
Abstract
Over the past two decades, compelling evidence has emerged indicating that immune mechanisms can contribute to the pathogenesis of major depressive disorder (MDD) and that drugs with primary immune targets can improve depressive symptoms. Patients with MDD are heterogeneous with respect to symptoms, treatment responses and biological correlates. Defining a narrower patient group based on biology could increase the treatment response rates in certain subgroups: a major advance in clinical psychiatry. For example, patients with MDD and elevated pro-inflammatory biomarkers are less likely to respond to conventional antidepressant drugs, but novel immune-based therapeutics could potentially address their unmet clinical needs. This article outlines a framework for developing drugs targeting a novel patient subtype within MDD and reviews the current state of neuroimmune drug development for mood disorders. We discuss evidence for a causal role of immune mechanisms in the pathogenesis of depression, together with targets under investigation in randomized controlled trials, biomarker evidence elucidating the link to neural mechanisms, biological and phenotypic patient selection strategies, and the unmet clinical need among patients with MDD.
Similar content being viewed by others

Introduction
Currently available therapeutics for major depressive disorder (MDD) lead to remission in approximately one third of patients and partial remission in another third1. This heterogeneity in treatment outcomes is thought to reflect the biological heterogeneity present within patients with MDD. Various forms of immune biomarker abnormalities characterize patients with poor treatment outcomes2,3,4, which has raised interest in targeting mechanisms in the immune system as precision medicine approaches in psychiatry.
The relationship between immune dysregulation and depression cuts across a fault line in the conventional diagnostic categorization of depression5. On one side of the line is a large group of patients diagnosed with MDD, defined by a clinical checklist comprising at least one of two core symptoms (low mood and anhedonia), and at least three of seven other defined symptoms, for at least 2 weeks, to a distressing or disabling degree. Formally, the diagnosis of MDD is excluded if the patient has a medical or physiological condition that could explain the depression. On the other side of the diagnostic fault line is a second large group of patients who manifest major depressive symptoms in the context of primary immune disorders, such as rheumatoid arthritis, Crohn’s disease or systemic lupus erythematosus. These patients are ineligible for a diagnosis of MDD. Instead, they are typically diagnosed with ‘comorbid depression’ and coded more formally using the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-5) category of “depressive disorder due to another medical condition”, based partly on the criterion of “evidence from the history, physical examination, or laboratory findings that the disturbance is the direct pathophysiological consequence of another medical condition”.
In this context, the immunology of depression is diagnostically disruptive, because immune dysfunction could contribute to both comorbid depression (associated with clinical inflammation) and MDD in a proportion of individuals who also have laboratory evidence of low-grade inflammation. Moreover, additional heterogeneity likely exists within this subset of patients with MDD and immune dysfunction. For example, one group accrued evidence for an ‘immunometabolic’ subtype of MDD by clustering immunological and metabolic dysregulations (such as elevated leptin levels and reduced insulin sensitivity) with specific behavioural symptoms that reflect an altered balance between energy intake and expenditure (hyperphagia, weight gain, hypersomnia and fatigue). This cluster of clinical features and biomarker abnormalities persisted in this subgroup of patients for several years6. Other MDD subgroups have also been described that have distinct or partly overlapping clusters of immune biomarker abnormalities, along with different phenotypic patterns of symptoms and non-responsiveness to conventional antidepressants, especially to selective serotonin reuptake inhibitors (SSRIs)4. Therefore, immune modulators could alter antidepressant efficacy for people with MDD and biomarker evidence of immune dysfunction related to the targeted pathway, as well as for individuals with comorbid depressive symptoms; these patients rank treatments for depressive symptoms as one of their greatest unmet medical needs7.
To pursue these ideas, we address the criteria required to develop a therapeutic for a novel subtype for patients within the broader MDD diagnostic class (Box 1). First, we consider evidence for a causal association between immune dysregulation and depression, and the potential targets implicated thus far. Second, we walk through the randomized, placebo-controlled trials (RCTs) that have been performed to date. We next examine emerging evidence of biomarkers associated with MDD for their potential as tools for patient stratification in clinical trials as well as in clinical treatment algorithms, and as end points in proof-of-concept studies of novel agents. In the final section, we discuss the unmet medical need among patients whose MDD seems to be mediated through an immune mechanism.
Evidence for causality
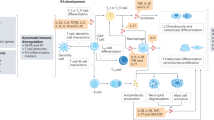
There is overwhelming evidence that immune dysregulation and depression are associated or correlated with each other8,9,10. MDD has been associated with elevated levels of pro-inflammatory cytokines and acute phase proteins in blood and cerebrospinal fluid (CSF), a decreased adaptive immune response, differences in the relative abundance of specific immune cell types, a bias towards autoimmunity, increased activation of microglia and other immune changes (Fig. 1). The most compelling rationale for developing immune-targeted drugs for depression, however, would be evidence that immune dysregulation can indeed cause depression in susceptible individuals, rather than merely evidence for an association between immune dysfunction and depression. We must therefore also understand how a peripheral or central immune target alters the functional organization of the central nervous system (CNS), thus distally causing depressive changes in emotion, cognition and behaviour.

a | In the periphery, a variety of immune markers have been associated with major depressive disorder (MDD). These include elevation of pro-inflammatory cytokines, increases in innate immune cell types and changes in differentiation of T cells and other immune cell types. Direction of change of expression or cell count in MDD indicated with an arrow. b | In the central nervous system (CNS), elevation of pro-inflammatory cytokines is observed in the cerebrospinal fluid (CSF), which may come from the periphery — entering the CNS through the blood–brain barrier (BBB; indicated in centre of figure) or the dural lymphatic system — or be produced from cells within the CNS, including glia. An increase in activated microglia, the resident macrophages in the CNS, in MDD has been implicated by imaging and post-mortem studies and is a current biological target of interest. Breg cell, regulatory B cell; HSC, haematopoietic stem cell; IFN, interferon; ILC, innate lymphoid cell; ROS, reactive oxygen species; Tc cell, conventional T cell; TH1 cell, T helper 1 cell; Treg cell, regulatory T cell; TGFβ, transforming growth factor-β; TNF, tumour necrosis factor.
In the rest of this section, we predominantly review human studies because these data are most directly relevant to the early clinical development of novel immunotherapeutics for patients with depression. Nevertheless, strong preclinical evidence also indicates that peripheral and central immune mechanisms can have adverse effects on neuronal function and cause anhedonia, social isolation and other depressive-like behaviours in animals11,12,13,14,15. Finally, we note that although immune pathways are complex and interconnected (Fig. 1), most studies in MDD have focused on the role of pro-inflammatory actors of the innate immune system and their therapeutic modulation. By contrast, the potential for harnessing mechanisms within the adaptive immune system to discover further therapeutic innovation in neuropsychiatry is relatively unexplored.
Longitudinal and challenge studies
Longitudinal studies have tested the hypothesis that inflammation can precede depression, which is necessary if inflammation is causal. There is supportive evidence for inflammation preceding depression or depressive symptoms over a wide range of timescales. A high concentration of IL-6 in blood at age 9 years predicted an elevated risk of depression at age 18 years in a UK birth cohort16. Middle-aged women who were not with depression but had C-reactive protein (CRP) levels higher than the upper limit of the normal range (3 mg/l) when tested in 2004 and 2008 were significantly more likely to present depression for the first time in 2012 than women who had normal CRP levels over the previous years17. Finally, among women with depression at baseline in the Netherlands Study of Depression and Anxiety (NESDA), higher IL-6 levels predicted a chronic course of MDD based on 2-year and 6-year follow-up evaluations18.
Challenge studies have demonstrated that an intervention causing immune activation can increase rates of depressive symptoms on short to medium timescales. For example, about a third of patients with hepatitis who were treated with the pro-inflammatory cytokine interferon-α (IFNα) had developed major depressive symptoms when reassessed 4–48 weeks later19,20. In many patients who manifested the new onset of a major depressive episode following IFNα therapy, the administration of SSRI antidepressants ameliorated these depressive symptoms21. As another example, the experimental administration of typhoid vaccine increased blood IL-6 levels and caused transient depressive or dysphoric symptoms within 24–48 h (ref.22).
Genetics and epigenetics
Genetic research is fundamental to target validation: an association between a disease and DNA variation in a particular gene implicates that gene in pathogenesis. Genetically validated targets have a higher probability of leading to successful drug development in many therapeutic areas23.
Genetic evidence that immune-related genes are causally implicated in depression is beginning to emerge. In the last few years, genome-wide association studies (GWAS) and meta-analyses have become sufficiently powerful, with sample sizes in the order of hundreds of thousands, to report statistically robust associations between allelic variations and MDD. In one of the largest studies to date, pathway analysis of 44 single-nucleotide polymorphisms (SNPs) from a MDD GWAS gene list indicated enrichment for 19 pathways, including cytokine and immune response pathways24. Many of the genetic variants that are significantly associated with MDD by GWAS are in non-coding regions of the genome and were enriched at epigenetically active regulatory sites in fetal and adult brain tissue, and in peripheral lymphoid cells, indicating that genetic risks for MDD are likely to be phenotypically expressed through both the adaptive immune system and the CNS25. In a candidate gene Mendelian randomization analysis, a functional SNP in the IL-6 receptor promoter region predicted levels of IL-6, CRP and depressive symptom severity in a population cohort26. In another candidate gene study of a distinct functional polymorphism in the IL-6 receptor promoter region (rs1800795), the ‘low IL-6’ genotype was associated with fewer depressive symptoms during IFNα treatment27. Furthermore, CRP levels and individual depressive symptoms are co-inherited28. Finally, genetic variation in P2RX7, which encodes the purinergic receptor P2X7, was implicated to cause depression in a study that integrated depression GWAS with human brain proteome data29. P2X7 is an ATP-gated cation channel that is abundantly expressed in myeloid cells, including microglia, and P2X7 stimulation mediates activation of the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome complex, which is a key mediator of innate immune activation30. This finding was replicated in an independent depression GWAS and proteome-wide association study data set and extended using cis-regulated mRNA levels associated with depression in an integrated depression GWAS plus human brain transcriptomic analysis29. In candidate gene studies, haplotypes containing gain-of-function SNPs of P2RX7 that increase P2X7 pore activity31 were associated with increased time spent with depression32 and increased depression severity among patients with bipolar disorder33,34, MDD and type 1 diabetes mellitus35,36,37.
Epigenetics will also be important to validate immune targets for depression and other stress-related disorders. In epigenetic studies in humans, as well as in animal models of MDD, DNA methylation of GR, which encodes the glucocorticoid receptor (GR), or of FKBP5, which encodes a GR-chaperone protein, reduced expression of glucocorticoid hormone-responsive signals to such an extent that it increased pro-inflammatory biomarkers in response to social and behavioural stress. This observation reflects the role of glucocorticoids in immune system regulation, as the activated GR–glucocorticoid complex upregulates the expression of anti-inflammatory proteins and suppresses the expression of pro-inflammatory proteins38. Therefore, epigenetic effects that reduce GR signalling putatively release some innate immune mechanisms from the negative feedback normally mediated by glucocorticoids via the hypothalamic–pituitary–adrenal axis39,40. Such epigenetic ‘memories’ of early life stress may reflect a persistent allostatic change related to the glucocorticoid resistance previously associated with MDD41 and with the clinically well-recognized increased risk of MDD in adult life following an episode of adversity in childhood42. These epigenetic data thus converge with the results of gene expression studies in patients with depression to provide evidence that immune system dysregulation and reduced GR signalling (which restrains the effects of some pro-inflammatory mediators, including P2X7 signalling) contribute to the pathophysiology of MDD43.
Genome-wide DNA methylation profiling in brain tissue obtained post-mortem from individuals with mood disorders who died of suicide and in blood sampled from patients with depression with suicide ideation associated suicide risk with additive epigenetic and genetic factors at a locus in SKA2, which regulates GR transactivation44. Another study of tissue obtained post-mortem from the prefrontal cortex of patients with depression who completed suicide found differentially methylated regions, versus non-psychiatric controls, in the psoriasis susceptibility gene, PSORS1C3, and in TAPBP, which is located in the major histocompatibility complex (MHC) and encodes a glycoprotein that mediates interaction between MHC class I molecules and the transporter associated with antigen processing43. Finally, genome-wide DNA methylation and gene expression analyses performed in patients prospectively defined as either responders or non-responders to treatment with the SSRI escitalopram identified two genes that exhibited increased DNA methylation and mRNA expression in the non-responders: JAK2, which encodes a protein that activates both innate and adaptive immunity; and CHN2, which may affect hippocampal neurogenesis45. Interestingly, whole methylome screening in a small case–control study of MDD demonstrated that methylation of peripheral white blood cells was highly correlated with the methylome of frontal cortex and other brain regions46, suggesting that stress disorder-related epigenetic changes may be coupled across peripheral immune cells, which are distal to the site of inflammation but easy to measure, and proximal neuronal and glial cells, which are found in the brain and, therefore, difficult to assess in the clinic.
In summary, whereas emerging genetic data support the hypothesis that immune mechanisms contribute to the pathogenesis of mood disorders, highly penetrant, clearly causal mutations have not yet been identified. These data instead corroborate the broader conclusion from genetic studies of psychiatric disorders that their aetiology involves daunting polygenicity and phenotypic complexity, and poses significant challenges for the elucidation of potential therapeutic targets47. Nevertheless, the genetic data converge with other evidence to implicate specific immune-related gene networks in the pathophysiology of MDD, which has guided early efforts to evaluate drugs targeting these immune pathways as potential therapeutics for depression.
Immune targets evaluated in depression
Several immune-targeted compounds have been evaluated for potential antidepressant effects. The studies assessing these effects use three approaches: meta-analyses of changes in depressive symptoms within aggregate data from clinical trials of primary immunological disorders; analyses of mood effects in immunology clinical trials for which patient-level data were available; and RCTs in patients with MDD (Table 1). The first two of those approaches were opportunistic insofar as each study’s primary outcome measure assessed the efficacy of the compound in improving key symptoms of an immunological disease, and the post hoc analyses of effects on depression ratings were exploratory. By contrast, the third approach is the most direct exploration of the effect of a compound, or evaluation of a mechanism of action, as a potentially viable antidepressant pharmacotherapy.
Numerous targets and mechanisms have been evaluated that modulate different points within complex immune networks. These targets include the production of prostaglandins, which is inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs); a variety of pro-inflammatory cytokines (tumour necrosis factor (TNF), IL-6, IL-12/23, IL-23), which have been targeted using monoclonal antibodies (mAbs); p38 MAP kinase (also known as MAPK14), which not only orchestrates innate immunity but also regulates the adaptive immune system; and B cell-related targets (such as CD20 and the soluble B lymphocyte stimulator (BLyS)). Additionally, several classes of compounds with anti-inflammatory effects among their pleiotropic actions (glucocorticoids, statins, minocycline, pioglitazone) have been evaluated. For example, statins, in addition to their lipid-lowering effects, have immunomodulatory and anti-inflammatory effects that include inhibition of pro-inflammatory cytokine expression in monocytes and reduction of CRP levels48,49.
However, in most of these studies of anti-inflammatory treatments in depression, target engagement was insufficiently assessed, therefore causality cannot be proven (proof requires that the decrease in inflammation is apparent and precedes the improvement in symptoms). Another limitation of this research is that it predominantly assessed modulation of inflammation and the innate immune response, leaving considerable room for innovation in targeting aspects of adaptive immunity, a wider range of immune cell types and autoantibody-driven pathologies.
Clinical trials
The ability of anti-inflammatory treatments to produce antidepressant effects was initially demonstrated in secondary analyses of studies in patients with immunological disorders that included depressive symptom ratings. These studies focused predominantly on NSAIDs and cytokine inhibitors, and are included in several meta-analyses50,51,52,53 (Fig. 2). Köhler et al. reported results for 6,262 participants across 14 trials, of which 10 evaluated the efficacy of NSAIDs and 4 evaluated cytokine inhibitors targeting TNF or IL-12/23 (ref.50). They found significant pooled effects for NSAIDs (standardized mean difference (SMD) 0.27, 95% confidence interval (CI) 0.08–0.45; a positive SMD indicates an effect that favours treatment) and cytokine inhibitors (SMD 0.38, 95% CI −0.12–0.88). Kappelmann et al. performed a random effect meta-analysis of 7 RCTs of 2,370 participants and found a significant effect of cytokine inhibitors on depressive symptoms compared with placebo (SMD 0.4, 95% CI 0.22–0.59), and specifically for TNF inhibitors (SMD 0.33, 95% CI 0.06–0.6)51. Köhler-Forsberg et al. evaluated a wider range of drug classes and combined studies evaluating treatment effects in comorbid depression as well as in MDD. Overall, they reported significant effects across pooled NSAIDs (SMD 0.4, 95% CI 0.18–1.62) and cytokine inhibitors (SMD 0.56, 95% CI 0.19–0.93), as well as glucocorticoids (SMD 0.9, 95% CI 0.36–1.44), statins (SMD 0.26, 95% CI 0.04–0.48) and minocycline (SMD 0.87, 95% CI 0.29–1.45). A sixth drug class composed of pioglitazone did not show a significant antidepressant effect (SMD 0.43, 95% CI −0.7 to 1.56)52 (Fig. 2).

Meta-analyses of placebo-controlled effects of various mechanistic classes of immunotherapeutics on ‘comorbid’ depressive symptoms, measured as secondary end points in randomized clinical trials of medical inflammatory disorders50,51,52,53, show that some immunomodulatory drugs may improve depressive symptoms. Standardized mean difference (SMD) and 95% confidence interval (CI) shown, aggregated by mechanism of action, across the four primary meta-analyses compiled. BLyS, B lymphocyte stimulator; COX2, cyclooxygenase 2; NSAID, nonsteroidal anti-inflammatory drug; TNF, tumour necrosis factor.
There are several challenges to interpreting the improvements in comorbid depressive symptoms in the context of a medical immune or inflammatory disorder that is responsive to the same treatment. First, most patients with primary immunological disorders enter studies with depression ratings in the non-depression range, so sensitivity for detecting an antidepressant effect is limited to the minority of patients with clinically significant depression at baseline. Second, improvement in primary disease symptoms, including pain and fatigue, could confound the improvements in mood51.
To address these concerns, Wittenberg et al. reported a mega-analysis of patient-level data combined from 18 RCTs conducted by Janssen or GlaxoSmithKline, each of which examined 1 of 9 disorders, for 9 drugs against 7 different molecular targets (n = 10,743 participants)53. Access to patient-level data enabled stratification of patients at baseline into a high-depressive symptom cohort and a low-depressive symptom cohort, the former being the appropriately enriched population for detecting an antidepressant effect. Access to patient-level data also enabled the statistical adjustment for potential confounding factors including changes in symptom severity for the primary indication (for example, changes in joint pain in trials of rheumatoid arthritis). The greatest effects were seen among anti-IL-6 antibodies (SMD 0.8, 95% CI 0.20–1.41) and an anti-IL-12/23 antibody (SMD 0.48, 95% CI 0.26–0.70) (Fig. 2). Antibodies targeting BLyS and TNF demonstrated a non-significant trend towards improved depressive symptoms, and small molecules targeting p38 MAP kinase or cyclooxygenase 2 (COX2) had no significant antidepressant effect (Fig. 2).
Although most of the attention on cytokine antagonists as antidepressants has focused on antibodies against TNF and IL-6, some studies noted a positive effect of the IL-12/23 antagonist ustekinumab on depressive symptoms53,54. Newer compounds such as guselkumab more selectively target IL-23. In patients with moderate-severe plaque psoriasis, guselkumab treatment was associated with a significant improvement in anxiety and depression symptoms relative to placebo55.
In summary, in seeking immune targets that might cause or contribute to MDD, a number of proteins have been suggested through longitudinal studies, inflammatory challenges, genetics and epigenetics, and studies in patients with comorbid immunological disorders. These include TNF, IL-6, IL-23, COX enzymes and P2X7. To date, only a subset of these has been tested in patients with primary mood disorders.
Immune drug development in MDD
Few studies have tested whether immunomodulatory drugs produce antidepressant efficacy in patients with MDD or bipolar disorder, and most of these studies used drugs that reportedly improved depressive symptoms arising comorbidly in patients with a primary immunological disorder or influenced immunological processes that affect synaptic function in preclinical models. For example, several cytokines — including TNF, IL-6 and IL-1β — are considered ‘neuroactive’ because they modulate synaptic plasticity mechanisms such as neurogenesis or dendritic remodelling, and/or mediate neuronal–glial interactions. The drugs assessed to date include mAbs that neutralize cytokines or cytokine receptors, compounds that alter molecular factors that are induced by cytokines (for example, cyclooxygenases, prostaglandins, kynurenine metabolites) and molecules that affect the production, maturation, processing, release and downstream signalling induced by cytokines (Fig. 3).

a | Anti-cytokine and cytokine receptor antibodies. b | Nonsteroidal anti-inflammatory drugs (NSAIDS) targeting cyclooxygenase 1 (COX1) and/or COX2. c | Minocycline exhibits indirect effects on multiple components of the kynurenine pathway and is hypothesized to induce a neuroprotective effect. d | P2X7, an ion channel on the surface of glial and neuronal cells, is opened by high concentrations of ATP, leading to activation of the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome and release of IL-1β. This pro-inflammatory signalling pathway is blocked by JNJ-54175446, an antagonist of the P2X7 receptor, and could also theoretically be blocked by an NLRP3 inflammasome inhibitor. BMP, bone morphogenic protein; IDO, indoleamine 2,3-dioxygenase; IL-6R, interleukin-6 receptor; KAT, kynurenine aminotransferase; KMO, kynurenine 3-monooxygenase; NMDAR, N-methyl-D-aspartate receptor; PGE2, prostaglandin E2; PGI2, prostaglandin I2; ROS, reactive oxygen species; TNF, tumour necrosis factor; TXA2, thromboxane A2.
Antibodies targeting cytokines or cytokine receptors
The first anti-cytokine antibody tested in patients with MDD was infliximab, an anti-TNF antibody, which was studied in two RCTs. Raison et al. focused on patients (n = 60) who were moderately treatment resistant, and stratified patients based on high-sensitivity CRP levels ≥2 mg l–1 (ref.56) (Table 1). Infliximab was studied as an augmentation therapy (n = 37) or in medication-free subjects (n = 23). The primary outcome measure (change in Hamilton Depression Rating Scale, 17-item version (HDRS-17) score at 12 weeks) did not differ between the treatment and placebo arms. However, a post hoc analysis suggested that for patients with baseline CRP levels ≥5 mg l–1, infliximab’s treatment effect exceeded that of placebo. Infliximab was also tested by McIntyre et al. in 60 participants who were in the depression phase of bipolar disorder and who had been non-responsive to conventional antidepressants57. Infliximab or placebo was administered adjunctively. The primary outcome measure (change in Montgomery–Åsberg Depression Rating Scale (MADRS) at 12 weeks) did not differ between treatment and placebo arms; however, a post hoc analysis suggested a treatment effect among patients with a childhood history of physical abuse. Notably, peripherally administered antibodies are expected to exert their effects predominantly outside the CNS because of the limited permeability of the blood–brain barrier (BBB) to large molecules. The ability of infliximab to affect CNS targets, directly or indirectly, remains unclear.
The anti-IL-6 antibody sirukumab was tested as an adjunctive treatment in patients with MDD who were inadequately responsive to conventional antidepressants, and had plasma CRP levels ≥3 mg l–1 at screening and baseline58. The change in HDRS-17 score at week 12 did not differ significantly between the sirukumab and placebo arms. However, secondary analyses showed that the mean Snaith–Hamilton Pleasure Scale (SHAPS) anhedonia rating improved in the sirukumab group versus the control group (nominal P = 0.014). Moreover, the mean HDRS-17 score decreased to a greater extent in patients treated with sirukumab than in those treated with placebo for participants with baseline CRP values ≥8 mg l–1 (although the subset of people with such CRP levels was relatively small).
The observation that sirukumab, a mAb that is poorly brain penetrant, may improve anhedonia has interesting implications. Potentially consistent with this observation, studies in animal models showed that anti-IL-6 antibodies, which directly neutralize IL-6 in the periphery, can reduce the amount of IL-6 in the brain, and have profound effects on anhedonia-like behaviours in rodent depression models14.
However, an open-label study in patients with rheumatoid arthritis using tocilizumab, a mAb that targets the IL-6 receptor, found no significant change in depression symptom ratings on the Hospital Anxiety and Depression Scale (HADS), which is sensitive to anhedonia59. Tocilizumab has poor brain penetrance, so these data suggest that blocking IL-6 receptors in the CNS rather than the periphery may be necessary. By contrast, by neutralizing IL-6 cytokines in the periphery, sirukumab may have reduced IL-6 receptor signalling in the CNS as well as in the periphery60.
Nonsteroidal anti-inflammatory agents
Several clinical trials in mood disorders have assessed the potential antidepressant effects of small-molecule drugs targeting molecular factors that interact with cytokines. The most commonly used agents have been NSAIDs that are known to be brain penetrant, including selective COX2 inhibitors (celecoxib), dual COX1 and COX2 inhibitors (naproxen, ibuprofen, high-dose aspirin) or predominantly COX1 inhibitors (low-dose aspirin). COX1 and COX2 inhibition can differentially affect neuroplasticity and neuroprotection mechanisms in some preclinical models61,62. These effects are mediated partly by modulatory effects on the arachidonic acid pathway, which is implicated in the pathophysiology of bipolar disorder63, and by preventing neurotoxicity associated with inflammation-driven activation of the kynurenine metabolic pathway (see below), which has been associated with reduced volumes of the hippocampus and other subcortical structures in MDD64,65.
As detailed elsewhere66,67, the sample sizes of these studies have been small, so meta-analyses have been conducted to increase statistical power (Fig. 2). Among four RCTs that evaluated celecoxib as an augmentation therapy in MDD, three found the celecoxib arm to be superior, although in one the beneficial effect at 4 weeks was no longer significant at the 8-week end point (Table 1). None of these studies stratified patients based on immune biomarkers, but in one study the baseline levels of serum IL-6 predicted changes in depression ratings at 6 weeks68. The results of studies assessing the antidepressant effects of adjunctive anti-inflammatory agents in bipolar depression are also mixed. A meta-analysis of eight RCTs that assessed adjunctive NSAIDs, omega-3 fatty acids, N-acetylcysteine and pioglitazone showed a moderate antidepressant effect compared with standard therapy; significant effects were observed for the omega-3 and N-acetylcysteine arms, but not for NSAIDs (SMD −0.02, 95% CI −0.56 to 0.52)69. These data showed a significant overall benefit of small-to-moderate effect size for combined results across NSAIDs, but no significant benefit for studies that evaluated selective COX2 inhibitors alone (Fig. 2).
Results of studies exploring more selective inhibition of COX1 in mood disorders are similarly mixed. A pharmacoepidemiological study found that low-dose aspirin showed benefits in stabilizing the illness course in bipolar disorder (the benefit was fewer medication changes), whereas high-dose aspirin, non-selective NSAIDs, selective COX2 inhibitors and glucocorticoids were associated with an increase in medication changes70. Similarly, a pharmacoepidemiological study of risk for developing MDD71 found that continued use of low-dose aspirin, as well as of statins, decreased the rate of incident depression, whereas continued use of non-aspirin NSAIDs or high-dose aspirin increased the rate of incident depression. Nevertheless, in a trial comparing health outcomes on low-dose aspirin versus placebo in older adults (n = 19,114, with 1,879 patients with depression at baseline), the depression symptoms increased to a greater extent in the aspirin arm versus the placebo arm across a median of 4.7 years67; among individuals with depression at baseline, aspirin was associated with a worsening in depression ratings. Finally, low-dose aspirin failed to reduce the incidence of new cases of depression72.
Minocycline and kynurenine metabolism
Another brain-penetrant, small-molecule drug tested for potential antidepressant effects is the tetracycline antibiotic, minocycline, which exhibits pleiotropic anti-inflammatory effects in the periphery and the brain73. Minocycline modulates immune function by inhibiting the activation, migration and/or proliferation of T cells, neutrophils and microglia, inhibiting the release of pro-inflammatory cytokines and increasing the release of anti-inflammatory and anti-apoptotic molecules74,75. In addition, minocycline indirectly reduces the pro-inflammatory activation of the kynurenine pathway by attenuating the expression of cytokines that are produced during a pro-inflammatory challenge, thereby reducing release of neuroactive kynurenine metabolites66,76.
In rodent models, the depression-like behaviours induced via inflammatory challenge with lipopolysaccharide (LPS) are blocked by genetic or pharmacological (using minocycline or the indoleamine 2,3-dioxygenase (IDO) antagonist 1-methyltryptophan) reduction in the activity of the enzyme, IDO, that converts tryptophan to kynurenine76,77. Cytokines such as IFNγ and TNF activate the kynurenine pathway by increasing IDO expression, thereby shunting tryptophan away from serotonin synthesis and towards kynurenine, increasing the production of neuroactive kynurenine metabolites. Depression has been associated with an imbalance in these metabolites: increased metabolism down the 3-hydroxykynurenine branch leads to increased levels of the putatively neurotoxic metabolite, quinolinic acid78. A post-mortem study of patients with MDD or bipolar disorder who died by suicide showed elevated concentrations of quinolinic acid in microglia in rostral and subgenual portions of the anterior cingulate cortex (ACC), a key region in the neurocircuitry of depression79. In MRI studies, the relative quantities of the putatively neuroprotective kynurenine metabolite, kynurenic acid, and the putatively neurotoxic kynurenine metabolites, 3-hydroxykynurenine and quinolinic acid, in blood correlated with reductions in grey matter thickness in this cortical region and a diagnosis of MDD; reductions in cortex thickness also correlated with elevated CRP levels80. The relative reduction in levels of kynurenic acid versus quinolinic acid also has been associated with reductions in hippocampal and amygdala volumes65, as well as with altered hippocampal function during emotion-dependent autobiographical memory recall in patients with MDD81.
Based on minocycline’s effects — reducing cytokine release and indirectly reducing IDO function — several RCTs have assessed its antidepressant efficacy in MDD and bipolar disorder. Husain et al. examined minocycline as an adjunctive therapy for patients who were resistant to conventional antidepressants82. In the minocycline arm (n = 21), patients had a greater decrease in HDRS-17 scores compared with those in the placebo arm (n = 20) at week 12 (standardized effect size 1.21, P < 0.001). By contrast, Dean et al. studied the efficacy of adjunctive minocycline treatment in patients selected from the general MDD population83, and found no significant change in MADRS scores, although improvements were observed in secondary outcomes related to global functioning and quality of life. Nevertheless, the negative primary outcome of the trial in participants from the general MDD population may highlight the importance of selecting patients based on biomarkers or treatment resistance to conventional antidepressants. Finally, as a passing comment, in an open-label study in which minocycline was added to SSRI antidepressants in patients with MDD with psychotic features, the antidepressant effect was profound, and this study population is generally non-responsive to SSRIs alone84.
Two studies assessed the efficacy of adjunctive minocycline for improving depressive symptoms in bipolar disorder. Husain et al. compared the efficacy of minocycline plus active celecoxib, minocycline alone and celecoxib alone versus placebo85. The mean change in HDRS-17 scores at 12 weeks did not differ across groups. By contrast, Savitz et al. tested the efficacy of low-dose aspirin and minocycline as augmentation therapy for bipolar depression86, in patients randomized to receive minocycline plus aspirin, minocycline alone, aspirin alone or placebo. The primary outcome measure was treatment response at week 6, defined conventionally as ≥50% decrease in the MADRS score. The minocycline plus aspirin group showed a significantly greater response rate than the placebo alone group. When all four arms were included in the analysis, the main effect of aspirin (but not minocycline) on treatment response was significant. Nevertheless, a significant interaction with serum IL-6 level was observed, indicating a greater response to minocycline versus placebo among participants with high IL-6 levels. Thus, although minocycline had a non-significant effect in the entire sample, consistent with the results of Husain et al., the preliminary observation that patients with bipolar disorder and depression, as well as elevated basal IL-6 levels, may benefit from minocycline warrants further testing14,87,88.
Signalling molecules involved in cytokine production
p38 MAP kinase plays a key role in pro-inflammatory cytokine synthesis. This gene was implicated as a hub in a network of white blood cell genes for which expression differed between individuals with MDD and healthy subjects in a meta-analysis89. A small-molecule inhibitor of p38 MAP kinase, losmapimod, was studied in two RCTs in participants with MDD90. Patients were selected for loss of energy and/or interest and psychomotor retardation but were excluded for previous antidepressant treatment resistance. Neither study showed a significant advantage for losmapimod versus placebo. The extent of target engagement within the CNS was not established in these studies.
Microglial activation
Recently, a focus in CNS drug development has turned towards targets that modulate microglial activation, especially via the P2X7–NLRP3 axis30. As an ATP-gated ion channel, P2X7 responds to extracellular ATP, which is released by stressed or dying cells or by psychosocial stress91. During innate immune responses, damage-associated molecular patterns or pathogen-associated molecular patterns activate pattern recognition receptors (namely the Toll-like receptors (TLRs)) that induce ATP release, thus activating P2X7. Two convergent signals are required for microglial activation. The TLR-mediated activation of the NF-κB pathway activation acts as a first signal, promoting the transcription of genes encoding inflammatory mediators, such as pro-IL-1β, and inflammasome components, such as NLRP3; P2X7 stimulation is the second signal, inducing inflammasome activation and the subsequent activation of caspase 1, which processes pro-IL-1β into its mature form, thereby enabling IL-1β release92. P2X7 activation also promotes IL-6 release and stimulates free radical production, phospholipase activation, cell cycle regulation and apoptosis. During adaptive immune responses, P2X7 stimulation participates in T cell activation, and ATP–P2X7 signalling decreases the suppressive activity and viability of regulatory T cells (Treg cells), favouring polarization of T cells into T helper 17 cells (TH17 cells)92.
P2X7 activation on microglia has been reported to have a pathological role in neuroinflammatory diseases (such as multiple sclerosis) and neuropsychiatric disorders, including MDD12,93,94,95,96. In patients with MDD, the P2RX7 mRNA levels were abnormally increased in individuals who were treatment resistant, irrespective of medication status, but reduced in medicated, treatment-responsive patients in remission97. Compatible with these data, abnormal increases in the expression of TLR proteins and mRNA transcripts were reported in patients with depression and who died by suicide98. Notably, in mice, stress-induced depression-like behaviours were reversed by brain-penetrant (but not by non-brain-penetrant) P2X7 antagonists30. An RCT is underway to test the antidepressant efficacy of a brain-penetrant P2X7 antagonist, JNJ-54175446, in MDD.
Anti-inflammatory cytokines
Another area of emerging interest in therapeutics discovery involves mechanisms that enhance the release or function of anti-inflammatory cytokines, such as IL-10, which participate in the immune system’s inherent compensatory mechanisms for restraining pro-inflammatory cytokine signalling. In preclinical models, the resolution of inflammation-induced depression-like behaviours required T lymphocytes, acting via an IL-10-dependent pathway to decrease the expression of IDO, suggesting that novel therapeutics targeting the T lymphocyte/IL-10 pathway could promote recovery from MDD15. The importance of inherent anti-inflammatory systems to recovery from depression was also suggested by a longitudinal study of MDD that assessed immune biomarkers before and after antidepressant pharmacotherapy; although treatment responders and non-responders both had elevated levels of pro-inflammatory cytokines pretreatment and increased levels of anti-inflammatory cytokines (IL-4, IL-5 and IL-10) post-treatment, the pro-inflammatory cytokine levels remained elevated only in the non-responders99. These data suggest that patients with MDD who respond poorly to conventional antidepressants manifest a defective anti-inflammatory response. Nevertheless, despite these encouraging leads, there has been no study yet in individuals with MDD with the objective of increasing anti-inflammatory activity.
Assessment of mechanism
Although most studies associating immune dysregulation and clinical depression have focused on the peripheral immune system, understanding mechanisms in the CNS whereby altered function mediates depressive symptoms is important to prioritize potential targets and characterize patient subgroups that are most likely to benefit from new treatments. Neuroimaging biomarkers assessed using positron emission tomography (PET), MRI (including functional MRI (fMRI)) and magnetic resonance spectroscopy (MRS) provide approaches to elucidate the effects of altered immune function on CNS function. Each of these neuroimaging modalities has a different set of trade-offs between immune specificity of the imaging signal, spatial resolution, cost and accessibility.
Positron emission tomography
PET can be used to visualize the most target-specific biomarker of human brain inflammation, but its use in research is limited by its comparatively high cost and low accessibility; PET also involves exposure to low levels of radioactivity, limiting its repeat use. To date, studies of neuroinflammation in MDD have relied on radioligands (such as [11C]PK11195 and [11C]PBR28) that bind to translocator protein (TSPO), which is expressed on mitochondrial membranes. The TSPO binding potential increases in multiple conditions involving neuroinflammation. However, TSPO is expressed not only on microglia but also on other glial cells and neurons, which limits the specificity of TSPO binding as a marker of microglial activation. Furthermore, a common SNP in TSPO (rs6971) causes an amino acid substitution in TSPO that alters the affinity of this protein for TSPO ligands other than [11C]PK11195 (ref.100), a limitation which must be addressed in the study design. Several case–control PET studies in patients with MDD reported small or moderately sized, but statistically significant, increases in TSPO binding in the ACC and other regions in which post-mortem studies had more specifically shown elevated activation of microglia in individuals with MDD or bipolar disorder who died by suicide79,101 (Fig. 4). Future development of ligands with greater specificity for microglia or other central immune targets is needed to optimize the potential of PET biomarkers of neuroinflammation in depression102.

Examples of neuroimaging results linking inflammation to brain states (clockwise from top left). Positron emission tomography (PET) using a radiotracer binding to translocator protein (TSPO) demonstrated increased TSPO binding, a putative marker of microglial activation, in anterior cingulate cortex (ACC) and other brain areas in case–control studies of major depressive disorder (MDD). Quantitative magnetization transfer (qMT), a microstructural MRI marker of cortical tissue composition, was significantly correlated with post-typhoid vaccine fatigue scores in posterior insular cortex (y axis, magnetization transfer exchange rate constant (Kf); x axis, fatigue visual analogue scale (fVAS), vaccine minus placebo (V–P)). Functional MRI (fMRI) data collected in the resting state demonstrated reduced functional connectivity between ventro-medial prefrontal cortex (vmPFC) and inferior ventral striatum (iVS) in patients with MDD with high blood concentrations of C-reactive protein (CRP). Macrostructural MRI metric of striatal volume scaled negatively with log of the ratio between kynurenine (Kyn) and tryptophan (TRP) blood concentrations. Magnetic resonance spectroscopy (MRS) measurements of brain glutamate concentration were increased in patients with depression with higher log blood CRP concentrations. fMRI measures of increased functional connectivity of subgenual ACC (sACC) were positively correlated with depressive mood changes after typhoid vaccination. SMD, standardized mean difference. Images in figure are adapted from refs104,105, Springer Nature Limited; with permission from ref.101, Elsevier; and from refs22,110, CC BY 4.0 (https://creativecommone.org/licenses/by/4.0/).
Functional MRI
fMRI data collected while patients are either performing simple cognitive tasks or in the ‘resting state’ can be used to measure functional activity in various brain regions, as well as the functional connectivity matrix (connectome) of dynamic coherence between the resting and active-state fMRI signals that are simultaneously measured in paired regions. This is a relatively safe, affordable and accessible imaging biomarker, and is suitable for repeated assessments. However, the haemodynamic signals measured via fMRI are non-specific and limited in the extent to which they can inform the neurobiological mechanisms underlying differences observed between case and control samples.
A review of human fMRI studies of the effects of inflammation on cerebral function defined two broad strategies across this literature: observational and experimental103. Observational studies measured the correlation between fMRI metrics and blood concentration of immune markers. Experimental studies measured fMRI metrics before and after a controlled pro-inflammatory stimulus (for example, typhoid vaccination). Meta-analysis of these data identified regions where functional activity was significantly changed in observational and experimental studies, including the amygdala, hippocampus, hypothalamus, striatum, insula, midbrain and brainstem, as well as prefrontal and temporal cortices. This observation is consistent with emerging evidence that systemic inflammation can be correlated with, or could cause, changes in functional connectivity between the areas of limbic, prefrontal and temporal cortices, and anatomically related subcortical nuclei104,105 that form the ‘medial prefrontal network’, which mediates and regulates emotional behaviour and comprises the neurocircuitry of mood disorders106. These data thus provide evidence that interactions of immune activation with brain interoceptive mechanisms can mediate discrete changes in brain and behaviour within the context of infection and inflammation107,108.
Microstructural MRI
Alternative magnetic resonance modalities may achieve greater neurobiological specificity than fMRI. Microstructural MRI enables measurements of physical parameters, such as T1 or T2 relaxation times and magnetization transfer, that can give insight into the levels of extracellular fluid, neurite density, myelination or other microscopic properties of brain tissue within an image volume element (voxel)109. Microstructural MRI metrics that are sensitive to the proportion of free water in a voxel seem intuitively more likely to be indicative of central inflammation than a macrostructural MRI metric such as cortical volume. Recent studies reported that changes in the microstructural MRI parameter of proton density (a measure of tissue water content) were associated with systemic inflammation, as indexed by CRP levels, and differed significantly in patients with MDD who manifested CRP levels >3 mg l–1 relative to patients with MDD and lower CRP levels, as well as relative to healthy controls with CRP levels >3 mg l–1 (refs110,111).
Magnetic resonance spectroscopy
MRS assesses the brain tissue concentration of molecules such as the neurotransmitters γ-aminobutyric acid (GABA) and glutamate. MRS studies have shown that peripheral administration of IFNα was associated with increased glutamate levels in the basal ganglia and cingulate cortex, which correlated with fatigue and anhedonia ratings. Basal ganglia glutamate concentration was also higher in patients with MDD who had high levels of CRP (>3 mg l–1) than in patients with MDD and low CRP levels (<1 mg l–1)105. Although the glutamate signal is predominantly intracellular, these data are compatible with evidence from preclinical models in which pro-inflammatory cytokines and microglial activation can alter glutamate release and transport, resulting in increased extracellular glutamate concentrations112. Neuroinflammation thus has been hypothesized to play a role in neurotoxicity via its downstream effects on glutamate metabolism113. However, many factors other than inflammation can alter glutamate levels, and MRS in vivo can only resolve the minor peak of glutamate resonance in relatively large (~1 cm3) volumes of tissue.
In summary, non-invasive biomarker measures of central immune status that can provide anatomically fine-grained information on specific molecular or cellular targets in the human brain are not yet available. The increasing use and technical refinement of fMRI, microstructural MRI and MRS may conceivably produce ‘bridging’ biomarkers that can be used to investigate relationships between immune mechanisms and depression. Further developments in PET radiochemistry, single-cell analysis of circulating immune cells, and greater assay sensitivity for studies of CSF and blood could provide more precise insights into immune mechanisms for small numbers of patients and prioritized immune targets. Nevertheless, peripheral blood biomarkers remain the most accessible and comprehensive platform for assessing the immune status in large numbers of patients.
Patient stratification
The discovery and development of novel antidepressants targeting immune dysregulation will require multidisciplinary partnerships between researchers with expertise in psychiatry, experimental medicine, biomarker and diagnostic development, and neuroimmunology, and success will likely depend on patient selection or stratification criteria. Diverse approaches have been taken to both inclusion criteria and within-trial stratification among the studies reviewed above, so stratification clearly remains an area of uncertainty in the field. A combination of varying levels of treatment resistance and CRP level thresholds have been used, along with symptoms related to anhedonia, in patients with either primary mood disorders or depression arising in the context of primary immunological diseases. Many other small studies focused on ‘all-comers’ with MDD or bipolar disorder, which might have resulted in a false negative, because an experimental treatment that was not statistically different from placebo in an all-comers trial may be effective in a patient subgroup (Table 1).
One approach to identifying potential options for patient stratification is to outline the different immune axes along which patients with depression are statistically distinct from individuals without depression. As potential diagnostics, blood biomarkers are more accessible than imaging-based markers and provide finely detailed mechanistic information about the circulating compartment of the peripheral immune system, although the extent to which central immune function can be inferred from peripheral immune markers remains unclear. Nevertheless, the output of immune-dysregulated brain circuits may manifest as observable symptoms and behaviours. Quantitatively characterizing phenotypic aspects of ‘immune-mediated depression’ may identify treatment cohorts. Too often, especially for molecular analyses, the focus is on analysing outcomes based on general clinical ratings or a diagnosis of MDD rather than on specific dimensional constructs, such as anhedonia. Ultimately, diagnostic refinement to enable precision medicine will require the collection of data from patient cohorts that are both phenotypically rich and biomarker dense.
Understanding case–control differences may prove helpful in generating hypotheses for candidate diagnostics and stratifying patients in early trial phases. But refinement of these diagnostics through clinical trials, in which treatment outcomes are characterized, will be needed to identify a diagnostic strategy that identifies the widest range of patients who are likely to benefit, while also enriching the target population for individuals with a high likelihood to benefit more from the novel therapeutic than from the current standard of care.
Biomarkers with diagnostic potential
A number of immune markers have some capacity to distinguish individuals with depression from those without, and to quantify the level of immune dysregulation. These markers include CRP114, cytokine levels115,116, genome-wide gene expression89,117,118 and quantitative PCR (qPCR) levels of specific mRNAs97,119, ex vivo LPS-stimulated cytokine or gene expression levels120,121,122,123 and quantitative immune cell counts124,125,126,127,128 (Fig. 1).
Studies that have derived CRP thresholds that can predict treatment response in the context of clinical trials have, to date, only identified putative ideal cut-off values post hoc56,58. However, these cut-off values identified subgroups that contained a small proportion of people with depression. Baseline IL-6 levels predicted response to both minocycline86 and celecoxib68. Cytokines have been increasingly measured using multiplex assays for dozens of proteins, and both aggregated summary scores121 and algorithmically derived scores119,129 have been considered as an approach to increase the robustness and precision of the measurements.
As diagnostic biomarkers, some cytokines are difficult to measure because levels that are considered elevated in the context of MDD are often near the limit of quantification of available assays. Furthermore, the intrinsic lability of cytokines poses a concern for their use as diagnostic biomarkers, as they fluctuate widely over the course of a day and in response to activity. Just as glycated haemoglobin A1c (HbA1c) provides average blood sugar control that is weeks long (whereas blood glucose levels provide a snapshot), a more stable measure of cytokine dysregulation may be more useful in the clinic.
Whole-genome mRNA expression has been measured in whole blood and in peripheral blood mononuclear cells in multiple MDD case–control studies — this analysis allows a more comprehensive assessment of peripheral immune pathways associated with depression89,117,118. Meta-analyses have identified that innate immune genes are overexpressed, and genes involved in adaptive immunity are under-expressed, in individuals with depression89. These depression-related impairments of adaptive immunity are consistent with previous findings130,131. Notably, the inverse relationship between innate and adaptive immune gene expression was evident at the level of individual subjects: individuals with MDD who had increased expression of innate immune genes also had decreased expression of adaptive immune genes, enabling the development of a single score to indicate immune dysregulation that could be used for patient selection89.
Studies focusing on the transcription of a few, preselected candidate genes have reported differential expression of pro-inflammatory, GR and neuroplasticity-related genes97,119. Genome-wide studies may guide the deliberate selection of a small number of stable mRNAs — representative of key immune processes, pathways and cell types — that can be assessed by qPCR-based assays; these may enable refinement of the immune compartments that should be prioritized for patient selection for an individual drug and/or its mechanism of action. For example, one study using this approach showed that patients with MDD who either were currently with depression and treatment resistant or currently with depression and drug-free could be differentiated by signatures of mRNA transcripts from healthy controls and drug-treated patients who had remitted during treatment97. These signatures putatively reflected inflammasome activation and glucocorticoid resistance. Moreover, a signature of the expression of six mRNAs (P2RX7, IL1B, IL6, TNF, CXCL12 and GR) distinguished patients who were antidepressant treatment resistant from those who were responsive. These data are similar to those from a longitudinal study in which drug-free patients with depression had increased mRNA levels of IL1B, IL6, TNF and FKBP5, together with reduced levels of GR. Furthermore, the pattern of mRNA expression of these and/or other inflammation-related genes predicted or correlated with responsiveness to antidepressant drug treatment119.
Beyond proteins and genes, another easily accessible snapshot of the state of the immune system is through immune cell counts. Individuals with depression have more white blood cells, more neutrophils, fewer T lymphocytes or B lymphocytes132, an increased neutrophil to lymphocyte ratio and an increased CD4+ to CD8+ T cell ratio relative to control individuals124,125,126,127,128. Depression may alter or result from altered CD4+ cell differentiation, as individuals with MDD have fewer Treg cells, and more TH1 cells and TH17 cells than those without133,134,135 (Fig. 1). In a recent study, based on absolute counts of 14 blood cell types in patients with MDD, it was possible to identify subgroups of individuals with ‘inflamed depression’, characterized by increased myeloid (neutrophil, monocyte) or lymphoid cell (CD4+ T cell) counts, and correlated with increased levels of CRP and IL-6 and increased depression severity136.
Single-cell sequencing can advance our understanding of the immune system at the cellular level, which may further elucidate key drivers of depressive symptoms and identify potential therapeutic targets. One study applying this approach in patients with bipolar disorder found remarkably robust biomarker abnormalities among mRNA transcripts measured specifically in monocytes. An inflammatory gene expression signature composed of these transcripts discriminated a large proportion of the patients as well as the offspring of parents with bipolar disorder from age-matched controls137.
The interplay between innate and adaptive systems modulates not only the protective immune response to microbial infection and other non-self-antigens but also the pathogenic generation of autoantibodies. For example, one study found that whereas patients with bipolar disorder showed both pro-inflammatory activation in monocytes and elevated percentages of anti-inflammatory T cells (specifically Treg cells), the two sets of abnormalities occurred independently from each other, and patients who manifested comorbid autoimmune thyroid disease (which is common in bipolar disorder) showed fewer anti-inflammatory T cells compared with patients without this condition138.
Furthermore, the potential for autoantibodies to play a pathogenic role in psychiatric syndromes has been seen in patients with autoimmune encephalitis resulting from autoantibodies against N-methyl-D-aspartate (NMDA) receptors139 or potassium channel complex proteins140,141. Although these antibodies are unlikely to drive symptoms in more than a small minority of patients, higher levels of overall autoantibody burden are seen among patients with depression and other psychiatric diseases compared with healthy patients, suggesting that this area merits further exploration as novel treatment options to regulate autoantibody levels become available142,143. Target-specific, rather than disease-specific, patient selection strategies may also emerge, and these could involve genetic mutations, epigenetic modifications, or even autoantibodies. The utility of the biomarker as a patient selection strategy, ultimately, will depend both on its predictive power and its prevalence within the population.
Phenotypes with diagnostic potential
Although no definitive standard exists to optimize the selection of patients with depression for immune-directed therapeutics, some patterns of common phenotypic traits in patients with elevated inflammatory markers are apparent within the more heterogeneous set of symptoms of MDD. Compelling evidence suggests that inflammation may have effects specifically on anhedonia and the cognitive processing of rewarding stimuli rather than an equally weighted impact across all DSM-5 symptoms. Similar to depressed mood and anxiety, anhedonia is observed among individuals with a variety of psychiatric, neurological or primary inflammatory disorders144. Ultimately, a patient selection approach based on a clinical phenotype would be more widely accessible and economical than a biological measurement, if sufficiently accurate.
Evidence suggesting a link between anhedonia, depression and the function of the mesolimbic dopaminergic circuits that underlie reward processing and motivated behaviour is converging across informatics, clinical and imaging studies. Inflammatory cytokines have been shown to exert a direct effect on mesolimbic dopamine transmission that is associated with a reduced willingness to expend effort for reward as well as with a reduced capacity for reward learning144. This effect, at least partly, involves a reduction in the availability of dopamine precursors, as neuroimaging and in vivo microdialysis studies in humans and/or non-human primates indicate that inflammatory cytokines such as IFNα reduce dopamine release in the ventral striatum, in association with depressive symptoms including anhedonia and psychomotor slowing113. In a non-human primate model, this effect could be reversed by administering the dopamine precursor, levodopa145.
By impacting motivated behaviour and psychomotor activity, the effects of inflammation on central dopaminergic transmission may contribute to the reduction in physical activity observed in patients with depression, which increases the risk for developing obesity and associated metabolic disturbances. Obesity further drives systemic inflammation, as adipose tissue actively secretes cytokines and obesity is associated with changes in the adipocyte secretome that increases the production of pro-inflammatory cytokines146. Notably, positive genetic correlations with body mass and Mendelian randomization analysis support the notion that body mass index (BMI) is causal or correlated with causal risk factors for depression24. Moreover, a longitudinal study of 3,809 adults without depression (age ≥50 years) found that participants with higher baseline levels of inflammatory markers were more likely to report low levels of physical activity 4 years later, and low activity partially mediated the relationship between systemic low-grade inflammation and subsequent depressive symptoms147. Interrupting the effects of inflammation on central dopaminergic pathways and physical activity via pharmacological and non-pharmacological (for example, exercise) approaches conceivably may reduce or prevent depressive symptoms and associated medical comorbidities.
A noteworthy study in this regard profiled drugs for their similarity across 4,201 side effects, and found that cytokine inhibitors were closest in the high-dimensional side effect space to drugs with a dopaminergic effect, but were distant from other antidepressants148. This finding suggests that dopaminergic compounds that influence reward-guided behaviours may be more effective in patients with an inflammatory component to their depression. Clinically, this hypothesis was confirmed in the Combining Medications to Enhance Depression Outcomes (CO-MED) study149, which showed that with increasing CRP levels, SSRI monotherapy became less effective, and augmentation with the dopamine and norepinephrine reuptake inhibitor, bupropion, became more effective150.
In clinical trials, IL-6 antagonists showed potential antidepressant activity in patients with rheumatoid arthritis (treated with sirukumab) and multicentric Castleman’s disease (treated with siltuximab), but notably these post hoc analyses relied upon a depressive symptom score which weighted anhedonia at 50%151. This may explain the findings in the sirukumab study in MDD discussed above58 in which significant improvement was not observed on the HDRS-17 (of which only 1 in 17 items assesses anhedonia) but benefit was observed on a scale that rated anhedonia severity (SHAPS).
Recently, in the NESDA study, the association of both basal and ex vivo LPS-stimulated inflammatory markers with individual depressive symptoms was explored in a large, longitudinal cohort. In both cases, inflammatory markers were most strongly associated with ‘sickness behaviour’ symptoms, including deficits in general interest and capacity for enjoying previously pleasurable activities or stimuli (both of which are anhedonic symptoms), at up to a 9-year follow-up123.
Convergently, multimodal neuroimaging studies of inflammation-related changes in brain structure, function and biochemistry have implicated components of the mesolimbic reward circuitry, including increased haemodynamic activity in the subgenual ACC (sACC)22, increased levels of basal ganglia glutamate105, decreased cortico-striatal functional connectivity105, decreased striatal volume64, increased markers of inflammation in the insular cortex110 and microglial activation in the sACC and other regions79,101 in individuals with depression (Fig. 4).
Although, in principle, a symptom-based patient selection approach should be simpler than a biological diagnostic, it remains unclear how patients with anhedonia and/or reward processing deficits can optimally be selected for clinical trials. Measurement of anhedonia is imprecise, and the available rating scales variably address different aspects of this complex phenomenon which includes deficits in motivation, consummatory pleasure and reward learning, taking into account both direct and indirect rewards152. New clinical scales which address these challenges are under development and validation, such as the Dimensional Anhedonia Rating Scale (DARS)152. As an alternative to symptom scales, functioning of the dopaminergic reward circuitry can be probed through cognitive testing that uses reward-related tasks, which may provide a more specific metric for patient selection144. These are currently being tested and validated in human studies, with different tasks measuring distinct aspects of reward learning153,154. Furthermore, with emerging trends in the digital health sector, both cognitive tests and symptom assessments could be deployed as digital diagnostics to provide online platforms to screen large numbers of potential participants for trials of new agents focusing on anhedonia or other specific dimensions of depression.
Unmet clinical need
In order to target an immune subtype of MDD as a viable strategy for drug development, this patient subpopulation should be demonstrably underserved by current therapeutic options, and the introduction of a new therapeutic should result in fewer patients with depression.
The unmet need for patients with high inflammatory burden has been demonstrated in several ways. For example, in the longitudinal NESDA study, patients with MDD recently starting an antidepressant and who had more than four indicators of chronic inflammation or metabolic dysregulation at baseline had a 6.85-fold (95% CI 1.95–24.06) increased odds of still having MDD at year 2 (ref.155). In addition, during antidepressant treatment, higher pretreatment levels of inflammation, as measured by levels of CRP, IL-6 and TNF, were associated with a poorer response to first-line antidepressants2,3,4, and after treatment the patients who were non-responsive had deficits in the ability of anti-inflammatory cytokine release to restrain further increases in pro-inflammatory cytokine release99. Furthermore, in longitudinal studies, patients with depression and who demonstrated suicidality had persistently elevated levels of CSF and kynurenine pathway metabolites (such as quinolinic acid), and IL-6 levels were elevated in the CSF of individuals who attempted suicide and these levels correlated with the severity of depression and suicide ideation156,157,158. Finally, in vitro evidence shows that inflammatory cytokines change expression of the tryptophan metabolic pathway enzymes and serotonin transporters, which conceivably may contribute to resistance to SSRI antidepressants159.
Beyond the impact of psychiatric symptoms on the morbidity and mortality associated with MDD, patients with depression also manifest a twofold to fourfold higher risk for cardiovascular disorders160, and have poorer outcomes during comorbid heart failure, stroke and peripheral artery disease161,162,163. Moreover, MDD is associated with exaggerated biological ageing: these individuals have premature decreases in leukocyte telomere length (a marker of cellular age that predicts several ageing-related diseases and early mortality) and increases in senescence-related gene transcription and molecular secretion patterns in physiological stress and inflammatory systems164,165,166. Dysregulation of the interactions among physiological stress systems that include inflammation, hypothalamic–pituitary–adrenal axis hyperactivity and metabolic dysregulation may partly underlie these associations167,168,169. In addition, early life stress, a risk factor for MDD, is associated with high levels of inflammation170 and increased risk of cardiovascular-related and all-cause mortality171. Breaking these links could yield positive outcomes for patients beyond mental health. For example, metformin (a drug used in the treatment of type 2 diabetes) has been proposed to have anti-senescence effects by modulating intracellular pathways (such as those involving NF-κB and p53) that reduce the production of senescence-associated secretory proteins, including pro-inflammatory cytokines, and mitigate negative health outcomes in individuals who are overweight with depression165.
Moreover, elevated pro-inflammatory cytokine levels, together with reactive oxygen species (ROS), putatively leads to the mitochondrial dysfunction and oxidative metabolism alterations that are evident in many patients with mood disorders, and which contribute to biological ageing172,173. Chronic oxidative stress, characterized by elevated ROS and reactive nitrogen species and/or reduced glutathione levels, is a particularly consistent finding in patients with bipolar disorder and other neuropsychiatric or neuroimmune disorders that are associated with elevated pro-inflammatory cytokine levels172. Activation of microglia by pro-inflammatory cytokines in turn leads to further release of reactive oxygen intermediates, pro-inflammatory cytokines, complement proteins and proteinases, driving a chronic inflammatory state that can trigger or maintain neurodegenerative processes174,175. Such processes may underlie progression in the cognitive impairment and morbidity in some people with bipolar disorder176. Elucidating the interactions between inflammation, oxidative stress, mitochondrial dysfunction and impaired neuroplasticity may be critical for identifying therapeutic targets that prevent illness progression and improve outcomes. Non-pharmacological treatments also counter the effects of inflammation and oxidative stress in mood disorders; for example, exercise (running therapy) reduces oxidative stress, inflammation, cortisol release and depressive symptoms, and has a beneficial impact on biological ageing (as measured by telomere length)177.
Among the unmet psychiatric needs associated with biomarkers of inflammation113, anhedonia itself predicts antidepressant non-response, as observed in the Genome-Based Therapeutic Drugs for Depression (GENDEP)2 and the Sequenced Treatment Alternatives to Relieve Depression (STAR*D)178 studies. Beyond anhedonia, other clinical phenotypes have been associated with different aspects of inflammation. For example, in patients with MDD the magnitude of pro-inflammatory cytokine release in response to LPS challenge was significantly greater in patients who manifested more severe anxiety symptoms, even though the basal levels of inflammatory cytokines were not associated with anxiety symptoms122.
Fatigue is associated with MDD and is a commonly comorbid and non-specific symptom in primary immunological disorders. In a meta-analysis of immune trials for effects on depressive symptoms53, treatment effects on fatigue did not remain significant after correction for concomitant improvement in primary illness. Changes in appetite in MDD have been mapped to the association between blood CRP and connectivity between the striatum and areas of the brain implicated in food hedonics179. Sleep changes have also been associated with inflammation in MDD180. These symptoms are troubling for patients, so these dimensions may be useful both as stratification approaches and as outcome measures for trials of immune-based therapies.
A somewhat different example of unmet need is provided by the recent emergence of depression and related symptoms (fatigue, anxiety, mild cognitive impairment) as part of the ‘long COVID-19’ syndrome of physical and mental health disorders experienced by many patients following infection with SARS-CoV-2 (ref.181). The role of immune mechanisms in causing post-COVID-19 depressive syndromes remains to be elucidated but is suggested by recent reports highlighting the association between MRI changes in brain structure and peripheral immune cell counts in patients following discharge from hospital after treatment for severe COVID-19 (ref.182).
Ultimately, the development and adoption of immune-targeted therapeutics for depression may disrupt the conventional, syndromic categorization of MDD and focus instead on transdiagnostic symptom dimensions, such as anhedonia, that may be particularly salient in some clinically defined subgroups of patients and more specifically related to tractable immune mechanisms113.
Additional considerations
Safety
In addition to addressing unmet medical need, new compounds targeting immune function to treat MDD will require a risk–benefit ratio that supports their differentiation from alternative treatments, increasing the need to identify patient subpopulations most likely to benefit. For example, an increased risk of infections, including reactivation of latent tuberculosis and opportunistic infections, has been cited in association with TNF antagonism183. Depending on the safety profile for an immune-targeted therapeutic, patient pre-screening for specific infections may be warranted184. Nevertheless, such risks must be balanced against the potential benefit of helping patients with treatment-resistant depression, which is associated with higher morbidity and mortality rates than MDD generally185.
In addition, most antidepressants include a black box warning indicating they may increase the risk of suicidal thinking in children and adolescents. Recently, the anti-IL-17 receptor mAb brodalumab was approved as a treatment for psoriasis with a warning that it has been linked to suicidal ideation186. Effects on suicidal ideation will need to be quantified for any new immune-targeted therapeutic in an at-risk population.
Treatment paradigms
Usually, novel therapeutics for MDD are clinically tested as adjunctive therapies to standard-of-care antidepressants. The expectation is that they will augment the response to available antidepressant therapies, which often are partially beneficial even in those patients who have an inadequate response. Nevertheless, for any immune-targeted therapeutic in MDD, the interaction with SSRIs may or may not be synergistic, and treatment with both therapies may indeed be worse than monotherapy. For example, although patients with elevated inflammatory markers respond more poorly to SSRIs or serotonin–norepinephrine reuptake inhibitors105, in the CO-MED trial150, patients with low CRP levels responded better to SSRIs alone than to a combination of bupropion and an SSRI, whereas in patients with higher CRP levels, the combination was superior to SSRIs alone. As another example, in a study testing the efficacy of infliximab as an adjunctive therapy in treatment-resistant MDD, patients whose CRP levels were in the lower part of the range pretreatment showed greater improvement on adjunctive placebo than on adjunctive infliximab56.
The development of a monotherapy is often considered to be more challenging from a regulatory and reimbursement perspective. However, ultimately the path for a compound will be paved by data from RCTs. A compound could be approved as a monotherapy if a well-defined subpopulation has a high likelihood of not responding to currently available monotherapies, and a high likelihood of response to the novel therapeutic.
Sex differences
As is true for MDD, most autoimmune disorders are more prevalent in women than in men. Inherent biological differences between sexes also may impact the relationship between immunity and MDD.
In the NESDA study, men, but not women, who were with depression had significantly higher CRP and IL-6 levels than healthy controls187. In a later study in the same cohort, IL-6 levels in patients with depression predicted a chronic course of depression in women, but not men18. In the EMBARC study, higher baseline CRP was predictive of poorer antidepressant therapy outcomes in women treated with the SSRI, sertraline, but this association was not observed in men188. In the GENDEP study, higher baseline CRP levels correlated with greater depression severity, but the effect was only significant in women189. The extant data highlight the need for further research to elucidate the complex relationships between immune dysregulation, depression and sex, which presumably will reflect complex interactions between endocrine, immune and neural function, along with lifestyle factors such as stress, diet and exercise. With the development of immune-targeted therapies for MDD, sex differences should be considered as they may play a role in the potential response rate and may be useful in patient selection strategies.
Outlook
Collectively, these data imply that immune mechanisms play a role in the pathophysiology of depressive symptoms for at least a subtype of patients with MDD and provide clues for the development of novel therapeutics and personalized medicine approaches in neuropsychiatry. Ultimately, the goal is to improve treatment outcomes by enabling patients with depressive episodes to achieve and maintain remission. Immune mechanisms could be promising because chronic inflammation is not unique to MDD but is found in patients with a variety of other medical conditions. Moreover, the immune system has a pathophysiological role in some other psychiatric conditions (such as schizophrenia), as well as in neurodegenerative and other neurological disorders. As biology cuts across diagnostic boundaries, the development of precision medicine approaches based on immune biomarkers may connect a medication to biological deficits and/or psychological dimensions that span multiple DSM-5 categories. Such approaches also hold the potential to redefine diagnostic boundaries in psychiatry, leading to a nosology based on pathophysiology rather than on symptom-based syndromes.
References
Warden, D., Rush, A. J., Trivedi, M. H., Fava, M. & Wisniewski, S. R. The STAR* D Project results: a comprehensive review of findings. Curr. Psychiatry Rep. 9, 449–459 (2007).
Uher, R. et al. An inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline. Am. J. Psychiatry 171, 1278–1286 (2014).
Haroon, E. et al. Antidepressant treatment resistance is associated with increased inflammatory markers in patients with major depressive disorder. Psychoneuroendocrinology 95, 43–49 (2018).
Arteaga-Henríquez, G. et al. Low-grade inflammation as a predictor of antidepressant and anti-inflammatory therapy response in MDD patients: a systematic review of the literature in combination with an analysis of experimental data collected in the EU-Moodinflame Consortium. Front. Psychiatry 10, 458 (2019).
DSM-5 American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders 5th edn (American Psychiatric Publishing, 2013).
Milaneschi, Y., Lamers, F., Berk, M. & Penninx, B. W. Depression heterogeneity and its biological underpinnings: toward immunometabolic depression. Biol. Psychiatry 88, 369–380 (2020). This paper introduces a proposed subtype of MDD — immunometabolic depression — associated with biological dysregulation of inflammatory, neuroendocrine and metabolic pathways, and clinically characterized by atypical depressive symptoms (for example, hyperphagia, hypersomnia, weight gain and fatigue).
National Rheumatoid Arthritis Society. Invisible disease: rheumatoid arthritis and chronic fatigue (NRAS, 2014).
Miller, A. H. & Raison, C. L. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 16, 22–34 (2016).
Bullmore, E. Inflamed depression. Lancet 392, 1189–1190 (2018).
Branchi, I. et al. Brain-immune crosstalk in the treatment of major depressive disorder. Eur. Neuropsychopharmacol. 45, 89–107 (2020).
Dantzer, R., O’Connor, J. C., Freund, G. G., Johnson, R. W. & Kelley, K. W. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56 (2008). This work is an early formative review of the neuroimmunology of sickness behaviours, illuminating both biological and phenotypic associations.
Bhattacharya, A., Derecki, N. C., Lovenberg, T. W. & Drevets, W. C. Role of neuro-immunological factors in the pathophysiology of mood disorders. Psychopharmacology 233, 1623–1636 (2016).
Felger, J. C. Role of inflammation in depression and treatment implications. Handb. Exp. Pharmacol. 250, 255–286 (2018).
Hodes, G. E. et al. Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc. Natl Acad. Sci. USA 111, 16136–16141 (2014). This paper demonstrates that inflammatory status precedes onset of depressive symptoms by replacing a stress-naive animal’s peripheral immune system with that of a stressed animal. This led to an increased susceptibility to social stress.
Laumet, G. et al. Resolution of inflammation-induced depression requires T lymphocytes and endogenous brain interleukin-10 signaling. Neuropsychopharmacology 43, 2597–2605 (2018).
Khandaker, G. M., Pearson, R. M., Zammit, S., Lewis, G. & Jones, P. B. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiatry 71, 1121–1128 (2014).
Bell, J., Kivimäki, M., Bullmore, E., Steptoe, A. & Carvalho, L. Repeated exposure to systemic inflammation and risk of new depressive symptoms among older adults. Transl. Psychiatry 7, e1208 (2017).
Lamers, F. et al. Longitudinal association between depression and inflammatory markers: results from the Netherlands Study of Depression and Anxiety. Biol. Psychiatry 85, 829–837 (2019).
Machado, M. O. et al. Biological mechanisms of depression following treatment with interferon for chronic hepatitis C: a critical systematic review. J. Affect. Disord. 209, 235–245 (2017).
Chiu, W., Su, Y., Su, K. & Chen, P. Recurrence of depressive disorders after interferon-induced depression. Transl. Psychiatry 7, e1026 (2017).
Baraldi, S., Hepgul, N., Mondelli, V. & Pariante, C. M. Symptomatic treatment of interferon-α–induced depression in hepatitis C: a systematic review. J. Clin. Psychopharmacol. 32, 531–543 (2012).
Harrison, N. A. et al. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol. Psychiatry 66, 407–414 (2009).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668 (2018).
Lynall, M.-E. et al. Genetic variants associated with cross-disorder and disorder-specific risk for psychiatric disorders are enriched at epigenetically active sites in peripheral lymphoid cells. Preprint at medRxiv https://doi.org/10.1101/2021.08.04.21261606 (2021).
Khandaker, G. M., Zammit, S., Burgess, S., Lewis, G. & Jones, P. B. Association between a functional interleukin 6 receptor genetic variant and risk of depression and psychosis in a population-based birth cohort. Brain Behav. Immun. 69, 264–272 (2018).
Bull, S. et al. Functional polymorphisms in the interleukin-6 and serotonin transporter genes, and depression and fatigue induced by interferon-α and ribavirin treatment. Mol. Psychiatry 14, 1095–1104 (2009).
Kappelmann, N. et al. Dissecting the association between inflammation, metabolic dysregulation, and specific depressive symptoms: a genetic correlation and 2-sample Mendelian randomization study. JAMA Psychiatry 78, 161–170 (2021). This study uses large-scale prior GWAS data for innovative analysis of the effects of CRP, IL-6 and BMI on depressive symptoms, putatively identifying BMI as causal for several depressive symptoms and genetically upregulated IL-6 signalling as causal for suicidality.
Wingo, T. S. et al. Brain proteome-wide association study implicates novel proteins in depression pathogenesis. Nat. Neurosci. 24, 810–817 (2021).
Bhattacharya, A. & Biber, K. The microglial ATP-gated ion channel P2X7 as a CNS drug target. Glia 64, 1772–1787 (2016).
Denlinger, L. C. et al. Human P2X7 pore function predicts allele linkage disequilibrium. Clin. Chem. 52, 995–1004 (2006).
Soronen, P. et al. P2RX7 gene is associated consistently with mood disorders and predicts clinical outcome in three clinical cohorts. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156, 435–447 (2011).
Halmai, Z. et al. Associations between depression severity and purinergic receptor P2RX7 gene polymorphisms. J. Affect. Disord. 150, 104–109 (2013).
Hejjas, K. et al. Association between depression and the Gln460Arg polymorphism of P2RX7 gene: a dimensional approach. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150, 295–299 (2009).
Vereczkei, A. et al. Association of purinergic receptor P2RX7 gene polymorphisms with depression symptoms. Prog. Neuropsychopharmacol. Biol. Psychiatry 92, 207–216 (2019).
Feng, W.-P., Zhang, B., Li, W. & Liu, J. Lack of association of P2RX7 gene rs2230912 polymorphism with mood disorders: a meta-analysis. PLoS ONE 9, e88575 (2014).
Czamara, D., Müller-Myhsok, B. & Lucae, S. The P2RX7 polymorphism rs2230912 is associated with depression: a meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 82, 272–277 (2018).
Rhen, T. & Cidlowski, J. A. Antiinflammatory action of glucocorticoids — new mechanisms for old drugs. N. Engl. J. Med. 353, 1711–1723 (2005).
Klengel, T., Pape, J., Binder, E. B. & Mehta, D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology 80, 115–132 (2014).
Menke, A. & Binder, E. B. Epigenetic alterations in depression and antidepressant treatment. Dialogues Clin. Neurosci. 16, 395 (2014).
Perrin, A. J., Horowitz, M. A., Roelofs, J., Zunszain, P. A. & Pariante, C. M. Glucocorticoid resistance: is it a requisite for increased cytokine production in depression? A systematic review and meta-analysis. Front. Psychiatry 10, 423 (2019).
Danese, A. et al. Adverse childhood experiences and adult risk factors for age-related disease: depression, inflammation, and clustering of metabolic risk markers. Arch. Pediatrics Adolesc. Med. 163, 1135–1143 (2009). This work is one of several major papers from the Dunedin longitudinal birth cohort, identifying significantly increased risk of depression and immunometabolic biomarkers in young adults (age 32 years) who had experienced one or more adverse childhood experiences in the first decade, drawing attention to the long memory of immune response to early social stress.
Murphy, T. et al. Methylomic profiling of cortex samples from completed suicide cases implicates a role for PSORS1C3 in major depression and suicide. Transl. Psychiatry 7, e989 (2017).
Guintivano, J. et al. Identification and replication of a combined epigenetic and genetic biomarker predicting suicide and suicidal behaviors. Am. J. Psychiatry 171, 1287–1296 (2014).
Ju, C. et al. Integrated genome-wide methylation and expression analyses reveal functional predictors of response to antidepressants. Transl. Psychiatry 9, 254 (2019).
Shimada, M. et al. An epigenome-wide methylation study of healthy individuals with or without depressive symptoms. J. Hum. Genet. 63, 319–326 (2018).
Hyman, S. E. The daunting polygenicity of mental illness: making a new map. Phil. Trans. R. Soc. B 373, 20170031 (2018).
Forbes, M. P. et al. Major depressive disorder in older patients as an inflammatory disorder: implications for the pharmacological management of geriatric depression. Drugs Aging 38, 451–467 (2021).
Köhler-Forsberg, O., Otte, C., Gold, S. M. & Østergaard, S. D. Statins in the treatment of depression: hype or hope? Pharmacol. Ther. 215, 107625 (2020).
Köhler, O. et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 71, 1381–1391 (2014).
Kappelmann, N., Lewis, G., Dantzer, R., Jones, P. B. & Khandaker, G. M. Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol. Psychiatry 23, 335–343 (2018).
Köhler-Forsberg, O. et al. Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: meta-analysis of clinical trials. Acta Psychiatr. Scand. 139, 404–419 (2019).
Wittenberg, G. M. et al. Effects of immunomodulatory drugs on depressive symptoms: a mega-analysis of randomized, placebo-controlled clinical trials in inflammatory disorders. Mol. Psychiatry 25, 1275–1285 (2020). This work analyses patient-level data on mood and anhedonia questionnaire items for 10,743 participants in 18 RCTs of immunomodulatory drugs for medical inflammatory disorders. There are significant treatment effects on depressive symptoms in the subgroup of patients with high symptom scores at baseline and after controlling for physical health benefits of treatment.
Langley, R. G. et al. Ustekinumab significantly improves symptoms of anxiety, depression, and skin-related quality of life in patients with moderate-to-severe psoriasis: results from a randomized, double-blind, placebo-controlled phase III trial. J. Am. Acad. Dermatol. 63, 457–465 (2010).
Gordon, K. B. et al. Anxiety and depression in patients with moderate-to-severe psoriasis and comparison of change from baseline after treatment with guselkumab vs. adalimumab: results from the Phase 3 VOYAGE 2 study. J. Eur. Acad. Dermatol. Venereol. 32, 1940–1949 (2018).
Raison, C. L. et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 70, 31–41 (2013). This early clinical study of peripheral anti-cytokine antibody treatment (anti-TNF infliximab) reports no effect on the primary end point (depressive symptom scale) in the whole sample, but a stronger antidepressant signal in patients with more inflammation with higher baseline CRP.
McIntyre, R. S. et al. Efficacy of adjunctive infliximab vs placebo in the treatment of adults with bipolar I/II depression: a randomized clinical trial. JAMA Psychiatry 76, 783–790 (2019).
Salvadore, G., Nash, A. & Bleys, C. A double-blind, placebo-controlled, multicenter study of sirukumab as adjunctive treatment to a monoaminergic antidepressant in adults with major depressive disorder. Neuropsychopharmacology 43, S292 (2018).
Traki, L. et al. Responsiveness of the EuroQol EQ-5D and Hospital Anxiety and Depression Scale (HADS) in rheumatoid arthritis patients receiving tocilizumab. Clin. Rheumatol. 33, 1055–1060 (2014).
Banks, W. A., Kastin, A. J. & Gutierrez, E. G. Penetration of interleukin-6 across the murine blood–brain barrier. Neurosci. Lett. 179, 53–56 (1994).
Choi, S., Aid, S., Choi, U. & Bosetti, F. Cyclooxygenases-1 and -2 differentially modulate leukocyte recruitment into the inflamed brain. Pharmacogenomics J. 10, 448–457 (2010).
Modi, K. K., Sendtner, M. & Pahan, K. Up-regulation of ciliary neurotrophic factor in astrocytes by aspirin: implications for remyelination in multiple sclerosis. J. Biol. Chem. 288, 18533–18545 (2013).
Rapoport, S. I. Lithium and the other mood stabilizers effective in bipolar disorder target the rat brain arachidonic acid cascade. ACS Chem. Neurosci. 5, 459–467 (2014).
Savitz, J. et al. Activation of the kynurenine pathway is associated with striatal volume in major depressive disorder. Psychoneuroendocrinology 62, 54–58 (2015).
Savitz, J. et al. Putative neuroprotective and neurotoxic kynurenine pathway metabolites are associated with hippocampal and amygdalar volumes in subjects with major depressive disorder. Neuropsychopharmacology 40, 463–471 (2015).
Roman, M. & Irwin, M. R. Novel neuroimmunologic therapeutics in depression: a clinical perspective on what we know so far. Brain Behav. Immun. 83, 7–21 (2020).
Berk, M. et al. Youth Depression Alleviation with Anti-inflammatory Agents (YoDA-A): a randomised clinical trial of rosuvastatin and aspirin. BMC Med. 18, 1–12 (2020).
Abbasi, S.-H., Hosseini, F., Modabbernia, A., Ashrafi, M. & Akhondzadeh, S. Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: randomized double-blind placebo-controlled study. J. Affect. Disord. 141, 308–314 (2012).
Rosenblat, J. D. et al. Anti-inflammatory agents in the treatment of bipolar depression: a systematic review and meta-analysis. Bipolar Disord. 18, 89–101 (2016).
Stolk, P. et al. Is aspirin useful in patients on lithium? A pharmacoepidemiological study related to bipolar disorder. Prostaglandins Leukot. Essent. Fatty Acids 82, 9–14 (2010).
Kessing, L. et al. New drug candidates for depression — a nationwide population-based study. Acta Psychiatr. Scand. 139, 68–77 (2019).
Berk, M. et al. Effect of aspirin vs placebo on the prevention of depression in older people: a randomized clinical trial. JAMA Psychiatry 77, 1012–1020 (2020).
Soczynska, J. K. et al. Novel therapeutic targets in depression: minocycline as a candidate treatment. Behav. Brain Res. 235, 302–317 (2012).
Möller, T. et al. Critical data-based re-evaluation of minocycline as a putative specific microglia inhibitor. Glia 64, 1788–1794 (2016).
Liu, X. et al. Microglia-derived IL-1β promoted neuronal apoptosis through ER stress-mediated signaling pathway PERK/eIF2α/ATF4/CHOP upon arsenic exposure. J. Hazard. Mater. 417, 125997 (2021).
O’Connor, J. C. et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2, 3-dioxygenase activation in mice. Mol. Psychiatry 14, 511–522 (2009).
Lawson, M. A. et al. Intracerebroventricular administration of lipopolysaccharide induces indoleamine-2, 3-dioxygenase-dependent depression-like behaviors. J. Neuroinflammation 10, 1–9 (2013).
Myint, A. M. & Kim, Y. K. Cytokine–serotonin interaction through IDO: a neurodegeneration hypothesis of depression. Med. Hypotheses 61, 519–525 (2003).
Steiner, J. et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflammation 8, 1–9 (2011).
Meier, T. B. et al. Relationship between neurotoxic kynurenine metabolites and reductions in right medial prefrontal cortical thickness in major depressive disorder. Brain Behav. Immun. 53, 39–48 (2016).
Young, K. D. et al. Kynurenine pathway metabolites are associated with hippocampal activity during autobiographical memory recall in patients with depression. Brain Behav. Immun. 56, 335–342 (2016).
Husain, M. I. et al. Minocycline as an adjunct for treatment-resistant depressive symptoms: a pilot randomised placebo-controlled trial. J. Psychopharmacol. 31, 1166–1175 (2017).
Dean, O. M. et al. Adjunctive minocycline treatment for major depressive disorder: a proof of concept trial. Aust. N. Z. J. Psychiatry 51, 829–840 (2017).
Miyaoka, T. et al. Minocycline as adjunctive therapy for patients with unipolar psychotic depression: an open-label study. Prog. Neuropsychopharmacol. Biol. Psychiatry 37, 222–226 (2012).
Husain, M. I. et al. Minocycline as adjunctive treatment for treatment-resistant depression: study protocol for a double blind, placebo-controlled, randomized trial (MINDEP2). BMC Psychiatry 20, 173 (2020).
Savitz, J. B. et al. Treatment of bipolar depression with minocycline and/or aspirin: an adaptive, 2 × 2 double-blind, randomized, placebo-controlled, phase IIA clinical trial. Transl. Psychiatry 8, 27 (2018).
Raison, C. L. et al. Activation of central nervous system inflammatory pathways by interferon-α: relationship to monoamines and depression. Biol. Psychiatry 65, 296–303 (2009).
Rizzo, S. S. et al. Evidence for sustained elevation of IL-6 in the CNS as a key contributor of depressive-like phenotypes. Transl. Psychiatry 2, e199 (2012).
Leday, G. G. et al. Replicable and coupled changes in innate and adaptive immune gene expression in two case–control studies of blood microarrays in major depressive disorder. Biol. Psychiatry 83, 70–80 (2018). This paper presents whole-genome differential expression analysis of whole blood, identifying overexpression of transcriptional network modules enriched for innate immune function, and coupled under-expression of adaptive immune gene expression, consistently in two independent samples of MDD compared with healthy controls.
Inamdar, A. et al. Evaluation of antidepressant properties of the p38 MAP kinase inhibitor losmapimod (GW856553) in major depressive disorder: results from two randomised, placebo-controlled, double-blind, multicentre studies using a Bayesian approach. J. Psychopharmacol. 28, 570–581 (2014).
He, Y., Taylor, N., Fourgeaud, L. & Bhattacharya, A. The role of microglial P2X7: modulation of cell death and cytokine release. J. Neuroinflammation 14, 135 (2017).
Savio, L. E., de Andrade Mello, P., da Silva, C. G. & Coutinho-Silva, R. The P2X7 receptor in inflammatory diseases: angel or demon? Front. Pharmacol. 9, 52 (2018).
Bhattacharya, A. & Drevets, W. C. Role of neuro-immunological factors in the pathophysiology of mood disorders: implications for novel therapeutics for treatment resistant depression. Curr. Top. Behav. Neurosci. 31, 339–356 (2017).
Jimenez-Pacheco, A. et al. Transient P2X7 receptor antagonism produces lasting reductions in spontaneous seizures and gliosis in experimental temporal lobe epilepsy. J. Neurosci. 36, 5920–5932 (2016).
Matute, C. et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 27, 9525–9533 (2007).
Beamer, E., Fischer, W. & Engel, T. The ATP-Gated p2x7 receptor as a target for the treatment of drug-resistant epilepsy. Front. Neurosci. 11, 21 (2017).
Cattaneo, A. et al. Whole-blood expression of inflammasome-and glucocorticoid-related mRNAs correctly separates treatment-resistant depressed patients from drug-free and responsive patients in the BIODEP study. Transl. Psychiatry 10, 232 (2020). This paper shows that candidate gene expression analysis by qPCR of 36 genes identified a profile of increased expression of pro-inflammatory genes, coupled with decreased expression of the GR gene, that was characteristic of treatment-resistant compared with treatment-responsive depressive disorder.
Pandey, G. N., Rizavi, H. S., Bhaumik, R. & Ren, X. Innate immunity in the postmortem brain of depressed and suicide subjects: role of Toll-like receptors. Brain Behav. Immun. 75, 101–111 (2019).
Syed, S. A. et al. Defective inflammatory pathways in never-treated depressed patients are associated with poor treatment response. Neuron 99, 914–924.e3 (2018).
Owen, D., Guo, Q., Rabiner, E. & Gunn, R. The impact of the rs6971 polymorphism in TSPO for quantification and study design. Clin. Transl. Imaging 3, 417–422 (2015).
Enache, D., Pariante, C. M. & Mondelli, V. Markers of central inflammation in major depressive disorder: a systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav. Immun. 81, 24–40 (2019).
Meyer J. H. et al. Neuroinflammation in psychiatric disorders: PET imaging and promising new targets. Lancet Psychiatry 7, 1064–1704 (2020) This review critically evaluates PET imaging studies using TSPO-binding radioligands as markers of inflammation across a range of psychiatric disorders, with a focus on development opportunities for new radiotracers that bind to more specific neuroinflammatory targets.
Kraynak, T. E., Marsland, A. L., Wager, T. D. & Gianaros, P. J. Functional neuroanatomy of peripheral inflammatory physiology: a meta-analysis of human neuroimaging studies. Neurosci. Biobehav. Rev. 94, 76–92 (2018).
Felger, J. C. et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol. Psychiatry 21, 1358–1365 (2016).
Haroon, E. et al. Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol. Psychiatry 21, 1351–1357 (2016).
Price, J. L. & Drevets, W. C. Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn. Sci. 16, 61–71 (2012).
Savitz, J. & Harrison, N. A. Interoception and inflammation in psychiatric disorders. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 3, 514–524 (2018). This paper reviews the role of humoral signals in communicating information about the bodily physiological state to the brain, perturbing neuronal structure, chemistry and function and driving behavioural changes. Implications for psychiatric disorders are outlined.
Aruldass, A. R. et al. Dysconnectivity of a brain functional network was associated with blood inflammatory markers in depression. Brain Behav. Immun. 98, 299–309 (2021).
Lerch, J. P. et al. Studying neuroanatomy using MRI. Nat. Neurosci. 20, 314–326 (2017).
Harrison, N. A. et al. Quantitative magnetization transfer imaging as a biomarker for effects of systemic inflammation on the brain. Biol. Psychiatry 78, 49–57 (2015).
Kitzbichler, M. G. et al. Peripheral inflammation is associated with micro-structural and functional connectivity changes in depression-related brain networks. Mol. Psychiatry https://doi.org/10.1038/s41380-021-01272-1 (2021).
Haroon, E. & Miller, A. H. Inflammation effects on brain glutamate in depression: mechanistic considerations and treatment implications. Curr. Top. Behav. Neurosci. 31, 173–198 (2016).
Lucido, M. J. et al. Aiding and abetting anhedonia: impact of inflammation on the brain and pharmacological implications. Pharmacol. Rev. 73, 1084–1117 (2021).
Wium-Andersen, M. K., Ørsted, D. D., Nielsen, S. F. & Nordestgaard, B. G. Elevated C-reactive protein levels, psychological distress, and depression in 73 131 individuals. JAMA Psychiatry 70, 176–184 (2013).
Haapakoski, R., Mathieu, J., Ebmeier, K. P., Alenius, H. & Kivimäki, M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav. Immun. 49, 206–215 (2015).
Dowlati, Y. et al. A meta-analysis of cytokines in major depression. Biol. Psychiatry 67, 446–457 (2010).
Mostafavi, S. et al. Type I interferon signaling genes in recurrent major depression: increased expression detected by whole-blood RNA sequencing. Mol. Psychiatry 19, 1267–1274 (2014).
Jansen, R. et al. Gene expression in major depressive disorder. Mol. Psychiatry 21, 339–347 (2016). This is one of several major papers from the NESDA study, identifying a set of MDD-associated gene expression clusters, enriched for IL-6 and natural killer cell pathways, and including an independent GWAS risk variant for MDD.
Cattaneo, A. et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology 38, 377–385 (2013). This work studies candidate gene expression analysis by qPCR of whole blood samples from patients with depression before and after antidepressant drug treatment. The study identifies two distinct transcriptional profiles predicting non-response to treatment at baseline (increased IL-1β, TNF, MIF) and associated with symptom improvement following treatment (decreased IL-6, FKBP5; increased BDNF, VGF).
Spijker, S. et al. Stimulated gene expression profiles as a blood marker of major depressive disorder. Biol. Psychiatry 68, 179–186 (2010).
Vogelzangs, N., De Jonge, P., Smit, J., Bahn, S. & Penninx, B. Cytokine production capacity in depression and anxiety. Transl. Psychiatry 6, e825 (2016).
Gaspersz, R. et al. The role of anxious distress in immune dysregulation in patients with major depressive disorder. Transl. Psychiatry 7, 1268 (2017).
van Eeden, W. A. et al. Basal and LPS-stimulated inflammatory markers and the course of individual symptoms of depression. Transl. Psychiatry 10, 235 (2020).
Darko, D. F. et al. Immune cells and the hypothalamic–pituitary axis in major depression. Psychiatry Res. 25, 173–179 (1988).
Maes, M. et al. Evidence for a systemic immune activation during depression: results of leukocyte enumeration by flow cytometry in conjunction with monoclonal antibody staining. Psychol. Med. 22, 45–53 (1992).
Tondo, L., Pani, P., Pellegrini-Bettoli, R. & Milia, G. T-lymphocytes in depressive disorder. Med. Sci. Res. 16, 867–568 (1988).
Maes, M. et al. Leukocytosis, monocytosis and neutrophilia: hallmarks of severe depression. J. Psychiatr. Res. 26, 125–134 (1992). This formative early paper of innate immunogenic (‘inflammatory’) mechanisms of depressive disorder analyses immune cell counts across leukocytes, monocytes and granulocytes (neutrophils, eosinophils and basophils). Depression severity is associated with the degrees of leukocytosis, neutrophilia and monocytosis.
Mazza, M. G. et al. Neutrophil/lymphocyte ratio and platelet/lymphocyte ratio in mood disorders: a meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 84, 229–236 (2018).
Cattaneo, A. et al. Absolute measurements of macrophage migration inhibitory factor and interleukin-1-β mRNA levels accurately predict treatment response in depressed patients. Int. J. Neuropsychopharmacol. 19, pyw045 (2016).
Miller, A. H. Depression and immunity: a role for T cells? Brain Behav. Immun. 24, 1–8 (2010). This formative early review of adaptive immunogenic mechanisms of depressive disorder outlines the role of activated T cells in stress and inflammation, and describes alterations of different T cell populations reported in patients with major depression, indicative of perturbed differentiation pathways.
Medina-Rodriguez, E. M., Lowell, J. A., Worthen, R. J., Syed, S. A. & Beurel, E. Involvement of innate and adaptive immune systems alterations in the pathophysiology and treatment of depression. Front. Neurosci. 12, 547 (2018).
Ahmetspahic, D., Brinker, D. & Alferink, J. in Inflammation and Immunity in Depression Ch. 1 (Elsevier, 2018).
Toben, C. & Baune, B. T. An act of balance between adaptive and maladaptive immunity in depression: a role for T lymphocytes. J. Neuroimmune Pharmacol. 10, 595–609 (2015).
Myint, A. M., Leonard, B. E., Steinbusch, H. W. & Kim, Y. K. TH1, TH2, and TH3 cytokine alterations in major depression. J. Affect. Disord. 88, 167–173 (2005).
Chen, Y. et al. Emerging tendency towards autoimmune process in major depressive patients: a novel insight from TH17 cells. Psychiatry Res. 188, 224–230 (2011).
Lynall, M.-E. et al. Peripheral blood cell-stratified subgroups of inflamed depression. Biol. Psychiatry 88, 185–196 (2020).
Padmos, R. C. et al. A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch. Gen. Psychiatry 65, 395–407 (2008).
Drexhage, R. C. et al. The activation of monocyte and T cell networks in patients with bipolar disorder. Brain Behav. Immun. 25, 1206–1213 (2011).
Kruse, J. L. et al. Psychiatric autoimmunity: N-methyl-D-aspartate receptor IgG and beyond. Psychosomatics 56, 227–241 (2015).
Prüss, H. & Lennox, B. R. Emerging psychiatric syndromes associated with antivoltage-gated potassium channel complex antibodies. J. Neurol. Neurosurg. Psychiatry 87, 1242–1247 (2016). This review describes immune impacts on neuropsychiatric symptoms mediated by autoantibodies to voltage-gated potassium channels and associated proteins, VGKCC, LGI1 and CASPR2. These are associated with limbic encephalitis and isolated psychiatric presentations.
Vincent, A. et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 127, 701–712 (2004).
Waldner, H. The role of innate immune responses in autoimmune disease development. Autoimmun. Rev. 8, 400–404 (2009).
Wittenberg, G. et al. Autoantibody burden in mood and psychotic disorders. Neuropsychopharmacology 44, 135–135 (2019).
Treadway, M. T., Cooper, J. A. & Miller, A. H. Can’t or won’t? Immunometabolic constraints on dopaminergic drive. Trends Cogn. Sci. 23, 435–448 (2019). This paper reviews the association between inflammatory cytokines and the mesolimbic dopamine system, hypothetically mediated by the metabolic demands of chronic low-grade inflammation, inducing a reduction in striatal dopamine.
Felger J. C., Hernandez C. R. & Miller A. H. Levodopa reverses cytokine-induced reductions in striatal dopamine release. Int. J. Neuropsychopharmacol. 18, pyu084 (2015).
Fuster, J. J., Ouchi, N., Gokce, N. & Walsh, K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ. Res. 118, 1786–1807 (2016).
Frank, P. et al. Systemic low-grade inflammation and subsequent depressive symptoms: is there a mediating role of physical activity? Brain Behav. Immun. 80, 688–696 (2019).
Sun, Y., Narayan, V. A. & Wittenberg, G. M. Side effect profile similarities shared between antidepressants and immune-modulators reveal potential novel targets for treating major depressive disorders. BMC Pharmacol. Toxicol. 17, 47 (2016).
Rush, A. J. et al. Combining Medications to Enhance Depression Outcomes (CO-MED): acute and long-term outcomes of a single-blind randomized study. Am. J. Psychiatry 168, 689–701 (2011).
Jha, M. K. et al. Can C-reactive protein inform antidepressant medication selection in depressed outpatients? Findings from the CO-MED trial. Psychoneuroendocrinology 78, 105–113 (2017).
Sun, Y. et al. The effects of interleukin-6 neutralizing antibodies on symptoms of depressed mood and anhedonia in patients with rheumatoid arthritis and multicentric Castleman’s disease. Brain Behav. Immun. 66, 156–164 (2017).
Rizvi, S. J. et al. Development and validation of the Dimensional Anhedonia Rating Scale (DARS) in a community sample and individuals with major depression. Psychiatry Res. 229, 109–119 (2015).
Pizzagalli, D. A. et al. Selective κ-opioid antagonism ameliorates anhedonic behavior: evidence from the Fast-fail Trial in Mood and Anxiety Spectrum Disorders (FAST-MAS). Neuropsychopharmacology 45, 1656–1663 (2020).
Bilderbeck, A. C. et al. Optimizing behavioral paradigms to facilitate development of new treatments for anhedonia and reward processing deficits in schizophrenia and major depressive disorder: study protocol. Front. Psychiatry 11, 1101 (2020).
Vogelzangs, N. et al. Inflammatory and metabolic dysregulation and the 2-year course of depressive disorders in antidepressant users. Neuropsychopharmacology 39, 1624–1634 (2014).
Lindqvist, D. et al. Interleukin-6 is elevated in the cerebrospinal fluid of suicide attempters and related to symptom severity. Biol. Psychiatry 66, 287–292 (2009).
Lindqvist, D. et al. CSF biomarkers in suicide attempters — a principal component analysis. Acta Psychiatr. Scand. 124, 52–61 (2011).
Bay-Richter, C. et al. A role for inflammatory metabolites as modulators of the glutamate N-methyl-D-aspartate receptor in depression and suicidality. Brain Behav. Immun. 43, 110–117 (2015).
Regan, T. et al. Effects of anti-inflammatory drugs on the expression of tryptophan-metabolism genes by human macrophages. J. Leukoc. Biol. 103, 681–692 (2018).
Baghai, T. C. et al. Classical risk factors and inflammatory biomarkers: one of the missing biological links between cardiovascular disease and major depressive disorder. Int. J. Mol. Sci. 19, 1740 (2018).
Pan, A., Sun, Q., Okereke, O. I., Rexrode, K. M. & Hu, F. B. Depression and risk of stroke morbidity and mortality: a meta-analysis and systematic review. JAMA 306, 1241–1249 (2011).
Rutledge, T., Reis, V. A., Linke, S. E., Greenberg, B. H. & Mills, P. J. Depression in heart failure: a meta-analytic review of prevalence, intervention effects, and associations with clinical outcomes. J. Am. Coll. Cardiol. 48, 1527–1537 (2006).
Grenon, S. M. et al. Association between depression and peripheral artery disease: insights from the heart and soul study. J. Am. Heart Assoc. 1, e002667 (2012).
Révész, D., Verhoeven, J. E., Milaneschi, Y. & Penninx, B. W. Depressive and anxiety disorders and short leukocyte telomere length: mediating effects of metabolic stress and lifestyle factors. Psychol. Med. 46, 2337–2349 (2016).
Diniz, B. S., Reynolds, C. F. III, Sibille, E., Bot, M. & Penninx, B. W. H. Major depression and enhanced molecular senescence abnormalities in young and middle-aged adults. Transl. Psychiatry 9, 198 (2019).
Cole, J. J. et al. No evidence for differential gene expression in major depressive disorder PBMCs, but robust evidence of elevated biological ageing. Transl. Psychiatry 11, 404 (2021).
Elderon, L. & Whooley, M. A. Depression and cardiovascular disease. Prog. Cardiovasc. Dis. 55, 511–523 (2013).
Nikkheslat, N. et al. Insufficient glucocorticoid signaling and elevated inflammation in coronary heart disease patients with comorbid depression. Brain Behav. Immun. 48, 8–18 (2015). This review explores the relationship between coronary heart disease and MDD, known to lead to poor outcomes in patients with cardiac disease. Impaired glucocorticoid signalling and high levels of inflammatory markers are implicated in mediating these effects.
Black, C. N., Bot, M., Scheffer, P. G., Cuijpers, P. & Penninx, B. W. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 51, 164–175 (2015).
Baumeister, D., Akhtar, R., Ciufolini, S., Pariante, C. M. & Mondelli, V. Childhood trauma and adulthood inflammation: a meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-α. Mol. Psychiatry 21, 642–649 (2016).
Kelly-Irving, M. et al. Adverse childhood experiences and premature all-cause mortality. Eur. J. Epidemiol. 28, 721–734 (2013).
Morris, G. & Berk, M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 13, 1–24 (2015).
Manji, H. et al. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 13, 293–307 (2012).
Missiroli, S. et al. The role of mitochondria in inflammation: from cancer to neurodegenerative disorders. J. Clin. Med. 9, 740 (2020).
de Oliveira et al. Unraveling the link between mitochondrial dynamics and neuroinflammation. Front. Immunol. 12, 752 (2021).
Wollenhaupt-Aguiar, B., Kapczinski, F. & Pfaffenseller, B. Biological pathways associated with neuroprogression in bipolar disorder. Brain Sci. 11, 228 (2021).
Lever-van Milligen, B. A. et al. The impact of depression and anxiety treatment on biological aging and metabolic stress: study protocol of the MOod treatment with antidepressants or running (MOTAR) study. BMC Psychiatry 19, 1–11 (2019).
Uher, R. et al. Depression symptom dimensions as predictors of antidepressant treatment outcome: replicable evidence for interest-activity symptoms. Psychol. Med. 42, 967 (2012).
Cosgrove, K. T. et al. Appetite change profiles in depression exhibit differential relationships between systemic inflammation and activity in reward and interoceptive neurocircuitry. Brain Behav. Immun. 83, 163–171 (2020).
Prather, A. A., Vogelzangs, N. & Penninx, B. W. Sleep duration, insomnia, and markers of systemic inflammation: results from the Netherlands Study of Depression and Anxiety (NESDA). J. Psychiatr. Res. 60, 95–102 (2015).
Taquet, M., Geddes, J. R., Husain, M., Luciano, S. & Harrison, P. J. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: a retrospective cohort study using electronic health records. Lancet Psychiatry 8, 416–427 (2021).
Raman, B. et al. Medium-term effects of SARS-CoV-2 infection on multiple vital organs, exercise capacity, cognition, quality of life and mental health, post-hospital discharge. EClinicalMedicine 31, 100683 (2021).
Murdaca, G. et al. Infection risk associated with anti-TNF-α agents: a review. Expert Opin. Drug Saf. 14, 571–582 (2015).
De Keyser, F. Choice of biologic therapy for patients with rheumatoid arthritis: the infection perspective. Curr. Rheumatol. Rev. 7, 77–87 (2011).
Bergfeld, I. O. et al. Treatment-resistant depression and suicidality. J. Affect. Disord. 235, 362–367 (2018).
Lebwohl, M. G. et al. Psychiatric adverse events during treatment with brodalumab: analysis of psoriasis clinical trials. J. Am. Acad. Dermatol. 78, 81–89.e5 (2018).
Vogelzangs, N. et al. Association of depressive disorders, depression characteristics and antidepressant medication with inflammation. Transl. Psychiatry 2, e79 (2012).
Jha, M. K. et al. Sex differences in the association of baseline c-reactive protein (CRP) and acute-phase treatment outcomes in major depressive disorder: findings from the EMBARC study. J. Psychiatr. Res. 113, 165–171 (2019).
Köhler-Forsberg, O. et al. Association between C-reactive protein (CRP) with depression symptom severity and specific depressive symptoms in major depression. Brain Behav. Immun. 62, 344–350 (2017).
Müller, N. et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry 11, 680–684 (2006).
Akhondzadeh, S. et al. Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depress Anxiety 26, 607–611 (2009).
Majd, M., Hashemian, F., Hosseini, S. M., Shariatpanahi, M. V. & Sharifi, A. A randomized, double-blind, placebo-controlled trial of celecoxib augmentation of sertraline in treatment of drug-naive depressed women: a pilot study. Iran. J. Pharm. Res. 14, 891 (2015).
Acknowledgements
The authors thank N. Derecki for assistance with early versions of this manuscript and artwork in Fig. 1. A collaboration between these authors was supported by the Wellcome Trust academic-industrial consortium for Neuroimmunology of Mood Disorders and Alzheimer’s Disease (NIMA).
Author information
Authors and Affiliations
Contributions
W.C.D., G.M.W., E.T.B. and H.K.M. discussed the content, reviewed the literature, contributed to writing the article, and edited and reviewed the manuscript prior to submission.
Corresponding author
Ethics declarations
Competing interests
G.M.W. and W.C.D. are employees of Janssen Research & Development and Johnson & Johnson, and own Johnson & Johnson stock. H.K.M. is an employee and shareholder of Johnson & Johnson. E.T.B. serves on the scientific advisory board of Sosei Heptares and as a consultant for GlaxoSmithKline.
Additional information
Peer review information
Nature Reviews Drug Discovery thanks Robert Dantzer, Hemmo Drexhage and the other, anonymous, reviewer for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Anhedonia
-
The inability to gain pleasure from normally pleasurable experiences.
- Hyperphagia
-
An abnormal condition of intense hunger and excessive eating.
- Microglia
-
A class of glial cells that function as the resident macrophages in the central nervous system (CNS), with roles in innate immune response and developmental synaptic pruning in the brain.
- NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome complex
-
A complex of proteins, including NLRP3, that functions as a pattern recognition receptor that recognizes pathogen-associated molecular patterns or products of damaged cells, and when activated, triggers an immune response.
- Glucocorticoid
-
A generic term for corticosteroid hormones, such as cortisol, involved in glucose metabolism and energy mobilization, which under many conditions also exert anti-inflammatory and immunosuppressive activity.
- Hypothalamic–pituitary–adrenal axis
-
A neuroendocrine system that controls reactions to stress and regulates a variety of body processes such as energy mobilization, immune function and emotional behaviour. It comprises a complex network of interactions among the hypothalamus, pituitary gland and adrenal gland.
- Nonsteroidal anti-inflammatory drugs
-
(NSAIDs). A class of drug with anti-inflammatory effects mediated by inhibition of the enzymes cyclooxygenase 1 (COX1) and/or COX2.
- Kynurenine
-
A metabolite of tryptophan whose regulation may be disrupted by inflammatory processes, and which is associated with psychiatric disorders.
- Regulatory T cells
-
(Treg cells). A specialized subpopulation of T cells that act to suppress immune response, inhibiting T cell proliferation, and play a role in maintaining self-tolerance, preventing autoimmunity.
- T helper 17 cells
-
(TH17 cells). A subset of pro-inflammatory T helper cells that produce IL-17.
- TH1 cells
-
A subset of pro-inflammatory T helper cells that produce interferon-γ (IFNγ) and IL-2, and lead to an increased cell-mediated immune response.
Rights and permissions
About this article

Cite this article
Drevets, W.C., Wittenberg, G.M., Bullmore, E.T. et al. Immune targets for therapeutic development in depression: towards precision medicine. Nat Rev Drug Discov 21, 224–244 (2022). https://doi.org/10.1038/s41573-021-00368-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-021-00368-1
This article is cited by
-
Genome-wide association study implicates the role of TBXAS1 in the pathogenesis of depressive symptoms among the Korean population
Translational Psychiatry (2024)
-
Bayesian network-based Mendelian randomization for variant prioritization and phenotypic causal inference
Human Genetics (2024)
-
Review of research progress on intestinal microbiota based on metabolism and inflammation for depression
Archives of Microbiology (2024)
-
Identification of mitophagy-related biomarkers and immune infiltration in major depressive disorder
BMC Genomics (2023)
-
New and emerging approaches to treat psychiatric disorders
Nature Medicine (2023)
