
Abstract
Enormous progress has been made in the last half-century in the management of diseases closely integrated with excess body weight, such as hypertension, adult-onset diabetes and elevated cholesterol. However, the treatment of obesity itself has proven largely resistant to therapy, with anti-obesity medications (AOMs) often delivering insufficient efficacy and dubious safety. Here, we provide an overview of the history of AOM development, focusing on lessons learned and ongoing obstacles. Recent advances, including increased understanding of the molecular gut–brain communication, are inspiring the pursuit of next-generation AOMs that appear capable of safely achieving sizeable and sustained body weight loss.
Similar content being viewed by others
Introduction
Control of excess body fat is one of the greatest healthcare challenges of our time1,2. The global obesity prevalence has nearly tripled since 1975 and, within the United States, excess body weight afflicts more than two thirds of the population, with more than one third of adults and 20% of adolescents having obesity (see Related links).
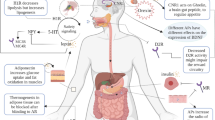
Obesity promotes the incidence of conditions such as type 2 diabetes (T2D)3 and cardiovascular diseases (CVD)4, and increases the risk of death due to cancer of the oesophagus, colon and rectum, liver, gallbladder, pancreas and kidney5,6,7. It complicates the management of multiple diseases, enhancing the prospect for unfavourable outcomes, as prominently noted in the current COVID-19 pandemic8. Compared with normal weight, individuals with a body mass index (BMI) of 30–34.9 kg m–2 carry a hazard ratio for overall mortality that is elevated by more than 40% and at a BMI > 40 kg m–2 the relative rate increases to 100%5. It is estimated that 4–9% of all cancer diagnoses are attributable to excess body fat9,10, and that obesity correlates with poorer prognosis for multiple malignant diseases6,7. Obesity is associated with decreased life expectancy of 5–20 years depending upon its duration, the magnitude of excess weight and the emergence of associated comorbid diseases5,11,12. Starting early in life, obesity increases the prevalence for psychological, neurological, pulmonary, gastrointestinal, renal, musculoskeletal and endocrine diseases13 (Fig. 1). Estimates of the financial burden of obesity upon modern healthcare systems are sizeable, with more than US$190 billion spent annually in the United States alone for obesity-related illnesses14.

Most prominent metabolic and psychological comorbidities associated with morbid obesity. ASVCD, atherosclerotic cardiovascular disease; COPD, chronic obstructive pulmonary disease; PCOS, polycystic ovary syndrome.
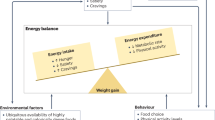
Commonly acknowledged environmental factors accounting for the steep increase in global obesity are increased access to energy-dense food coupled with reduced physical activity15. Sleep deprivation16, circadian desynchronization17, chronic stress18 and the use of anti-epileptic and psychotropic drugs19 may further propel weight gain. Genetic and environmental factors each appreciably contribute to the variance in BMI20. With an estimated heritability of ∼40–70%20,21, the contribution of genetic factors to BMI is comparable with that reported for Tourette syndrome (58–77%)22, psoriasis (66%)23, heart disease (34–53%)24 or breast cancer (25–56%)25.
Increased recognition of obesity as a chronic, degenerative disease26,27 serves to destigmatize the common belief that obesity results from insufficient self-discipline (see Related links). This further provides the framework for healthcare providers and insurance companies to establish obesity management programmes, promotes funding for basic and clinical research, and encourages pharmaceutical companies to develop strategies for body weight management. The central argument defining obesity as a chronic illness rather than a risk factor is the distinct pathophysiology that leads to excess fat accumulation and serves to defend it, coupled with homeostatic mechanisms that hinder weight loss and promote further weight gain28. These altered biological mechanisms may explain why short-term behavioural interventions are frequently insufficient for long-term weight loss.
As lifestyle and behavioural interventions provide moderate efficacy, obesity treatment strategies should be escalated by adding pharmacological and/or surgical interventions. Bariatric surgery represents the most effective approach to weight loss, leading to decreased mortality from CVD or cancer by 30% and 23%, respectively29. With steadily improving laparoscopic procedures, hospitalization time decreases and bariatric surgery increases overall life expectancy by as much as 3 years29, with notable and sustainable improvements in blood pressure, glucose and lipid metabolism30. Nonetheless, surgical interventions are incapable of meeting the global magnitude of medical need.
The pursuit of anti-obesity medications (AOMs) has been tremendously challenging for technical and societal reasons. Only in the last two decades has the definition of the molecular mechanisms that control appetite (Box 1; Fig. 2) advanced to a point where drug discovery can be rationally pursued31. Historically, there has been a collection of AOM failures that have occurred after regulatory approval. Most of these pertain to adverse cardiovascular effects (sibutramine, fenfluramine, dexfenfluramine, rainbow pills), increased suicidal risk (rimonabant) or enhanced likelihood of drug dependence and abuse (methamphetamine) (Table 1). As such, certain drugs are recommended only for short-term use, due to addictive potential or emergence of tachyphylaxis (phentermine, amfepramone, cathin hydrochloride)32,33. Nonetheless, phentermine has not shown adverse cardiovascular outcomes in real-life studies and remains a commonly prescribed long-term AOM.

Peripheral hormones integrate in central control of homeostatic and hedonic eating behaviour. α-MSH, α-melanocyte-stimulating hormone; AgRP, agouti-related peptide; AP, area postrema; ARC, arcuate nucleus; CART, cocaine- and amphetamine-regulated transcript; CCK, cholescystokinin; CPu, caudate putamen; DMH, dorsomedial hypothalamus; DMV, dorsal motor nucleus of the vagus; FGF21, fibroblast growth factor 21; GIP, glucose-dependent insulinotropic polypeptide; GLP1, glucagon-like peptide 1; LH, lateral hypothalamus; MC4R, melanocortin 4 receptor; NAcc, nucleus accumbens; NPY, neuropeptide Y; NTS, nucleus tractus solitarius; OXM, oxyntomodulin; PFC, prefrontal cortex; POMC, pro-opiomelanocortin; PVN, paraventricular nucleus; PYY, peptide tyrosine tyrosine; VMH, ventromedial hypothalamus; VTA, ventral tegmental area; Y1R, neuropeptide Y receptor type 1.
Until recently, long-term pharmacotherapy to achieve body weight normalization along with suitable tolerability and safety remained an insurmountable challenge34. However, recent clinical trials with advanced therapeutic candidates including glucagon-like peptide 1 receptor (GLP1R) agonism are promoting the belief that breakthrough, drug-based management of obesity may be possible. On 4 June 2021, the US Food and Drug Administration (FDA) approved semaglutide 2.4 mg for chronic weight management in adults with obesity or overweight with at least one weight-related condition (such as high blood pressure or cholesterol, or T2D), for use in addition to a reduced-calorie diet and increased physical activity (see Related links). This now constitutes the second GLP1R agonist registered for body weight management, as liraglutide 3 mg was approved by the FDA in 2014 for treatment of adult obesity and in 2020 for obesity in adolescents aged 12–17 years (see Related links).
With the exception of semaglutide 2.4 mg (refs35,36,37,38), the average percent body weight reduction for currently registered drug treatments varies in the single-digit range, with only a small fraction of subjects capable of achieving and maintaining >10% loss at tolerable doses39,40 (Fig. 3). Although such weight loss is clinically meaningful41,42, and serves to improve the severity of comorbid diseases43, it is paltry when viewed against the efficacy of bariatric surgery41,44. An ideal AOM should sizeably and sustainably correct excess weight while reducing the risk of CVD and other comorbidities, devoid of the potential for abuse, tachyphylaxis and other adverse effects that have historically plagued this field41. It is a lofty goal and, at times, still challenged by the question of whether obesity itself constitutes a disease worthy of chronic drug therapy45,46.

Body weight loss achieved through lifestyle changes, currently approved anti-obesity medications (AOMs) and bariatric surgery (part a) and correlation of drug-induced body weight loss in rodents and humans (part b). Data in panel a refer to liraglutide 3 mg (ref.176), orlistat289, naltrexone/bupropion292, phentermine/topiramate291, semaglutide 1 mg (ref.125), semaglutide 2.4 mg (ref.38) and tirzepatide (5 and 15 mg)126. Data in panel b refer to naltrexone/bupropion39,295, orlistat39,296, lorcaserin39,297, sibutramine154,298, liraglutide39,299, phentermine121,145, semaglutide38,123 and tirzepatide122,127.
This article reviews the history of obesity drug therapy and discusses ongoing challenges and recent advances in the development of AOMs. Although mechanistic understanding of energy homeostasis has dramatically progressed since the discovery of leptin just over 25 years ago47, the translation to targeted therapies has largely been empirical, with rodent models remaining of seminal importance, but of variable value for drug candidate selection. This is prominently witnessed in the ongoing debate pertaining to the gut hormone glucose-dependent insulinotropic polypeptide (GIP), where, based on rodent pharmacology studies, both GIPR agonism or antagonism can provide supplemental pharmacology to GLP1 agonism48. Lifelong pharmacological management of chronic diseases such as hypertension might offer relevant benchmarks for obesity treatment strategies. In these diseases, it is common practice to target multiple mechanisms to achieve optimal disease management. It seems inevitable, and with good precedent, that such a conceptual approach to lowering body weight will eventually prevail40.
Regulation of body weight
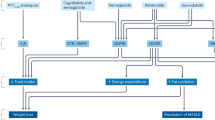
Throughout human evolution, the environmental pressure for survival has likely included a drive to preserve body fat. With increased industrialization and ready access to high-fat foods, this acquired benefit has emerged as a liability. Physiologically, we defend body weight by peripheral and central mechanisms within a surprisingly small range, to protect against a broad array of conditions that include chronic overfeeding at one extreme and starvation at the other. Even the less well controlled long-term outcomes are associated with body weight change of rarely more than 20%, in either direction. The brain controls both hunger and systemic energy metabolism (Box 1; Fig. 2), and harbours most gene products and pathways that have been linked to obesity in hundreds of genetics studies49,50,51. However, direct modulation with central nervous system (CNS) signalling pathways requires selective targeting of cellular circuits, which remains a technological stretch, as historic attempts have shown more than once. For optimal weight loss efficacy, it seems apparent that drug therapy would have to target both energy intake and expenditure. However, intervention in central ‘survival’ mechanisms is a delicate endeavour that has led to withdrawal of many AOMs (Table 1). Striking a balance in striving for efficacy that promotes metabolic health and is psychologically meaningful to a patient, but of suitable chronic tolerability and safety, constitutes the medicinal challenge. Most currently registered medicines fulfil only a mere fraction of the performance that is desired, but there is reason for optimism as late-stage drug candidates hold much more promise52,53. A recurrent question is whether pharmacology can ever be as efficacious in lowering body fat at tolerable doses as bariatric surgery, or alternatively might it in time prove superior.
Unquestionably, advances in understanding the molecular elements that control appetite and energy utilization have provided a road map for more informed AOM development (Box 1; Fig. 2). The sizeable and rapid lowering of body weight achieved by bariatric surgery that results in much improved long-term mortality29 has further provided a vision of what might be pharmacologically possible. Indeed, mimicking the effects of bariatric surgery has become one vision for discovery of future AOMs.
History of AOMs
Pharmacotherapy of obesity has a long and chequered history that is constituted by promising drugs that were withdrawn due to safety concerns (Box 2). In the last century, the pharmacological management of obesity has included amphetamines, thyroid hormones, dinitrophenol and various drug combinations (rainbow pills) that were withdrawn shortly after regulatory approval due to serious adverse effects34 (Table 1). Several centrally acting sympathomimetics such as phentermine, cathine and diethylpropion continue in short‐term use. Medicines that have been investigated in obesity include agents as diverse as mitochondrial uncouplers54,55,56, sympathomimetics33,34, serotonergic agonists57,58,59,60,61,62,63,64,65, lipase inhibitors64,66, cannabinoid receptor antagonists67,68,69 and a growing family of gastrointestinal-derived peptides chemically optimized for pharmaceutical use34. A sobering realization across most of these approaches is the common inability to achieve placebo-adjusted mean weight loss greater than 10% of initial body weight when chronically administered at tolerable doses. As greater weight loss is achieved, it is typically accompanied by various serious acute or chronic adverse effects34 (Table 1). A notable exception is the recently approved GLP1R agonist semaglutide 2.4 mg, which in phase III clinical trials decreased body weight in individuals with obesity or overweight without diabetes after 68 weeks of treatment by −14.9% relative to −2.4% in placebo-treated controls38.
AOMs predominantly function by peripheral or central pathways governing energy balance, but rarely both70,71. Orlistat, for example, acts as a lipase inhibitor to reduce the uptake of dietary fat from the gastrointestinal tract. AOMs that act centrally to increase satiety often function by modulating serotonergic, noradrenergic or dopaminergic action. These AOMs block catecholamine reuptake or directly stimulate satiety receptors in the hypothalamus and limbic system34. In addition, certain AOMs increase energy expenditure by inducing thermogenesis or lipolysis through actions at peripheral or central sites34. Sympathomimetic agents, such as phentermine, operate in the CNS, where they increase norepinephrine in the synaptic cleft and directly stimulate β-adrenergic receptors. The sympathomimetic agent phentermine has been combined with topiramate, an anti-epileptic carbonic anhydrase inhibitor that potentially affects energy metabolism through modulation of GABAergic neurotransmission72. Sympathomimetics may also increase thermogenesis73, but α‐adrenergic and β‐adrenergic receptor stimulation is also associated with vasoconstriction and increased sympathetic tone that can result in increased blood pressure and heart rate.
Despite prominent failures of AOMs (Box 2), more recently approved drugs for obesity management are accessible for use in addition to behaviour modifications. In the United States and Europe, orlistat, naltrexone/bupropion, liraglutide 3 mg and, most recently, semaglutide 2.4 mg are registered and promoted. In addition, in the United States, phentermine/topiramate is even available for long-term use40.
Bupropion is a reuptake inhibitor of dopamine and norepinephrine. Although naltrexone, an opioid antagonist, does not cause weight loss in monotherapy, it blocks the inhibitory effects of opioid receptors activated by β-endorphin released in the hypothalamus, which stimulates feeding. In combination with bupropion, it reduces food intake74. Although naltrexone/bupropion may increase blood pressure and should therefore not be used in patients with uncontrolled hypertension, no adverse signal for increased cardiovascular events was found in the interim analysis of a cardiovascular outcome trial75.
In 2014, liraglutide 3 mg became the first GLP1-based AOM to be introduced to the US market for treatment of obesity in adults, and in 2020 was approved for weight management in adolescents aged 12 years and older with obesity (see Related links). Prior to this (since 2010), liraglutide was used as a subcutaneous injection for treatment of T2D in daily doses of up to 1.8 mg, demonstrating a lower incidence of major adverse cardiovascular events compared with best standard of care in the LEADER trial76. The most common complaints in patients treated with subcutaneous liraglutide 1.8 mg are gastrointestinal side effects including nausea, diarrhoea, vomiting and constipation77. The more recently FDA-approved semaglutide at a dose of 2.4 mg lowers mean body weight to ~15% after 68 weeks of treatment (relative to ~2.4% in placebo controls)38. The drug is generally well tolerated although the typical GLP1-related adverse effects (primarily nausea, diarrhoea, vomiting and constipation) still prevail38.
Challenges confronting AOM development
Heterogeneity of patient cohorts
Obesity is a heterogeneous condition constituted by rare monogenetic49,78 and, more commonly, polygenic aetiology associated with neurobehavioural, endocrine and metabolic causes51,79,80,81,82,83,84,85,86. Obesity-related risk factors and/or quantitative trait loci are found on nearly every chromosome87,88,89,90. Epigenetic processes may yet account for additional factors predisposing to obesity91,92,93. Further scientific dissection of the heterogeneity in genetic, epigenetic and environmental risk factors is of major importance as these may not only explain the variance in BMI but also affect the individual response to certain pharmacotherapies82,94.
Rare chromosomal abnormalities are observed in >10% of children with severe obesity51. Monogenetic obesity is observed in individuals with loss-of-function mutations in genes encoding for leptin95,96,97, the leptin receptor (LEPR)98, pro-opiomelanocortin (POMC)99 and the melanocortin 4 receptor (MC4R)50,100. The most common polygenic risk factors for obesity include mutations in the fat mass and obesity-associated gene (FTO)101 and MC4R102.
A more thorough metabolic and genetic characterization in combination with detailed disease aetiology and response to different mechanisms in drug action should lead to an improvement in patient care. Additionally, this can also potentially foster the next generation of AOMs by advancing a deeper understanding into the molecular pharmacology of body weight regulation. It remains to be determined whether one, two or more mechanisms in drug action will prove successful in treatment of most patients with obesity, or whether far more diverse customization will be required to optimally tackle the obesity pandemic.
Neuroendocrine considerations
Various peripherally derived endocrine factors regulate food intake by jointly acting on defined neurocircuits in the hypothalamus and other brain regions103,104,105,106 (Box 1; Fig. 2). Although this tightly controlled system is pivotal for survival, it has emerged as a major obstacle to achieving sizeable body weight reduction, as it progressively defends against negative energy balance and undernutrition107,108,109,110. One of the likely relevant underlying mechanisms is a decrease in peripheral adiposity signals (leptin, insulin) following weight loss, and prolonged fasting leads to increased expression and sensitization to orexigenic neuropeptides in the hypothalamus and the hindbrain. Simultaneously, the expression of and sensitivity to anorexigenic neuropeptides decrease in these same areas to constitute a double-barrelled defence of body weight111,112,113. Concurrently, the density and strength of the orexigenic agouti-related peptide (AgRP)/neuropeptide Y (NPY) fibres that project from the arcuate nucleus (ARC) to the paraventricular hypothalamic nuclei increase in response to prolonged fasting. This remodelling of the ARCAgRP/NPY projections correlates with increased activation of paraventricular hypothalamic nuclei neurons with the goal to restore food intake114. Another obstacle in weight loss pharmacology is that persistent elevation of adiposity signals such as leptin and insulin results in desensitization, leading to an impaired responsiveness of this homeostatic system115,116,117. A striking finding supporting this perspective is that leptin supplementation shows remarkable efficacy in lowering body weight in individuals with congenital leptin deficiency96,118,119, but is largely ineffective in more common polygenetic forms of obesity115,116,117.
Translation of pharmacology from animals to humans
Effects on body weight
Several studies have shown high correlation between rodents and humans in the weight-lowering properties of phentermine/topiramate, sibutramine, rimonabant, topiramate, phentermine and orlistat120,121. Meta-analyses confirmed that results from animal models predicted human effects of the more recently approved naltrexone/bupropion39. Incretin-based therapy with peptides such as exendin 4, liraglutide, semaglutide and the GIP/GLP1 dual agonist tirzepatide lower body weight in rodents122,123 and humans38,53,124. Overall, with the exception of semaglutide 2.4 mg (ref.38), the mean placebo-corrected body weight loss achievable through therapy with a registered AOM resides in a relatively narrow range of 3–7% after 6–12 months of treatment, with a finite fraction of subjects surpassing weight loss of 10% and much fewer 15% relative to placebo39,41 (Fig. 3). Of special merit, semaglutide 2.4 mg and tirzepatide (10 or 15 mg once weekly) have recently reported a mean weight loss >10% in phase II and III clinical studies of subjects without diabetes38,53,124,125,126,127. Weight loss is considerably lower in patients with T2D, indicating that insulin resistance and chronic hyperglycaemia correlate with diminished efficacy of GLP1R agonists35,36,37,38.
However, whereas weight loss effects generally translate from rodents to humans, maximal efficacy is historically two to four times lower in humans relative to rodents (Fig. 3). It can be argued that greater relative weight loss in rodents is expected as mice possess a higher mass-specific energy expenditure than humans, with a greater contribution of brown adipose tissue to metabolic rate128. Consequently, mice may be more susceptible to drugs that affect energy expenditure. The high mass-specific metabolic rate requires sufficiently high caloric intake to protect against a chronic deficit in energy balance. It is consequently logical that mice can ingest food matching more than 10% of their body weight in a single day. Therefore, pharmacological inhibition of food intake offers a larger dynamic range and more immediate impact on weight loss in rodents relative to humans.
Glucose and lipid metabolism
A decrease in body weight of 5–10% can provide a clinically meaningful improvement in HbA1c, blood pressure, serum triglycerides and HDL cholesterol. These cardiometabolic improvements are progressively enhanced with further weight loss129. Decreased abdominal and hepatic fat deposition with improvement of β-cell function and insulin sensitivity are observed with modest degrees of weight loss. Certain AOMs are also capable of directly improving glycaemic control, which provides supplemental benefit to cardiometabolic outcomes. In particular, GLP1R and GIPR agonists improve glycaemia via their ability to enhance insulin secretion130 and by inhibiting gastric emptying to slow glucose entry to general circulation131.
In a large-scale meta-analysis comprising 29,018 participants, low to moderate improvement of glucose metabolism was demonstrated after 1 year of treatment with orlistat, naltrexone/bupropion, phentermine/topiramate and liraglutide132. All of these medicines also provided low to moderately improved LDL cholesterol and, except for orlistat, increased HDL cholesterol132. A recent placebo-controlled 26-week phase II study of tirzepatide dramatically improved HbA1c, fasting blood glucose and triglycerides with superior efficacy relative to treatment with the GLP1R selective agonist dulaglutide124. In phase III clinical trials, tirzepatide, at all tested doses, lowered HbA1c, fasting glucose and body weight with greater efficacy relative to a 1 mg dose of semaglutide125. Remarkably, 40 weeks of treatment with tirzepatide reduced HbA1c <5.7% in 29–51% of patients depending on the dose, relative to 20% in patients treated with semaglutide 1 mg. Weight loss ≥15% was observed in 15–40% of patients treated with tirzepatide relative to 9% of patients treated with semaglutide125.
Curiously, not all weight-lowering agents improve glycaemia. In particular, fibroblast growth factor 21 (FGF21) agonists have proven enigmatic. As a class, they potently lower body weight and improve metabolism in preclinical studies. As one example of several FGF21 analogues clinically tested, the specific FGF21 agonist PF-05231023 demonstrated improvements in body weight, lipid metabolism and glycaemia in rodent and non-human primates133,134. Although there was a large and highly significant improvement in lipid metabolism in humans, PF-05231023 failed to improve glucose to any appreciable degree134,135. The effect on body weight is less certain, but considerably less than what has been preclinically reported or, certainly, in comparison with incretin-based therapy. Longer duration studies, or further increases in dose, may lead to clinically significant weight reduction or improvements in glucose metabolism of the type witnessed prominently in mice, but this remains to be demonstrated. However, at this point, it serves as a notable example where the pharmacological profile observed in preclinical studies has proven disappointingly different in clinical study.
Safety aspects
The search for greater efficacy in next-generation AOMs must inevitably be anchored by the critical challenge of safety. Whether employing well-understood and more specific mechanisms of action, or pursued through adjunctive agents proven to be independently safe, the risk for toxicity must be fully assessed. To overcome this challenge, AOM development strategies need to increasingly reflect the heterogeneity of the human condition where diversity is far greater than can be encompassed in animal models. Initial AOM development and registration studies are influenced by commercial considerations, and as such specific patient populations, often of greatest need and risk, are under-represented. Clinical studies assessing different drug candidates are typically more alike than different and are directed at large patient populations of common severity, typically individuals who are middle-aged with a body weight near to or slightly above 100 kg.
Undoubtedly, patients with extreme obesity, patients with multiple comorbidities and those at younger age confronting a lifelong struggle with excess body weight require special attention. In these instances, the importance of safety is paramount and yet the need for efficacy is equally enhanced. Certain AOMs unsuitable for the broader population with obesity might still hold promise in special circumstances and when carefully administered and monitored by a specialist. As an example, therapy with leptin in patients with congenital deficiency or with setmelanotide in patients deficient in POMC is highly effective82,117,136, yet currently of little (leptin) or uncertain (setmelanotide) value in other more common forms of obesity115,116,137,138. In any case of weight loss pharmacotherapy, the initial priority should be to safely achieve maximal weight reduction, followed by sustained therapy with AOMs and lifestyle changes that might require less supervision to maintain reduced body weight. Such an approach aims to reduce the risks of intensified therapy by scheduled migration to less forceful forms of therapy. Aggressive use of glucocorticoid therapy in severe inflammatory diseases followed by dose reduction seems a suitable example, where careful patient management and specific drugs can suitably provide efficacy and safety139. Each patient managed by an informed caregiver might progress through a schedule of different drugs in combination with lifestyle modification to eventually achieve an optimal outcome.
Most obesity-related deaths are due to CVD1,140, and therefore improving cardiovascular health constitutes a primary objective for weight loss therapies. Although the risk of a major adverse cardiovascular event is generally lower in individuals who are lean relative to individuals with obesity4, the manner in which body weight is reduced by pharmacotherapy can result in significantly different outcomes, with some lessening and some worsening cardiovascular health. The cosmetic appeal for reduced body weight constitutes an independent risk for abuse as subjects strive for more rapid and larger reductions despite the potential for harmful effects. Importantly, there are no prospective cardiovascular outcome trial results for patients with obesity devoid of significant cardiometabolic comorbidities. The SELECT trial, designed to assess major adverse cardiovascular event reduction for selected AOMs, will clarify whether targeting obesity may result in improved cardiovascular outcomes141.
Amphetamine-induced release of norepinephrine can result in increased blood pressure, heart rate, cardiac contractility, conduction velocity and cardiac excitability via binding to vasculature and heart adrenergic receptors142. Amphetamines also carry a certain risk for abusiveness due to their action on the brain reward system. Cardiovascular effects ensuing from amphetamine abuse can present as chest pain, tachycardia, dyspnoea, primary pulmonary hypertension (PPH), dysrhythmias, acute myocardial infarction and, even, sudden cardiac arrest142. The fears of such toxicity led to discontinuation of methamphetamine (desoxyephedrine) for lowering body weight in the 1940s34. Phentermine and diethylpropion were designed to retain the anorectic activity, but with much reduced effects on the cardiovascular and brain reward system143. Several clinical studies report the absence of major adverse effects of phentermine144,145 or diethylpropion146,147,148,149 on blood pressure and heart rate. Nonetheless, rare occurrences of PPH and/or valvular heart disease have been reported and, therefore, their use is contraindicated in patients who are hypertensive or otherwise elevated in risk for CVD (see Related links). Distribution of fenfluramine and dexfenfluramine was discontinued in 1997 due to the risk of PPH and valvular heart disease150,151,152, whereas sibutramine was withdrawn due to the increased risk of stroke and non-fatal myocardial infarction, particularly in patients with CVD142,153,154,155 (see Box 2). Sibutramine has been associated with increased pulse rate156, blood pressure157,158,159,160 and cardiac arrhythmia154,159. Improvement in blood pressure has been reported in a meta-analysis for naltrexone/bupropion and orlistat, with greater cardiovascular beneficial effects reported for phentermine/topiramate132. Better yet, liraglutide (1.8 mg once daily)76 and injectable semaglutide 1 mg (ref.161) have been documented to improve cardiovascular outcomes in patients with T2D, notably with decreased rates of cardiovascular death, non-fatal myocardial infarction and non-fatal stroke.
An important question is why so many agents designed to decrease food intake eventually failed in clinical trials due to insufficient cardiovascular safety. The most common responses include species-related differences and the lack of preclinical models that reliably predict human cardiovascular safety. Although rodents and other animals are an essential tool to study drug effects on body weight and glucose control162, they are relatively resistant to adverse drug effects pertaining to the cardiovascular and pulmonary systems, rendering them less capable of predicting human cardiovascular safety. To date, there are no animal models that can predict drug-induced PPH and valvulopathy in humans163. Additionally, it is difficult to capture in otherwise healthy, inbred animals the heterogeneity of subjects that constitute human use. Most patients with obesity are older, afflicted with cardiovascular and associated diseases such as diabetes. It is near impossible to preclinically capture the full risk for use of AOMs in such a diverse patient population. Cardiovascular outcome trials such as the SELECT trial are needed to evaluate cardiovascular safety and potential cardiovascular risk reduction in patients with obesity but without major cardiovascular risk factors. The prominent factors that have collectively contributed to drug failure due to adverse cardiovascular effects have made themselves known in such clinical studies. The increased awareness has led to an emphasis on cardiovascular pharmacology and a demonstration for favourable cardiovascular outcomes as part of the process in AOM approval and broader distribution.
Novel and emerging obesity therapies
Despite numerous disappointments, several prominent therapeutic targets have captured the attention of the scientific community34,164,165,166 (Table 2). They reflect the state of the art in how novel drug candidates have been identified and advanced to human study. Four target areas (leptin, ghrelin, mitochondrial uncouplers and growth differentiation factor 15 (GDF15)) were initiated and advanced with obesity constituting the primary therapeutic purpose (Table 2). By contrast, the research pertaining to incretins and, most notably, GLP1, as well as amylin, was predominately focused on diabetes that evolved through concurrent empirical observations of body weight lowering. However, the maturation of incretin biology has led to late-phase AOM candidates that potently activate GLP1R and/or GIPR to establish a much elevated, new benchmark for performance. These subjects are discussed in the following subsections.
Incretin-based therapy
GLP1-related drug candidates
Advancement in incretin biology over the last decades has resulted in a family of registered GLP1R agonists167. Their development was partially triggered by the success of oral DPP4 inhibitors that indirectly raise circulating concentrations of endogenous GLP1 and GIP to improve glycaemic control without risk of hypoglycaemia168,169,170,171,172,173,174. The parenteral administration of bioactive hormone paralogs and synthetic analogues provided increased circulating drug concentrations that resulted in enhanced glycaemic control and an increased appreciation for the inherent body weight-lowering properties of GLP1R agonism.
The magnitude of weight lowering in initial clinical studies employing GLP1R agonists was modest and largely consistent with that previously observed with other gut hormones175. The pharmacology reports were associated with profound effects on gastrointestinal motility that complicated the assessment of how much of the weight lowering was a function of adverse local gastrointestinal effects that served to minimize appetite. The emergence of peptide analogues that extended and flattened pharmacokinetics in concert with dose titrations that lessened the frequency of adverse gastrointestinal effects, collectively enabled more sustained and intense treatment that solidified the metabolic and weight-lowering effects of GLP1R agonism. The specific mechanism of action is multifactorial, with gut, brain and systemic improvements in insulin sensitivity each contributing a finite fraction to the total efficacy123 (Fig. 4).

Glucagon-like peptide 1 receptor (GLP1R) agonism exerts both direct and indirect effects on energy and glucose metabolism in key peripheral organs as well as the brain.
At the end of 2014, liraglutide 3 mg became the first GLP1R agonist to be approved for the treatment of obesity, at approximately twice the highest dose employed in the treatment of T2D. Following 1 year of treatment there was a reported mean decrease of 8% body weight in subjects treated with liraglutide relative to 2.6% in vehicle-treated controls176, with approximately two thirds of patients treated with liraglutide achieving >5% body weight reduction and one third experiencing >10%. This compares favourably with 27% and 11%, respectively, achieving similar outcomes in control subjects. The reduced body weight was associated with improvements in insulin sensitivity, circulating lipids and blood pressure, but with mean heart rate increasing by 2.4 bpm. The ability of liraglutide to lower body weight is competitive in magnitude to other conventional oral AOMs165. These results established that GLP1R agonism could be used to improve metabolism and modestly lower body weight in patients with obesity while reducing cardiovascular risk, building upon the previously demonstrated success in improving cardiovascular outcomes in chronic treatment of T2D (refs76,177). Whether liraglutide also improves cardiovascular outcomes in obesity without T2D warrants clarification. In addition, questions remain as to whether the degree of weight loss justifies the financial cost of the drug and regarding the obstacles associated with achieving compliance to a chronic, daily injection.
Semaglutide at 2.4 mg, a dose much higher than registered for treatment of T2D, gained approval in June 2021 for chronic weight management in adults with obesity or overweight. In a 1-year phase II study employing daily doses ~10% that of high-dose liraglutide, body weight loss was approximately doubled53. Daily dosing achieved >15% weight loss in half of the study participants, whereas one third experienced more than a 20% reduction. In a recent phase III clinical trial in patients of excess weight without diabetes, once-weekly treatment with semaglutide 2.4 mg decreased body weight after 68 weeks of treatment by −14.9% relative to −2.4% in placebo-treated controls38. In patients with diabetes and obesity, semaglutide decreased body weight by −9.6% relative to −3.4% in placebo controls35. These transformative results, particularly in the patients with obesity without diabetes, establish a new benchmark for efficacy and the FDA recently approved semaglutide 2.4 mg for the treatment of obesity as an adjunct to caloric restriction and increased physical activity (see Related links). Not long ago, achievement of this degree of weight loss was thought not to be safely possible, and weekly administration of semaglutide 2.4 mg is a notable virtue relative to liraglutide 3 mg, just as it proved in the treatment of T2D relative to liraglutide 1.8 mg. Whether comparable weight reduction can eventually be achieved by administration of the recently introduced oral form of semaglutide remains to be demonstrated178.
Several other peptide and small-molecule GLP1R agonists are currently in clinical development, including formulations designed for oral administration. Another oral GLP1R agonist (GLPR-NPA) is currently in phase II clinical trials at Eli Lilly (Table 2) (see Related links).
GIP-related drug candidates
Engagement of GIPR agonism for the treatment of obesity and T2D is regarded with notable scepticism, as the insulinotropic effect of GIP is diminished in patients with T2D179. In addition, appreciable preclinical evidence indicates that GIPR antagonism can improve systemic energy and glucose metabolism180,181,182,183, possibly through improvement of central leptin sensitivity180. However, long-acting (acyl) GIPR agonists decrease body weight in obese wild-type and GLP1R knockout mice184,185 and GIP affects body weight through signalling via the GIPR in the CNS. In line with this notion, GIPR is expressed in neurons of the hypothalamus and the hindbrain186,187 and DREADD-mediated activation of hypothalamic GIPR cells decreases food intake186. Consistent with this, single central administration of a fatty acyl-GIP decreases body weight and food intake in DIO mice and increases cFOS neuronal activity in the hypothalamus185. When peripherally administered, fatty acyl-GIP decreases body weight and food intake in obese wild-type and GLP1R knockout mice, but shows blunted weight loss in CNS GIPR-deficient mice185. In summary, long-acting GIPR agonists have been shown to decrease body weight and to improve glucose handling in a series of preclinical studies184,185 and a long-acting GIPR agonist is in phase I clinical trials for the treatment of T2D (Table 2) (see Related links).
Incretin-based poly-agonists
Simultaneous to the structural optimization of selective GLP1R and GIPR mono-agonists has been research to pharmacologically harness the fact that mammalian organisms govern energy balance through much more than a single hormone. The most notable breakthrough in that direction has been the discovery of poly-agonists that simultaneously target the GLP1, GIP and/or glucagon receptors188,189. Multiple drug candidates have advanced to clinical development (Table 2). The most prominent approaches pertain to unimolecular combination of GIP and/or glucagon receptor (GcgR) agonism with highly potent, complementary GLP1R agonism. GIPR agonists, once chemically integrated with GLP1R agonism, have demonstrated metabolic benefits and reduced body weight in mice when compared with pharmacokinetically matched GLP1R agonists122,189. There are multiple reasons why GIP agonism might provide supplemental metabolic benefits to GLP1 therapy, apart from lowering body weight and food intake via GLP1R-independent mechanisms184,185. GIP blocks the emetic effects of GLP1R agonism in musk shrews190 and near-normalization of blood glucose has been reported to restore the insulinotropic effect of GIP in patients with T2D191. Furthermore, GIP agonism enhances adipocyte storage capacity to protect from adipocyte lipid spill over and ectopic lipid deposition192. Nonetheless, as discussed in the preceding subsection, the use of GIPR agonists for the treatment of obesity and T2D is controversial.
Importantly, phase II results for two unimolecular, long-acting GIPR/GLP1R co-agonists have been reported. The first, NN9709 (formerly MAR709 and RG7697) (Table 2), is suited for once-daily subcutaneous injection and demonstrates balanced high potency at human GLP1R and GIPR193. NN9709 reduced blood glucose, body weight and total cholesterol in a 12-week phase II study of T2D as compared with placebo193. However, the improvement in body weight was not statistically different relative to dose-titrated liraglutide. Development of this specific co-agonist was discontinued in 2020 given the efficacy of semaglutide 2.4 mg in phase III clinical trials (see Related links). More recently, in mice with CNS deletion of GIPR, MAR709 was shown to lose its superior ability to lower body weight and food intake relative to a pharmacokinetically matched GLP1 (ref.185). This observation underscores the contribution of central GIPR agonism to the body weight-lowering mechanism of this AOM.
Tirzepatide (formerly LY3298176) possesses fivefold increased relative potency at human GIPR as compared with GLP1R and is designed for once-weekly subcutaneous injection122. In a phase II trial in patients with T2D, 26 weeks of treatment with tirzepatide demonstrated dramatically superior results relative to a specific once-weekly GLP1R agonist in both HbA1c and body weight lowering194. At the highest doses employed, glucose control was unusually strong with nearly one third of patients achieving HbA1c <5.7%, and weight loss in these patients with diabetes exceeded 10%. Collectively, these results have generated great interest in GIPR/GLP1R co-agonists, while deepening the debate as to the relative direct and indirect contribution of GIPR agonism192,195,196. In a recent phase III trial in patients with T2D of excess weight, tirzepatide showed superior ability to decrease HbA1c and body weight at all doses tested, relative to treatment with semaglutide 1 mg (ref.125). Forty weeks of treatment with tirzepatide decreased HbA1c <5.7% in 29–51% of patients relative to 20% treated with semaglutide, and decreased body weight ≥15% in 15–40% of patients relative to 9% with semaglutide125. A subsequent phase III trial in patients with obesity or overweight with diabetes confirmed that treatment with tirzepatide for 40 weeks similarly decreased HbA1c <5.7% in 34–52% of patients and lowered body weight ≥15% in 13–27% of patients127. Consistent with this, in patients with T2D who are insulin-dependent, 1 year of treatment with tirzepatide improved glycaemic control with much greater efficacy relative to comparative treatment with insulin (degludec)126. How the more recently approved semaglutide 2.4 mg compares with tirzepatide remains to be determined35,36,37,38.
Co-therapy of GLP1R agonism with glucagon (GcgR) agonists is designed to employ more than a single mechanism in body weight reduction (appetite suppression, thermogenesis and lipolysis, respectively), while minimizing the risk of hyperglycaemia186,197. Clinical results have been reported for two GLP1R/GcgR co-agonists (cotadutide, formerly MEDI0382 and SAR425899). Each of them is palmitoylated, with once-daily time action notably more potent at GLP1R relative to GcgR. In a 54-week phase IIb study in patients with overweight and obesity with T2D, cotadutide reduced body weight and hepatic fat content and improved glucose tolerance relative to placebo198. Mean weight loss was ~5%, with 15.5% of patients achieving weight loss greater than 10% relative to 5.8% receiving liraglutide 1.8 mg. SAR425899 has completed phase I trials in healthy volunteers and patients with T2D199,200,201. Body weight loss of ~7% was reported after 4 weeks of treatment, with improvements in glucose tolerance. Whether additional unimolecular GLP1R/GcgR co-agonists with greater relative glucagon activity or more extended duration of action prove more effective, and sufficiently safe for chronic use, remains to be determined202.
Given the power of the approach, multi-agonism therapy has been repeatedly employed in preclinical treatment of obesity, typically but not exclusively in combination with some form of GLP1 agonism. Representative co-therapies include leptin with pramlintide180,181,182,203,204, leptin with exendin 4 or FGF21 (ref.205), salmon calcitonin with exendin 4 (ref.206), GLP1 with PYY207, exenatide with CCK208 and liraglutide with setmelanotide209.
Further development specific to glucagon-like peptides has been anchored by the enhanced performance demonstrated for GLP1 co-agonists with GIP or glucagon agonism. These results have promoted integration of the three activities into a single-molecule tri-agonist that includes balanced and full agonism at GLP1R, GIPR and GcgR. Such a tri-agonist has shown great promise in animal testing and advanced to clinical studies210,211. The presence of both GLP1 and GIP components within the same molecule is reported to more effectively minimize the risk of glucagon-mediated hyperglycaemia, and thereby permit more aggressive dosing to achieve additional weight reduction.
In 2015, the first report of superior reductions in body weight and plasma cholesterol in DIO mice as compared with placebo, a GLP1R mono-agonist and a matched GLP1R/GIPR co-agonist, were disclosed for a specific tri-agonist210. The contribution of each individual receptor activity within the tri-agonist was further identified through testing in receptor knockout mice, and with selective chemical antagonism at each receptor. NN9423, a peptide tri-agonist, has advanced to clinical study, but outcomes have yet to be reported. Additional drug candidates include a series of fatty acylated unimolecular GLP1R/GIPR/GcgR tri-agonists (see Related links) and an Fc fusion183. The latter of these candidates (HM15211) is currently in early clinical trials for treatment of nonalcoholic steatohepatitis (Table 2). LY3437943 (GGG) is a GIP/GcG/GLP1 tri-agonist suitable for once-weekly dosing. In phase I clinical trials, 12 weeks of treatment in patients with T2D revealed substantially greater weight loss relative to treatment with tirzepatide but, importantly, equal glycaemic efficacy212.
Leptin, leptin sensitizers and MC4 agonists
The discovery of leptin in 1994 (ref.47) forged our understanding of how peripheral hormones signal to the brain to regulate energy balance (Box 1; Fig. 2). The loss of leptin leads to severe metabolic disturbances, which include extreme hyperphagia, lipodystrophy and hypothalamic amenorrhoea136,213. Several clinical studies confirmed the effectiveness of rDNA-derived human leptin for the treatment of hypothalamic amenorrhoea214,215 and leptin supplementation in ob/ob mice is sufficient to restore fertility216. Metreleptin (Amylin Pharmaceuticals, now AstraZeneca) was approved by the FDA in 2014, and by the European Medicines Agency (EMA) in 2018, for the treatment of lipodystrophy, and leptin supplementation largely normalizes metabolic and neuroendocrine alterations in humans with congenital leptin deficiency95,118,136,217 and in patients with anorexia nervosa218. However, although leptin supplementation is effective in individuals with congenital leptin deficiency, the hormone shows little ability to lower body weight under conditions of common, polygenetic, obesity115,116,137,138. Also, despite not being correlative to lower efficacy or safety, the development of antibodies against metreleptin constitutes an obstacle for its clinical use219. Whereas leptin appears not to hold promise as a stand-alone therapy for the treatment of common obesity, its combination with pramlintide (Amylin Pharmaceuticals) induces greater body weight loss in individuals of excess weight relative to treatment with either drug alone181,220. Improvement of leptin responsiveness has also been confirmed preclinically following co-therapy with either exendin 4 (ref.205), FGF21 (ref.205) or GLP1/glucagon221. Also, plant-derived small molecules such as celastrol222 and withaferin A223 have been shown to decrease body weight through improvement in leptin sensitivity (Table 2).
Leptin regulates energy metabolism via activation of POMC neurons in the ARC while, at the same time, inhibiting AgRP neurons in the same area (Box 1; Fig. 2). POMC neurons project to the paraventricular nucleus (PVN), where they induce satiety through activation of the brain MC4R. Although the brain MC4R is an acknowledged target for the treatment of obesity, the development of selective and safe MC4R agonists imposes notable challenges. Over the last 30 years, a series of MC4R agonists have been developed and shown to decrease body weight and food intake in experimental DIO animals224. However, MC4R agonists are prone to cross-stimulate the structurally related MC1, MC3 and MC5 receptors, which serve important roles in various neuroendocrine processes including hair and skin pigmentation, energy homeostasis and erythrocyte differentiation. Furthermore, activation of MC4R can elevate blood pressure and heart rate through activation of the sympathetic nervous system and induce sexual arousal in males224,225. MC4R agonists that were clinically tested but stopped for insufficient weight loss or the aforementioned adverse effects include LY2112688 (Eli Lilly), MC4-NN-0453 (Novo Nordisk), MK-0493 (Merck) and AZD2820 (AstraZeneca)224. By contrast, setmelanotide is a structurally related MC4R agonist developed by Rhythm Pharmaceuticals. Unlike previous MC4R agonists, setmelanotide did not affect heart rate and blood pressure in monkeys and humans224,226,227. This peptide exhibited profound weight loss in humans with congenital deficiency of either POMC228 or LEPR229 and was well tolerated without any major adverse effects in phase III clinical trials230. The FDA approved setmelanotide in November 2020 for the treatment of obesity in patients with POMC, PCSK1 or LEPR deficiency. Future studies are warranted to investigate whether setmelanotide can decrease body weight in more common, polygenetic forms of obesity. Studies in patients with Prader–Willi syndrome have demonstrated that setmelanotide can decrease body weight in individuals where the primary source of obesity is not directly attributable to the melanocortin system.
Amylin
Amylin (also known as IAPP) is a peptide that is co-secreted with insulin and reduces food intake through central control of satiety pathways231,232 (Box 1; Fig. 2). Amylin activates specific receptors including those of the calcitonin gene-related peptide (CGRP). Although the major effect of amylin on energy metabolism is mediated through increasing satiety, amylin has also been shown to affect hedonic control of eating, including a reduction in feeding reward neurocircuits233. However, the clinical application of native amylin in treating obesity has been shadowed by physical aggregates related to pancreatic islet death in humans234, a finding not observed with rat amylin235. The anorexigenic potential of amylin promoted the development of pramlintide, a rat-based synthetic analogue of amylin236.
Pramlintide is approved by the FDA for use in patients with T1D and T2D who are using mealtime insulin alone, or in combination with an oral agent such as metformin or a sulfonylurea165,237. Importantly, effects of pramlintide on reducing food intake and body weight are not limited to patients with impaired glucose metabolism233. Therefore, other amylin analogues with improved pharmacokinetics are being considered as AOMs. Amylin agonists seem to be particularly useful for weight loss in combination with other agents, such as leptin181,220 or calcitonin receptor agonists238.
The human amylin receptor subtypes are complexes of the calcitonin receptor with receptor activity-modifying proteins239. Recently, dual-acting amylin and calcitonin receptor agonists (DACRAs) have been developed as potential AOMs (Table 2). Several DACRAs (for example, davalintide (AC2307), KBP-088, KBP-089, KBP-042) have been shown to induce weight loss in animal models of obesity165,240,241,242. In addition, a long-acting amylin analogue, cagrilintide, suitable for once-weekly treatment has successfully completed a phase Ib trial (Table 2) and is favourably progressing in subsequent studies in combination with semaglutide to what might constitute enhanced chronic efficacy243.
Ghrelin
As a peptide hormone secreted from x/a-like cells (P/D1 cells in humans) of the gastric fundus, ghrelin acts on hypothalamic feeding centres to stimulate food intake244 (Fig. 2). Independent of its orexigenic effect, ghrelin promotes adiposity and elevates blood glucose through inhibition of insulin secretion245. Envisioned strategies to harness ghrelin biology for potential treatment of obesity include suppression of active circulating hormone and antagonism of signalling at its receptor, the growth hormone secretagogue receptor (GHSR). The latter can be achieved through GHSR antagonists and inverse agonists, such as the liver-enriched antimicrobial peptide 2 (LEAP2), or the des-acyl form of ghrelin (DAG). Therapeutic interest has been spurred by observations in rodents, where neutralization of acyl-ghrelin246, inhibition of ghrelin O-acyltransferase (GOAT) as the activating fatty acylation enzyme247 or direct antagonism of GHSR248 have demonstrated decreases in body weight and food intake.
In patients with Prader–Willi syndrome, circulating levels of acyl-ghrelin are increased249 and 14-day treatment with a UAG analogue (AZP-531) (Table 2) improved food-related behaviour, body fat mass and postprandial levels of blood glucose, without any major sign of intolerability250. Nonetheless, ghrelin is a disputed target for treatment of obesity251,252, where the circulating concentrations of acyl-ghrelin are reported to be elevated in individuals who are lean and those with anorexia, and low in certain conditions of obesity. Furthermore, excess body fat is associated with ghrelin unresponsiveness. This is potentially mediated by a LEAP2-associated increase in obesity that serves to competitively bind GHSR and inhibit biological signalling253.
Approaches to decrease acyl-ghrelin include a therapeutic peptide vaccine that ameliorated body weight gain in rodents, interestingly without affecting food intake. The efficacy was reported to be specific to the plasma binding of the acyl form of ghrelin254. A similar acting vaccine, CYT009-GhrQb (Table 2), was developed by Cyto Biotechnology. The vaccine advanced to early clinical trials (phase I/II) in which it showed no effect on body weight or food intake255. Separately, no long-term beneficial effects on body weight or food intake were reported when a specific anti-ghrelin monoclonal antibody was tested in DIO mice at Amgen256. A comparable outcome resulted in the use of anti-ghrelin Spiegelmers developed at NOXXON Pharma that only moderately improved metabolism in preclinical studies, with no effect on food intake after 8 days of treatment246.
In summary, pharmacotherapies targeting the ghrelin pathway so far have yet to reveal a clinically validated AOM candidate. Targeting the ghrelin pathway, however, warrants further investigation as ghrelin remains the only known circulating signal to increase hunger and potently activate hypothalamic AGRP neurons that drive appetite244.
Targeted mitochondrial uncouplers
The tissues most involved in thermogenesis are skeletal muscle and adipose tissue, most notably brown adipose tissue. Energy derived from dietary substrates is captured by TCA-mediated catabolism in the mitochondria in association with an electron transport chain leading to ATP synthesis257. UCP1, localized in the inner mitochondrial membrane of brown and beige adipocytes, catalyses the transport of protons across the mitochondrial membrane and, thereby, induces mitochondrial uncoupling of oxygen consumption from ATP synthesis258,259. Pharmacologically, UCP1 activity can be induced by catecholamines with subsequent activation of β3-adrenergic receptors of brown adipose tissue257. Thyroid hormone (T3) is an endogenous entity with uncoupling capability mediated by several different mechanisms260.
Enhancement in mitochondrial uncoupling can have beneficial health effects. Mitochondrial uncouplers, such as 2,4-dinitrophenol (DNP), increase mitochondrial inefficiency, rendering metabolism and production of ATP less efficient261. Although DNP was welcomed for obesity treatment in 1934 (ref.55), it was later banned from therapeutic use due to multiple adverse effects and numerous reports of DNP-associated deaths261. Nonetheless, the substance has continued to be used by bodybuilders and others. The UK Food Standard Agency issued a warning in 2003, given increasing concern for toxicity associated with unregulated use that DNP was ‘not fit for human consumption’ (see Related links).
Mitochondrial uncouplers are cytotoxic at high concentrations, an effect resulting from a drop in ATP concentration and on plasma and lysosomal membrane depolarization and permeabilization. However, the effect is concentration-dependent, and at doses that are not toxic, mitochondrial uncoupling can protect cells against death262. Consequently, the development of mitochondria-specific and safer uncoupling agents suitable for human use might yet result in a powerful and differentiated approach to treating these diseases263. Recent studies using a controlled-release oral formulation of DNP, called CRMP (controlled-release mitochondrial protonophore), is one prominent attempt to achieve an enhanced therapeutic index. In rats, CRMP was employed to achieve low-level hepatic mitochondrial uncoupling that reversed hypertriglyceridemia, insulin resistance, hepatic steatosis and diabetes264.
BAM15 ((2-fluorophenyl){6-[(2-fluorophenyl)amino](1,2,5-oxadiazolo [3,4e] pyrazin-5-yl)} amine) (Table 2) is a novel mitochondria-specific protonophore uncoupler that demonstrates similar potency to DNP to increase energy expenditure265. BAM15 is an orally administered drug that can increase nutrient oxidation, and decreases body fat mass without altering food intake, lean body mass, body temperature or haematological markers of toxicity. Mice treated with BAM15 were reported to be resistant to weight gain265,266. BAM15 improves insulin sensitivity in multiple tissues and in vitro enhanced mitochondrial respiration, improved insulin action and stimulated nutrient uptake by sustained activation of AMPK. These results collectively illustrate that mitochondrial uncoupling with BAM15 has robust anti-obesity and insulin-sensitizing effects, without compromising lean mass or affecting food intake265,266. However, it remains too early to say with confidence whether BAM15 or another related approach will provide much-enhanced therapeutic safety for treatment of obesity-related comorbidities or excess body weight itself.
GDF15
Macrophage inhibitory cytokine 1 (MIC1; also known as GDF15) has gained attention as a target for obesity treatment267. GDF15 is a divergent member of the transforming growth factor-β (TGFβ) superfamily267. Physiologically, GDF15 is expressed in multiple tissues at a low concentration, but increases in response to or association with tissue injury, cancer, metabolic disease, CVD and inflammation267,268. GDF15 has also been proposed to act as an anti-inflammatory cytokine in the infarcted heart269.
Exogenous administration of rDNA-derived GDF15 and analogues decreases body weight in diet-induced obese mice and non-human primates, suggesting a homeostatic role in energy homeostasis267,270. Recently, GDF15 was shown to physiologically regulate energy homeostasis and body weight — primarily via appetite suppression — through activation of the receptor, GDNF family receptor α-like (GFRAL)270. Some studies suggested that the anorectic effect of GDF15 is mediated through induction of nausea and engagement of emetic neurocircuitries271,272, but this has not been confirmed by all studies270. Nonetheless, its depletion results in increased body weight273,274, whereas GDF15 overexpression has the opposite effect274,275,276. Chronic study demonstrating sustained efficacy, sufficiently devoid of safety risks such as nausea/vomiting, tumorigenicity and cachectic lean body mass reduction, needs to be thoughtfully considered. Ultimately, only in human study can the assessment of whether GDF15 analogues will prove efficacious and safe for weight loss management be determined267.
Peptide tyrosine tyrosine
Peptide tyrosine tyrosine (PYY) is a member of the NPY family that is co-secreted from the intestinal L cells as PYY1–36, along with GLP1. After being released, PYY1–36 is rapidly cleaved by DPP-IV to its major active form, PYY3–36, a high-affinity agonist at the NPY receptor type 2 (Y2R). This receptor is highly expressed in parasympathetic and sympathetic neurons of the periphery as well as in several regions of the CNS, including the limbic and cortical areas and the brainstem277. In the hypothalamus, Y2R is highly expressed on NPY neurons of the ARC278 and PYY3–36 decreases food intake and body weight in rodents278,279,280 and humans207,281,282, at least in part through its ability to silence Npy neurons and, hence, to indirectly activate Pomc neurons278. Additional mechanisms that may be implicated in the regulation of food intake by PYY3–36 include Y2R-mediated activation of the mesolimbic dopaminergic system as well as of GABAergic and glutamatergic neurons in cortical and subcortical regions and the brainstem277. Consistent with the relevance of dopaminergic signalling in mesolimbic brain regions, PYY3–36 has effects that go well beyond the regulation of food intake, and include memory and learning, central information processing and behavioural response to dopamine-stimulating drugs277. The ability of PYY3–36 to decrease food intake in rodents and humans has stimulated the development of PYY3–36 analogues for the treatment of obesity283. In line with this notion, several long-acting PYY3–36 analogues (NN9748 and NNC0165-1875) have completed phase I trials for the treatment of obesity (see Related links) and NNC0165-1875 is now being assessed in a phase II combination study with semaglutide (see Related links). Additionally, Lilly Research Laboratories announced a phase I trial with a PYY analogue for the treatment of T2D (see Related links).
Outlook and future directions
The pursuit of AOMs has been a long-standing endeavour propelled in recent years by several concurrent developments. These include the dramatic increase in the global prevalence of obesity, the significant advances in molecular understanding of appetite homeostasis along with the identification of several novel drug targets, as well as the success in developing incretins as drugs for T2D that has provided unprecedented efficacy in body weight management. It seems plausible that a 20% or greater reduction in body weight may yet be possible based on late-phase clinical reports. If so, it is interesting to ponder whether patients of far higher initial body weight might find the next 20% reduction to be easier or harder to achieve in a relative sense, as these are the individual subjects of greatest need.
GLP1R agonism is establishing a heightened foundation for measuring performance with other entities, and the full depth of its efficacy and the ability to chronically sustain weight loss in multiple populations, many distinct from those in which initial drug registration has occurred, remains to be determined. As with any rapidly advancing field, there are more questions than answers. Of primary interest is why GLP1R agonism works so well and how GIP might synergize with GLP1 to enhance weight loss. Short of the results that have been achieved in vivo, most notably the 6-month and 1-year clinical studies that appear to indicate significant additional benefits of semaglutide when compared with liraglutide, it is difficult to ascribe a molecular basis for that difference. These two agents are both highly potent and selective GLP1R agonists, similarly fatty acylated, that provide sustained drug plasma concentrations when used as prescribed. The difference is not simply a matter of extended time action as even a long-action Fc agonist such as dulaglutide does not match the body weight lowering of semaglutide284. Initial study suggests increased activity in central locations of importance to weight control123. However, this is just a beginning and a deeper molecular understanding might lead to even further improvements in GLP1R agonists, or other agents that might act by an independent mechanism at similar anatomical sites.
Unquestionably, the clinical results with tirzepatide have captured great attention and fuelled interest in GIP-based dual agonists and other combinatorial approaches. However, is this interest justified by these clinical results? The situation appears to exemplify that despite the enormous advance in our molecular understanding of obesity, we remain relatively primitive in ascribing in vivo efficacy to mechanism. It remains to be demonstrated in mechanistic detail how GIPR agonism serves as the basis for the heightened efficacy of tirzepatide relative to dulaglutide. Very recently, it was shown that CNS loss of GIPR renders mice resistant to GIP-induced body weight loss, indicating that GIP regulates energy metabolism via CNS GIPR signalling185. Substantiating the relevance of this finding, it is noteworthy that the superior weight-lowering effect of MAR709 relative to a GLP1 monotherapy of matched structure and pharmacokinetics vanished in CNS Gipr knockout mice185. The central mechanisms and target regions for GIP synergy with GLP1 remain to be determined, and notably there are conflicting preclinical results that promote GIPR antagonism as a therapeutic option for treating obesity184. In time, these questions and uncertainties will eventually be answered.
Next-generation discoveries are heavily influenced by current clinical performance and limitations in our ability to successfully translate in vitro and animal pharmacology to human experiments. High-dose semaglutide and tirzepatide are reporting sustained reduction in body weight of approximately 0.5 kg per week. This is a breakthrough performance relative to registered AOMs that begs the question of what the highest next priority is, and whether we have the skills necessary to properly achieve it. Clearly, additional mechanisms of action that can match the performance of these two drugs would be welcomed, but to document this requires appreciably long studies. Underpowered 4-week, 6-week, 8-week and, even, 12-week studies without suitable registered drugs as controls have largely failed to document relative efficacy.
Efficacy studies struggle with the question of how much additional weight reduction is advisable in a finite period, and the duration necessary for documenting it with confidence. Given the efficacy that is being achieved and the chronic nature of obesity, it is arguable that maintaining the rate in weight loss for subjects of continued excess weight is the primary objective. These studies are lengthy and rarely undertaken until there is great confidence for success. Shortening the studies with the objective of accelerating the relative rate of weight reduction may not prove advisable for the patient and could lead to adverse effects that eliminate approaches that otherwise would prove viable, if applied less aggressively. This is a point of particular importance in the assessment of glucagon-based tri-agonists that aim to outperform GLP1–GIPR co-agonists, as glucagon is likely an agonist of reduced therapeutic index relative to the two incretins.
In a related manner, might drug candidates that fail in monotherapy prove successful when added to the best-in-class incretins either at initiation of therapy or after sizeable weight loss? The clinical success of GLP1 with GIP raises the question of whether adjunctive therapy of semaglutide with another weight-lowering agent such as amylin, PYY or FGF21 can safely lower body weight beyond what is possible with either drug alone. In this regard, it should be noted that leptin therapy proved successful in reducing body weight when used following sizeable weight loss in obese mice181,205,221. Might the same prove true in selected patients with obesity now that comparable percent reductions in body weight with what has proven successful preclinically are being achieved with semaglutide and tirzepatide?
Finally, there is the question of what is most needed to accelerate the realization of the next leap forward in safely normalizing body weight. Next-generation multi-omics have provided some novel targets, but, overall, rapidly evolving enabling technologies have been more useful in characterizing preclinical mechanism of action than in discovery of clinically successful drug candidates. Iterative rodent testing largely using diet-induced obese mice and rats has been the primary screen to assess body weight lowering. Genetic models and, even more so, engineered mice where specific receptors have been deleted, and increasingly so in a target-specific manner, have proven of indispensable value to investigation of mechanism of action.
The clinical situation is more challenging, where there is infrequent access to individuals homozygous-deficient in a specific biological mechanism. In those rare instances, the nature of the obesity and the response to therapy differ from the general population. Additionally, selective antagonists suitable for pharmacological use are seldom available to selectively silence a single mechanism of action to explore its relation to endogenous control of body weight, or to block the action of a specific drug or a single element in a multi-action peptide, such as the incretin co-agonists. Lastly, the simultaneous comparison of peptides matched in structure and pharmacokinetics, but otherwise devoid of a single biological activity, constitutes a prohibitive investment when the length of study is measured in months. Consequently, what we most need to speed drug discovery and optimization is correlative diagnostic means to complement a body weight scale. If we could serologically or non-invasively predict with increased confidence those patients and mechanisms that are likely to succeed long term, this would promote better outcomes and increase exploratory clinical research to identify molecular entities and combinations that most justify assessment in long-term studies. In analogy, it is readily recognized what plasma glucose monitoring and HbA1c have meant to diabetes care and drug discovery relative to urine testing or monitoring of longer-term microvascular outcomes. If a predictive correlate between metabolic profiling and propensity to weight loss can be established, this could have a profound influence on the future of healthcare in obesity.
Summary
Pharmacological management of obesity has a lengthy history populated with multiple prominent disappointments. The basis of failure has been multifactorial and pertains to the limited translational value of animal models to predict cardiovascular safety coupled with considerable patient heterogeneity. Patients with obesity are often at high risk for vascular diseases and afflicted with comorbidities that complicate assessment of drug safety. Long-term, large-scale clinical trials in heterogeneous patients with obesity are expensive to conduct and difficult to justify when success has been so elusive and failures so prominent.
The recent precedent-setting results with semaglutide and tirzepatide, in which each reported mean weight loss well in excess of 10%, employing a GLP1 mechanism that has separately proven to improve cardiovascular outcomes in T2D studies, inspires confidence for the future. Clinical application will continue and focus on relative efficacy and safety, which is difficult to ascribe when best-in-class candidates are simultaneously rapidly advancing and not immediately accessible for direct comparative clinical study125. Independently, setmelanotide and leptin have proven successful in obesity management of individuals with congenital deficiency in genes of the leptinergic–melanocortinergic pathway. These successes illuminate the paths for future research targeting other monogenetic forms of the disease and the possibility for additive pharmacology in broader populations of patients with obesity. A more thorough characterization of patients should serve to increase the near-term likelihood for success and provide informed direction for advancing the next generation of AOMs. Ongoing clinical studies will determine whether more efficacious drugs than semaglutide and tirzepatide might achieve efficacy comparable with bariatric surgery. The many prospects currently being considered suggest that one or more might achieve this lofty objective.
References
Afshin, A. et al. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 377, 13–27 (2017). This article summarizes the prevalence rates and health consequences of obesity from 1990 until 2015.
Bauer, U. E., Briss, P. A., Goodman, R. A. & Bowman, B. A. Prevention of chronic disease in the 21st century: elimination of the leading preventable causes of premature death and disability in the USA. Lancet 384, 45–52 (2014).
Bjerregaard, L. G. et al. Change in overweight from childhood to early adulthood and risk of type 2 diabetes. N. Engl. J. Med. 378, 1302–1312 (2018).
Twig, G. et al. Body-mass index in 2.3 million adolescents and cardiovascular death in adulthood. N. Engl. J. Med. 374, 2430–2440 (2016).
Berrington de Gonzalez, A. et al. Body-mass index and mortality among 1.46 million white adults. N. Engl. J. Med. 363, 2211–2219 (2010).
Calle, E. E. Obesity and cancer. BMJ 335, 1107–1108 (2007).
Calle, E. E., Rodriguez, C., Walker-Thurmond, K. & Thun, M. J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 348, 1625–1638 (2003).
Belanger, M. J. et al. Covid-19 and disparities in nutrition and obesity. N. Engl. J. Med. 383, e69 (2020).
Arnold, M. et al. Global burden of cancer attributable to high body-mass index in 2012: a population-based study. Lancet Oncol. 16, 36–46 (2015).
Arnold, M. et al. Obesity and cancer: an update of the global impact. Cancer Epidemiol. 41, 8–15 (2016).
Fontaine, K. R., Redden, D. T., Wang, C., Westfall, A. O. & Allison, D. B. Years of life lost due to obesity. JAMA 289, 187–193 (2003).
Prospective Studies, C. et al. Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet 373, 1083–1096 (2009).
Ebbeling, C. B., Pawlak, D. B. & Ludwig, D. S. Childhood obesity: public-health crisis, common sense cure. Lancet 360, 473–482 (2002).
Cawley, J. & Meyerhoefer, C. The medical care costs of obesity: an instrumental variables approach. J. Health Econ. 31, 219–230 (2012).
McAllister, E. J. et al. Ten putative contributors to the obesity epidemic. Crit. Rev. Food Sci. Nutr. 49, 868–913 (2009).
Cappuccio, F. P. et al. Meta-analysis of short sleep duration and obesity in children and adults. Sleep 31, 619–626 (2008).
Noh, J. The effect of circadian and sleep disruptions on obesity risk. J. Obes. Metab. Syndr. 27, 78–83 (2018).
Scott, K. A., Melhorn, S. J. & Sakai, R. R. Effects of chronic social stress on obesity. Curr. Obes. Rep. 1, 16–25 (2012).
Ness-Abramof, R. & Apovian, C. M. Drug-induced weight gain. Drugs Today 41, 547–555 (2005).
Maes, H. H., Neale, M. C. & Eaves, L. J. Genetic and environmental factors in relative body weight and human adiposity. Behav. Genet. 27, 325–351 (1997).
Bray, M. S. et al. NIH working group report — using genomic information to guide weight management: from universal to precision treatment. Obesity 24, 14–22 (2016).
Davis, L. K. et al. Partitioning the heritability of Tourette syndrome and obsessive compulsive disorder reveals differences in genetic architecture. PLoS Genet. 9, e1003864 (2013).
Grjibovski, A. M., Olsen, A. O., Magnus, P. & Harris, J. R. Psoriasis in Norwegian twins: contribution of genetic and environmental effects. J. Eur. Acad. Dermatol. Venereol. 21, 1337–1343 (2007).
Katzmarzyk, P. T. et al. Familial resemblance for coronary heart disease risk: the HERITAGE Family Study. Ethn. Dis. 10, 138–147 (2000).
Czene, K., Lichtenstein, P. & Hemminki, K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int. J. Cancer 99, 260–266 (2002).
Bray, G. A., Kim, K. K., Wilding, J. P. H. & World Obesity, F. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes. Rev. 18, 715–723 (2017).
Burki, T. European Commission classifies obesity as a chronic disease. Lancet Diabetes Endocrinol. 9, 418 (2021).
Bluher, M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 15, 288–298 (2019).
Carlsson, L. M. S. et al. Life expectancy after bariatric surgery in the Swedish Obese Subjects Study. N. Engl. J. Med. 383, 1535–1543 (2020).
Adams, T. D. et al. Weight and metabolic outcomes 12 years after gastric bypass. N. Engl. J. Med. 377, 1143–1155 (2017).
Farooqi, I. S. Defining the neural basis of appetite and obesity: from genes to behaviour. Clin. Med. 14, 286–289 (2014).
Hauner, H. et al. Efficacy and safety of cathine (nor-pseudoephedrine) in the treatment of obesity: a randomized dose-finding study. Obes. Facts 10, 407–419 (2017).
Lucchetta, R. C. et al. Systematic review and meta-analysis of the efficacy and safety of amfepramone and mazindol as a monotherapy for the treatment of obese or overweight patients. Clinics 72, 317–324 (2017).
Muller, T. D., Clemmensen, C., Finan, B., DiMarchi, R. D. & Tschop, M. H. Anti-obesity therapy: from rainbow pills to polyagonists. Pharmacol. Rev. 70, 712–746 (2018).
Davies, M. et al. Semaglutide 2.4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet 397, 971–984 (2021). This article shows the metabolic effects of semaglutide 2.4 mg in patients with obesity and diabetes.
Rubino, D. et al. Effect of continued weekly subcutaneous semaglutide vs placebo on weight loss maintenance in adults with overweight or obesity: the STEP 4 randomized clinical trial. JAMA 325, 1414–1425 (2021). This article shows the effects of continuous versus withdrawing treatment with semaglutide 2.4 mg in people with overweight and obesity.
Wadden, T. A. et al. Effect of subcutaneous semaglutide vs placebo as an adjunct to intensive behavioral therapy on body weight in adults with overweight or obesity: the STEP 3 randomized clinical tTrial. JAMA 325, 1403–1413 (2021).
Wilding, J. P. H. et al. Once-weekly semaglutide in adults with overweight or obesity. N. Engl. J. Med. 384, 989 (2021). This article shows the metabolic effects of semaglutide 2.4 mg in patients with obesity without diabetes.
Tak, Y. J. & Lee, S. Y. Anti-obesity drugs: long-term efficacy and safety: an updated review. World J. Mens Health 39, 208–221 (2020).
Bray, G. A., Fruhbeck, G., Ryan, D. H. & Wilding, J. P. Management of obesity. Lancet 387, 1947–1956 (2016).
Heymsfield, S. B. & Wadden, T. A. Mechanisms, pathophysiology, and management of obesity. N. Engl. J. Med. 376, 254–266 (2017).
Magkos, F. et al. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab. 23, 591–601 (2016).
Knowler, W. C. et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 346, 393–403 (2002).
Sjostrom, L. et al. Effects of bariatric surgery on mortality in Swedish obese subjects. N. Engl. J. Med. 357, 741–752 (2007).
Bluher, M. Metabolically healthy obesity. Endocr. Rev. 41, 405–420 (2020).
Stefan, N., Haring, H. U. & Schulze, M. B. Metabolically healthy obesity: the low-hanging fruit in obesity treatment? Lancet Diabetes Endocrinol. 6, 249–258 (2018). This article shows the heterogeneity of obesity.
Zhang, Y. et al. Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432 (1994). This article describes the discovery of leptin.
Adriaenssens, A. E., Gribble, F. M. & Reimann, F. The glucose-dependent insulinotropic polypeptide signaling axis in the central nervous system. Peptides 125, 170194 (2020). This article comprehensively summarizes GIP effects in the brain and periphery.
Farooqi, I. S. Monogenic human obesity. Front. Horm. Res. 36, 1–11 (2008).
Farooqi, I. S. et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 348, 1085–1095 (2003). This article gives a sophisticated overview of the polygenic nature of MC4R mutations.
Farooqi, I. S. & O’Rahilly, S. The Genetics of Obesity in Humans (Endotext, 2000).
Kushner, R. F. et al. Semaglutide 2.4 mg for the treatment of obesity: key elements of the STEP Trials 1 to 5. Obesity 28, 1050–1061 (2020).
O’Neil, P. M. et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet 392, 637–649 (2018).
Tainter, M. L., Cutting, W. C. & Stockton, A. B. Use of dinitrophenol in nutritional disorders: a critical survey of clinical results. Am. J. Public. Health Nations Health 24, 1045–1053 (1934).
Tainter, M. L., Stockton, A. B. & Cutting, W. C. Use of dinitrophenol in obesity and related conditions. JAMA 101, 1472–1475 (1933).
Dunlop, D. M. The use of 2:4-dinitrophenol as a metabolic stimulant. Br. Med. J. 3820, 524 (1934).
Alphin, R. S. & Ward, J. W. Anorexigenic effects of fenfluramine hydrochloride in rats, guinea pigs, and dogs. Toxicol. Appl. Pharmacol. 14, 182–191 (1969).
Pinder, R. M., Brogden, R. N., Sawyer, P. R., Speight, T. M. & Avery, G. S. Fenfluramine: a review of its pharmacological properties and therapeutic efficacy in obesity. Drugs 10, 241–323 (1975).
Weintraub, M. et al. Long-term weight control study. I (weeks 0 to 34). The enhancement of behavior modification, caloric restriction, and exercise by fenfluramine plus phentermine versus placebo. Clin. Pharmacol. Ther. 51, 586–594 (1992).
Finer, N., Craddock, D., Lavielle, R. & Keen, H. Effect of 6 months therapy with dexfenfluramine in obese patients: studies in the United Kingdom. Clin. Neuropharmacol. 11, S179–186 (1988).
Geyer, G. et al. Effect of dexfenfluramine on eating behavior and body weight of obese patients: results of a field study of isomeride in Austrian general practice [German]. Acta Med. Austriaca 22, 95–101; 104–109 (1995).
Holdaway, I. M., Wallace, E., Westbrooke, L. & Gamble, G. Effect of dexfenfluramine on body weight, blood pressure, insulin resistance and serum cholesterol in obese individuals. Int. J. Obes. Relat. Metab. Disord. 19, 749–751 (1995).
Andersen, T., Astrup, A. & Quaade, F. Dexfenfluramine as adjuvant to a low-calorie formula diet in the treatment of obesity: a randomized clinical trial. Int. J. Obes. Relat. Metab. Disord. 16, 35–40 (1992).
Khera, R. et al. Association of pharmacological treatments for obesity with weight loss and adverse events: a systematic review and meta-analysis. JAMA 315, 2424–2434 (2016).
Smith, S. R. et al. Multicenter, placebo-controlled trial of lorcaserin for weight management. N. Engl. J. Med. 363, 245–256 (2010).
Davidson, M. H. et al. Weight control and risk factor reduction in obese subjects treated for 2 years with orlistat: a randomized controlled trial. JAMA 281, 235–242 (1999).
Despres, J. P., Golay, A. & Sjostrom, L., Rimonabant in Obesity-Lipids Study Group. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N. Engl. J. Med. 353, 2121–2134 (2005).
Pi-Sunyer, F. X. et al. Effect of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: RIO-North America: a randomized controlled trial. JAMA 295, 761–775 (2006).
Van Gaal, L. F. et al. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet 365, 1389–1397 (2005).
Bray, G. A. A concise review on the therapeutics of obesity. Nutrition 16, 953–960 (2000).
Ioannides-Demos, L. L., Proietto, J. & McNeil, J. J. Pharmacotherapy for obesity. Drugs 65, 1391–1418 (2005).
Antel, J. & Hebebrand, J. Weight-reducing side effects of the antiepileptic agents topiramate and zonisamide. Handb. Exp. Pharmacol. 209, 433–466 (2012).
Walter, C. P., Bleske, B. E. & Dorsch, M. P. Pharmacotherapy for weight loss: the cardiovascular effects of the old and new agents. J. Clin. Pharm. Ther. 39, 475–484 (2014).
Apovian, C. M. et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity 21, 935–943 (2013).
Nissen, S. E. et al. Effect of naltrexone–bupropion on major adverse cardiovascular events in overweight and obese patients with cardiovascular risk factors: a randomized clinical trial. JAMA 315, 990–1004 (2016).
Marso, S. P. et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 375, 311–322 (2016). This article shows the beneficial effects of liraglutide on cardiovascular health.
Astrup, A. et al. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int. J. Obes. 36, 843–854 (2012).
Farooqi, I. S. & O’Rahilly, S. Monogenic human obesity syndromes. Recent. Prog. Horm. Res. 59, 409–424 (2004).
Hebebrand, J., Sommerlad, C., Geller, F., Gorg, T. & Hinney, A. The genetics of obesity: practical implications. Int. J. Obes. Relat. Metab. Disord. 25, S10–18 (2001).
Li, L. et al. Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci. Transl Med. 7, 311ra174 (2015).
Lyssenko, V. et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N. Engl. J. Med. 359, 2220–2232 (2008).
O’Rahilly, S. Human genetics illuminates the paths to metabolic disease. Nature 462, 307–314 (2009). This article summarizes how human genetics influenced our understanding of the pathophysiological mechanisms leading to obesity and T2D.
Xue, A. et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 9, 2941 (2018).
Ahlqvist, E. et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 6, 361–369 (2018).
Chatterjee, S. & Davies, M. J. Accurate diagnosis of diabetes mellitus and new paradigms of classification. Nat. Rev. Endocrinol. 14, 386–387 (2018).
Udler, M. S. et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes:a soft clustering analysis. PLoS Med. 15, e1002654 (2018).
Fall, T. & Ingelsson, E. Genome-wide association studies of obesity and metabolic syndrome. Mol. Cell Endocrinol. 382, 740–757 (2014).
Farooqi, S. & O’Rahilly, S. Genetics of obesity in humans. Endocr. Rev. 27, 710–718 (2006).
Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
Rankinen, T. et al. The human obesity gene map: the 2005 update. Obesity 14, 529–644 (2006).
Huypens, P. et al. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat. Genet. 48, 497–499 (2016). This article shows how epigenetic factors influence the predisposition to obesity and diabetes.
Dong, C. et al. Possible genomic imprinting of three human obesity-related genetic loci. Am. J. Hum. Genet. 76, 427–437 (2005).
Hoggart, C. J. et al. Novel approach identifies SNPs in SLC2A10 and KCNK9 with evidence for parent-of-origin effect on body mass index. PLoS Genet. 10, e1004508 (2014).
Melvin, A., O’Rahilly, S. & Savage, D. B. Genetic syndromes of severe insulin resistance. Curr. Opin. Genet. Dev. 50, 60–67 (2018).
Farooqi, I. S. et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Invest. 110, 1093–1103 (2002).
Gibson, W. T. et al. Congenital leptin deficiency due to homozygosity for the Δ133G mutation: report of another case and evaluation of response to four years of leptin therapy. J. Clin. Endocrinol. Metab. 89, 4821–4826 (2004).
Montague, C. T. et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387, 903–908 (1997).
Farooqi, I. S. et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N. Engl. J. Med. 356, 237–247 (2007).
Krude, H. et al. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 19, 155–157 (1998).
Farooqi, I. S. et al. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J. Clin. Invest. 106, 271–279 (2000).
Frayling, T. M. et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894 (2007). This article discovers that genetic variation in the FTO gene is associated with obesity.
Hinney, A., Volckmar, A. L. & Knoll, N. Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis. Prog. Mol. Biol. Transl. Sci. 114, 147–191 (2013).
Friedman, J. M. A war on obesity, not the obese. Science 299, 856–858 (2003).
Friedman, J. M. Modern science versus the stigma of obesity. Nat. Med. 10, 563–569 (2004).
Friedman, J. M. Obesity: causes and control of excess body fat. Nature 459, 340–342 (2009).
Friedman, J. M. & Halaas, J. L. Leptin and the regulation of body weight in mammals. Nature 395, 763–770 (1998).
Kraschnewski, J. L. et al. Long-term weight loss maintenance in the United States. Int. J. Obes. 34, 1644–1654 (2010).
Leibel, R. L. et al. Biologic responses to weight loss and weight regain: report from an American Diabetes Association Research Symposium. Diabetes 64, 2299–2309 (2015).
Lowe, M. R., Miller-Kovach, K. & Phelan, S. Weight-loss maintenance in overweight individuals one to five years following successful completion of a commercial weight loss program. Int. J. Obes. Relat. Metab. Disord. 25, 325–331 (2001).
Maclean, P. S., Bergouignan, A., Cornier, M. A. & Jackman, M. R. Biology’s response to dieting: the impetus for weight regain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R581–R600 (2011).
Korner, J., Savontaus, E., Chua, S. C. Jr., Leibel, R. L. & Wardlaw, S. L. Leptin regulation of Agrp and Npy mRNA in the rat hypothalamus. J. Neuroendocrinol. 13, 959–966 (2001).
Levin, B. E. & Dunn-Meynell, A. A. Chronic exercise lowers the defended body weight gain and adiposity in diet-induced obese rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R771–R778 (2004).
Yu, Y., South, T., Wang, Q. & Huang, X. F. Differential expression of hypothalamic CART mRNA in response to body weight change following different dietary interventions. Neurochem. Int. 52, 1422–1430 (2008).
Cabral, A. et al. Fasting induces remodeling of the orexigenic projections from the arcuate nucleus to the hypothalamic paraventricular nucleus, in a growth hormone secretagogue receptor-dependent manner. Mol. Metab. 32, 69–84 (2020).
Heymsfield, S. B. et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA 282, 1568–1575 (1999).
Hukshorn, C. J. et al. Weekly subcutaneous pegylated recombinant native human leptin (PEG-OB) administration in obese men. J. Clin. Endocrinol. Metab. 85, 4003–4009 (2000).
Scarpace, P. J. & Zhang, Y. Leptin resistance: a prediposing factor for diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R493–R500 (2009).
Farooqi, I. S. et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 341, 879–884 (1999).
von Schnurbein, J. et al. Rapid improvement of hepatic steatosis after initiation of leptin substitution in a leptin-deficient girl. Horm. Res. Paediatr. 79, 310–317 (2013).
Heal, D., Gosden, J. & Smith, S. The 5-HT6 receptor as a target for developing novel antiobesity drugs. Int. Rev. Neurobiol. 96, 73–109 (2011).
Vickers, S. P., Jackson, H. C. & Cheetham, S. C. The utility of animal models to evaluate novel anti-obesity agents. Br. J. Pharmacol. 164, 1248–1262 (2011).
Coskun, T. et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol. Metab. 18, 3–14 (2018).
Gabery, S. et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight 5, e133429 (2020).
Frias, J. P. et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 392, 2180–2193 (2018).
Frias, J. P. et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N. Engl. J. Med. 385, 503–515 (2021). This article compares the metabolic effects of tirzepatide and semaglutide 1 mg in patients with obesity and diabetes.
Ludvik, B. et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet 398, 583–598 (2021). This article compares the metabolic effects of tirzepatide and insulin degludec in patients with T2D.
Rosenstock, J. et al. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet 398, 143–155 (2021).
Tschop, M. H. et al. A guide to analysis of mouse energy metabolism. Nat. Methods 9, 57–63 (2011).
Wing, R. R. et al. Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care 34, 1481–1486 (2011).
Muller, T. D. et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 30, 72–130 (2019). This article gives a comprehensive overview of the metabolic effects of GLP1.
Meier, J. J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 8, 728–742 (2012).
Khera, R. et al. Effects of weight-loss medications on cardiometabolic risk profiles: a systematic review and network meta-analysis. Gastroenterology 154, 1309–1319.e7 (2018).
Dostalova, I., Haluzikova, D. & Haluzik, M. Fibroblast growth factor 21: a novel metabolic regulator with potential therapeutic properties in obesity/type 2 diabetes mellitus. Physiol. Res. 58, 1–7 (2009).
Talukdar, S. et al. A long-acting FGF21 molecule, PF-05231023, decreases body weight and improves lipid profile in non-human primates and type 2 diabetic subjects. Cell Metab. 23, 427–440 (2016).
Kim, A. M. et al. Once-weekly administration of a long-acting fibroblast growth factor 21 analogue modulates lipids, bone turnover markers, blood pressure and body weight differently in obese people with hypertriglyceridaemia and in non-human primates. Diabetes Obes. Metab. 19, 1762–1772 (2017).
Farooqi, I. S. & O’Rahilly, S. Monogenic obesity in humans. Annu. Rev. Med. 56, 443–458 (2005).
Lejeune, M. P., Hukshorn, C. J., Saris, W. H. & Westerterp-Plantenga, M. S. Effect of dietary restraint during and following pegylated recombinant leptin (PEG-OB) treatment of overweight men. Int. J. Obes. Relat. Metab. Disord. 27, 1494–1499 (2003).
Zelissen, P. M. et al. Effect of three treatment schedules of recombinant methionyl human leptin on body weight in obese adults: a randomized, placebo-controlled trial. Diabetes Obes. Metab. 7, 755–761 (2005).
Liu, D. et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin. Immunol. 9, 30 (2013).
Global, B. M. I. M. C. et al. Body-mass index and all-cause mortality: individual-participant-data meta-analysis of 239 prospective studies in four continents. Lancet 388, 776–786 (2016).
Ryan, D. H. et al. Semaglutide Effects on Cardiovascular Outcomes in People With Overweight or Obesity (SELECT) rationale and design. Am. Heart J. 229, 61–69 (2020).
Mladenka, P. et al. Comprehensive review of cardiovascular toxicity of drugs and related agents. Med. Res. Rev. 38, 1332–1403 (2018).
Colman, E. Anorectics on trial: a half century of federal regulation of prescription appetite suppressants. Ann. Intern. Med. 143, 380–385 (2005). This article gives a comprehensive overview of AOMs used in the twentieth century.
Hendricks, E. J., Greenway, F. L., Westman, E. C. & Gupta, A. K. Blood pressure and heart rate effects, weight loss and maintenance during long-term phentermine pharmacotherapy for obesity. Obesity 19, 2351–2360 (2011).
Kang, J. G., Park, C. Y., Kang, J. H., Park, Y. W. & Park, S. W. Randomized controlled trial to investigate the effects of a newly developed formulation of phentermine diffuse-controlled release for obesity. Diabetes Obes. Metab. 12, 876–882 (2010).
Hadden, D. R. & Lucey, C. Diethylpropion in the treatment of obesity. A cross-over trial of a long-acting preparation. Ulst. Med. J. 30, 109–113 (1961).
Nash, J. The treatment of obesity by control of appetite with diethylproprion (Tenuate). J. Ir. Med. Assoc. 48, 15–17 (1961).
Seaton, D. A., Duncan, L. J., Rose, K. & Scott, A. M. Diethyl-propion in the treatment of “refractory” obesity. Br. Med. J. 1, 1009–1011 (1961).
Wilson, R. & Long, C. A clinical evaluation of Tenuate — a new anti-appetite compound. J. Ir. Med. Assoc. 46, 86–88 (1960).
Cannistra, L. B., Davis, S. M. & Bauman, A. G. Valvular heart disease associated with dexfenfluramine. N. Engl. J. Med. 337, 636 (1997).
Centers for Disease Control and Prevention. Cardiac valvulopathy associated with exposure to fenfluramine or dexfenfluramine: U.S. Department of Health and Human Services interim public health recommendations, November 1997. MMWR Morb. Mortal. Wkly. Rep. 46, 1061–1066 (1997).
Connolly, H. M. et al. Valvular heart disease associated with fenfluramine–phentermine. N. Engl. J. Med. 337, 581–588 (1997).
Hayes, J. F., Bhaskaran, K., Batterham, R., Smeeth, L. & Douglas, I. The effect of sibutramine prescribing in routine clinical practice on cardiovascular outcomes: a cohort study in the United Kingdom. Int. J. Obes. 39, 1359–1364 (2015).
James, W. P. et al. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N. Engl. J. Med. 363, 905–917 (2010).
Zhou, Y. H. et al. Effect of anti-obesity drug on cardiovascular risk factors: a systematic review and meta-analysis of randomized controlled trials. PLoS ONE 7, e39062 (2012).
Seimon, R. V. et al. Changes in body weight and pulse: outcome events in overweight and obese subjects with cardiovascular disease in the SCOUT trial. Int. J. Obes. 39, 849–857 (2015).
Fujioka, K. et al. Weight loss with sibutramine improves glycaemic control and other metabolic parameters in obese patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2, 175–187 (2000).
McMahon, F. G. et al. Efficacy and safety of sibutramine in obese white and African American patients with hypertension: a 1-year, double-blind, placebo-controlled, multicenter trial. Arch. Intern. Med. 160, 2185–2191 (2000).
Wooltorton, E. Obesity drug sibutramine (Meridia): hypertension and cardiac arrhythmias. CMAJ 166, 1307–1308 (2002).
Yanovski, S. Z. & Yanovski, J. A. Obesity. N. Engl. J. Med. 346, 591–602 (2002).
Marso, S. P. et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 375, 1834–1844 (2016).
Kleinert, M. et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 14, 140–162 (2018).
Elangbam, C. S. Review paper: Current strategies in the development of anti-obesity drugs and their safety concerns. Vet. Pathol. 46, 10–24 (2009).
Arch, J. R. Horizons in the pharmacotherapy of obesity. Curr. Obes. Rep. 4, 451–459 (2015).
Srivastava, G. & Apovian, C. Future pharmacotherapy for obesity: new anti-obesity drugs on the horizon. Curr. Obes. Rep. 7, 147–161 (2018).
Wharton, S. & Serodio, K. J. Next generation of weight management medications: implications for diabetes and CVD risk. Curr. Cardiol. Rep. 17, 35 (2015).
Nauck, M. A., Quast, D. R., Wefers, J. & Meier, J. J. GLP-1 receptor agonists in the treatment of type 2 diabetes — state-of-the-art. Mol. Metab. 46, 101102 (2021). This article gives a comprehensive overview of the use of GLP1R agonists for the treatment of T2D.
Seino, Y., Rasmussen, M. F., Zdravkovic, M. & Kaku, K. Dose-dependent improvement in glycemia with once-daily liraglutide without hypoglycemia or weight gain: a double-blind, randomized, controlled trial in Japanese patients with type 2 diabetes. Diabetes Res. Clin. Pract. 81, 161–168 (2008).
Vilsboll, T. et al. Liraglutide, a long-acting human glucagon-like peptide-1 analog, given as monotherapy significantly improves glycemic control and lowers body weight without risk of hypoglycemia in patients with type 2 diabetes. Diabetes Care 30, 1608–1610 (2007).
Ponzani, P., Scardapane, M., Nicolucci, A. & Rossi, M. C. Effectiveness and safety of liraglutide after three years of treatment. Minerva Endocrinol. 41, 35–42 (2016).
Vilsboll, T., Krarup, T., Madsbad, S. & Holst, J. J. No reactive hypoglycaemia in type 2 diabetic patients after subcutaneous administration of GLP-1 and intravenous glucose. Diabet. Med. 18, 144–149 (2001).
Nauck, M. A. et al. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J. Clin. Endocrinol. Metab. 87, 1239–1246 (2002).
Goldsmith, F. et al. Induction of energy expenditure by sitagliptin is dependent on GLP-1 receptor. PLoS ONE 10, e0126177 (2015).
Gallwitz, B. Sitagliptin: profile of a novel DPP-4 inhibitor for the treatment of type 2 diabetes. Drugs Today 43, 13–25 (2007).
Huthmacher, J. A., Meier, J. J. & Nauck, M. A. Efficacy and safety of short- and long-acting glucagon-like peptide 1 receptor agonists on a background of basal insulin in type 2 diabetes: a meta-analysis. Diabetes Care 43, 2303–2312 (2020).
Pi-Sunyer, X. et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N. Engl. J. Med. 373, 11–22 (2015).
Marso, S. P. et al. Effects of liraglutide on cardiovascular outcomes in patients with diabetes with or without heart failure. J. Am. Coll. Cardiol. 75, 1128–1141 (2020).
Buckley, S. T. et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl Med. 10, eaar7047 (2018).
Nauck, M. A. et al. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Invest. 91, 301–307 (1993). This article shows that the insulinotropic effect of GIP is impaired in patients with T2D.
Chan, J. L., Roth, J. D. & Weyer, C. It takes two to tango: combined amylin/leptin agonism as a potential approach to obesity drug development. J. Investig. Med. 57, 777–783 (2009).
Roth, J. D. et al. Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc. Natl Acad. Sci. USA 105, 7257–7262 (2008).
Trevaskis, J. L. et al. Amylin-mediated restoration of leptin responsiveness in diet-induced obesity: magnitude and mechanisms. Endocrinology 149, 5679–5687 (2008).
Cavaco, M., Castanho, M. A. R. B. & Neves, V. Peptibodies: an elegant solution for a long-standing problem. Pept. Sci. 110, e23095 (2018).
Mroz, P. A. et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol. Metab. 20, 51–62 (2019).
Zhang, Q. et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. 33, 833–844.e5 (2021). This article shows that long-acting GIP decreases body weight and food intake via CNS GIPR signalling.
Adriaenssens, A. E. et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. 30, 987–996.e6 (2019). This article shows that activation of hypothalamic GIPR cells/neurons decreases food intake.
Dowsett, G. K. C. et al. A survey of the mouse hindbrain in the fed and fasted states using single-nucleus RNA sequencing. Mol. Metab. 53, 101240 (2021).
Day, J. W. et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 5, 749–757 (2009). This article shows that unimolecular GLP1/glucagon co-agonism corrects obesity in mice.
Finan, B. et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci. Transl Med. 5, 209ra151 (2013). This article shows that unimolecular GLP1/GIP co-agonism corrects obesity and glucose intolerance in obese rodents and non-human primates.
Hayes, M. R., Borner, T. & De Jonghe, B. C. The role of GIP in the regulation of GLP-1 satiety and nausea. Diabetes 70, 1956–1961 (2021). This article shows that GIP attenuates GLP1-mediated effects on emesis in rodents and shrews.
Hojberg, P. V. et al. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 52, 199–207 (2009). This article shows that an insulin-mediated decrease of hyperglycaemia restores the insulinotropic effects of GIP.
Samms, R. J., Coghlan, M. P. & Sloop, K. W. How may GIP enhance the therapeutic efficacy of GLP-1? Trends Endocrinol. Metab. 31, 410–421 (2020).
Frias, J. P. et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. 26, 343–352.e2 (2017).
Frias, J. P. et al. Efficacy and tolerability of tirzepatide, a dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist in patients with type 2 diabetes: a 12-week, randomized, double-blind, placebo-controlled study to evaluate different dose-escalation regimens. Diabetes Obes. Metab. 22, 938–946 (2020).
DiMarchi, R. D. “Let’s Stay Together”; GIP and GLP-1 dual agonism in the treatment of metabolic disease. Mol. Metab. 18, 1–2 (2018).
Min, T. & Bain, S. C. The role of tirzepatide, dual GIP and GLP-1 receptor agonist, in the management of type 2 diabetes: the SURPASS clinical trials. Diabetes Ther. 12, 143–157 (2021).
Kleinert, M., Sachs, S., Habegger, K. M., Hofmann, S. M. & Muller, T. D. Glucagon regulation of energy expenditure. Int. J. Mol. Sci. 20, 5407 (2019).
Nahra, R. et al. Effects of cotadutide on metabolic and hepatic parameters in adults with overweight or obesity and type 2 diabetes: a 54-week randomized phase 2b study. Diabetes Care 44, 1433–1442 (2021).
Eriksson, O. et al. Receptor occupancy of dual glucagon-like peptide 1/glucagon receptor agonist SAR425899 in individuals with type 2 diabetes. Sci. Rep. 10, 16758 (2020).
Tillner, J. et al. A novel dual glucagon-like peptide and glucagon receptor agonist SAR425899: results of randomized, placebo-controlled first-in-human and first-in-patient trials. Diabetes Obes. Metab. 21, 120–128 (2019).
Visentin, R. et al. Dual glucagon-like peptide-1 receptor/glucagon receptor agonist SAR425899 improves β-cell function in type 2 diabetes. Diabetes Obes. Metab. 22, 640–647 (2020).
Kleinert, M. et al. Emerging poly-agonists for obesity and type 2 diabetes. Obesity 25, 1647–1649 (2017).
Trevaskis, J. L. et al. Interaction of leptin and amylin in the long-term maintenance of weight loss in diet-induced obese rats. Obesity 18, 21–26 (2010).
Turek, V. F. et al. Mechanisms of amylin/leptin synergy in rodent models. Endocrinology 151, 143–152 (2010).
Muller, T. D. et al. Restoration of leptin responsiveness in diet-induced obese mice using an optimized leptin analog in combination with exendin-4 or FGF21. J. Pept. Sci. 18, 383–393 (2012).
Bello, N. T., Kemm, M. H., Ofeldt, E. M. & Moran, T. H. Dose combinations of exendin-4 and salmon calcitonin produce additive and synergistic reductions in food intake in nonhuman primates. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R945–R952 (2010).
Neary, N. M. et al. Peptide YY3–36 and glucagon-like peptide-17–36 inhibit food intake additively. Endocrinology 146, 5120–5127 (2005).
Trevaskis, J. L. et al. Synergistic metabolic benefits of an exenatide analogue and cholecystokinin in diet-induced obese and leptin-deficient rodents. Diabetes Obes. Metab. 17, 61–73 (2015).
Clemmensen, C. et al. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol. Med. 7, 288–298 (2015).
Finan, B. et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 21, 27–36 (2015).
Tschop, M. H. et al. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab. 24, 51–62 (2016).
Coskun, T. et al. The novel GIP, GLP-1, and glucagon triple receptor agonist LY3437943 exhibits robust efficacy in preclinical models of obesity and diabetes. In American Diabetes Association 81st Scientific Sessions (American Diabetes Association, 2021).
Hebebrand, J., Muller, T. D., Holtkamp, K. & Herpertz-Dahlmann, B. The role of leptin in anorexia nervosa: clinical implications. Mol. Psychiatry 12, 23–35 (2007).
Chou, S. H. et al. Leptin is an effective treatment for hypothalamic amenorrhea. Proc. Natl Acad. Sci. USA 108, 6585–6590 (2011).
Welt, C. K. et al. Recombinant human leptin in women with hypothalamic amenorrhea. N. Engl. J. Med. 351, 987–997 (2004).
Chehab, F. F., Lim, M. E. & Lu, R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat. Genet. 12, 318–320 (1996).
Licinio, J. et al. Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc. Natl Acad. Sci. USA 101, 4531–4536 (2004).
Milos, G. et al. Short-term metreleptin treatment of patients with anorexia nervosa: rapid on-set of beneficial cognitive, emotional, and behavioral effects. Transl. Psychiatry 10, 303 (2020).
Chan, J. L. et al. Immunogenicity associated with metreleptin treatment in patients with obesity or lipodystrophy. Clin. Endocrinol. 85, 137–149 (2016).
Ravussin, E. et al. Enhanced weight loss with pramlintide/metreleptin: an integrated neurohormonal approach to obesity pharmacotherapy. Obesity 17, 1736–1743 (2009).
Clemmensen, C. et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes 63, 1422–1427 (2014).
Liu, J., Lee, J., Salazar Hernandez, M. A., Mazitschek, R. & Ozcan, U. Treatment of obesity with celastrol. Cell 161, 999–1011 (2015).
Lee, J. et al. Withaferin A is a leptin sensitizer with strong antidiabetic properties in mice. Nat. Med. 22, 1023–1032 (2016).
Sharma, S. et al. Current mechanistic and pharmacodynamic understanding of melanocortin-4 receptor activation. Molecules 24, 1892 (2019).
Van der Ploeg, L. H. et al. A role for the melanocortin 4 receptor in sexual function. Proc. Natl Acad. Sci. USA 99, 11381–11386 (2002).
Collet, T. H. et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (setmelanotide) in MC4R deficiency. Mol. Metab. 6, 1321–1329 (2017).
Kievit, P. et al. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes 62, 490–497 (2013).
Kuhnen, P. et al. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N. Engl. J. Med. 375, 240–246 (2016).
Clement, K. et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat. Med. 24, 551–555 (2018).
Clement, K. et al. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: single-arm, open-label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 8, 960–970 (2020).
Lutz, T. A. Control of food intake and energy expenditure by amylin — therapeutic implications. Int. J. Obes. 33 (Suppl. 1), S24–S27 (2009).
Young, A. & Denaro, M. Roles of amylin in diabetes and in regulation of nutrient load. Nutrition 14, 524–527 (1998).
Boyle, C. N., Lutz, T. A. & Le Foll, C. Amylin — its role in the homeostatic and hedonic control of eating and recent developments of amylin analogs to treat obesity. Mol. Metab. 8, 203–210 (2018).
Ling, W., Huang, Y. M., Qiao, Y. C., Zhang, X. X. & Zhao, H. L. Human amylin: from pathology to physiology and pharmacology. Curr. Protein Pept. Sci. 20, 944–957 (2019).
Bower, R. L. & Hay, D. L. Amylin structure–function relationships and receptor pharmacology: implications for amylin mimetic drug development. Br. J. Pharmacol. 173, 1883–1898 (2016).
Hollander, P. et al. Effect of pramlintide on weight in overweight and obese insulin-treated type 2 diabetes patients. Obes. Res. 12, 661–668 (2004).
Srivastava, G. & Apovian, C. M. Current pharmacotherapy for obesity. Nat. Rev. Endocrinol. 14, 12–24 (2018).
Gydesen, S. et al. Optimization of tolerability and efficacy of the novel dual amylin and calcitonin receptor agonist KBP-089 through dose escalation and combination with a GLP-1 analog. Am. J. Physiol. Endocrinol. Metab. 313, E598–E607 (2017).
Bailey, R. J. et al. Pharmacological characterization of rat amylin receptors: implications for the identification of amylin receptor subtypes. Br. J. Pharmacol. 166, 151–167 (2012).
Gydesen, S. et al. KBP-088, a novel DACRA with prolonged receptor activation, is superior to davalintide in terms of efficacy on body weight. Am. J. Physiol. Endocrinol. Metab. 310, E821–E827 (2016).
Hjuler, S. T., Andreassen, K. V., Gydesen, S., Karsdal, M. A. & Henriksen, K. KBP-042 improves bodyweight and glucose homeostasis with indices of increased insulin sensitivity irrespective of route of administration. Eur. J. Pharmacol. 762, 229–238 (2015).
Mack, C. M. et al. Davalintide (AC2307), a novel amylin-mimetic peptide: enhanced pharmacological properties over native amylin to reduce food intake and body weight. Int. J. Obes. 34, 385–395 (2010).
Enebo, L. B. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2.4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet 397, 1736–1748 (2021).
Tschop, M., Smiley, D. L. & Heiman, M. L. Ghrelin induces adiposity in rodents. Nature 407, 908–913 (2000).
Muller, T. D. et al. Ghrelin. Mol. Metab. 4, 437–460 (2015).
Shearman, L. P. et al. Ghrelin neutralization by a ribonucleic acid-SPM ameliorates obesity in diet-induced obese mice. Endocrinology 147, 1517–1526 (2006).
Barnett, B. P. et al. Glucose and weight control in mice with a designed ghrelin O-acyltransferase inhibitor. Science 330, 1689–1692 (2010).
Kjeldsen-Kragh, J., Hvatum, M., Haugen, M., Forre, O. & Scott, H. Antibodies against dietary antigens in rheumatoid arthritis patients treated with fasting and a one-year vegetarian diet. Clin. Exp. Rheumatol. 13, 167–172 (1995).
Haqq, A. M. et al. Ghrelin concentrations in Prader–Willi syndrome (PWS) infants and children: changes during development. Clin. Endocrinol. 69, 911–920 (2008).
Allas, S. et al. AZP-531, an unacylated ghrelin analog, improves food-related behavior in patients with Prader–Willi syndrome: a randomized placebo-controlled trial. PLoS ONE 13, e0190849 (2018).
Sun, Y., Ahmed, S. & Smith, R. G. Deletion of ghrelin impairs neither growth nor appetite. Mol. Cell Biol. 23, 7973–7981 (2003).
Sun, Y., Butte, N. F., Garcia, J. M. & Smith, R. G. Characterization of adult ghrelin and ghrelin receptor knockout mice under positive and negative energy balance. Endocrinology 149, 843–850 (2008).
Mani, B. K. et al. LEAP2 changes with body mass and food intake in humans and mice. J. Clin. Invest. 129, 3909–3923 (2019).
Zorrilla, E. P. et al. Vaccination against weight gain. Proc. Natl Acad. Sci. USA 103, 13226–13231 (2006).
Patterson, M., Bloom, S. R. & Gardiner, J. V. Ghrelin and appetite control in humans — potential application in the treatment of obesity. Peptides 32, 2290–2294 (2011).
Lu, S. C. et al. An acyl-ghrelin-specific neutralizing antibody inhibits the acute ghrelin-mediated orexigenic effects in mice. Mol. Pharmacol. 75, 901–907 (2009).
Cannon, B. & Nedergaard, J. Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359 (2004).
Xu, Z., Liu, J., You, W., Wang, Y. & Shan, T. Cold exposure induces nuclear translocation of CRTC3 in brown adipose tissue. J. Cell Biochem. 120, 9138–9146 (2019).
Cao, W. et al. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell Biol. 24, 3057–3067 (2004).
Villarroya, F., Peyrou, M. & Giralt, M. Transcriptional regulation of the uncoupling protein-1 gene. Biochimie 134, 86–92 (2017).
Grundlingh, J., Dargan, P. I., El-Zanfaly, M. & Wood, D. M. 2,4-dinitrophenol (DNP): a weight loss agent with significant acute toxicity and risk of death. J. Med. Toxicol. 7, 205–212 (2011).
Demine, S., Renard, P. & Arnould, T. Mitochondrial uncoupling: a key controller of biological processes in physiology and diseases. Cells 8, 795 (2019).
Geisler, J. G. 2,4 Dinitrophenol as medicine. Cells 8, 280 (2019).
Perry, R. J., Zhang, D., Zhang, X. M., Boyer, J. L. & Shulman, G. I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256 (2015).
Axelrod, C. L. et al. BAM15-mediated mitochondrial uncoupling protects against obesity and improves glycemic control. EMBO Mol. Med. 12, e12088 (2020).
Alexopoulos, S. J. et al. Mitochondrial uncoupler BAM15 reverses diet-induced obesity and insulin resistance in mice. Nat. Commun. 11, 2397 (2020).
Hale, C. & Veniant, M. M. Growth differentiation factor 15 as a potential therapeutic for treating obesity. Mol. Metab. 46, 101117 (2021).
Assadi, A., Zahabi, A. & Hart, R. A. GDF15, an update of the physiological and pathological roles it plays: a review. Pflug. Arch. 472, 1535–1546 (2020).
Kempf, T. et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat. Med. 17, 581–588 (2011).
Mullican, S. E. et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 23, 1150–1157 (2017).
Borner, T. et al. GDF15 induces anorexia through nausea and emesis. Cell Metab. 31, 351–362.e5 (2020).
Borner, T. et al. GDF15 induces an aversive visceral malaise state that drives anorexia and weight loss. Cell Rep. 31, 107543 (2020).
Strelau, J. et al. Progressive postnatal motoneuron loss in mice lacking GDF-15. J. Neurosci. 29, 13640–13648 (2009).
Tsai, V. W. W., Husaini, Y., Sainsbury, A., Brown, D. A. & Breit, S. N. The MIC-1/GDF15–GFRAL pathway in energy homeostasis: implications for obesity, cachexia, and other associated diseases. Cell Metab. 28, 353–368 (2018).
Baek, S. J. et al. Nonsteroidal anti-inflammatory drug-activated gene-1 over expression in transgenic mice suppresses intestinal neoplasia. Gastroenterology 131, 1553–1560 (2006).
Bagdade, J. D., Bierman, E. L. & Porte, D. Jr. The significance of basal insulin levels in the evaluation of the insulin response to glucose in diabetic and nondiabetic subjects. J. Clin. Invest. 46, 1549–1557 (1967).
Stadlbauer, U., Woods, S. C., Langhans, W. & Meyer, U. PYY3-36: beyond food intake. Front. Neuroendocrinol. 38, 1–11 (2015).
Batterham, R. L. et al. Gut hormone PYY(3–36) physiologically inhibits food intake. Nature 418, 650–654 (2002).
Challis, B. G. et al. Acute effects of PYY3–36 on food intake and hypothalamic neuropeptide expression in the mouse. Biochem. Biophys. Res. Commun. 311, 915–919 (2003).
Halatchev, I. G., Ellacott, K. L., Fan, W. & Cone, R. D. Peptide YY3–36 inhibits food intake in mice through a melanocortin-4 receptor-independent mechanism. Endocrinology 145, 2585–2590 (2004).
Batterham, R. L. et al. Inhibition of food intake in obese subjects by peptide YY3–36. N. Engl. J. Med. 349, 941–948 (2003). This article shows how PYY affects energy metabolism in humans.
Degen, L. et al. Effect of peptide YY3–36 on food intake in humans. Gastroenterology 129, 1430–1436 (2005).
Poulsen, C. et al. Rational development of stable PYY3–36 peptide Y2 receptor agonists. Pharm. Res. 8, 1369–1385 (2021).
Pratley, R. E. et al. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. 6, 275–286 (2018).
Carvajal, A., Garcia del Pozo, J., Martin de Diego, I., Rueda de Castro, A. M. & Velasco, A. Efficacy of fenfluramine and dexfenfluramine in the treatment of obesity: a meta-analysis. Methods Find. Exp. Clin. Pharmacol. 22, 285–290 (2000).
Aronne, L. J. et al. Evaluation of phentermine and topiramate versus phentermine/topiramate extended-release in obese adults. Obesity 21, 2163–2171 (2013).
Cohen, P. A., Goday, A. & Swann, J. P. The return of rainbow diet pills. Am. J. Public. Health 102, 1676–1686 (2012).
Van Gaal, L., Pi-Sunyer, X., Despres, J. P., McCarthy, C. & Scheen, A. Efficacy and safety of rimonabant for improvement of multiple cardiometabolic risk factors in overweight/obese patients: pooled 1-year data from the Rimonabant in Obesity (RIO) program. Diabetes Care 31, S229–240 (2008).
Sjostrom, L. et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet 352, 167–172 (1998).
Gadde, K. M. et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet 377, 1341–1352 (2011).
Garvey, W. T. et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am. J. Clin. Nutr. 95, 297–308 (2012).
Greenway, F. L. et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 376, 595–605 (2010).
Pires, N. et al. Safety and pharmacokinetics of withaferin-A in advanced stage high grade osteosarcoma: a phase I trial. J. Ayurveda Integr. Med. 11, 68–72 (2020).
Cascao, R., Fonseca, J. E. & Moita, L. F. Celastrol: a spectrum of treatment opportunities in chronic diseases. Front. Med. 4, 69 (2017).
Clapper, J. R. et al. Effects of amylin and bupropion/naltrexone on food intake and body weight are interactive in rodent models. Eur. J. Pharmacol. 698, 292–298 (2013).
Ji, W. et al. Effects of canagliflozin on weight loss in high-fat diet-induced obese mice. PLoS ONE 12, e0179960 (2017).
Thomsen, W. J. et al. Lorcaserin, a novel selective human 5-hydroxytryptamine2C agonist: in vitro and in vivo pharmacological characterization. J. Pharmacol. Exp. Ther. 325, 577–587 (2008).
Hansen, H. H. et al. The novel triple monoamine reuptake inhibitor tesofensine induces sustained weight loss and improves glycemic control in the diet-induced obese rat: comparison to sibutramine and rimonabant. Eur. J. Pharmacol. 636, 88–95 (2010).
Simonds, S. E. et al. Determining the effects of combined liraglutide and phentermine on metabolic parameters, blood pressure, and heart rate in lean and obese male mice. Diabetes 68, 683–695 (2019).
Andermann, M. L. & Lowell, B. B. Toward a wiring diagram understanding of appetite control. Neuron 95, 757–778 (2017).
Lutter, M. & Nestler, E. J. Homeostatic and hedonic signals interact in the regulation of food intake. J. Nutr. 139, 629–632 (2009).
Skibicka, K. P., Hansson, C., Alvarez-Crespo, M., Friberg, P. A. & Dickson, S. L. Ghrelin directly targets the ventral tegmental area to increase food motivation. Neuroscience 180, 129–137 (2011).
Woods, S. C., Lotter, E. C., McKay, L. D. & Porte, D. Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282, 503–505 (1979). This article shows how central insulin action controls food intake and body weight.
Woods, S. C. & Porte, D. Jr. in Hunger: Basic Mechanisms and Clinical Implications (eds Novin, D., Bray, G. A. & Wyrwichka, W.) 273–280 (Raven, 1976).
Becskei, C., Grabler, V., Edwards, G. L., Riediger, T. & Lutz, T. A. Lesion of the lateral parabrachial nucleus attenuates the anorectic effect of peripheral amylin and CCK. Brain Res. 1162, 76–84 (2007).
Lutz, T. A. et al. Lesion of the area postrema/nucleus of the solitary tract (AP/NTS) attenuates the anorectic effects of amylin and calcitonin gene-related peptide (CGRP) in rats. Peptides 19, 309–317 (1998).
Riediger, T., Schmid, H. A., Lutz, T. & Simon, E. Amylin potently activates AP neurons possibly via formation of the excitatory second messenger cGMP. Am. J. Physiol. Regulatory, Integr. Comp. Physiol. 281, R1833–R1843 (2001).
Riediger, T., Zuend, D., Becskei, C. & Lutz, T. A. The anorectic hormone amylin contributes to feeding-related changes of neuronal activity in key structures of the gut–brain axis. Am. J. Physiol. Regulatory, Integr. Comp. Physiol. 286, R114–R122 (2004).
Mietlicki-Baase, E. G. et al. Amylin modulates the mesolimbic dopamine system to control energy balance. Neuropsychopharmacology 40, 372–385 (2015).
Coskun, T. et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 149, 6018–6027 (2008).
Kharitonenkov, A. et al. FGF-21 as a novel metabolic regulator. J. Clin. Invest. 115, 1627–1635 (2005). This article shows how FGF21 regulates systemic energy metabolism.
Sarruf, D. A. et al. Fibroblast growth factor 21 action in the brain increases energy expenditure and insulin sensitivity in obese rats. Diabetes 59, 1817–1824 (2010).
Chau, M. D., Gao, J., Yang, Q., Wu, Z. & Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK–SIRT1–PGC-1α pathway. Proc. Natl Acad. Sci. USA 107, 12553–12558 (2010).
Gibbs, J., Young, R. C. & Smith, G. P. Cholecystokinin decreases food intake in rats. J. Comp. physiological Psychol. 84, 488–495 (1973).
Kissileff, H. R., Pi-Sunyer, F. X., Thornton, J. & Smith, G. P. C-terminal octapeptide of cholecystokinin decreases food intake in man. Am. J. Clin. Nutr. 34, 154–160 (1981).
Muurahainen, N., Kissileff, H. R., Derogatis, A. J. & Pi-Sunyer, F. X. Effects of cholecystokinin-octapeptide (CCK-8) on food intake and gastric emptying in man. Physiol. Behav. 44, 645–649 (1988).
Moran, T. H., Baldessarini, A. R., Salorio, C. F., Lowery, T. & Schwartz, G. J. Vagal afferent and efferent contributions to the inhibition of food intake by cholecystokinin. Am. J. Physiol. 272, R1245–R1251 (1997).
Moran, T. H., Robinson, P. H., Goldrich, M. S. & McHugh, P. R. Two brain cholecystokinin receptors: implications for behavioral actions. Brain Res. 362, 175–179 (1986).
Alhadeff, A. L., Rupprecht, L. E. & Hayes, M. R. GLP-1 neurons in the nucleus of the solitary tract project directly to the ventral tegmental area and nucleus accumbens to control for food intake. Endocrinology 153, 647–658 (2012).
Dickson, S. L. et al. The glucagon-like peptide 1 (GLP-1) analogue, exendin-4, decreases the rewarding value of food: a new role for mesolimbic GLP-1 receptors. J. Neurosci. 32, 4812–4820 (2012).
Dossat, A. M., Lilly, N., Kay, K. & Williams, D. L. Glucagon-like peptide 1 receptors in nucleus accumbens affect food intake. J. Neurosci. 31, 14453–14457 (2011).
Terrill, S. J. et al. Role of lateral septum glucagon-like peptide 1 receptors in food intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 311, R124–R132 (2016).
Muller, T. D., Finan, B., Clemmensen, C., DiMarchi, R. D. & Tschop, M. H. The new biology and pharmacology of glucagon. Physiol. Rev. 97, 721–766 (2017).
Weintraub, M. et al. Long-term weight control study. V (weeks 190 to 210). Follow-up of participants after cessation of medication. Clin. Pharmacol. Ther. 51, 615–618 (1992).
Weintraub, M. et al. Long-term weight control study. IV (weeks 156 to 190). The second double-blind phase. Clin. Pharmacol. Ther. 51, 608–614 (1992).
Weintraub, M., Sundaresan, P. R., Schuster, B., Moscucci, M. & Stein, E. C. Long-term weight control study. III (weeks 104 to 156). An open-label study of dose adjustment of fenfluramine and phentermine. Clin. Pharmacol. Ther. 51, 602–607 (1992).
Cambon, D. & Leclercq, F. Clinical and echographic characteristics of patients exposed to fenfluramin or its derivatives: results of a prospective, single-centre, observational study. Arch. Cardiovasc. Dis. 108, 172–180 (2015).
Kurz, X. & Van Ermen, A. Valvular heart disease associated with fenfluramine–phentermine. N. Engl. J. Med. 337, 1772–1773; author reply 1775 (1997).
Rasmussen, S., Corya, B. C. & Glassman, R. D. Valvular heart disease associated with fenfluramine–phentermine. N. Engl. J. Med. 337, 1773; author reply 1775 (1997).
Astrup, A. et al. Effect of tesofensine on bodyweight loss, body composition, and quality of life in obese patients: a randomised, double-blind, placebo-controlled trial. Lancet 372, 1906–1913 (2008).
Maggioni, A. P. et al. Relation between weight loss and causes of death in patients with cardiovascular disease: finding from the SCOUT trial. J. Cardiovasc. Med. 18, 144–151 (2017).
Harrison-Woolrych, M., Ashton, J. & Herbison, P. Fatal and non-fatal cardiovascular events in a general population prescribed sibutramine in New Zealand: a prospective cohort study. Drug. Saf. 33, 605–613 (2010).
Horn, H., Bohme, B., Dietrich, L. & Koch, M. Endocannabinoids in body weight control. Pharmaceuticals 11, 55 (2018).
Taylor, D. Withdrawal of rimonabant — walking the tightrope of 21st century pharmaceutical regulation? Curr. Drug. Saf. 4, 2–4 (2009).
Acknowledgements
This work was supported, in part, by funding from the Alexander von Humboldt Foundation, the European Research Council (ERC) AdG HypoFlam no. 695054 and the German Center for Diabetes Research (DZD e.V.). T.D.M. further received research funding from the German Research Foundation (TRR152 and TRR296). Figures were made using material provided by either Motifolio or Servier Medical Art (Servier), the latter under consideration of a Creative Commons Attribution 3.0 Unported License.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
M.H.T. is a member of the scientific advisory board of ERX Pharmaceuticals, Cambridge, MA, USA; was a member of the Research Cluster Advisory Panel (ReCAP) of the Novo Nordisk Foundation between 2017 and 2019; attended a scientific advisory board meeting of the Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, in 2016; received funding for his research projects from Novo Nordisk (2016–2020) and Sanofi-Aventis (2012–2019); was a consultant for Bionorica SE (2013–2017), Menarini Ricerche S.p.A. (2016) and Bayer Pharma AG Berlin (2016); and, as former Director of the Helmholtz Diabetes Center and the Institute for Diabetes and Obesity at Helmholtz Zentrum München (2011–2018), and, since 2018, as CEO of Helmholtz Zentrum München, has been responsible for collaborations with a multitude of companies and institutions worldwide — in this capacity, he discussed potential projects with and has signed/signs contracts for his institute(s) and for the staff for research funding and/or collaborations with industry and academia worldwide, including but not limited to pharmaceutical corporations such as Boehringer Ingelheim, Eli Lilly, Novo Nordisk, Medigene, Arbormed, BioSyngen and others; in this role, was/is further responsible for commercial technology transfer activities of his institute(s), including diabetes-related patent portfolios of Helmholtz Zentrum München as, for example, WO/2016/188932 A2 or WO/2017/194499 A1; and confirms that, to the best of his knowledge, none of the above funding sources were involved in the preparation of this paper. M.B. received honoraria as a consultant and speaker from Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Lilly, Novo Nordisk, Novartis, Pfizer and Sanofi. T.D.M. receives research funding from Novo Nordisk, but these funds are unrelated to the work here described, and further received speaking fees from Eli Lilly, Novo Nordisk, Mercodia, AstraZeneca, Berlin Chemie and Sanofi-Aventis. R.D.D. is a co-inventor on intellectual property owned by Indiana University and licensed to Novo Nordisk, and was recently employed by Novo Nordisk and, previously, Lilly Research Laboratories.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Amylin and Takeda Discontinue Development of Pramlintide/Metreleptin Combination Treatment for Obesity: https://www.takeda.com/en-us/newsroom/news-releases/2011/amylin-and-takeda-discontinue-development-of-pramlintidemetreleptin-combination-treatment-for-obesity-following-commercial-reassessment-of-the-program/
Apidex prescribing information: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/085128s065lbl.pdf
Centers for Disease Control and Prevention — Adult Obesity Facts: https://www.cdc.gov/obesity/data/adult.html
Center for Disease Control and Prevention — Childhood Obesity Facts: https://www.cdc.gov/obesity/data/childhood.html
Davalintide completed Phase 2 Trials for Obesity: https://go.drugbank.com/drugs/DB14956/clinical_trials?conditions=DBCOND0023883%2CDBCOND0023881&phase=2&purpose=treatment&status=completed&__cf_chl_jschl_tk__=pmd_a94RcAw_79CQDm7dLpCrob5g4UpoTRCdTnCA21OADus-1633088489-0-gqNtZGzNAqWjcnBszQiR
Eli Lilly — Medicines in Development: https://www.lilly.com/discovery/clinical-development-pipeline
FDA Approves New Drug Treatment for Chronic Weight Management, First Since 2014: https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-treatment-chronic-weight-management-first-2014
FDA approves weight management drug for patients aged 12 and older: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-weight-management-drug-patients-aged-12-and-older
FDA requests the withdrawal of the weight-loss drug Belviq, Belviq XR (lorcaserin) from the market: https://www.fda.gov/drugs/fda-drug-safety-podcasts/fda-requests-withdrawal-weight-loss-drug-belviq-belviq-xr-lorcaserin-market
National Institute of Health — Overweight & Obesity Statistics: https://www.niddk.nih.gov/health-information/health-statistics/overweight-obesity
Novo Nordisk axes experimental obesity drugs after success with late-stage meds: https://www.fiercebiotech.com/biotech/novo-nordisk-cans-early-experimental-obesity-tests-after-success-later-stage-meds
Obesity vaccine enters clinical trial: https://newatlas.com/obesity-vaccine-enters-clinical-trial/4052/
Public Health England (PHE) supports FSA warnings over deadly weight loss supplement: https://www.gov.uk/government/news/phe-supports-fsa-warnings-over-deadly-weight-loss-supplement
Research programme: obesity therapy — NOXXON Pharma: https://adisinsight.springer.com/drugs/800022779
Tenuate prescribing information: https://www.accessdata.fda.gov/drugsatfda_ docs/label/2004/11722s029,12546s032lbl.pdf
World Health Organization (WHO) — Obesity and overweight: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight
World Health Organization — Obesity: https://www.who.int/news-room/facts-in-pictures/detail/6-facts-on-obesity
Zealand Pharma — Significant pipeline evolution and a commitment to continue to deliver: https://static1.squarespace.com/static/58983777d1758e28995640b4/t/604784234098312195a1215c/1615299619955/Zealand+Pipeline+slide+3.8.21.png
Glossary
- Obesity
-
Excess body fat mass as a result of a chronic surplus of energy intake over energy expenditure.
- Diabetes
-
Chronically elevated blood glucose as a result of insufficient action or production of insulin.
- Anti-obesity medications
-
(AOMs). Pharmacotherapy aiming to decrease excess body fat.
- Incretin
-
An intestinal peptide that stimulates the release of insulin.
- Poly-agonists
-
Unimolecular peptides capable of acting on multiple receptors.
Rights and permissions
About this article

Cite this article
Müller, T.D., Blüher, M., Tschöp, M.H. et al. Anti-obesity drug discovery: advances and challenges. Nat Rev Drug Discov 21, 201–223 (2022). https://doi.org/10.1038/s41573-021-00337-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-021-00337-8
This article is cited by
-
Electro-acupuncture for central obesity: a patient-assessor blinded, randomized sham-controlled clinical trial
BMC Complementary Medicine and Therapies (2024)
-
Facile adipocyte uptake and liver/adipose tissue delivery of conjugated linoleic acid-loaded tocol nanocarriers for a synergistic anti-adipogenesis effect
Journal of Nanobiotechnology (2024)
-
The impact of selected xanthophylls on oil hydrolysis by pancreatic lipase: in silico and in vitro studies
Scientific Reports (2024)
-
The emergence of obesity in type 1 diabetes
International Journal of Obesity (2024)
-
Common and ethnic-specific derangements in skeletal muscle transcriptome associated with obesity
International Journal of Obesity (2024)

{kind=link}