to map hepatic macrophage niches99. Disadvantages of this approach are that not all assays are compatible with all sample types (for example, fresh-frozen versus FFPE), that it suffers from sample heterogeneity, as even adjacent sections will differ slightly as to structure and cellular composition, and that different spatial assays may have different resolutions. Although computational tools exist that can compensate for these challenges (see below), the concordance between sections is never unambiguous. Hence, innovative methods enabling spatial multi-omics measurements on the same tissue sections have started to emerge.

Spatial multi-ome profiling of tissue samples can be achieved by applying spatial mono-omics assays separately on adjacent or serial tissue sections (part a) or in a combined way on the same tissue section (parts b–e). a, Serial fresh-frozen or formalin-fixed paraffin-embedded (FFPE) tissue sections can be analysed using different spatial mono-omic assays, potentially also combining with morphological stainings and annotations on the same or adjacent sections, followed by computational data integration. b, Microfluidic deterministic barcoding strategies in tissue allow next-generation sequencing (NGS)-based spatial multi-omics profiling of transcriptome-plus-proteins, as in DBiT-seq103 and Spatial-CITE-seq122, and epigenome-plus-transcriptome, as in ATAC&RNA-seq and CUT&Tag-RNAseq100. Using dual microfluidic chip-based spatial barcoding of poly(A) RNAs together with proteins or epigenome information at the crossroads of chip channels, a spatially barcoded 2D pixel map of the tissue is created. c, Advanced fluorescence in situ hybridization (FISH)-based methods, including MERFISH107,108,111 and seqFISH+109,110,112, allow microscopy-based identification of thousands of transcripts together with genomic loci in single cells, in addition to being compatible with limited protein readouts using fluorescent or DNA-conjugated antibody readout strategies. These high-resolution imaging methods leverage predefined optical barcoding schemes and complex encoding and readout probe designs. d, Array-based assays, including Spatial Transcriptomics170 (ST) and 10x Genomics Visium115, make use of slides with arrayed oligo-dT spots for capturing and spatial barcoding of poly(A) RNAs followed by NGS profiling. This can be combined with upfront haematoxylin and eosin (H&E) staining or limited protein antibody staining and tissue imaging for spatial mapping. In SM-Omics117 and SPOTS116, these technologies have also been shown to be compatible with antibody-derived tag (ADT)-conjugated antibody-based co-profiling of a larger number of proteins. e, NanoString GeoMx digital spatial profiling (DSP)118,119,121 allows quantification of RNAs and proteins in specific regions of interest (ROIs) by counting uniquely barcoded oligonucleotides that are covalently linked through a UV-photocleavable linker with probes or antibodies. Tissue marker staining, imaging, ROI selection and illumination by directed UV light causes disintegration of the photocleavable linkers that are collected and profiled by NGS, followed by spatial mapping to the ROIs. cDNA, complementary DNA; gDNA, genomic DNA; OCT, optimal cutting temperature compound; UMI, unique molecular identifier.
Spatial (epi)genomics-plus-transcriptomics
The possibility for simultaneous unbiased profiling of chromatin accessibility or specific histone modifications and gene expression on the same tissue cryosections has been described for spatial ATAC&RNA-seq and spatial CUT&Tag-RNA-seq, respectively100. These methods are based on combining microfluidic deterministic barcoding in tissue (DBiT) strategies (Box 1) for spatial-ATAC-seq101 or spatial-CUT&Tag102 with DBiT-seq poly(A) transcript profiling103 (Fig. 5b). For capturing chromatin accessibility, accessible gDNA is first tagmented in situ with a universal ligation linker. For capturing specific histone modifications, the tissue is first incubated with primary antibodies against the epigenetic marks, followed by a secondary antibody that allows protein A-transposome tethering for tagmentation of the DNA at these specific locations. Combined mapping of the transcriptome is achieved by hybridizing a biotinylated oligo-dT that also contains a universal ligation linker and primes RT in situ100 (Fig. 5b). DBiT-seq barcoding with serial attachment to the tissue of two microfluidics chips with equidistant channels perpendicular to each other is then used to administer two sets of channel-specific barcodes (Box 1), with the first set ‘A’ ligating with the universal ligation linkers present on the tagmented DNA and poly(A) mRNA-derived cDNA, and the second set ‘B’ ligating to the set ‘A’ barcodes, resulting in an in situ 2D grid with uniquely barcoded AB crossroads (20–25-µm pixels)100. The spatially barcoded gDNA and cDNA fragments are collected by reverse crosslinking, cDNA is enriched with streptavidin-coated magnetic beads, gDNA is retained in the supernatant, and NGS libraries are constructed separately for sequencing (Fig. 5b). Based on the spatial barcodes, sequencing reads are combined with microscopy images of the tissue section, allowing the multi-omics sequence information to be mapped spatially100. Limitations of these assays are the near-single-cell resolution (20–25-µm pixel size), the small analysable area (2,500–10,000 pixels), the uncharacterized spaces in between adjacent pixels (depending on channel distances) and the expertise that is required in fabricating and handling microfluidics chips for implementation. Nevertheless, these methods were successfully applied to developing mouse and adult human brains, revealing how epigenetic states or modifications regulate cell type, states and dynamics100.
Alternatively, microscopy-based methods can enable spatial profiling of genome or epigenome information together with gene expression by directly imaging DNA loci, chromosomal and nuclear structures, and transcripts within single cells at up to subcellular resolution. Several methods showing imaging-based multi-omic measurements for a limited number of combined analytes exist104,105,106, but we focus on those that can characterize at least one analyte layer more comprehensively. Multiplex single-molecule fluorescent in situ hybridization (smFISH) methods, such as MERFISH107,108 and seqFISH109,110 approaches (Box 1), originally designed for targeted high-sensitivity spatial profiling of thousands of transcripts at subcellular resolution, were adapted to allow genome-scale chromatin tracing as in DNA-MERFISH111 and DNA-seqFISH+112. Combined RNA, chromatin and nuclear body imaging in the same fibroblast cells was enabled by sequentially staining for >1,100 nascent transcripts using RNA-MERFISH, followed by DNA-MERFISH for >1,000 genomic loci, and finally fluorescent antibody readouts for cell-cycle-state determination and landmark nuclear structures, including nuclear speckles and nucleoli111 (Fig. 5c). This approach allows for the characterization of chromatin domains, compartments and trans-chromosomal interactions and their relationship to transcription in single cells111. Similarly, DNA-seqFISH+ imaging of up to 3,660 chromosomal loci in mouse embryonic stem cells was shown to be compatible with RNA-seqFISH of 70 mRNAs and intron-seqFISH of 1,000 genes at their nascent transcription active sites, as well as sequential immunofluorescence targeting of 17 nuclear structures, including the nuclear lamina, nucleolus and histone modification marks, using primary antibodies conjugated with DNA-oligonucleotides detectable by fluorescently labelled readout probes112 (Fig. 5c). In addition to identifying heterogeneity in chromosome structure, this study found that many active gene loci reside at the surface of nuclear bodies and the presence of persistent global chromatin states112. Also, OligoFISSEQ methods allow rapid in situ sequencing-based visualization (Box 1) of multiple genomic loci in single cells, with the potential for genome-wide application and being compatible with immunofluorescence and other FISSEQ-based methods for protein and RNA characterization, respectively113. Although these microscopy-based methods are important for studying chromosomal and nuclear structures and organization linked to gene expression regulation at a single-cell level, they require expertise in complex optical barcoding schemes and high-resolution imaging modalities, are challenging to apply in complex tissue samples, are costly and usually can only characterize a limited area.
Alternatively, by using laser capture microdissection (LCM)-based isolation of specific (single) cells from tissue sections, single-cell sequencing-based genome-plus-transcriptome or epigenome-plus-transcriptome profiling methods as described above can be applied at spatial resolution, as was done for analysing tumour development, metastasis and prognosis in patients with triple-negative breast cancer114.
Spatial transcriptomics-plus-proteomics
Methodologies allowing parallel spatial interrogation of both the transcriptome and proteome are currently still limited, are often based on serial characterization of both modalities, mostly allow co-characterization of only a limited number of proteins and often lack single-cell resolution. For example, the commercial array-based 10x Genomics Visium technology for poly(A) RNA capture and spatial barcoding at 55-µm resolution followed by NGS identification (Box 1) currently supports immunofluorescence protein detection of one or two targets on the same fresh-frozen or FFPE tissue section, although at the cost of the otherwise applied H&E staining used for spatial mapping115 (Fig. 5d). However, Spatial PrOtein and Transcriptome Sequencing (SPOTS) demonstrates that Visium is also compatible with co-profiling of a larger number of proteins using polyadenylated antibody-derived tag (ADT)-conjugated antibodies (Box 1), as was shown for 21 proteins in mouse spleen and breast cancer tissue cryosections116. Similarly, Spatial Multi-Omics (SM-Omics) shows Spatial Transcriptomics (the forerunner of Visium with 100-µm resolution) to be compatible with DNA-barcoded antibody and/or immunofluorescence co-mapping of six proteins, with the added concept of full automation using liquid-handling robotics achieving increased throughput and performance117.
Alternatively, NanoString GeoMx Digital Spatial Profiling (DSP) allows quantification of the abundance of RNAs and/or proteins by counting unique indexing oligonucleotides, which are covalently linked via a UV-photocleavable linker with probes or antibodies that target transcripts or proteins of interest, respectively118,119 (Fig. 5e). Whole-transcriptome DSP assays or DSP assays for >100 protein targets are possible, although coupled to image-based selection of specific regions of interest following fluorescent antibody and/or RNAscope120 marker stainings. Selected regions of interest are illuminated by directed UV light, which cleaves the photocleavable linkers and releases oligonucleotide indices, enabling their collection and identification using NGS. Achieving single-cell resolution is challenging with this technology, but the combination of both transcriptome and protein readouts on the same sample was shown in FFPE cell pellet array sections stained with the GeoMx Human Whole Transcriptome Atlas (WTA) probe set and a 59 GeoMx antibody panel121.
Chip-based DBiT-seq approaches (Box 1) also allow co-mapping of poly(A) mRNAs with proteins in tissue cryosections, as shown in the original DBiT-seq method with ~10-μm pixel resolution and tens of proteins co-profiled103, but improved upon in spatial-CITE-seq (spatial co-indexing of transcriptomes and epitopes), in which whole-transcriptome and co-mapping of ~200–300 proteins was achieved at 20-μm pixel resolution122 (Fig. 5b). Both these methods employ cocktails of ADT-conjugated antibodies (Box 1) each bearing a poly(A) oligonucleotide with an antibody-specific barcode. The channel-specific set ‘A’ barcodes, delivered by the first chip with equidistant channels placed on the tissue, consist of barcoded poly(T) DNA adapters also containing a universal ligation linker that hybridizes to both poly(A) ADT and mRNA molecules, and are then incorporated during in situ RT. The second chip introduces perpendicularly a set of channel-specific set ‘B’ barcodes that are ligated to the ‘A’ barcodes and also contain a PCR handle functionalized with biotin. The specific ‘AB’ barcoded cDNA and ADTs at the channel crossroads (pixels) are collected, purified with streptavidin-coated magnetic beads and prepared for NGS, after which reads for both mRNAs and proteins can be superimposed on an image of the section103 (Fig. 5b). Spatial-CITE-seq was applied for improved tissue mapping to several mouse tissue types, as well as human tonsillar and skin biopsy tissue at the COVID-19 mRNA-vaccine injection site122. Although limited by the same aspects as previously mentioned for DBiT-based methods, including lack of single-cell resolution, to date spatial-CITE-seq is the method that allows for the highest number of proteins to be simultaneously spatially profiled together with the poly(A) transcriptome, with potential for further expansion.
As to microscopy-based approaches, several of the established seqFISH-based, MERFISH-based and in situ sequencing (ISS)-based methods for spatial transcript profiling of hundreds to thousands of targeted genes (Box 1) are also (sequentially) compatible with immunofluorescence or DNA-conjugated antibody protein readouts in the same sample (Fig. 1). For the latter, oligonucleotide sequences compatible with the specific fluorescent readout approaches of the transcriptome profiling methods are mostly used (Fig. 5c)107,111,113,123,124,125. Staining and localization of cell boundaries or other cellular, nuclear or subnuclear markers in these ways can enable more accurate cell segmentation, transcript allocation or resolution of nuclear organization. Additionally, combined spatial transcriptome and protein readouts on the same tissue section are crucial to correlate transcript with protein expression, localization and interactions, to help unravel the cellular mechanisms that govern specific cell types and states. Several platforms that recently became commercially available for automated imaging-based spatial transcriptome profiling of tissue sections at single-cell resolution, including the NanoString CosMx (smFISH-based)126, Vizgen MERSCOPE (MERFISH-based)127 and 10x Genomics Xenium (ISS-based)128 platforms, will allow for imaging-based co-profiling of a few to tens of proteins, making these technologies more accessible.
Data integration
A wide variety of algorithms have been described for the tailored data analysis of individual analytes in single cells. Here, we focus on computational advances that allow the integration of measurements across several modalities, as these algorithms leverage the multi-omic nature of these technologies to enhance our understanding of complex cellular states. Each modality covers different aspects of cellular identity and has its strengths and weaknesses. A principal goal of multi-omics data integration is to achieve robust and sensitive cell type or cell state identification129. This integrated multi-omics view of cellular identity can improve our understanding of differentiation trajectories, their underlying gene regulatory networks, cell–cell interactions, microenvironmental spatial organization, cellular lineages and clonal dynamics. Ultimately, a holistic view of cellular identity disentangles causal relationships between the different molecular layers that give rise to the observed cellular phenotypes. However, to achieve meaningful integration of high-dimensional data modalities, computational and statistical models need to be developed that consider the technical and biological intricacies of these technologies.
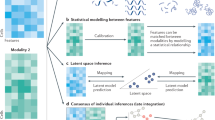
Recently, Argelaguet et al.130 defined three categories of data integration strategies depending on the anchor used to link the different data modalities. Horizontal integration strategies use common data features measured across different datasets to integrate independently assayed groups of cells, such as when assaying different batches with the same technology or when integrating across different technologies measuring the same molecular analyte. Inversely, vertical integration strategies use the cell as the anchoring unit to integrate non-overlapping data features, such as when measuring multiple omics layers of the same cells in parallel (Fig. 6a). The hardest integration problem emerges when neither cells nor common data features can be used as anchors. In this case, diagonal integration strategies are used to map separate groups of cells profiled by different molecular assays (Fig. 6b). Many computational methods have already been developed to tackle horizontal integration problems, as cell atlas efforts require the integration of many batches of single-cell data. For a comparison of these approaches, we refer to recent reviews and benchmark studies130,131,132,133. Here, we focus primarily on vertical integration approaches as these are applicable to most multi-omics technologies in which parallel measurements are made of individual cells.

Examples of different scenarios in which various types of data integration strategies can be used. a, Vertical integration strategies aim to integrate information from paired molecular layers to obtain holistic representations of biological systems, at the single-cell level or at the tissue region level. Here, we illustrate an example of a spatial multi-omics experiment in which mirrored tissue slices have been assayed by two different spatial modalities (yellow and blue). To integrate both data sources, haematoxylin and eosin (H&E) staining images of each modality are first registered to account for deformations during sample preparation. Subsequently, to account for differences in resolutions across modalities, data points are averaged in windows of a predetermined region size154. For every region the averaged regional profiles can be used as paired inputs for either linear or nonlinear vertical integration approaches. In this example, we illustrate integration through the use of a multi-view autoencoder neural network143. Each modality is used as input into a dedicated encoder–decoder network that learns a shared data representation, effectively integrating both data modalities. This shared representation can be used for downstream analysis and/or visualization. The terms gi and mi correspond to the measurements of region i for modality 1 and modality 2, respectively, with g’i and m’i being the molecular profiles reconstructed from the shared representation by the decoder networks. b, Integration of datasets from different experiments with independent observations of individual cells and non-overlapping molecular features is the hardest integration problem and requires diagonal integration approaches. Here, we illustrate this problem for the integration of independently acquired single-cell RNA sequencing (scRNA-seq) and single-cell assay for transposase-accessible chromatin (scATAC-seq) datasets through the use of autoencoder neural networks with a probabilistic coupling to map the different data modalities to a shared latent space144. Although single-cell resolution is lost in this coupling, clusters obtained in this shared latent space can be used to ascertain correlations between molecular layers, discover multimodal biomarkers and/or translate between the different modalities.
Linking molecular layers
When paired data modalities can be unambiguously assigned to individual cells, several integration strategies can be applied depending on the end goal of the analysis. For finding significant cross-modal associations between specific features (coined local integration methods by Argelaguet et al.130), classic regression-based statistical models can be fitted in a supervised way134. These models can be expanded to handle common confounders (such as population stratification biases, sample-specific effects and sequence context-dependent effects), which might inflate the resulting association statistics, by explicitly modelling these confounders using linear hierarchical mixed models135. However, hypothesis testing the very large number of possible correlations of analytes between different molecular layers would require unattainable sample sizes after multiple testing correction. Therefore, the hypothesis search space is often constrained by previous biological information (for example, the distance from the epigenetic marker or accessible regulatory element to the expressed gene). These approaches can be used to identify, for example, cell-type-specific allelic effects of genetic variation on gene expression (expression quantitative trait loci (eQTLs)) in differentiating induced pluripotent stem cells (iPS cells)136 or the effect of genetic perturbations on gene expression using multiplexed CRISPR technologies to identify cis-acting enhancer–gene pairs137. Local integration approaches can also be used to facilitate the inference of gene regulatory networks (GRNs). Classic GRN inference approaches, using only scRNA-seq data, identify sets of co-expressed genes enriched with particular upstream regulatory motifs138. However, this approach is prone to false-positive gene associations as the presence of these motifs does not necessarily guarantee the activity of the associated TF. Using methods for simultaneous analysis of open chromatin and gene expression with sequencing, it has been shown that chromatin accessibility at regulatory domains precedes gene expression38 and may thus outperform methods for GRN inference from scRNA-seq alone.
In contrast to local approaches, global integration strategies aim to identify larger-scale patterns (hundreds to thousands of features) of covariation across modalities to identify global shifts in the cellular state in an unsupervised way. To this end, linear matrix decomposition approaches such as principal component analysis (PCA), canonical correlation analysis (CCA)139 and non-negative matrix factorization (NMF)140,141 have proven successful to identify latent factors underlying cellular heterogeneity. For example, MOFA+142 extends on this principle and builds on the Bayesian Group Factor Analysis framework to jointly model variation across spatial and temporal covariates and employs a sparse hierarchical prior architecture that handles both shared sources of variation and private variation that is particular to individual modalities. This approach was used to integrate the different modalities in scNMT-seq data to reveal lineage-specific enhancers during embryo development68. Although the linearity of these approaches makes them inherently robust and results in readily interpretable factors of variation, they suffer from a lack of explanatory power when dealing with non-linearities that are commonly found in complex biological systems. Nonlinear multi-view neural network architectures143,144 such as multimodal autoencoders have been proposed to tackle this limitation. These neural networks aim to learn simple (low-dimensional) representations of the data, also known as an encoding, from complex (high-dimensional) input datasets. Instead of learning individual representations for each modality, these representations can be forced to be shared across different data modalities, thus forcing the neural network to implicitly learn common links between them. Several alternative architectures have been proposed that optimize for different criteria such as robustness to dropouts and batch effects, disentanglement of the learned latent factors for improved interpretability and cross-modal translation for missing modality imputation145,146. For further details we refer the reader to a recent overview of proposed deep learning approaches147.
Data integration for spatial multi-omics
Spatial mono-omics and multi-omics technologies also benefit from various data integration strategies. Before applying vertical integration, horizontal integration algorithms can be used to correct for technical variability between different sections of the same or different samples, and can also be expanded to fully utilize spatial correlations between sections148. Additionally, for spatial omics technologies that do not achieve single-cell resolution, deconvolution methods can be used to integrate non-spatial single-cell data to identify the cellular constituents of a particular tissue region149,150,151.
Vertical integration of non-spatial single-cell multi-omics data followed by spatial deconvolution using the RNA modality allows other molecular modalities to be indirectly spatially mapped. This approach was used, for example, by Foster et al., using ArchR152 for scRNA-seq and scATAC-seq integration, to identify spatial fibroblast epigenome patterns during tissue repair153. Spatial multi-omics technologies also allow for vertical integration, although few currently available technologies allow for true multimodal assaying at single-cell resolution on the same tissue section. Hence, generating spatial multi-omics data often still relies on the analysis of adjacent tissue sections and is thus limited to studying correlations across modalities of tissue regions (Fig. 6a). Linear approaches are suitable for the inference of these correlations, although special care needs to be taken in the statistical analysis as spatial autocorrelation can violate independence assumptions of the observed datapoints154,155. Although more spatial multi-omics technologies are emerging, spatial mono-omics assays can often also be combined with simple histological stains, such as H&E staining, allowing for the integration of cellular morphology and gene expression data. Nonlinear neural networks have been proposed to map discrete spot-based expression profiles onto high-resolution morphology images156 or to learn a joint cell morphology-plus-expression representation for the identification of novel cellular subpopulations missed by individual modalities157. As more spatial multi-omics technologies become available that can characterize many analytes at single-cell resolution, we expect rapid developments of vertical integration strategies based on adaptations of currently available non-spatial single-cell methodologies. By leveraging spatial information linking a cellular state to its respective micro-environments and macro-environments (for example, through the use of graph neural networks), more fine-grained multimodal representations of cellular state should be obtainable.
Finally, approaches for the harder problem of diagonal integration of unpaired spatial and non-spatial data modalities have also been proposed using autoencoder neural network architectures with a probabilistic coupling between the different data modalities. For these approaches, however, previous knowledge is generally required to constrain the resulting shared representations to biologically accurate alignments144.
Current challenges in data integration
Despite extensive research, several challenges remain for computational data integration. These strategies make implicit assumptions about the expected similarity of cellular states captured across different experiments and/or modalities. In the case of horizontal data integration across different batches, this can lead to overcorrection of true biological variation158, especially when studying subtle shifts in cellular state in different experimental conditions. This problem is further exacerbated in vertical integration strategies when correspondence between features is not immediately obvious and/or when non-linearities between the different layers might be biologically relevant132. For example, gene expression changes have been shown to be foreshadowed by chromatin priming, as seen in cell cycling via SHARE-seq38. Also, other biological phenomena such as alternative splicing and/or PTMs are likely to influence the correlation structure in cell state across the different molecular layers. Therefore, integration methods need to be developed that can not only identify the common anchors between the different samples and/or molecular layers but can also account for sample-specific and modality-specific variation in a readily interpretable way. Such models have already been proposed in other fields of multi-domain learning but have not yet been adapted to cellular multi-omics datasets159. The expansion of these models to integrate paired, unpaired and multimodal spatial assays will require robust data standards160,161 for scalable analysis of multimodal data in addition to well-established benchmarks to evaluate their efficacy.
Perspectives
In the decades to come, multi-omics at single-cell and spatial resolution will innovate further, leading to a more holistic understanding of cell biology. Advances can be anticipated on multiple fronts, including improvements in throughput, reduction of cost and the incorporation of more modalities in a single assay. Additionally, we expect improvements of sensitivity and specificity in the detection and characterization of each modality as part of multi-omic measurements. For example, on the genome level, full and error-free characterization of all genetic variants is still a challenge, which currently limits opportunities for comprehensive somatic mutation profiling at single-cell resolution and reconstructing phylogenetic cell lineages from naturally acquired mutations. Similarly, measurements of the epigenome are severely limited for co-detection of the range of epigenomic features co-regulating gene expression and other DNA-related processes. For instance, histone PTMs can currently be detected only a single or a few marks at a time; hence, these methods would benefit from drastic increments in the number of PTMs that can be co-detected, also together with other epigenomic features. The characterization of the transcriptome is often limited to poly(A) RNA rather than total RNA measurements; hence, it would be beneficial to include both coding and (small) non-coding RNAs as well as concurrent isoform detection. Proteome assays are still antibody-based and thus limited in how many proteins can be profiled simultaneously. Unbiased low-input methods such as mass-spectrometry-based approaches may circumvent this, but cannot currently be combined with assays for other molecular layers. This lack of multimodal integration also holds true for metabolome and lipidome assays. In addition to such improvements for characterizing modalities in multi-omics methods, we also anticipate the development of multimodal assays that incorporate entirely new modalities that currently remain uncharted, such as the epitranscriptome (consisting of base modifications to transcripts, some of which are known to affect gene expression)162. Furthermore, the field is likely to see a continued strong technological push for spatial multi-omics, avoiding the need for tissue dissociation and enabling concurrent multi-omic profiling of cell-intrinsic and -extrinsic molecular features defining cell types and states. Moreover, integration of phylogenetic cell lineages reconstructed from naturally acquired or artificially induced DNA mutations with other spatial or single-cell multi-ome information will transform our understanding of organismal development, cell migration routes and stem cell biology in health and disease. Finally, it will be necessary to develop methods that capture not only transient phenotypes but also ancestral states, apply multi-omic technologies to serial measurements in live cells163, and computationally improve the accuracy of data extraction from each molecular layer, as well as undertake integrative analyses across modalities to unravel dependencies within and across different data sources.
To arrive at such holistic multi-ome profiling of single cells dissociated from tissue or at spatial resolution will require many challenges to be overcome in the following years. For a view on how challenges associated with each modality specifically could be tackled, we refer to previous reviews on single-cell and spatial mono-omic profiling11,86,164,165,166,167,168. Optimization of low-input bulk molecular analysis tools to single-cell and/or spatial resolution, like amplification-free long-read single-molecule sequencing approaches, will enable the incorporation of novel molecular readouts. In parallel, to enable integration of more modalities per assay will require innovation in the principles presented in Fig. 2, including expansion and combination of the different principles. Making various single-cell and spatial multi-omics assays commercially available will also make them more accessible and applicable for the wide research community, but we are already starting to see this evolution169.
All these technological and computational developments will translate into a better understanding of development, organismal function and functional decline of organs with ageing, and will be key in unravelling the cellular pathogenesis of diseases, identifying more effective stratification strategies of disease processes, devising novel therapeutic modalities and guiding precision medicine.
References
Regev, A. et al. The Human Cell Atlas. eLife 6, e27041–e27041 (2017).
Kolodziejczyk, A. A., Kim, J. K., Svensson, V., Marioni, J. C. & Teichmann, S. A. The technology and biology of single-cell RNA sequencing. Mol. Cell 58, 610–620 (2015).
Grün, D. & van Oudenaarden, A. Design and analysis of single-cell sequencing experiments. Cell 163, 799–810 (2015).
Elmentaite, R., Domínguez Conde, C., Yang, L. & Teichmann, S. A. Single-cell atlases: shared and tissue-specific cell types across human organs. Nat. Rev. Genet. 23, 395–410 (2022).
Wen, L. & Tang, F. Recent advances in single-cell sequencing technologies. Precis. Clin. Med. 5, pbac002 (2022).
Macaulay, I. C. et al. Separation and parallel sequencing of the genomes and transcriptomes of single cells using G&T-seq. Nat. Protoc. 11, 2081–2103 (2016).
Macaulay, I. C. et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 12, 519–522 (2015). This paper presents one of the first demonstrations of physically separating DNA and poly(A) RNA from a single cell for whole-genome sequencing and full-length transcript sequencing.
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).
Li, W., Calder, R. B., Mar, J. C. & Vijg, J. Single-cell transcriptogenomics reveals transcriptional exclusion of ENU-mutated alleles. Mutat. Res. Mol. Mech. Mutagen. 772, 55–62 (2015).
Macaulay, I. C. & Voet, T. Single cell genomics: advances and future perspectives. PLoS Genet. 10, e1004126 (2014).
Gawad, C., Koh, W. & Quake, S. R. Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17, 175–188 (2016).
Svensson, V., Vento-Tormo, R. & Teichmann, S. A. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 13, 599–604 (2018).
Han, K. Y. et al. SIDR: simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells. Genome Res. 28, 75–87 (2018).
Zachariadis, V., Cheng, H., Andrews, N. & Enge, M. A highly scalable method for joint whole-genome sequencing and gene-expression profiling of single cells. Mol. Cell 80, 541–553.e5 (2020). This paper presents the feasibility of separating cytoplasmic RNA from nuclear DNA, enabling separate RNA-seq and DNA-seq library construction through direct tagmentation of the DNA, thus avoiding upfront WGA.
Valecha, M. & Posada, D. Somatic variant calling from single-cell DNA sequencing data. Comput. Struct. Biotechnol. J. 20, 2978–2985 (2022).
Han, L. et al. Co-detection and sequencing of genes and transcripts from the same single cells facilitated by a microfluidics platform. Sci. Rep. 4, 6485 (2014).
van Strijp, D. et al. Complete sequence-based pathway analysis by differential on-chip DNA and RNA extraction from a single cell. Sci. Rep. 7, 11030 (2017).
Dey, S. S., Kester, L., Spanjaard, B., Bienko, M. & van Oudenaarden, A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 33, 285–289 (2015). This paper presents one of the first demonstrations of joint preamplification of single-cell gDNA and poly(A) RNA-derived cDNA, followed by splitting of the reaction for DNA-seq and RNA-seq library preparation.
Rodriguez-Meira, A. et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol. Cell 73, 1292–1305.e8 (2019).
Yu, L. et al. scONE-seq: a single-cell multi-omics method enables simultaneous dissection of phenotype and genotype heterogeneity from frozen tumors. Sci. Adv. 9, eabp8901 (2023).
Yin, Y. et al. High-throughput single-cell sequencing with linear amplification. Mol. Cell 76, 676–690.e10 (2019). This paper presents one of the first demonstrations of combinatorial indexing for joint single-cell DNA and RNA sequencing, enabling massive throughput.
Rambow, F. et al. Toward minimal residual disease-directed therapy in melanoma. Cell 174, 843–855.e19 (2018).
Miller, M. B. et al. Somatic genomic changes in single Alzheimer’s disease neurons. Nature 604, 714–722 (2022).
Bizzotto, S. & Walsh, C. A. Genetic mosaicism in the human brain: from lineage tracing to neuropsychiatric disorders. Nat. Rev. Neurosci. 23, 275–286 (2022).
Proukakis, C. Somatic mutations in neurodegeneration: an update. Neurobiol. Dis. 144, 105021 (2020).
Voet, T. & Vermeesch, J. R. Mutational processes shaping the genome in early human embryos. Cell 168, 751–753 (2017).
van Neerven, S. M. & Vermeulen, L. Cell competition in development, homeostasis and cancer. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-022-00538-y (2022).
Ren, P., Dong, X. & Vijg, J. Age-related somatic mutation burden in human tissues. Front. Aging 3, 1018119 (2022).
Bekaert, B. et al. CRISPR/Cas gene editing in the human germline. Semin. Cell Dev. Biol. 131, 93–107 (2022).
Alanis-Lobato, G. et al. Frequent loss of heterozygosity in CRISPR-Cas9–edited early human embryos. Proc. Natl Acad. Sci. USA 118, e2004832117 (2021).
Fogarty, N. M. E. et al. Genome editing reveals a role for OCT4 in human embryogenesis. Nature 550, 67–73 (2017).
Sheth, R. U. & Wang, H. H. DNA-based memory devices for recording cellular events. Nat. Rev. Genet. 19, 718–732 (2018).
Bhattarai-Kline, S. et al. Recording gene expression order in DNA by CRISPR addition of retron barcodes. Nature 608, 217–225 (2022).
Choi, J. et al. A time-resolved, multi-symbol molecular recorder via sequential genome editing. Nature 608, 98–107 (2022).
Liu, L. et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat. Commun. 10, 470 (2019).
Cheng, H. et al.Smart3-ATAC: a highly sensitive method for joint accessibility and full-length transcriptome analysis in singlecells. Preprint at bioRxiv https://doi.org/10.1101/2021.12.02.470912 (2021).
Cao, J. et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018).
Ma, S. et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183, 1103–1116.e20 (2020). SHARE-seq is a powerful method to profile gene expression and chromatin accessibility in the same cell; combining both layers of information shows that changes in enhancer activity can precede gene activation.
Plongthongkum, N., Diep, D., Chen, S., Lake, B. B. & Zhang, K. Scalable dual-omics profiling with single-nucleus chromatin accessibility and mRNA expression sequencing 2 (SNARE-seq2). Nat. Protoc. 16, 4992–5029 (2021).
Zhu, C. et al. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat. Struct. Mol. Biol. 26, 1063–1070 (2019).
Chen, S., Lake, B. B. & Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 37, 1452–1457 (2019).
Xing, Q. R. et al. Parallel bimodal single-cell sequencing of transcriptome and chromatin accessibility. Genome Res. 30, 1027–1039 (2020).
Xu, W. et al. ISSAAC-seq enables sensitive and flexible multimodal profiling of chromatin accessibility and gene expression in single cells. Nat. Methods 19, 1243–1249 (2022).
No authors listed. Sensitive, flexible and modular single-cell multi-omics profiling with ISSAAC-seq. Nat. Methods 19, 1183–1184 (2022).
Tedesco, M. et al. Chromatin Velocity reveals epigenetic dynamics by single-cell profiling of heterochromatin and euchromatin. Nat. Biotechnol. 40, 235–244 (2022).
Pan, L. et al. scPCOR-seq enables co-profiling of chromatin occupancy and RNAs in single cells. Commun. Biol. 5, 678 (2022).
Xiong, H., Luo, Y., Wang, Q., Yu, X. & He, A. Single-cell joint detection of chromatin occupancy and transcriptome enables higher-dimensional epigenomic reconstructions. Nat. Methods 18, 652–660 (2021).
Zhu, C. et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat. Methods 18, 283–292 (2021).
Sun, Z. et al. Joint single-cell multiomic analysis in Wnt3a induced asymmetric stem cell division. Nat. Commun. 12, 5941 (2021).
Janssens, D. H. et al. CUT&Tag2for1: a modified method for simultaneous profiling of the accessible and silenced regulome in single cells. Genome Biol. 23, 81 (2022).
Gopalan, S., Wang, Y., Harper, N. W., Garber, M. & Fazzio, T. G. Simultaneous profiling of multiple chromatin proteins in the same cells. Mol. Cell 81, 4736–4746.e5 (2021).
Liu, Y. et al. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 37, 424–429 (2019).
Vaisvila, R. et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 31, 1280–1289 (2021).
Hu, Y. et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 17, 88 (2016).
Angermueller, C. et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 13, 229–232 (2016).
Hou, Y. et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 26, 304–319 (2016).
Bian, S. et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 362, 1060–1063 (2018).
Gu, H. et al. Smart-RRBS for single-cell methylome and transcriptome analysis. Nat. Protoc. 16, 4004–4030 (2021).
Clark, S. J. et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun. 9, 781 (2018).
Li, L. et al. Single-cell multi-omics sequencing of human early embryos. Nat. Cell Biol. 20, 847–858 (2018).
Gu, C., Liu, S., Wu, Q., Zhang, L. & Guo, F. Integrative single-cell analysis of transcriptome, DNA methylome and chromatin accessibility in mouse oocytes. Cell Res. 29, 110–123 (2019).
Pott, S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife 6, e23203 (2017).
Wang, Y. et al. Single-cell multiomics sequencing reveals the functional regulatory landscape of early embryos. Nat. Commun. 12, 1247 (2021).
Yan, R. et al. Decoding dynamic epigenetic landscapes in human oocytes using single-cell multi-omics sequencing. Cell Stem Cell 28, 1641–1656.e7 (2021).
Luo, C. et al. Single nucleus multi-omics identifies human cortical cell regulatory genome diversity. Cell Genomics 2, 100107 (2022).
Lee, D.-S. et al. Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat. Methods 16, 999–1006 (2019).
Li, G. et al. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods 16, 991–993 (2019).
Argelaguet, R. et al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature 576, 487–491 (2019). By applying scNMT-seq to the early mouse development, this paper highlights the potential of this multi-omic method to disentangle hierarchies of transcriptional and epigenetic changes beyond mono-omic analyses.
Liu, X. et al. N6-methyladenine is incorporated into mammalian genome by DNA polymerase. Cell Res. 31, 94–97 (2021).
Rooijers, K. et al. Simultaneous quantification of protein–DNA contacts and transcriptomes in single cells. Nat. Biotechnol. 37, 766–772 (2019).
Borsos, M. et al. Genome–lamina interactions are established de novo in the early mouse embryo. Nature 569, 729–733 (2019).
Aughey, G. N., Estacio Gomez, A., Thomson, J., Yin, H. & Southall, T. D. CATaDa reveals global remodelling of chromatin accessibility during stem cell differentiation in vivo. eLife 7, e32341 (2018).
Rang, F. J. et al. Single-cell profiling of transcriptome and histone modifications with EpiDamID. Mol. Cell 82, 1956–1970.e14 (2022). EpiDamID profiles histone modifications and transcription in the same cell; histone modification profiling occurs orthogonally to classic antibody-based approaches, and as the presence of modifications can be written on DNA over a longer period, such methods may generate time records.
Stergachis, A. B., Debo, B. M., Haugen, E., Churchman, L. S. & Stamatoyannopoulos, J. A. Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science 368, 1449–1454 (2020).
Peterson, V. M. et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 35, 936–939 (2017).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
Chung, H. et al. Joint single-cell measurements of nuclear proteins and RNA in vivo. Nat. Methods 18, 1204–1212 (2021).
Reimegård, J. et al. A combined approach for single-cell mRNA and intracellular protein expression analysis. Commun. Biol. 4, 624 (2021).
Mimitou, E. P. et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 16, 409–412 (2019).
Gerlach, J. P. et al. Combined quantification of intracellular (phospho-)proteins and transcriptomics from fixed single cells. Sci. Rep. 9, 1469 (2019).
Mimitou, E. P. et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol. 39, 1246–1258 (2021).
Swanson, E. et al. Simultaneous trimodal single-cell measurement of transcripts, epitopes, and chromatin accessibility using TEA-seq. eLife 10, e63632 (2021).
Zhang, B. et al. Characterizing cellular heterogeneity in chromatin state with scCUT&Tag-pro. Nat. Biotechnol. 40, 1220–1230 (2022).
Chen, A. F. et al. NEAT-seq: simultaneous profiling of intra-nuclear proteins, chromatin accessibility and gene expression in single cells. Nat. Methods 19, 547–553 (2022).
Fiskin, E. et al. Single-cell profiling of proteins and chromatin accessibility using PHAGE-ATAC. Nat. Biotechnol. 40, 374–381 (2022).
Mund, A., Brunner, A.-D. & Mann, M. Unbiased spatial proteomics with single-cell resolution in tissues. Mol. Cell 82, 2335–2349 (2022).
Hu, W. et al. Single-cell transcriptome and translatome dual-omics reveals potential mechanisms of human oocyte maturation. Nat. Commun. 13, 5114 (2022).
Datlinger, P. et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301 (2017).
Jaitin, D. A. et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-Cell RNA-Seq. Cell 167, 1883–1896.e15 (2016).
Dixit, A. et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17 (2016).
Rubin, A. J. et al. Coupled single-cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell 176, 361–376.e17 (2019).
Liscovitch-Brauer, N. et al. Profiling the genetic determinants of chromatin accessibility with scalable single-cell CRISPR screens. Nat. Biotechnol. 39, 1270–1277 (2021).
Pierce, S. E., Granja, J. M. & Greenleaf, W. J. High-throughput single-cell chromatin accessibility CRISPR screens enable unbiased identification of regulatory networks in cancer. Nat. Commun. 12, 2969 (2021).
Replogle, J. M. et al. Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559–2575.e28 (2022). By providing a first description of the full compendium of transcriptional changes induced by inactivating any expressed gene, this study demonstrates the potential of single-cell multi-omic methods for hypothesis-free genome-wide functional screening.
Norman, T. M. et al. Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science 365, 786–793 (2019).
Eisenstein, M. Seven technologies to watch in 2022. Nature 601, 658–661 (2022).
Yagnik, G., Liu, Z., Rothschild, K. J. & Lim, M. J. Highly multiplexed immunohistochemical MALDI-MS imaging of biomarkers in tissues. J. Am. Soc. Mass. Spectrom. 32, 977–988 (2021).
Truong, J. X. M. et al. Removal of optimal cutting temperature (O.C.T.) compound from embedded tissue for MALDI imaging of lipids. Anal. Bioanal. Chem. 413, 2695–2708 (2021).
Guilliams, M. et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 185, 379–396.e38 (2022).
Fan, R. et al. Spatially resolved epigenome-transcriptome co-profiling of mammalian tissues at the cellular level. Res. Sq. https://doi.org/10.21203/rs.3.rs-1728747/v1 (2022).
Deng, Y. et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 609, 375–383 (2022).
Deng, Y. et al. Spatial-CUT&Tag: spatially resolved chromatin modification profiling at the cellular level. Science 375, 681–686 (2022).
Liu, Y. et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 183, 1665–1681.e18 (2020). This is the first paper showing the possibility for spatial multi-omics (transcriptome+protein) profiling of tissue sections using a microfluidics-based deterministic barcoding-in-tissue approach.
Cardozo Gizzi, A. M. et al. Microscopy-based chromosome conformation capture enables simultaneous visualization of genome organization and transcription in intact organisms. Mol. Cell 74, 212–222.e5 (2019).
Mateo, L. J. et al. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54 (2019).
Liu, M. et al. Multiplexed imaging of nucleome architectures in single cells of mammalian tissue. Nat. Commun. 11, 2907 (2020).
Xia, C., Fan, J., Emanuel, G., Hao, J. & Zhuang, X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl Acad. Sci. USA 116, 19490–19499 (2019).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
Shah, S. et al. Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell 174, 363–376.e16 (2018).
Shah, S., Lubeck, E., Zhou, W. & Cai, L. seqFISH accurately detects transcripts in single cells and reveals robust spatial organization in the hippocampus. Neuron 94, 752–758.e1 (2017).
Su, J.-H., Zheng, P., Kinrot, S. S., Bintu, B. & Zhuang, X. Genome-scale imaging of the 3D organization and transcriptional activity of chromatin. Cell 182, 1641–1659.e26 (2020). This paper shows combined RNA, chromatin and nuclear body imaging in the same single cells by applying RNA-MERFISH and DNA-MERFISH for >1,000 nascent transcripts and genomic loci together with fluorescent antibody readouts for cell-cycle-state markers and nuclear structures.
Takei, Y. et al. Integrated spatial genomics reveals global architecture of single nuclei. Nature 590, 344–350 (2021). This paper shows combined RNA, chromatin and nuclear body imaging in the same single cells by applying DNA-seqFISH+ to thousands of genomic loci with RNA-seqFISH of 70 mRNAs and nascent transcriptionally active sites of 1,000 genes, together with antibody targeting of 17 nuclear structures.
Nguyen, H. Q. et al. 3D mapping and accelerated super-resolution imaging of the human genome using in situ sequencing. Nat. Methods 17, 822–832 (2020).
Zhu, Z. et al. Genome profiles of pathologist-defined cell clusters by multiregional LCM and G&T-seq in one triple-negative breast cancer patient. Cell Rep. Med. 2, 100404 (2021).
10x Genomics. Spatial proteogenomics. 10x Genomics https://www.10xgenomics.com/products/spatial-proteogenomics (2023).
Ben-Chetrit, N. et al. Integrated protein and transcriptome high-throughput spatial profiling. Preprint at bioRxiv https://doi.org/10.1101/2022.03.15.484516 (2022).
Vickovic, S. et al. SM-Omics is an automated platform for high-throughput spatial multi-omics. Nat. Commun. 13, 795 (2022).
NanoString. GeoMx DSP overview. NanoString https://nanostring.com/products/geomx-digital-spatial-profiler/geomx-dsp-overview/ (2023).
Merritt, C. R. et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 38, 586–599 (2020).
Wang, F. et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 14, 22–29 (2012).
Bonnett A. S. et al. The development and performance of a GeoMx® based proteogenomic workflow for the detection of both RNA and protein on a single FFPE slide (nanostring.com, 2022).
Fan, R. et al. Spatial-CITE-seq: spatially resolved high-plex protein and whole transcriptome co-mapping. Res. Sq. https://doi.org/10.21203/rs.3.rs-1499315/v1 (2022). The spatial-CITE-seq method shows the highest level of spatial proteomics multiplexing in combination with unbiased transcriptome profiling to date using a microfluidics-based deterministic barcoding-in-tissue approach.
Wang, G., Moffitt, J. R. & Zhuang, X. Multiplexed imaging of high-density libraries of RNAs with MERFISH and expansion microscopy. Sci. Rep. 8, 4847 (2018).
Kohman, R. E. & Church, G. M. Fluorescent in situ sequencing of DNA barcoded antibodies. Preprint at bioRxiv https://doi.org/10.1101/2020.04.27.060624 (2020).
Alon, S. et al. Expansion sequencing: spatially precise in situ transcriptomics in intact biological systems. Science 371, eaax2656 (2021).
He, S. et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat. Biotechnol. 40, 1794–1806 (2022).
Liu, J. et al. Concordance of MERFISH spatial transcriptomics with bulk and single-cell RNA sequencing. Life Sci. Alliance 6, e202201701 (2022).
Janesick, A. et al. High resolution mapping of the breast cancer tumor microenvironment using integrated single cell, spatial and in situ analysis of FFPE tissue. Preprint at bioRxiv https://doi.org/10.1101/2022.10.06.510405 (2022).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Argelaguet, R., Cuomo, A. S. E., Stegle, O. & Marioni, J. C. Computational principles and challenges in single-cell data integration. Nat. Biotechnol. 39, 1202–1215 (2021).
Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2022).
Rautenstrauch, P., Vlot, A. H. C., Saran, S. & Ohler, U. Intricacies of single-cell multi-omics data integration. Trends Genet. 38, 128–139 (2022).
Efremova, M. & Teichmann, S. A. Computational methods for single-cell omics across modalities. Nat. Methods 17, 14–17 (2020).
Packer, J. & Trapnell, C. Single-cell multi-omics: an engine for new quantitative models of gene regulation. Trends Genet. TIG 34, 653–665 (2018).
Vallejos, C. A., Marioni, J. C. & Richardson, S. BASiCS: Bayesian analysis of single-cell sequencing data. PLoS Comput. Biol. 11, e1004333 (2015).
Cuomo, A. S. E. et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat. Commun. 11, 810 (2020).
Gasperini, M. et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 377–390.e19 (2019).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019).
Welch, J. D. et al. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177, 1873–1887.e17 (2019).
Liu, J. et al. Jointly defining cell types from multiple single-cell datasets using LIGER. Nat. Protoc. 15, 3632–3662 (2020).
Argelaguet, R. et al. MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. 21, 111 (2020).
Lotfollahi, M., Litinetskaya, A. & Theis, F. J. Multigrate: single-cell multi-omic data integration. Preprint a bioRxiv https://doi.org/10.1101/2022.03.16.484643 (2022).
Yang, K. D. et al. Multi-domain translation between single-cell imaging and sequencing data using autoencoders. Nat. Commun. 12, 31 (2021).
Wu, K. E., Yost, K. E., Chang, H. Y. & Zou, J. BABEL enables cross-modality translation between multiomic profiles at single-cell resolution. Proc. Natl Acad. Sci. USA 118, e2023070118 (2021).
Martinez-de-Morentin, X. et al. Adaptative machine translation between paired single-cell multi-omics data. Preprint at bioRxiv https://doi.org/10.1101/2021.01.27.428400 (2022).
Stanojevic, S., Li, Y., Ristivojevic, A. & Garmire, L. X. Computational methods for single-cell multi-omics integration and alignment. Genomics Proteomics Bioinformatics https://doi.org/10.1016/j.gpb.2022.11.013 (2022).
Zeira, R., Land, M., Strzalkowski, A. & Raphael, B. J. Alignment and integration of spatial transcriptomics data. Nat. Methods 19, 567–575 (2022).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 40, 661–671 (2022).
Elosua-Bayes, M., Nieto, P., Mereu, E., Gut, I. & Heyn, H. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 49, e50 (2021).
Biancalani, T. et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 18, 1352–1362 (2021).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Foster, D. S. et al. Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc. Natl Acad. Sci. USA 118, e2110025118 (2021).
Ravi, V. M. et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 40, 639–655.e13 (2022).
Velten, B. et al. Identifying temporal and spatial patterns of variation from multimodal data using MEFISTO. Nat. Methods 19, 179–186 (2022).
Bergenstråhle, L. et al. Super-resolved spatial transcriptomics by deep data fusion. Nat. Biotechnol. 40, 476–479 (2022).
Bao, F. et al. Integrative spatial analysis of cell morphologies and transcriptional states with MUSE. Nat. Biotechnol. 40, 1200–1209 (2022).
Büttner, M., Miao, Z., Wolf, F. A., Teichmann, S. A. & Theis, F. J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 16, 43–49 (2019).
Lee, M. & Pavlovic, V. Private-shared disentangled multimodal VAE for learning of hybrid latent representations. Preprint at https://doi.org/10.48550/arXiv.2012.13024 (2020).
Bredikhin, D., Kats, I. & Stegle, O. MUON: multimodal omics analysis framework. Genome Biol. 23, 42 (2022).
Palla, G. et al. Squidpy: a scalable framework for spatial omics analysis. Nat. Methods 19, 171–178 (2022).
Wiener, D. & Schwartz, S. The epitranscriptome beyond m6A. Nat. Rev. Genet. 22, 119–131 (2021).
Chen, W. et al. Live-seq enables temporal transcriptomic recording of single cells. Nature 608, 733–740 (2022).
Preissl, S., Gaulton, K. J. & Ren, B. Characterizing cis-regulatory elements using single-cell epigenomics. Nat. Rev. Genet. 24, 21–43 (2022).
Moffitt, J. R., Lundberg, E. & Heyn, H. The emerging landscape of spatial profiling technologies. Nat. Rev. Genet. 23, 741–759 (2022).
Bouwman, B. A. M., Crosetto, N. & Bienko, M. The era of 3D and spatial genomics. Trends Genet. 38, 1062–1075 (2022).
Lundberg, E. & Borner, G. H. H. Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 20, 285–302 (2019).
Taylor, M. J., Lukowski, J. K. & Anderton, C. R. Spatially resolved mass spectrometry at the single cell: recent innovations in proteomics and metabolomics. J. Am. Soc. Mass. Spectrom. 32, 872–894 (2021).
Zawistowski, J. S. et al. Unifying genomics and transcriptomics in single cells with ResolveOME amplification chemistry to illuminate oncogenic and drug resistance mechanisms. Preprint at bioRxiv https://doi.org/10.1101/2022.04.29.489440 (2022).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Moses, L. & Pachter, L. Museum of spatial transcriptomics. Nat. Methods 19, 534–546 (2022).
Liao, J., Lu, X., Shao, X., Zhu, L. & Fan, X. Uncovering an organ’s molecular architecture at single-cell resolution by spatially resolved transcriptomics. Trends Biotechnol. 39, 43–58 (2021).
Asp, M., Bergenstråhle, J. & Lundeberg, J. Spatially resolved transcriptomes — next generation tools for tissue exploration. BioEssays 42, 1900221 (2020).
Xu, J. & Liu, Y. A guide to visualizing the spatial epigenome with super-resolution microscopy. FEBS J. 286, 3095–3109 (2019).
Ganesh, S. et al. Spatially resolved 3D metabolomic profiling in tissues. Sci. Adv. 7, eabd0957 (2021).
Salmén, F. et al. Barcoded solid-phase RNA capture for spatial transcriptomics profiling in mammalian tissue sections. Nat. Protoc. 13, 2501–2534 (2018).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Vickovic, S. et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990 (2019).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792 (2022).
Wählby, C., Erlandsson, F., Bengtsson, E. & Zetterberg, A. Sequential immunofluorescence staining and image analysis for detection of large numbers of antigens in individual cell nuclei. Cytometry 47, 32–41 (2002).
Lin, J.-R., Fallahi-Sichani, M., Chen, J.-Y. & Sorger, P. K. Cyclic Immunofluorescence (CycIF), a highly multiplexed method for single-cell imaging. Curr. Protoc. Chem. Biol. 8, 251–264 (2016).
Cattoretti, G., Bosisio, F. M., Marcelis, L. & Bolognesi, M. M. Multiple iterative labeling by antibody neodeposition (MILAN). Protoc. Exch. https://doi.org/10.1038/protex.2018.106 (2018).
Black, S. et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc. 16, 3802–3835 (2021).
Saka, S. K. et al. Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat. Biotechnol. 37, 1080–1090 (2019).
Eng, C.-H. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568, 235–239 (2019).
Ke, R. et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 10, 857–860 (2013).
Lee, J. H. et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc. 10, 442–458 (2015).
Lee, J. H. et al. Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014).
Zhu, X., Xu, T., Peng, C. & Wu, S. Advances in MALDI mass spectrometry imaging single cell and tissues. Front. Chem. 9, 782432 (2022).
Acknowledgements
T.V., K.V., A.S. and B.T. are supported by Aligning Science Across Parkinson’s (ASAP-000430) through the Michael J. Fox Foundation for Parkinson’s Research (MJFF). T.V., K.V. and A.S. are supported by a grant from The Leona M. and Harry B. Helmsley Charitable Trust to KU Leuven (1903-03785) and by KU Leuven (IDN/19/039; IDN/21/006). T.V. and K.V. are supported by KU Leuven (C14/18/092 and C14/22/125), the Research Foundation Flanders (FWO: G081318N, G0C6120N, G088621N and I001818N), the Flemish government (iBOF/21/043), the Chan Zuckerberg Foundation (CZF, CZF2019-002436, 6000511-5500001380) and Opening the Future (OtF). B.T. is supported by FWO (G0C7519N and S003422N), the Foundation against Cancer (F/2020/1544) and KU Leuven (METH/21/06). A.S. is supported by KU Leuven (C24M/19/075) and FWO (G005923N).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
T.V. is co-inventor on licensed patents WO/2011/157846 (Methods for haplotyping single cells), WO/2014/053664 (High-throughput genotyping by sequencing low amounts of genetic material) and WO/2015/028576 (Haplotyping and copy number typing using polymorphic variant allelic frequencies).
Peer review
Peer review information
Nature Reviews Genetics thanks Martin Enge, Kun Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- 3C or Hi-C
-
A class of methods uncovering the higher-order chromosome conformation and chromatin interactions in the nucleus.
- Autoencoders
-
A class of architectures of neural networks that take a highly dimensional input and encode into a low-dimensional representation via an encoder network. These networks are trained in a self-supervised way by reconstructing the original input from the encoding using a decoder network.
- Bisulfite sequencing
-
A method in which bisulfite treatment of DNA before sequencing converts unmethylated cytosines to uracil, while 5-methylcytosines are protected from conversion. This method hence discloses sites in the genome where DNA methylation is found.
- Cell plasticity
-
The ability of a cell to remodel its epigenomic, transcriptomic and proteomic landscape, leading to new phenotypic features.
- Cell state
-
A specific stable or unstable functional condition of a cell belonging to a particular cell type.
- Cell type
-
A categorization of cells by specific morphological and/or phenotypic characteristics.
- Chromatin accessibility
-
The structures of DNA wrapped around histone octamer proteins (nucleosomes) that make up chromosomes. Chromatin can be highly condensed with DNA inaccessible or open with DNA accessible.
- Displacement preamplification followed by PCR
-
(DA-PCR). A class of methods for whole-genome amplification of single-cell DNA using multiple rounds of strand displacement amplification, generating amplicons with PCR-handles at both ends, enabling molecules to be further amplified by PCR.
- DNA methylation
-
The deposition of methyl groups onto DNA. In mammals, methylation is predominantly found at cytosines as 5-methylcytosine, and mostly in a cytosine–guanine dinucleotide (CpG) context.
- Epigenome
-
The ensemble of modifications to DNA and DNA-associated proteins that signal and regulate gene expression and other DNA-related processes. Examples include DNA methylation, histone post-translational modifications and chromatin remodelling proteins.
- Exome sequencing
-
A targeted next-generation sequencing approach that enables sequencing the coding exons, which can be supplemented with flanking intronic sequences as well as (part of) the gene regulatory sequences, approximating 1.1% or more of the human genome.
- FFPE
-
Formalin-fixed paraffin-embedded (FFPE) tissues are preserved for long-term archival storage by formalin fixation followed by embedding in paraffin wax blocks.
- Genome
-
The genetic blueprint or DNA of an organism established at fertilization, which for humans normally consists of 23 pairs of chromosomes contained within the nucleus, plus the mitochondrial DNA molecules present at multiple copies in the mitochondria of the cell.
- Hierarchical mixed models
-
Statistical regression models that model nested hierarchies in the data taking into account complex covariate data structures.
- Human Cell Atlas
-
The Human Cell Atlas (HCA) aims to create a reference map of all human cells, providing a basis for understanding human health and for diagnosing, monitoring and treating diseases.
- Linear matrix decomposition
-
Numerical analysis algorithms that factorize observations into a product of smaller matrices. These matrices usually represent factors or components of variation that are easier to interpret (such as biological processes) and their presence within the observations (cells).
- Metabolome
-
The ensemble of metabolites present within a cell or tissue.
- Multiple displacement amplification
-
(MDA). An isothermal DNA amplification method that applies DNA polymerase(s) with strand displacement activity.
- Neural networks
-
A class of algorithms that use densely connected networks of artificial neurons that are non-linearly activated given a combination of input values from connected input neurons, mimicking the human brain.
- OCT compound
-
Optimal cutting temperature (OCT) compound is a standard type of sample-embedding medium used to embed fresh-frozen tissue samples for optimal storage and cryosectioning.
- Phenome
-
The set of traits or characteristics expressed by an organism or cell.
- Proteome
-
The ensemble of proteins translated from transcribed genes contained within the genome, which can be highly variable among different cell types.
- Smart-seq2
-
Switching mechanism at the end of the 5′ end of the RNA transcript (Smart)-based method for single-cell cDNA generation, amplification and sequencing library conversion.
- Tagmentation
-
A method in which double-stranded DNA is cleaved and tagged with adapter sequences in a single step by using a transposase complex loaded with these adapter sequences.
- Template switching
-
A method in which Moloney murine leukaemia virus (MMLV)-type reverse transcriptases add non-templated nucleotides (CCC) to first-strand cDNA near the 5′ end of the transcript, enabling annealing of an rGrGrG-containing template-switching oligonucleotide and the reverse transcriptase to switch templates and copy the template-switching oligonucleotide sequence to the cDNA.
- Transcriptome
-
The ensemble of transcripts or RNA molecules transcribed from genes contained within the genome, which can be highly variable among different cell types.
- Unique molecular identifier
-
Short sequences that barcode each molecule individually and are added as tags to DNA fragments in next-generation sequencing-based approaches to identify molecules of interest with increased accuracy.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vandereyken, K., Sifrim, A., Thienpont, B. et al. Methods and applications for single-cell and spatial multi-omics. Nat Rev Genet 24, 494–515 (2023). https://doi.org/10.1038/s41576-023-00580-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41576-023-00580-2
