
Abstract
Cholangiocarcinoma (CCA) is a highly lethal adenocarcinoma of the hepatobiliary system, which can be classified as intrahepatic, perihilar and distal. Each anatomic subtype has distinct genetic aberrations, clinical presentations and therapeutic approaches. In endemic regions, liver fluke infection is associated with CCA, owing to the oncogenic effect of the associated chronic biliary tract inflammation. In other regions, CCA can be associated with chronic biliary tract inflammation owing to choledocholithiasis, cholelithiasis, or primary sclerosing cholangitis, but most CCAs have no identifiable cause. Administration of the anthelmintic drug praziquantel decreases the risk of CCA from liver flukes, but reinfection is common and future vaccination strategies may be more effective. Some patients with CCA are eligible for potentially curative surgical options, such as resection or liver transplantation. Genetic studies have provided new insights into the pathogenesis of CCA, and two aberrations that drive the pathogenesis of non-fluke-associated intrahepatic CCA, fibroblast growth factor receptor 2 fusions and isocitrate dehydrogenase gain-of-function mutations, can be therapeutically targeted. CCA is a highly desmoplastic cancer and targeting the tumour immune microenvironment might be a promising therapeutic approach. CCA remains a highly lethal disease and further scientific and clinical insights are needed to improve patient outcomes.
Similar content being viewed by others
Introduction
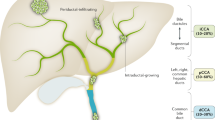
Cholangiocarcinoma (CCA) is a highly lethal, epithelial cell malignancy that occurs anywhere along the biliary tree and/or within the hepatic parenchyma. CCA displays features of cholangiocyte differentiation and probably arises predominantly from the epithelial cells lining the bile ducts, which are termed cholangiocytes; however, the cancers may also develop from peribiliary glands and hepatocytes, depending on the underlying liver disease and location1,2,3,4. These cancers are heterogeneous and are best classified according to the primary, anatomic subtype as intrahepatic CCA (iCCA), perihilar CCA (pCCA) or distal CCA (dCCA)5,6 (Fig. 1). iCCA is located proximally to the second-order bile ducts within the liver parenchyma, pCCA is localized between the second-order bile ducts and the insertion of the cystic duct into the common bile duct, and dCCA is confined to the common bile duct below the cystic duct insertion. The true incidence of pCCA and iCCA is unclear owing to the extensive misclassification of pCCA as iCCA in national databases6,7. In addition, enhanced diagnostic capabilities have enabled increased clinical distinction between carcinoma of unknown primary and iCCA8,9. These factors have, in part, contributed to the reported increase in incidence of iCCA over the past two or three decades. Each of the anatomic subtypes is characterized by unique genetic aberrations, clinical presentations and management options10. However, many databases categorize both pCCA and dCCA as extrahepatic CCA. Most CCAs are adenocarcinomas and other histological subtypes, such as adenosquamous carcinoma or clear cell carcinoma, are encountered rarely11. These cancers are highly desmoplastic and are enmeshed in dense networks of inflammatory cells and matrix termed the tumour immune microenvironment12,13,14.

Cholangiocarcinoma (CCA) is best classified according to the primary, anatomic subtype as intrahepatic CCA (iCCA), perihilar CCA (pCCA) and distal CCA (dCCA). iCCA is located proximally to the second-order bile ducts within the liver parenchyma. pCCA is localized between the second-order bile ducts and the insertion of the cystic duct into the common bile duct. dCCA is confined to the common bile duct below the cystic duct insertion.
The epidemiology of these cancers varies worldwide. Infections with specific trematodes (flatworm parasites, commonly called flukes) are a major cause of CCA in some regions. For example, in Southeast Asia, the liver fluke Opisthorchis viverrini is the leading cause of CCA15. CCA occurring secondary to fluke infestation can arise anywhere within the biliary tree and present as any one of the three anatomic subsets. Fluke-related CCA may have a specific pathogenesis, especially genetic aberrations, but the diagnosis and management are not different from non-fluke-related CCA. In the Western world, most patients with CCA do not have an identifiable risk factor, except for some with primary sclerosing cholangitis (PSC)7,10. Further insights into the epidemiology, risk factors and biology of CCA are needed to improve its prevention and therapy.
In this Primer, we discuss the epidemiology and pathophysiological mechanisms of liver-fluke-related and non-liver-fluke-related CCA and associated risk factors and summarize diagnosis and management of CCA depending on aetiology and anatomic subtype. We highlight the patient experience and future directions for control and treatment of this disease. Gallbladder carcinoma and ampullary carcinoma are not discussed in this Primer, owing to differing pathogenesis, treatment options and prognosis compared with CCA.
Epidemiology
The epidemiology of CCA is diverse across the globe, and it is likely that further subsets will be identified in the future. Currently, liver flukes are a major risk factor in Southeast Asia, whereas the risk factors outside this region are largely unknown. Thus, the epidemiology of CCA can be discussed as fluke-related and non-fluke-related CCA.
Fluke-related CCA
Infections with specific trematodes are a major cause of CCA in some regions, and these flatworm parasites are group 1 biological carcinogens, that is, definite causes of cancer, including several fish-borne liver flukes of the Opisthorchiidae family16. The importance of fish-borne parasites Clonorchis sinensis, O. viverrini and Opisthorchis felineus to human health is due to their infection-associated morbidity and risk of CCA16,17,18,19. Among them, only C. sinensis and O. viverrini have so far been shown to cause CCA in animal studies20. The incidence of CCA associated with liver fluke infection, calculated as an age‐standardized rate, varies by geographical region and other risk factors but has exceeded 100 per 100,000 in men and 40 per 100,000 in women in hotspots in northeast Thailand21. At least 700 million people are at risk of infection with these liver flukes16,17,19,21,22,23. Infection follows the consumption of undercooked freshwater cyprinid fish carrying the larval parasite, termed the metacercaria24,25,26. The adult liver flukes inhabit the biliary tract, from where the parasite eggs are shed into the bile and passed with the faeces to the external environment. The life cycle includes an obligate intermediate host snail, the freshwater fish and the human host. Following ingestion of infected fish flesh, gastric and intestinal juices digest the encysted metacercariae, whereupon excysted juvenile flukes migrate through the ampulla of Vater into the common bile duct and into the intrahepatic bile ducts. Here they mature, reproduce sexually, and can live for many years27.
Infection caused by C. sinensis (clonorchiasis) is endemic on the Korean peninsula (particularly along the drainage of the major southern rivers, including Nakdong-gang, Seomjin-gang, Youngsan-gang and Geum-gang); in far-eastern Russia; in Taiwan and several regions of the Chinese mainland (including Liaoning, Jilin and Heilongjiang in the northeast, and Guangdong, Guanxi, Hunan and Sichuan in the south); in northern Vietnam; and occasionally in Japan18,27,28,29,30,31,32. Infection with O. felineus (opisthorchiasis) is prevalent in western Siberia, including Surgut, Tyumen and Tomsk oblast, and Kazakhstan along the drainage of the Ob and Irtysh rivers25,26,33. As many as 100 million people are at risk of infection with O. viverrini in the lower Mekong River drainage, with ~10 million infected in the northern and Isan region of Thailand (including Roi Et, Yasothon, Nakhon Phanom, Mukdahan, Nong Khai and Khon Kaen, and provinces that are drained by the Mun River and Chi River), and 2 million infected in the Lao People’s Democratic Republic (mainly in the central and southern provinces of Khammouane, Savannakhet and Champasack); many infections also occur in southern Vietnam, Cambodia and Myanmar34,35,36,37.
Non-fluke-related CCA
In most countries, liver flukes are not endemic and CCA is a relatively rare malignancy; however, incidence and mortality rates have been consistently increasing in the past few decades. Multiple studies have shown a consistent pattern of rising rates of iCCA and stable or decreasing rates of pCCA and/or dCCA7,38,39,40,41,42,43,44,45. The most recent and comprehensive study examined WHO and Pan American Health Organization databases of 32 countries in Europe, the American continents, Asia and Oceania between 1995 and 2016 (ref.40). Overall, global age-standardized mortality rates (ASMRs) for iCCA increased. The highest incremental changes occurred in Norway, Croatia, Denmark, the UK, Germany and Portugal among European countries; in Argentina, Chile and Brazil among American countries; and Australia. ASMRs for iCCA in men were 1–2 per 100,000 in most countries, with the highest rates in Hong Kong (2.5 per 100,000), followed by France, Austria, Spain, the UK and Australia (1.5–1.8 per 100,000). By contrast, ASMRs for pCCA and/or dCCA decreased in most countries. Between 2010 and 2014, ASMRs for pCCA and/or dCCA were <1 per 100,000 men in all countries, except Japan (2.8 per 100,000 men). The lowest ASMRs (<0.1 per 100,000) were observed in central and northern European regions, Canada, Latin American regions (Mexico, Chile, Brazil, Venezuela and Argentina), Israel and Australia. These patterns were similar to those in women but differences in the ASMRs in men and women between countries were notable. Overall CCA mortality is higher in older patients than in younger patients, in men than in women, and in Asian countries than in Western countries40,41,42. In countries with different large ethnic groups, mortality rates can vary between them. For example, in the USA, the largest increases in mortality between 2004 and 2014 occurred in individuals of African–American descent (45%), whereas the increases were 22% in those of Asian descent and 20% in white individuals46.
Controversy around iCCA and pCCA
Interpretation of rising iCCA and declining pCCA and/or dCCA rates is hampered by the fact that historical versions of the International Classification of Diseases (ICD) did not include a separate code for pCCA, and previous versions of ICD–Oncology (ICD–O) cross-referenced pCCA to iCCA7,45,47. In addition, studies refer to Klatskin tumour, hilar CCA or perihilar CCA — different terms referring to the same fundamental entity. Some have suggested that this subgroup of CCA accounts for only 1–7% of all CCAs, but this is a much smaller proportion than seen in real-world clinical practice, where the perihilar area is the commonest site of CCA45,48,49. Thus, the lack of a specific code for pCCA may have led to systematic errors, particularly with miscoding of pCCA as iCCA7,45,47.
In a study in the UK published in 2021, retrospective review of 625 hepatobiliary cancers from three independent regional centres by two clinicians found that only 98 of 226 (43%) CCAs that were originally coded according to ICD-10 as iCCA (C22.1) were true iCCA, whereas 76 iCCAs (34%) were actually pCCA50. Conversely, 92% of those that were truly pCCA were incorrectly coded as iCCA. Thus, miscoding of pCCA — the commonest form of CCA — as iCCA may be contributing to the apparent rise in iCCA rates. The next iterations of both ICD and ICD–O (ICD-11 and ICD–O-4, respectively) will, for the first time, have separate codes to record iCCA, pCCA and dCCA51, facilitating more accurate epidemiological data.
Risk factors for non-fluke-related CCA
Differences in risk factors account, at least in part, for the geographical variations in the incidence of CCA. In Western countries, primary sclerosing cholangitis is the most well-known risk factor for CCA7. Several risk factors are associated with all three subtypes, whereas others are subtype-specific. For example, Caroli disease and choledochal cysts (congenital disorders of the bile ducts) have a strong association with all three CCA subtypes52,53. By contrast, cirrhosis, non-alcoholic fatty liver disease (NAFLD) and hepatitis B have a stronger association with iCCA, whereas choledocholithiasis (bile duct stones) has a stronger correlation with pCCA and/or dCCA52. The global rise in obesity and NAFLD may be contributing to the rising rates of iCCA. Although multiple risk factors for CCA exist, most CCAs do not have an identifiable risk factor.
Mechanisms/pathophysiology
Genetic and epigenetic aberrations
Extensive genomic and epigenomic studies have shown that the molecular landscapes of CCAs differ considerably by aetiology, highlighting how cancer subtypes in the same organ may arise through different extrinsic and intrinsic carcinogenic processes54,55,56. The findings also illustrate the importance of conducting these molecular studies in diverse populations, as differences between their genomic and epigenomic profiles point to the need for distinct biomarkers and therapies. Among CCAs, genetic aberrations differ depending on their anatomic locations; for example, FGFR2 fusions are almost exclusively found in iCCAs, whereas PRKCA–PRKCB fusions are observed in pCCAs and dCCAs56. For CCAs of other causes, such as those related to herbal carcinogen aristolochic acid, primary biliary cirrhosis and choledochal cysts, current knowledge of their molecular landscape is sparse owing to insufficient numbers of samples studied.
Common mutations and related molecular pathways
According to studies of ~500 fluke-related and non-fluke-related CCAs, some of the most commonly mutated genes (mutation frequencies 10–26%) in CCA include TP53, ARID1A, KRAS, SMAD4, BAP1 and APC, followed by at least another 20 genes with lower mutation frequencies of 1–6%54,55,56,57,58,59,60,61,62. Interestingly, the mutation frequencies of driver genes differ between CCA aetiologies. For example, TP53 and ARID1A mutations are highly enriched in fluke-related CCAs, whereas BAP1 and IDH1 and IDH2 mutations are highly enriched in non-fluke-related CCAs (Table 1). These gene mutations are known to be associated with key cancer-related molecular pathways, such as RAS–RAF–MAPK (for example, MAP2K4 and PTEN), WNT (for example, APC and RNF43), DNA repair (for example, BRCA2 and MSH3) and epigenetic modulation (for example, ARID1A and BAP1). Some of these mutated genes may be related to the same biological pathway and their mutations are usually mutually exclusive but may occasionally occur in combination. For example, members of the Wnt signalling pathway, including APC, RNF43, AXIN1 and different forms of catenin (CTNNA2, CTNND2 and CTNNB1), were found to be mutated and, when the mutations occurred in combination, they contributed to a higher percentage of CCAs than mutation of a single gene55.
Epigenetically, CCA tumours exhibit DNA hypermethylation and distinct DNA hypermethylation patterns are found that differentiate fluke-related CCA (predominantly in CpG islands) and non-fluke-related CCA (predominantly in CpG island shores)55. Integrative analysis of somatic mutations and DNA methylation led to the proposal that fluke-related CCAs are probably caused by early exposure to external carcinogenic agents that induce a chronic inflammatory milieu, which results in genome-wide epigenetic dysregulation that drives tumour development; by contrast, in non-fluke-related CCAs, an initial genetic driver mutation causes tumorigenesis and epigenetic changes occur during this process49,54.
Fluke-related cholangiocarcinoma
Generally, this group of CCAs exhibit substantially more somatic mutations than non-fluke-related CCAs55, probably reflecting their underlying aetiology associated with fluke-related chronic inflammation. Inactivating mutations that are more prevalent in this subgroup than in non-fluke-related CCAs include TP53, ARID1A, ARID2, BRCA1 and BRCA2 (refs54,55,61,63,64). Non-coding mutations in promoters associated with H3K7me3 have also been found to be enriched55. Copy number analysis also detected more frequent ERBB2 amplification in fluke-related CCAs, which may have considerable clinical implications, as these tumours may be more sensitive to ERBB2 inhibitor treatment. In addition, the expression of several genes, including TET1, encoding a DNA demethylation enzyme, and EZH2, encoding a histone methyltransferase, has been found to be aberrant, implying that these genes may have a role in the hypermethylation phenotype in CCA55. Another study identified two fluke-related CCA subtypes: the C1 subtype that is enriched with mutations in genes, such as ECT2, that lead to mitotic checkpoint defects, and the C2 subtype that is related to bile acid metabolism, T cell infiltration and obesity63.
Non-fluke-related cholangiocarcinoma
Inactivating mutations in PBRM1, BAP1, PIK3CA and ELF3, and gain-of-function mutations in IDH1 and IDH2 are predominantly found in this group of CCAs54,55,56,57,58. Chromosome translocation involving mainly FGFR and to a lesser extent PKARC is another key genetic alteration55,56,65,66. FGFR2 translocations in CCAs were first discovered through a clinical sequencing programme for advanced CCA and are rarely found in fluke-associated CCA67. PRKACA and PRKACB are part of the cAMP-dependent protein kinase signalling pathway, and are enriched in non-fluke-related CCA56. Epigenetically, this group of CCAs is dominated by hypermethylation in promoter CpG shores55, with prevalent C>T and/or G>A substitutions at CpG sites56. Of note, the distribution of mutations between non-fluke subtypes seems to vary according to the anatomic site of the CCA; for example, FGFR2 translocations are exclusively found in iCCAs50,56,63.
Fluke pathophysiology
Chronic liver fluke infection is associated with numerous hepatobiliary diseases, including inflammation of the gallbladder and bile ducts (cholecystitis and cholangitis, respectively), periductal fibrosis and, ultimately, CCA. Liver fluke infection is thought to drive CCA via multiple distinct but interacting pathways: mechanical damage to the bile duct epithelium caused by adult flukes grazing on the resident cells, notably cholangiocytes; immunopathology driven by chronic infection-related inflammation; and effects of parasite excretory–secretory molecules, including secreted vesicles, proteins and small molecules68. The interplay of these mechanisms, in addition to a traditional diet in disease-endemic areas that is rich in nitrosamine-containing foods, such as fermented fish contaminated with liver fluke metacercariae, is in keeping with current knowledge of carcinogenesis (Fig. 2).

a | Resected liver from a patient with cholangiocarcinoma (CCA) showing numerous adult Opisthorchis viverrini flukes (arrows) in the bile ducts (BD). A CCA tumour mass can be seen in the upper left part of the image. b | Transverse section of an O. viverrini fluke (Ov) attached via its oral sucker (S) to the bile duct epithelium (E) of an experimentally infected hamster. Cellular infiltrate in response to the feeding fluke is evident. c | Proliferating cholangiocytes in the biliary epithelium (BE) producing 8-oxo-deoxyguanosine (arrows, brown stain) as a marker of DNA damage in the bile duct of a hamster infected with an O. viverrini fluke (Ov). d | Uptake of O. viverrini-secreted extracellular vesicles (green dots) by human H69 primary cholangiocytes in culture. e | Thickened and disordered biliary epithelium (BE) in a hamster infected with wild-type O. viverrini flukes (Ov; left panel) compared with the BE in a hamster infected with O. viverrini in which Ov-grn-1, which encodes a secreted granulin-like growth factor, has been edited using CRISPR–Cas9 (right panel); editing of Ov-grn-1 has resulted in substantial reduction of biliary hyperplasia as highlighted by the square brackets. L, liver. Part a courtesy of B. Sripa, Khon Kaen University. Part b reprinted with permission from ref.209, Royal Society of Chemistry. Part d courtesy of S. Chaiyadet, Khon Kaen University. Part e reprinted from ref.78, CC BY 4.0.
The attachment of liver flukes to the biliary wall results in ulceration and formation of precancerous lesions69. This process is accompanied by immune cell infiltration and persistent secretion of inflammatory cytokines, such as IL-6, which is well known as a link between inflammation and carcinogenesis, particularly in liver tissue70. Elevated IL-6 levels are strongly associated with advanced and persistent periductal fibrosis in O. viverrini infections, which is thought to contribute to the pathogenesis of fluke-induced CCA71. Similar to other helminth infections, flukes induce local recruitment of type 2 macrophages, eosinophils, mast cells and T cells. Parasite-specific B lymphocyte and T lymphocyte responses occur to a diverse array of antigens15,71; however, despite this robust response, sterile immunity does not develop, and older people living in endemic areas are often heavily infected72,73, necessitating a vaccine that induces long-term anti-parasite immunity and protects against the onset of fluke-induced CCA (Box 1).
The hamster model of liver fluke-induced CCA is a powerful tool for investigating the aetiology and immunopathogenesis of fluke-infection-associated liver pathologies. Hamsters infected with O. viverrini fed a diet high in nitrosamines develop CCA within 6 months74. Soon after flukes arrive in the biliary tree, proliferating cholangiocytes can be detected, highlighting a process of constant wounding and repair that occurs over decades in infected people. Opisthorchis spp. secrete several molecular entities that contribute to this process, including a glutathione-dependent prostaglandin synthase that drives formation of precancerous lesions75, a granulin-like growth factor76 and extracellular vesicles (EVs)77, the latter two of which drive cholangiocyte proliferation and IL-6 secretion in vitro. Indeed, O. viverrini flukes that had undergone CRISPR–Cas9-induced editing of the granulin gene still colonized the biliary tract of hamsters and developed into adult flukes, but the resulting pathological changes were reduced to biliary hyperplasia and fibrosis78.
Molecular biology of progression and invasion
CCA cells gradually adopt invasive phenotypes to metastasize, for example by changing to a mesenchymal-like phenotype, which increases their migratory and invasion capabilities, and eventually deposit at distant sites. Various alterations related to cancer hallmarks occur to gain these invasive properties, including those during the invasion process79,80 and in angiogenesis and lymphangiogenesis81.
An integrated and in-depth understanding of the molecular mechanisms in CCA progression could aid in developing precision therapy for advanced CCA. Several pathways are dysregulated and represent potential therapeutic targets, including the inflammation-related IL-6–JAK–STAT3 pathway82, oestrogen and oestrogen receptors83, epithelial–mesenchymal transition84, EGFR activating the MAPK–ERK pathway85, hepatocyte growth factor–AKT–ERK signalling86 and others. Further transcription factors involving morphogenetic signalling pathways, for example, Hedgehog, Wnt, and Notch, as well as microRNAs are dysregulated to support the invasiveness of CCA. The PI3K–AKT–mTOR, HIF1α and MYC pathways are stimulated to support metabolic shifts in CCA cells87.
Post-translational modifications, O-GlcNAcylation and glycosylation have also been shown to mediate CCA invasiveness (Fig. 3). O-GlcNAcylation is a reversible process, in which a single GlcNAc residue is added to proteins, modulating protein function, stability and localization with or without coordinating phosphorylation. In human CCA tissues, high levels of O-GlcNAcylation have been observed and are associated with a poor prognosis88. Upregulation of O-GlcNAcylation enhances the stability of the structural protein vimentin and increases the nuclear translocation of proteins that activate expression of downstream genes involved in epithelial–mesenchymal transition, cell migration and invasion89,90. Epidemiological studies have indicated that diabetes mellitus is a risk factor and possibly a promoting factor for CCA91. Increased levels of O-GlcNAcylation and STAT3 activation have been reported to be key processes in the aggressiveness of CCA cells enhanced by high glucose levels92,93. Increased initial O-GalNAcylation94 and terminal fucosylation94 have been implicated in CCA development in the hamster model and in tissues from inpatients with CCA. Modulation of either process substantially affects the metastatic potential of CCA cells. Upregulation of specific high-mannose N-glycans facilitates the progression of highly metastatic CCA cells95,96, some of which can be detected in the serum from patients with CCA97. The collective evidence suggests that certain glycans and/or enzymes involved in glycan synthesis might serve as new biomarkers and targets to manage CCA metastasis.

Several pathways are dysregulated to transform phenotypes and functions of cholangiocarcinoma (CCA) cells. Cancer-associated fibroblasts (CAFs) and tumour-associated macrophages (TAMs) in the tumour microenvironment produce autocrine and paracrine signals that enhance CCA metastasis. The crosstalk between CCA cells, CAFs and TAMs progressively remodels the tumour stroma to facilitate invasion of tumour cells from the primary site to the secondary site. In addition, dysregulation of intracellular O-GlcNAcylation of proteins by adding or removing N-acetylglucosamine (GlcNAc) influences function, stability and localization of several proteins associated with metastasis. Modulation of extracellular glycosylation, for example, fucosylation and O-GalNAcylation of surface glycoproteins or secretory proteins, has an important role in enhancing the metastatic activity of CCA cells. ER, endoplasmic reticulum.
Pathology, inflammation and tumour microenvironment
CCA has three predominant macroscopic growth patterns: mass-forming lesions; periductal infiltrating lesions; and intraductal papillary lesions. The histopathology may also be classified as small bile duct type (which may derive from septal and interlobular bile ducts, progenitor cells, and possibly hepatocytes), large bile duct type (potentially arising from segmental bile ducts or associated peribiliary glands), and rare variants11,98,99,100. For iCCA, patients with mass-forming and periductal infiltrating subtypes have the poorest prognosis, whereas those with intraductal papillary lesions have the most favourable outcomes following curative surgical resection101.
The mass-forming growth pattern is most common in iCCA and is generally seen at presentation as a single, nodular solid mass. Advanced mass-forming iCCA may also have satellite or multifocal tumour growth within the liver. The periductal infiltrating growth pattern of CCA does not form a nodular mass but grows longitudinally along the walls of the large bile ducts and spreads along the portal tracts, resulting in strictures of the affected bile ducts and dilation of the smaller proximal bile ducts. This is the growth pattern most frequently observed in pCCA. The intraductal papillary type of CCA is seen at presentation as a slow-growing polypoid or papillary tumour growing within the lumen of a dilated bile duct11,98,99,100.
Histologically, 90–95% of CCAs are adenocarcinomas, which may be well, moderately or poorly differentiated. They can be small bile duct or large bile duct type lesions. The small bile duct types usually show no or minimal mucin production, whereas the large bile duct types are mucin-producing adenocarcinomas. pCCA, dCCA and large bile duct iCCA share similar pathological and molecular features11,102,103. Whether precursor lesions for mass-forming iCCA exist is unknown, but the presence of premalignant dysplastic precursor lesions for pCCA, dCCA and large bile duct type iCCA has been established11.
Unique rare variants of CCA include intestinal-type CCA, combined hepatocellular carcinoma and CCA (cHCC–CCA), and lymphoepithelioma-like CCA11,104,105. Cholangiolocellular carcinoma (CLC) is a rare primary liver cancer in which the epithelial component resembles cholangioles (canals of Hering)105. It is recommended that CLC is categorized as a histological subtype of well-differentiated iCCA106.
Unlike conventional HCCs, CCAs frequently have a prominent desmoplastic microenvironment characterized by a dense collagen-fibre-enriched tumour stroma and matricellular proteins (for example, periostin and tenascin C), an abundance of cancer-associated fibroblasts (Fig. 4) and, to a lesser extent, tumour-associated macrophages and varying numbers of innate immune cells107. CCAs are also dissimilar to HCC by often being hypovascular108, although CCAs formed in cirrhosis can display increased vascularity109.

a | Masson trichrome staining of a moderately to poorly differentiated small duct type mass-forming intrahepatic cholangiocarcinoma (iCCA) largely comprising prominent desmoplastic stroma strongly stained for collagen (blue staining). Arrows point to representative small clusters of cholangiocarcinoma. b | The vast majority of cancer-associated fibroblasts (CAFs) accumulated within the desmoplastic stroma of CCA are strongly immunoreactive for α-smooth muscle actin (αSMA), a biomarker of myofibroblast differentiation, whereas cholangiocarcinoma cells (CC) do not show αSMA staining. c | Picrosirius red (PR) staining for collagen (orange staining under polarized light) typically reveals the extracellular matrix of desmoplastic CCA to consist of thick collagen fibres (predominantly comprising collagen type I). d | Immunostaining for matricellular periostin (POSTN), produced by αSMA+ CAFs, which has a binding site for collagen, is exclusively localized to the desmoplastic stroma of iCCA. It is now generally believed that αSMA+ CAFs in iCCA are principally derived from activated portal fibroblasts and hepatic stellate cells and are phenotypically and functionally heterogeneous107. High expression levels of αSMA and POSTN, both of which are induced by transforming growth factor-β, have been associated with poor iCCA prognosis following surgical resection. Parts a–d, x33. Parts a, b and d reprinted with permission from ref.210, Elsevier.
This evolving and complex desmoplastic tumour microenvironment has a pre-eminent role in promoting CCA progression, therapeutic resistance and immunosuppression14,110,111,112. Cancer-associated myofibroblasts are a major source of secreted stromal components, including multiple growth factors, cytokines, metabolites, extracellular matrix (ECM) proteins and modifying enzymes that facilitate CCA growth and invasiveness, cell survival, ECM remodelling, and metabolic reprogramming. Indeed, in preclinical models, deletion of cancer-associated fibroblasts limits tumour progression113. Stromal matrix stiffness in CCA can trigger signalling pathways regulating malignant behaviour and mechanically collapse blood microvessels, causing hypoxia and limiting drug and immune cell bioavailability114.
Diagnosis, screening and prevention
Diagnosis
Diagnosis and management of CCA vary by anatomic subtype of disease (Table 2).
iCCA
iCCA is often seen at presentation as an intrahepatic mass and is incidentally found in 25–30% of patients115. Patients with iCCA are often asymptomatic during early disease stages and develop symptoms or signs, such as abdominal pain or less commonly jaundice, during disease progression to an advanced stage. Carbohydrate antigen 19-9 (CA19-9) is the primary serum biomarker used in CCA diagnosis, although it has subpar specificity and can be elevated in various conditions, such as biliary obstruction or pancreatic cancer. Nonetheless, a levels of CA19-9 >1,000 U/ml are concerning for the presence of metastatic CCA116. Imaging modalities used for iCCA diagnosis include conventional ultrasonography, CT, MRI and contrast-enhanced ultrasonography (CEUS). MRI may provide enhanced assessment of the primary mass, whereas CT imaging has superior detection of vascular enhancement and is, therefore, important in determining resectability117. In patients with cirrhosis, HCC surveillance may facilitate earlier iCCA diagnosis118. However, distinguishing HCC and iCCA can be difficult in this patient population. iCCAs are characterized by an initial arterial contrast enhancement at the tumour periphery and progressive homogeneous contrast enhancement119,120. HCCs are characterized by arterial hyper-enhancement and washout in the portal venous phase or delayed phase. In a cirrhotic liver, gadolinium-enhanced MRI has an increased specificity, but lower sensitivity, for diagnosing HCC and distinguishing it from iCCA when portal venous phase washout rather than conventional delayed phase washout is used121. Compared with CT or MRI, CEUS is more likely to misdiagnose iCCA as HCC122. In CEUS imaging, iCCAs have an earlier contrast washout from the vascularized portions of the lesions, whereas HCCs have delayed portal venous washout118. Hence, CEUS is not reliable as the sole imaging technique to differentiate iCCA from HCC but may be useful in scenarios with inconclusive CT or MRI. PET scanning is typically not used in the diagnosis of iCCA owing to limited accuracy123. 18F-Fluorodeoxyglucose (18F-FDG) PET imaging has reasonable performance in detection of lymph node and distant metastasis and, therefore, may have a role in CCA staging124. Histopathological analysis of a biopsy specimen remains the mainstay for confirmation of an iCCA diagnosis. If a patient is eligible for resection, then a biopsy need not be performed.
pCCA and dCCA
Patients with pCCA and dCCA typically present with painless jaundice owing to underlying biliary obstruction. Following initial CT that may be concerning for pCCA and/or dCCA, a specific type of MRI termed magnetic resonance cholangiopancreatography (MRCP) is employed for CCA detection125. The sensitivity and specificity of MRCP to distinguish benign and malignant causes of hilar obstruction are 87% and 85%, respectively126. MRCP can also delineate the biliary anatomy before endoscopic intervention with endoscopic retrograde cholangiography (ERC). ERC has a diagnostic and therapeutic role in pCCA and dCCA as it enables detection of malignant strictures and acquisition of biliary brushings for cytology and fluorescence in situ hybridization (FISH) analysis. Biliary cytology or a biopsy sample positive for adenocarcinoma are diagnostic of CCA127. However, the sensitivity of biliary cytology for pCCA detection is low (43% according to one meta-analysis127). FISH analysis has a higher sensitivity than cytology (65% versus 19% in one series) and similar specificity for detection of CCA128. Endoscopic techniques such as cholangioscopy and confocal laser endomicroscopy can be used to visualize indeterminate biliary strictures129. However, whether these advanced techniques can improve tissue diagnostic yield through targeted biliary biopsies remains unclear.
Endoscopic ultrasonography (EUS) is useful in the diagnosis and staging of pCCA, as it enables a detailed visual assessment of the extrahepatic bile duct as well as tissue acquisition via fine-needle aspiration (FNA). EUS–FNA has a higher sensitivity for detection of dCCA than detection of pCCA (81% versus 59%)130. In addition, EUS-guided tissue acquisition of pCCA is controversial owing to the potential risk of tumour dissemination131. PET scanning is typically not utilized in the diagnosis of pCCA owing to limited accuracy123. 18F-FDG PET has subpar performance for detection of the primary tumour123 but may have a role in the assessment of lymph node and distant metastasis124.
Prevention and screening of fluke-related CCA
Populations at high risk of clonorchiasis and opisthorchiasis include those who prefer meals that include considerable amounts of raw, undercooked, fermented or dried freshwater fish in endemic areas18,132,133. Current or past infection with liver fluke can be diagnosed through faecal, blood and/or urine examinations for the presence of the eggs, fluke antigens, antibodies and/or nucleic acids27,34,134,135. Infection is treated and cured with the anthelmintic drug praziquantel taken by mouth136. For prevention, health education is necessary and increasingly employed and supported by public health authorities; for example, the Lawa model used in northeast Thailand aims to change food consumption behaviours away from dishes that include raw fish, which are important elements of local and traditional culture137. Health education for schoolchildren and the wider community in endemic regions addresses the biology and modes of infection, highlights freshwater fish as the infection source, safe handling of food, safe cooking procedures, and improved personal and community sanitation to avoid the entry of unprocessed sewage into the local freshwater environment. Preventive measures also address screening, diagnosis and treatment, in particular with praziquantel, at both the individual and population levels27,132,137,138,139,140,141.
Given the high prevalence of liver fluke infection in diverse geographical regions25, large-scale screening has been undertaken, involving stool examination for fluke ova coupled with abdominal ultrasonography or other radiological imaging33,132,133,142,143,144 (Fig. 5). Serological assays are used to supplement stool examination as a screening tool in the clinic for the diagnosis of opisthorchiasis and clonorchiasis, and may be useful in screening populations at risk of CCA associated with liver fluke infection131. Imaging can reveal periductal, fibrosis-induced, chronic inflammation, which can progress to CCA145. Although praziquantel adequately treats the infection, the periductal fibrosis rarely resolves following parasite clearance71,142. Regarding population-level public health screening, the CCA screening and care programme at Khon Kaen University, Thailand, is noteworthy in its population reach and impact, reporting the screening of >200,000 at-risk residents, with radiological diagnosis indicating a ~1% incidence of iCCA. The programme incorporates teleconsultation ultrasonography in its follow-up protocols143,146,147.

a | Egg of Opisthorchis viverrini in human stool (egg dimensions 19–30 µm long and 10–20 µm wide). b | Suspected cholangiocarcinoma (CCA) imaged using abdominal sonography during community screening in liver fluke endemic regions of northeast Thailand; the ultrasonographic image shows a mass (green circle), calcification (thin arrow) and dilated intrahepatic duct (large arrow) c | Immunochromatographic device for the serodiagnosis of opisthorchiasis and clonorchiasis. Part a adapted from ref.211, CC BY 4.0. Part b reprinted with permission from ref.142, Elsevier. Part c reprinted with permission from ref.135, The American Journal of Tropical Medicine and Hygiene.
Surveillance in primary sclerosing cholangitis
PSC is a premalignant biliary tract disease that confers a considerable risk of CCA development, usually pCCA. The incidence of hepatobiliary pancreatic malignancy, of which most are CCA, is 1.43 cases per 100 patient years, and the cumulative incidence at 20 years is 20–25%148. Although the incidence of CCA is high in patients with PSC, surveillance strategies in asymptomatic patients are not universally endorsed because of several diagnostic and therapeutic concerns149. First, the diagnosis of CCA in PSC is challenging because inflammation-related dominant biliary tract strictures mimic CCA on radiography. Second, conventional cytology and advanced cytological techniques, such as FISH for polysomy, lack sensitivity and require invasive endoscopic procedures129. Finally, curative treatment options rely on the availability of liver transplantation and neoadjuvant chemoradiation therapy150. Despite these caveats, retrospective data from a multicentre review suggest that surveillance of asymptomatic patients with PSC with annual MRCP and CA19-9 assessment is life-saving151. In patients with PSC and asymptomatic CCA, MRI detection was associated with a reduction in mortality compared with ultrasonography (HR 0.10, 95% CI 0.01–0.96). Whether these data can be confirmed by others remains to be seen. In the meantime, societal guidelines remain ambiguous regarding surveillance for CCA in patients with PSC152.
Management
Like diagnosis, the management of CCA varies by anatomic subtype of disease (Table 2).
iCCA
Surgical resection
Surgery with intent to attain a margin-negative (R0) resection is a potentially curative treatment option for iCCA. However, most patients with iCCA present with large, unresectable tumours153. Staging laparoscopy is recommended particularly in patients with high-risk features, such as multicentric disease, high CA19-9 levels, suspicion of vascular invasion or peritoneal disease154. Diagnostic laparoscopy can detect occult metastatic disease not apparent on preoperative cross-sectional staging imaging155. In patients undergoing surgical resection, a positive resection margin (R1) is associated with an increased risk of recurrence and shortened overall survival156. Multifocal disease is a prognostic factor that is associated with poor long-term outcomes157,158. Another important prognostic factor in patients undergoing surgical resection is lymph node status. Accordingly, porta hepatis lymphadenectomy is performed routinely during surgical resection of iCCA. Patients with lymph node metastasis have worse disease-specific survival159. Moreover, the median survival in patients with lymph node metastasis undergoing surgical resection is similar to that in patients who have been treated with chemotherapy alone, indicating that resection in these patients does not offer a survival benefit160.
Liver transplantation
Emerging data suggest that liver transplantation may be an option in patients with iCCA with small tumours. In a multicentre retrospective study, the 5-year actuarial survival in patients with cirrhosis and small (<2 cm), incidental iCCA tumours was 65% following liver transplantation161. These patients probably underwent liver transplantation for presumed HCC; it is uncommon to diagnose small iCCA tumours unless a patient with liver cirrhosis is undergoing surveillance for HCC. In a follow-up study in a larger, multinational cohort of patients with cirrhosis and incidental iCCA, the 1-year, 3-year and 5-year actuarial survival in patients with very early iCCA (≤2 cm) was 93%, 84% and 65%, respectively162. Factors associated with tumour recurrence were presence of microvascular invasion and poor tumour differentiation. Liver transplantation in patients with large iCCA tumours is associated with a high risk of recurrence163.
Locoregional therapy
Locoregional therapy is an option in patients with locally advanced iCCA who are not eligible for surgical options. A multicentre retrospective analysis assessed the efficacy of intra-arterial therapy (conventional transarterial chemoembolization, drug-eluting beads or transarterial bland embolization) in 198 patients with locally advanced iCCA (median tumour size 8.1 cm; 47.5% of patients with a solitary lesion)164. The median overall survival was 13.2 months and the type of intra-arterial therapy did not affect patient responses. A phase II clinical trial of hepatic arterial infusion of floxuridine in combination with systemic gemcitabine and oxaliplatin in 38 patients with locally advanced iCCA demonstrated promising efficacy165. The median overall survival was 25.0 months and 1-year overall survival was 89.5%. Advanced external beam radiation therapy (EBRT) techniques, such as charged-particle (proton or carbon) beam techniques, can be used to deliver high-dose EBRT to patients while sparing adjacent non-malignant tissues. A single-arm, phase II, multicentre study evaluated the efficacy and safety of high-dose, hypofractionated proton beam therapy in 92 patients with HCC or iCCA (37 with iCCA only)166. Proton beam therapy achieved a local disease control rate of 94.1% at 2 years.
pCCA and dCCA
Surgical resection
Surgery is an option in patients who present with early-stage pCCA or dCCA. Staging laparoscopy before planned resection for pCCA can detect radiologically occult metastatic disease. In one series including 116 patients, staging laparoscopy had an all-cause yield of 27.2% for the detection of unresectable disease167. Although the surgical approach has become more aggressive over time with more patients with locally advanced pCCA undergoing resection, long-term survival following resection has also increased168,169. The incidence of major hepatectomy, bile duct resection, and vascular resection and/or reconstruction has increased over time170,171, whereas operative complications have decreased. In a series of 574 patients with pCCA undergoing surgical resection, the 5-year disease-specific survival was 32.5%169. Over the period 2001–2010, 243 patients with R0 disease and no lymph node metastasis after resection had a 5-year survival of 67.1%. The tumours of patients with locally advanced pCCA with bilateral involvement of the second-order bile ducts have traditionally been considered unresectable. However, advances in surgical techniques have enabled resection of locally advanced tumours, and patients with bilateral involvement of the second-order bile ducts who undergo resection have an improved overall survival compared with those who do not undergo resection (5-year survival 32.8% versus 1.5% in one series)172. Unilateral and main hepatic artery involvement are independent poor prognostic factors, as patients with these features do not have a survival benefit following resection173. Lymph node metastasis is associated with poor prognosis in patients undergoing resection for pCCA169.
Surgical resection of dCCA typically involves a pancreaticoduodenectomy or Whipple procedure with removal of the gallbladder and bile duct, the head of the pancreas, and the first part of the duodenum. The 5-year survival of patients with dCCA following surgical resection ranges from 20% to 40% depending on disease extent48,174. Predictors of survival following resection of dCCA include resection margin status, lymph node status, tumour size and degree of tumour differentiation48.
Liver transplantation
Liver transplantation following neoadjuvant chemoradiation therapy is an option in patients with early-stage, unresectable pCCA or dCCA in the setting of PSC regardless of resectability150,175,176. The treatment protocol includes EBRT with concomitant 5-fluorouracil followed by bile duct brachytherapy, and subsequent capecitabine maintenance treatment until the time of transplantation. Operative staging is performed before transplantation. Data from 12 large-volume centres in the USA showed that patients with early-stage, unresectable pCCA who underwent neoadjuvant chemoradiation followed by liver transplantation had a 5-year post-transplant recurrence-free survival of 65%, and an intent-to-treat survival of 53%150. Predictors of a patient dropping out before liver transplantation, for example owing to disease progression include CA19-9 levels of ≥500 U/ml, malignant brushing or biopsy, and a model for end-stage liver disease (a measure of liver disease severity) score of ≥20 (ref.177). Predictors of recurrence following liver transplantation include high pretransplant CA19-9 levels of ≥500 U/ml, portal vein encasement and residual tumour on explant.
Systemic therapy for CCA
Cytotoxic therapy
In patients with advanced-stage CCA who are ineligible for surgical or locoregional options, chemotherapy with first-line cisplatin and gemcitabine is an option. The ABC-02 study demonstrated a median overall survival of 11.7 months for gemcitabine in combination with cisplatin compared with 8.1 months for gemcitabine alone178. In patients whose disease has progressed on gemcitabine and cisplatin, the ABC-06 study demonstrated benefit of folinic acid, 5-fluorouracil and oxaliplatin (FOLFOX) in the second-line setting179. FOLFOX has become the standard second-line therapy for advanced CCA. A phase III randomized trial of gemcitabine plus S-1 (also known as Teysuno, an oral combination of the 5-fluorouracil prodrug tegafur with gimeracil and oteracil to increase efficacy and reduce adverse effects) demonstrated non-inferiority of this combination to gemcitabine and cisplatin180. Preliminary results from an open-label, randomized phase II–III trial show that the combination of 5-fluorouracil, oxaliplatin and irinotecan (FOLFIRINOX) does not result in improved progression-free survival compared with gemcitabine and cisplatin181. Chemotherapy combinations currently under investigation as first-line therapy for CCA include gemcitabine plus cisplatin combined with albumin-bound paclitaxel182.
In patients with advanced disease, well-designed, stratified studies examining specifically systemic therapy, targeted therapy or immunotherapy in those with iCCA, pCCA or dCCA are lacking. In those studies with post hoc analyses, the numbers of patients with dCCA have been insufficient to draw conclusions. In the original trial that investigated cisplatin plus gemcitabine the confidence intervals for pCCA crossed 1, suggesting a lack of benefit178. Indeed, post hoc analysis of the ABC-01, ABC-02 and ABC-03 studies demonstrated that patients with iCCA had a longer overall survival than those with non-iCCA biliary cancers (HR 0.58, 95% CI 0.35–0.95; P = 0.03)183.
Targeted therapy
Each CCA subtype has a distinct genetic landscape. iCCAs are characterized by mutations of IDH1 and IDH2 (~15% of iCCAs), which encode isocitrate dehydrogenase, genetic alterations in fibroblast growth factor receptors, BAP1 mutations and others56. Early results of treatment with inhibitors of IDH or fibroblast growth factor receptor (FGFR) in human CCA were promising. In a phase III multicentre, double-blind, randomized controlled trial with a crossover design in 185 patients with chemotherapy-refractory iCCA with IDH1 mutations, there was a statistically significant improvement in progression-free survival in patients receiving ivosidenib, an inhibitor of mutant IDH1, compared with those receiving placebo (2.7 months versus 1.4 months; HR 0.37, 95% CI 0.25–0.54)184. Several FGFR inhibitors have been investigated in human CCA185,186,187. In a phase II trial, ~35% of patients with CCA and FGFR2 gene fusions or rearrangements had an objective response to pemigatinib, a potent selective inhibitor of FGFR1–3 (ref.185). Pemigatinib subsequently received accelerated FDA approval for the treatment of adults with previously treated, unresectable, locally advanced or metastatic CCA with FGFR2 gene fusions or rearrangements. Of note, pemigatinib treatment may not be suitable for fluke-related CCA, owing to the low incidence of FGFR2 fusions found in these tumours59. BRAF mutations occur in 3–5% of CCAs, primarily in iCCAs188,189. A phase II, open-label, single-arm basket trial of dabrafenib, a BRAF inhibitor, and trametinib, a MEK inhibitor, demonstrated promising efficacy of this combination in patients with CCA with BRAFV600E mutations190.
Immunotherapy
Emerging clinical data of immune-directed therapies, such as immune checkpoint blockade, suggest modest efficacy in CCA. In KEYNOTE-158, a phase II trial in patients with advanced biliary tract cancer, the objective response rate with pembrolizumab, a PD1 inhibitor, was a subpar 5.8%191. Although the data are limited in CCA, pembrolizumab has enhanced activity against mismatch repair-deficient tumours192. Two subsequent trials of immune checkpoint blockade in patients with CCA have demonstrated improved efficacy. In a phase II trial in 54 patients with previously treated biliary tract cancer, treatment with the PD1 inhibitor nivolumab resulted in an objective response rate of 22%193. In the intention-to-treat population, median overall survival was 14 months. Phase II trial data for the combination of nivolumab and ipilimumab, a cytotoxic T lymphocyte associated protein 4 (CTLA4) inhibitor, demonstrated an overall survival of 5.7 months and progression-free survival of 2.9 months194; notably, the responses were exclusively observed in patients with iCCA or gallbladder CCA. Characterization of the immune landscape of each CCA subtype will be essential in the effort to develop effective immune-directed therapies. A phase III clinical trial of pembrolizumab in combination with gemcitabine and cisplatin (KEYNOTE-966) is currently ongoing195.
Adjuvant therapy
Overall survival following surgical resection for CCA remains suboptimal with a high risk of recurrent disease even with R0 resection, prompting interest in prospective trials of adjuvant therapies196,197. The PRODIGE trial of adjuvant gemcitabine and oxaliplatin in all biliary tract cancers did not demonstrate a clear benefit of adjuvant therapy198. In addition, adjuvant gemcitabine following resection of pCCA or dCCA did not demonstrate a benefit in the BCAT trial199. The BILCAP study, a phase III randomized controlled trial of capecitabine versus observation following surgical resection, included 447 patients with biliary tract cancer of whom 43 patients had iCCA and 65 patients had pCCA. Although a protocol-specified sensitivity analysis adjusted for nodal status, disease grade and patient sex showed a significant difference in overall survival with capecitabine (HR 0.71, 95% CI 0.55–0.92), the unadjusted intention-to-treat analysis did not demonstrate a significant benefit in overall survival (HR 0.81, 95% CI 0.63–1.04)200. On the basis of these data, practice guidelines recommend capecitabine for 6 months following surgical resection of CCA201.
Quality of life
Robust studies and data on the quality of life of patients with CCA are sparse. This paucity of information is due to the low incidence of the disease, its high lethality, the limited number of patients enrolling in clinical trials until very recently, and the lack of dedicated quality-of-life measurement tools. quality of life following liver resection surgery or a pancreaticoduodenectomy has been well documented202,203. Also, quality of life is highly dependent on the systemic therapy regimen employed. In the absence of robust data, the voice of the patient is a powerful narrative that can help us reflect on and guide our research mission. The patient’s experience is unique in its highly successful outcome given the high lethality of advanced CCA, but the medical odyssey described, including the decision to undergo invasive procedures and engage in clinical trials, is a shared experience common to many patients with CCA.
Perspective of a patient and advocate
As an 11-year survivor of advanced stage iCCA, I have experienced multiple surgeries, disease recurrence, various chemotherapy regimens, numerous adverse effects, and participated in a clinical trial. I found my perspective and approach to this journey similar to other patients with CCA who I have interacted with during the past 10 years. I resonate with other patients and advocates I have connected and collaborated with through various forums sponsored by the Cholangiocarcinoma Foundation (CCF). This foundation sponsors an annual meeting attended by equal numbers of patients and professionals (physicians and scientists) with expertise in CCA. Patients share their stories from the podium, which is beneficial to patients, caregivers, physicians and scientists alike. This relationship is a powerful incentive to continue enhancing the collaborative network of CCA stakeholders and improving the lives of patients with CCA.
Surgery is a potentially curative treatment option for patients with tumours that have not grown too large. Surgeries may be performed to remove both the primary and recurrent disease and to obtain malignant tissue to guide other therapies. However, surgery can affect many organs, and serious complications can occur. I had a total of five operations and I would have another if it were necessary for my treatment plan. Imaging showed disease recurrence 3 months after my initial liver resection; however, it is likely that metastases were already present at the time of surgery. Even so, the surgical resection bought me time to find the clinical trial that has kept me alive and healthy for 11 years. The four other surgeries consisted of three lung wedges with video-assisted thoracoscopic surgery (VATS) procedures and one thoracotomy. The first VATS was to confirm metastasis, whereas the others were to remove tumours to harvest tumour-infiltrating lymphocytes for treatment in a clinical trial. The thoracotomy was to remove malignant tissue again for the clinical trial and to guide next steps. All of these surgeries were worth the risk and contributed to my survival time. Other patients have also confirmed their willingness to undergo invasive surgical procedures to extend survival or buy time to explore additional therapies.
Especially for treatments that are not surgical, molecular profiling is an essential and necessary part of treatment plans. Patients with CCA are proactive about their medical care and want their treatments personalized. I was willing to undergo treatments with severe adverse effects for 3–12 months. Because there is no proven treatment and minimal options for CCA, patients are willing to try treatments that have no guarantee of working. I enrolled in a clinical trial with the thought that I had nothing to lose and everything to gain, especially as there were no other options for me. All patients, including myself, desire to increase our survival time. However, I came to a point in my journey in which quality of life became more important than the quantity of life, and my fellow patients agree with this sentiment. I came to this decision because my quality of life declined after 2 years of undergoing chemotherapy, and I had no desire to continue. I did not want my children to think I was giving up, but I wished to spend what time I had left with a good quality of life.
Clinical trials are a significant component of our treatment plan. I knew that to survive this diagnosis, I would have to find a clinical trial. My oncologist found a clinical trial for me as first-line therapy. Unfortunately, my insurance at that time would not cover the standard-of-care costs that go along with being in a clinical trial. I chose not to join the trial because I did not want to put my family in financial distress. Two years later, after chemotherapy failed to control my disease, I stumbled upon the clinical trial that successfully prolonged my life. My husband and I made phone calls to be evaluated for this trial and ultimately did all the work to be enrolled. Most patients struggle with understanding how to find and qualify for a clinical trial. They rely on their oncologist to provide the information.
Access to clinical trials is the primary barrier patients face to enrolling and researchers face in accruing participants. Patients most likely will have to travel to be able to participate in a trial and that, in turn, causes other issues, such as financial burden. Patients with a diagnosis as grim as CCA are deterred from clinical trials with a placebo arm unless there is an opportunity for crossover after a reasonable amount of time. There is no time to waste on a placebo. In addition to improved access, technologies are needed to help patients identify clinical trials and determine whether they match the inclusion and exclusion criteria. Furthermore, data on patient-reported outcomes should be better documented in these trials. For example, quantitative information from the patient’s perspective on adverse effects, reasons for foregoing and dropping out of trials, and the overall patient experience need to be incorporated into the clinical trial design. It is imperative to consider the patient perspective when planning future studies. Patient-reported outcomes and quality of life instruments provide information that is critical in optimizing research and therapeutic endeavours. As we move into an era of virtual visits and remote monitoring, I am hopeful that engaging in clinical trials will become less burdensome.
Outlook
Despite the recent progress in our knowledge of CCA mechanisms and management, many critical scientific and clinical questions remain, and prevention and treatment of this devastating disease still need to be better developed and defined, including identification of preventable risk factors. From an experimental perspective, preclinical models of pCCA and dCCA are still lacking. These models will be required if we are to better understand and develop rational therapies for these diseases. Although several preclinical models of iCCA have been developed204, many of the mutations commonly present in human iCCA, such as ARID1A, BAP1 and PBRM1, have not been examined as oncogenic drivers in these preclinical models. Given the unpredictability of extrapolating the information obtained from animal models, the use of human CCA organoids and patient-derived xenografts needs to be more deeply explored for disease mechanisms and response to new therapeutic agents. The relationships between coding and non-coding genetic and epigenetic alterations will require intense investigation and bioinformatics interrogation.
Even in the clinic, better diagnostic modalities are necessary for pCCA and dCCA, including molecular diagnostics in bile and blood129. Chemopreventive strategies in populations at high risk of CCA require further study, such as the use of statins205. In addition, vaccine development for liver-fluke-associated CCA requires extensive work before these agents can be used in humans.
The tumour immune microenvironment is a potential target for the treatment of CCA. Targeting cancer-associated fibroblasts, T cells and myeloid-derived suppressor cells are emerging therapeutic strategies with a high potential for the treatment of this highly desmoplastic cancer14,111,206. Although exciting, the effectivity of targeted therapies with pemigatinib in patients with FGFR2 fusion aberrations and ivosidenib in patients with isocitrate dehydrogenase gain-of-function mutations is hampered by primary treatment resistance and a short durability of response185,207. Combinatorial therapies will be necessary to improve outcomes in these patient populations.
The advances in identifying targetable genetic aberrations in CCA highlight the emerging role of precision medicine for this disease. Analysis of circulating cell-free DNA will probably aid in this approach208 and constitutes an important advance that will continue to be refined. The use of neoadjuvant and adjuvant therapeutic strategies in combination with surgery and locoregional therapies to improve outcomes is also an active area of clinical investigation. Finally, selection of patients who will benefit from surgery in the long term remains a challenge and further insight into prognostic biomarkers is needed. Thus, considerably more information on this enigmatic cancer is required if we are to adequately minimize its current devastating human impact.
References
Fan, B. et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Invest. 122, 2911–2915 (2012).
Nakagawa, H. et al. Biliary epithelial injury-induced regenerative response by IL-33 promotes cholangiocarcinogenesis from peribiliary glands. Proc. Natl Acad. Sci. USA 114, E3806–E3815 (2017).
Razumilava, N. & Gores, G. J. Cholangiocarcinoma. Lancet 383, 2168–2179 (2014).
Zhu, Y. & Kwong, L. N. Insights into the origin of intrahepatic cholangiocarcinoma from mouse models. Hepatology 72, 305–314 (2020).
Blechacz, B., Komuta, M., Roskams, T. & Gores, G. J. Clinical diagnosis and staging of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 8, 512–522 (2011). The first paper that describes the three anatomic subtypes of cholangiocarcinoma.
Cardinale, V. Classifications and misclassification in cholangiocarcinoma. Liver Int. 39, 260–262 (2019).
Khan, S. A., Tavolari, S. & Brandi, G. Cholangiocarcinoma: epidemiology and risk factors. Liver Int. 39, 19–31 (2019).
Hainsworth, J. D. et al. Molecular gene expression profiling to predict the tissue of origin and direct site-specific therapy in patients with carcinoma of unknown primary site: a prospective trial of the Sarah Cannon Research Institute. J. Clin. Oncol. 31, 217–223 (2013).
Varadhachary, G. R. & Raber, M. N. Cancer of unknown primary site. N. Engl. J. Med. 371, 757–765 (2014).
Rizvi, S., Khan, S. A., Hallemeier, C. L., Kelley, R. K. & Gores, G. J. Cholangiocarcinoma – evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 15, 95–111 (2018).
Kendall, T. et al. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 39, 7–18 (2019).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 (2018).
Sirica, A. E. et al. Intrahepatic cholangiocarcinoma: continuing challenges and translational advances. Hepatology 69, 1803–1815 (2019).
Fabris, L. et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int. 39, 63–78 (2019).
Sripa, B., Tangkawattana, S. & Brindley, P. J. Update on pathogenesis of opisthorchiasis and cholangiocarcinoma. Adv. Parasitol. 102, 97–113 (2018).
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 100, 1–441 (2012).
Sripa, B. et al. Opisthorchiasis and opisthorchis-associated cholangiocarcinoma in Thailand and Laos. Acta Trop. 120, S158–168 (2011).
Qian, M. B., Utzinger, J., Keiser, J. & Zhou, X. N. Clonorchiasis. Lancet 387, 800–810 (2016).
Sripa, B. et al. Liver fluke induces cholangiocarcinoma. PLoS Med. 4, e201 (2007).
Schwartz, D. A. Helminths in the induction of cancer: Opisthorchis viverrini, Clonorchis sinensis and cholangiocarcinoma. Trop. Geogr. Med. 32, 95–100 (1980).
Sithithaworn, P., Yongvanit, P., Duenngai, K., Kiatsopit, N. & Pairojkul, C. Roles of liver fluke infection as risk factor for cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 21, 301–308 (2014).
Ogorodova, L. M. et al. Opisthorchiasis: an overlooked danger. PLoS Negl. Trop. Dis. 9, e0003563 (2015).
Choi, B. I., Han, J. K., Hong, S. T. & Lee, K. H. Clonorchiasis and cholangiocarcinoma: etiologic relationship and imaging diagnosis. Clin. Microbiol. Rev. 17, 540–552 (2004).
Keiser, J. & Utzinger, J. Food-borne trematodiases. Clin. Microbiol. Rev. 22, 466–483 (2009).
Petney, T. N., Andrews, R. H., Saijuntha, W., Wenz-Mucke, A. & Sithithaworn, P. The zoonotic, fish-borne liver flukes Clonorchis sinensis, Opisthorchis felineus and Opisthorchis viverrini. Int. J. Parasitol. 43, 1031–1046 (2013).
Pakharukova, M. Y. & Mordvinov, V. A. The liver fluke Opisthorchis felineus: biology, epidemiology and carcinogenic potential. Trans. R. Soc. Trop. Med. Hyg. 110, 28–36 (2016).
Na, B. K., Pak, J. H. & Hong, S. J. Clonorchis sinensis and clonorchiasis. Acta Trop. 203, 105309 (2020).
Lai, Y. S., Zhou, X. N., Pan, Z. H., Utzinger, J. & Vounatsou, P. Risk mapping of clonorchiasis in the People’s Republic of China: a systematic review and Bayesian geostatistical analysis. PLoS Negl. Trop. Dis. 11, e0005239 (2017).
Qian, M. B., Chen, Y. D., Liang, S., Yang, G. J. & Zhou, X. N. The global epidemiology of clonorchiasis and its relation with cholangiocarcinoma. Infect. Dis. Poverty 1, 4 (2012).
Doanh, P. N. & Nawa, Y. Clonorchis sinensis and Opisthorchis spp. in Vietnam: current status and prospects. Trans. R. Soc. Trop. Med. Hyg. 110, 13–20 (2016).
Cho, S. H. et al. Prevalence of clonorchiasis in southern endemic areas of Korea in 2006. Korean J. Parasitol. 46, 133–137 (2008).
Sohn, W. M. et al. High endemicity with Clonorchis sinensis metacercariae in fish from yongjeon-cheon (Stream) in Cheongsong-gun, Gyeongsangbuk-do, Korea. Korean J. Parasitol. 59, 97–101 (2021).
Fedorova, O. S. et al. Opisthorchis felineus infection, risks, and morbidity in rural Western Siberia, Russian Federation. PLoS Negl. Trop. Dis. 14, e0008421 (2020).
Sripa, B., Kaewkes, S., Intapan, P. M., Maleewong, W. & Brindley, P. J. Food-borne trematodiases in Southeast Asia epidemiology, pathology, clinical manifestation and control. Adv. Parasitol. 72, 305–350 (2010).
Sithithaworn, P. et al. The current status of opisthorchiasis and clonorchiasis in the Mekong Basin. Parasitol. Int. 61, 10–16 (2012).
Sohn, W. M. et al. Low-grade endemicity of opisthorchiasis, Yangon, Myanmar. Emerg. Infect. Dis. 25, 1435–1437 (2019).
Namsanor, J. et al. Infection dynamics of opisthorchis viverrini metacercariae in cyprinid fishes from two endemic areas in Thailand and Lao PDR. Am. J. Trop. Med. Hyg. 102, 110–116 (2020).
Taylor-Robinson, S. D. et al. Increase in mortality rates from intrahepatic cholangiocarcinoma in England and Wales 1968-1998. Gut 48, 816–820 (2001).
Patel, T. Worldwide trends in mortality from biliary tract malignancies. BMC Cancer 2, 10 (2002).
Bertuccio, P. et al. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J. Hepatol. 71, 104–114 (2019).
Saha, S. K., Zhu, A. X., Fuchs, C. S. & Brooks, G. A. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 21, 594–599 (2016).
Lepage, C. et al. Trends in the incidence and management of biliary tract cancer: a French population-based study. J. Hepatol. 54, 306–310 (2011).
Khan, S. A. et al. Changing international trends in mortality rates for liver, biliary and pancreatic tumours. J. Hepatol. 37, 806–813 (2002).
Jepsen, P., Vilstrup, H., Tarone, R. E., Friis, S. & Sorensen, H. T. Incidence rates of intra- and extrahepatic cholangiocarcinomas in Denmark from 1978 through 2002. J. Natl Cancer Inst. 99, 895–897 (2007).
Banales, J. M. et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 17, 557–588 (2020).
Yao, K. J., Jabbour, S., Parekh, N., Lin, Y. & Moss, R. A. Increasing mortality in the United States from cholangiocarcinoma: an analysis of the National Center for Health Statistics Database. BMC Gastroenterol. 16, 117 (2016).
Khan, S. A. et al. Rising trends in cholangiocarcinoma: is the ICD classification system misleading us? J. Hepatol. 56, 848–854 (2012).
DeOliveira, M. L. et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann. Surg. 245, 755–762 (2007).
Nakeeb, A. et al. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann. Surg. 224, 463–473 (1996). discussion 473-465.
Selvadurai, S. et al. Cholangiocarcinoma miscoding in hepatobiliary centres. Eur. J. Surg. Oncol. 47, 635–639 (2021).
Bosman, F. T., Carneiro, F., Hruban, R. H. & Theise, N. D. WHO Classification of Tumours: Digestive System Tumours (IARC, 2019).
Clements, O., Eliahoo, J., Kim, J. U., Taylor-Robinson, S. D. & Khan, S. A. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: a systematic review and meta-analysis. J. Hepatol. 72, 95–103 (2020). This paper is an updated and comprehensive review and meta-analysis of cholangiocarcinoma risk factors.
Petrick, J. L. et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: a population-based study in SEER-Medicare. PLoS ONE 12, e0186643 (2017).
Chan-On, W. et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 45, 1474–1478 (2013). This paper links aetiological exposures to distinct somatic mutations.
Jusakul, A. et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 7, 1116–1135 (2017).
Nakamura, H. et al. Genomic spectra of biliary tract cancer. Nat. Genet. 47, 1003–1010 (2015). This paper provides a comprehensive molecular characterization of the different subtypes of biliary tract cancers.
Farshidfar, F. et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 18, 2780–2794 (2017).
Jiao, Y. et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 45, 1470–1473 (2013).
Liu, Z. H. et al. Whole-exome mutational and transcriptional landscapes of combined hepatocellular cholangiocarcinoma and intrahepatic cholangiocarcinoma reveal molecular diversity. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2360–2368 (2018).
Lowery, M. A. et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin. Cancer Res. 24, 4154–4161 (2018).
Ong, C. K. et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 44, 690–693 (2012).
Zou, S. et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 5, 5696 (2014).
Chaisaingmongkol, J. et al. Common molecular subtypes among Asian hepatocellular carcinoma and cholangiocarcinoma. Cancer Cell 32, 57–70.e3 (2017).
Montal, R. et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 73, 315–327 (2020). This paper is the most comprehensive, integrated genomic analysis of perihilar/distal cholangiocarcinoma to date.
Sia, D. et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 6, 6087 (2015).
Wu, Y. M. et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 3, 636–647 (2013).
Kongpetch, S. et al. Lack of targetable FGFR2 fusions in endemic fluke-associated cholangiocarcinoma. JCO Glob. Oncol. 6, 628–638 (2020).
Suttiprapa, S. et al. Opisthorchis viverrini proteome and host–parasite interactions. Adv. Parasitol. 102, 45–72 (2018).
Siripongsakun, S. et al. Premalignant lesions of cholangiocarcinoma: characteristics on ultrasonography and MRI. Abdom. Radiol. 44, 2133–2146 (2019).
Wu, M. Y., Yiang, G. T., Cheng, P. W., Chu, P. Y. & Li, C. J. Molecular targets in hepatocarcinogenesis and implications for therapy. J. Clin. Med. 7, 213 (2018).
Sripa, B. et al. Advanced periductal fibrosis from infection with the carcinogenic human liver fluke Opisthorchis viverrini correlates with elevated levels of interleukin-6. Hepatology 50, 1273–1281 (2009).
Forrer, A. et al. Spatial distribution of, and risk factors for, Opisthorchis viverrini infection in southern Lao PDR. PLoS Negl. Trop. Dis. 6, e1481 (2012).
Lun, Z. R. et al. Clonorchiasis: a key foodborne zoonosis in China. Lancet Infect. Dis. 5, 31–41 (2005).
Thamavit, W., Bhamarapravati, N., Sahaphong, S., Vajrasthira, S. & Angsubhakorn, S. Effects of dimethylnitrosamine on induction of cholangiocarcinoma in Opisthorchis viverrini-infected Syrian golden hamsters. Cancer Res. 38, 4634–4639 (1978).
Pakharukova, M. Y., Zaparina, O. G., Kovner, A. V. & Mordvinov, V. A. Inhibition of Opisthorchis felineus glutathione-dependent prostaglandin synthase by resveratrol correlates with attenuation of cholangiocyte neoplasia in a hamster model of opisthorchiasis. Int. J. Parasitol. 49, 963–973 (2019).
Smout, M. J. et al. Carcinogenic parasite secretes growth factor that accelerates wound healing and potentially promotes neoplasia. PLoS Pathog. 11, e1005209 (2015).
Chaiyadet, S. et al. Carcinogenic liver fluke secretes extracellular vesicles that promote cholangiocytes to adopt a tumorigenic phenotype. J. Infect. Dis. 212, 1636–1645 (2015).
Arunsan, P. et al. Programmed knockout mutation of liver fluke granulin attenuates virulence of infection-induced hepatobiliary morbidity. eLife 8, e41463 (2019).
Brivio, S., Cadamuro, M., Fabris, L. & Strazzabosco, M. Molecular mechanisms driving cholangiocarcinoma invasiveness: an overview. Gene Expr. 18, 31–50 (2018).
Labib, P. L., Goodchild, G. & Pereira, S. P. Molecular pathogenesis of cholangiocarcinoma. BMC Cancer 19, 185 (2019).
Roy, S., Glaser, S. & Chakraborty, S. Inflammation and progression of cholangiocarcinoma: role of angiogenic and lymphangiogenic mechanisms. Front. Med 6, 293 (2019).
Servais, F. A. et al. Modulation of the IL-6-signaling pathway in liver cells by miRNAs targeting gp130, JAK1, and/or STAT3. Mol. Ther. Nucleic Acids 16, 419–433 (2019).
Alvaro, D. et al. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am. J. Pathol. 169, 877–888 (2006).
Zavadil, J. & Bottinger, E. P. TGF-β and epithelial-to-mesenchymal transitions. Oncogene 24, 5764–5774 (2005).
Claperon, A. et al. EGF/EGFR axis contributes to the progression of cholangiocarcinoma through the induction of an epithelial-mesenchymal transition. J. Hepatol. 61, 325–332 (2014).
Miyamoto, M. et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br. J. Cancer 105, 131–138 (2011).
Pant, K., Richard, S., Peixoto, E. & Gradilone, S. A. Role of glucose metabolism reprogramming in the pathogenesis of cholangiocarcinoma. Front. Med. 7, 113 (2020).
Phoomak, C. et al. Overexpression of O-GlcNAc-transferase associates with aggressiveness of mass-forming cholangiocarcinoma. Asian Pac. J. Cancer Prev. 13, 101–105 (2012).
Phoomak, C. et al. Mechanistic insights of O-GlcNAcylation that promote progression of cholangiocarcinoma cells via nuclear translocation of NF-κB. Sci. Rep. 6, 27853 (2016).
Phoomak, C. et al. O-GlcNAc-induced nuclear translocation of hnRNP-K is associated with progression and metastasis of cholangiocarcinoma. Mol. Oncol. 13, 338–357 (2019).
Saengboonmee, C., Seubwai, W., Wongkham, C. & Wongkham, S. Diabetes mellitus: possible risk and promoting factors of cholangiocarcinoma: association of diabetes mellitus and cholangiocarcinoma. Cancer Epidemiol. 39, 274–278 (2015).
Phoomak, C. et al. High glucose levels boost the aggressiveness of highly metastatic cholangiocarcinoma cells via O-GlcNAcylation. Sci. Rep. 7, 43842 (2017).
Saengboonmee, C., Seubwai, W., Pairojkul, C. & Wongkham, S. High glucose enhances progression of cholangiocarcinoma cells via STAT3 activation. Sci. Rep. 6, 18995 (2016).
Indramanee, S. et al. Terminal fucose mediates progression of human cholangiocarcinoma through EGF/EGFR activation and the Akt/Erk signaling pathway. Sci. Rep. 9, 17266 (2019).
Phoomak, C. et al. O-GlcNAcylation mediates metastasis of cholangiocarcinoma through FOXO3 and MAN1A1. Oncogene 37, 5648–5665 (2018).
Park, D. D. et al. Metastasis of cholangiocarcinoma is promoted by extended high-mannose glycans. Proc. Natl Acad. Sci. USA 117, 7633–7644 (2020).
Talabnin, K., Talabnin, C., Ishihara, M. & Azadi, P. Increased expression of the high-mannose M6N2 and NeuAc3H3N3M3N2F tri-antennary N-glycans in cholangiocarcinoma. Oncol. Lett. 15, 1030–1036 (2018).
Nakanuma, Y. et al. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J. Hepatol. 2, 419–427 (2010).
Banales, J. M. et al. Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 13, 261–280 (2016).
Vijgen, S., Terris, B. & Rubbia-Brandt, L. Pathology of intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 6, 22–34 (2017).
Sirica, A. E. et al. Intrahepatic cholangiocarcinoma progression: prognostic factors and basic mechanisms. Clin. Gastroenterol. Hepatol. 7, S68–S78 (2009).
Bragazzi, M. C. et al. New insights into cholangiocarcinoma: multiple stems and related cell lineages of origin. Ann. Gastroenterol. 31, 42–55 (2018).
Akita, M. et al. Histological and molecular characterization of intrahepatic bile duct cancers suggests an expanded definition of perihilar cholangiocarcinoma. HPB 21, 226–234 (2019).
Bae, J. Y. et al. Intestinal type cholangiocarcinoma of intrahepatic large bile duct associated with hepatolithiasis–a new histologic subtype for further investigation. Hepatogastroenterology 49, 628–630 (2002).
Brunt, E. et al. cHCC-CCA: Consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentation. Hepatology 68, 113–126 (2018).
Balitzer, D. et al. Immunohistochemical and molecular features of cholangiolocellular carcinoma are similar to well-differentiated intrahepatic cholangiocarcinoma. Mod. Pathol. 32, 1486–1494 (2019).
Brivio, S., Cadamuro, M., Strazzabosco, M. & Fabris, L. Tumor reactive stroma in cholangiocarcinoma: the fuel behind cancer aggressiveness. World J. Hepatol. 9, 455–468 (2017).
Bosmuller, H. et al. Microvessel density and angiogenesis in primary hepatic malignancies: differential expression of CD31 and VEGFR-2 in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Pathol. Res. Pract. 214, 1136–1141 (2018).
Xu, J. et al. Intrahepatic cholangiocarcinomas in cirrhosis are hypervascular in comparison with those in normal livers. Liver Int. 32, 1156–1164 (2012).
Cadamuro, M. et al. The deleterious interplay between tumor epithelia and stroma in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1435–1443 (2018).
Loeuillard, E., Conboy, C. B., Gores, G. J. & Rizvi, S. Immunobiology of cholangiocarcinoma. JHEP Rep. 1, 297–311 (2019).
Vaquero, J., Aoudjehane, L. & Fouassier, L. Cancer-associated fibroblasts in cholangiocarcinoma. Curr. Opin. Gastroenterol. 36, 63–69 (2020).
Affo, S. et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell. 39, 866–882.e11 (2021). The first paper to describe subtypes of cancer-associated fibroblasts in cholangiocarcinoma.
Sahai, E. et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186 (2020).
Alvaro, D. et al. Cholangiocarcinoma in Italy: a national survey on clinical characteristics, diagnostic modalities and treatment. Results from the “Cholangiocarcinoma” committee of the Italian Association for the Study of Liver disease. Dig. Liver Dis. 43, 60–65 (2011).
Patel, A. H., Harnois, D. M., Klee, G. G., LaRusso, N. F. & Gores, G. J. The utility of CA 19-9 in the diagnoses of cholangiocarcinoma in patients without primary sclerosing cholangitis. Am. J. Gastroenterol. 95, 204–207 (2000).
Kim, M. J., Choi, J. Y. & Chung, Y. E. Evaluation of biliary malignancies using multidetector-row computed tomography. J. Comput. Assist. Tomogr. 34, 496–505 (2010).
Wildner, D. et al. CEUS in hepatocellular carcinoma and intrahepatic cholangiocellular carcinoma in 320 patients–early or late washout matters: a subanalysis of the DEGUM multicenter trial. Ultraschall Med. 36, 132–139 (2015).
Iavarone, M. et al. Contrast enhanced CT-scan to diagnose intrahepatic cholangiocarcinoma in patients with cirrhosis. J. Hepatol. 58, 1188–1193 (2013).
Kim, S. H. et al. Typical and atypical imaging findings of intrahepatic cholangiocarcinoma using gadolinium ethoxybenzyl diethylenetriamine pentaacetic acid-enhanced magnetic resonance imaging. J. Comput. Assist. Tomogr. 36, 704–709 (2012).
Choi, S. H. et al. Intrahepatic cholangiocarcinoma in patients with cirrhosis: differentiation from hepatocellular carcinoma by using gadoxetic acid-enhanced MR imaging and dynamic CT. Radiology 282, 771–781 (2017).
Vilana, R. et al. Intrahepatic peripheral cholangiocarcinoma in cirrhosis patients may display a vascular pattern similar to hepatocellular carcinoma on contrast-enhanced ultrasound. Hepatology 51, 2020–2029 (2010).
Petrowsky, H. et al. Impact of integrated positron emission tomography and computed tomography on staging and management of gallbladder cancer and cholangiocarcinoma. J. Hepatol. 45, 43–50 (2006).
Lamarca, A. et al. (18)F-fluorodeoxyglucose positron emission tomography ((18)FDG-PET) for patients with biliary tract cancer: systematic review and meta-analysis. J. Hepatol. 71, 115–129 (2019).
Jhaveri, K. S. & Hosseini-Nik, H. MRI of cholangiocarcinoma. J. Magn. Reson. Imaging 42, 1165–1179 (2015).
Saluja, S. S., Sharma, R., Pal, S., Sahni, P. & Chattopadhyay, T. K. Differentiation between benign and malignant hilar obstructions using laboratory and radiological investigations: a prospective study. HPB 9, 373–382 (2007).
Trikudanathan, G., Navaneethan, U., Njei, B., Vargo, J. J. & Parsi, M. A. Diagnostic yield of bile duct brushings for cholangiocarcinoma in primary sclerosing cholangitis: a systematic review and meta-analysis. Gastrointest. Endosc. 79, 783–789 (2014).
Barr Fritcher, E. G. et al. An optimized set of fluorescence in situ hybridization probes for detection of pancreatobiliary tract cancer in cytology brush samples. Gastroenterology 149, 1813–1824.e1 (2015). Fluorescence in situ hybridization has become an essential tool in cholangiocarcinoma diagnosis.
Rizvi, S., Eaton, J., Yang, J. D., Chandrasekhara, V. & Gores, G. J. Emerging technologies for the diagnosis of perihilar cholangiocarcinoma. Semin. Liver Dis. 38, 160–169 (2018).
Mohamadnejad, M. et al. Role of EUS for preoperative evaluation of cholangiocarcinoma: a large single-center experience. Gastrointest. Endosc. 73, 71–78 (2011).
Heimbach, J. K., Sanchez, W., Rosen, C. B. & Gores, G. J. Trans-peritoneal fine needle aspiration biopsy of hilar cholangiocarcinoma is associated with disease dissemination. HPB 13, 356–360 (2011).
Lim, J. H. Liver flukes: the malady neglected. Korean J. Radiol. 12, 269–279 (2011).
Khuntikeo, N. et al. The socioeconomic burden of cholangiocarcinoma associated with Opisthorchis viverrini sensu lato infection in northeast Thailand: a preliminary analysis. Adv. Parasitol. 102, 141–163 (2018).
Saijuntha, W. et al. Recent advances in the diagnosis and detection of Opisthorchis viverrini sensu lato in human and intermediate hosts for use in control and elimination programs. Adv. Parasitol. 101, 177–214 (2018).
Sadaow, L. et al. Development of an immunochromatographic point-of-care test for serodiagnosis of opisthorchiasis and clonorchiasis. Am. J. Trop. Med. Hyg. 101, 1156–1160 (2019).
Sayasone, S. et al. Efficacy and safety of tribendimidine versus praziquantel against Opisthorchis viverrini in Laos: an open-label, randomised, non-inferiority, phase 2 trial. Lancet Infect. Dis. 18, 155–161 (2018).
Sripa, B., Tangkawattana, S. & Sangnikul, T. The Lawa model: a sustainable, integrated opisthorchiasis control program using the EcoHealth approach in the Lawa Lake region of Thailand. Parasitol. Int. 66, 346–354 (2017).
Phimpraphai, W., Tangkawattana, S., Kasemsuwan, S. & Sripa, B. Social influence in liver fluke transmission: application of social network analysis of food sharing in Thai Isaan culture. Adv. Parasitol. 101, 97–124 (2018).
Tang, Z. L., Huang, Y. & Yu, X. B. Current status and perspectives of Clonorchis sinensis and clonorchiasis: epidemiology, pathogenesis, omics, prevention and control. Infect. Dis. Poverty 5, 71 (2016).
Shin, H. R. et al. Descriptive epidemiology of cholangiocarcinoma and clonorchiasis in Korea. J. Korean Med. Sci. 25, 1011–1016 (2010).
Tangkawattana, S. & Sripa, B. Integrative EcoHealth/One Health approach for sustainable liver fluke control: the Lawa model. Adv. Parasitol. 102, 115–139 (2018).
Mairiang, E. et al. Ultrasonography assessment of hepatobiliary abnormalities in 3359 subjects with Opisthorchis viverrini infection in endemic areas of Thailand. Parasitol. Int. 61, 208–211 (2012).
Khuntikeo, N. et al. Cohort profile: cholangiocarcinoma screening and care program (CASCAP). BMC Cancer 15, 459 (2015).
Khuntikeo, N. et al. A comparison of the proportion of early stage cholangiocarcinoma found in an ultrasound-screening program compared to walk-in patients. HPB 22, 874–883 (2020).
Chamadol, N. et al. Histological confirmation of periductal fibrosis from ultrasound diagnosis in cholangiocarcinoma patients. J. Hepatobiliary Pancreat. Sci. 21, 316–322 (2014).
Khuntikeo, N., Loilome, W., Thinkhamrop, B., Chamadol, N. & Yongvanit, P. A comprehensive public health conceptual framework and strategy to effectively combat cholangiocarcinoma in Thailand. PLoS Negl. Trop. Dis. 10, e0004293 (2016).
Chamadol, N. et al. Teleconsultation ultrasonography: a new weapon to combat cholangiocarcinoma. ESMO Open 2, e000231 (2017).
Weismuller, T. J. et al. Patient age, sex, and inflammatory bowel disease phenotype associate with course of primary sclerosing cholangitis. Gastroenterology 152, 1975–1984.e8 (2017).
Rizvi, S., Eaton, J. E. & Gores, G. J. Primary sclerosing cholangitis as a premalignant biliary tract disease: surveillance and management. Clin. Gastroenterol. Hepatol. 13, 2152–2165 (2015).
Darwish Murad, S. et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 143, 88–98 e83 quiz e14 (2012). This multicentre study established neoadjuvant chemoradiation plus liver transplantation as an effective option for perihilar cholangiocarcinoma.
Eaton, J. E. et al. Early cholangiocarcinoma detection with magnetic resonance imaging versus ultrasound in primary sclerosing cholangitis. Hepatology 73, 1868–1881 (2021).
Chapman, R. et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 51, 660–678 (2010).
Doussot, A. et al. Outcomes after resection of intrahepatic cholangiocarcinoma: external validation and comparison of prognostic models. J. Am. Coll. Surg. 221, 452–461 (2015).
Weber, S. M. et al. Intrahepatic cholangiocarcinoma: expert consensus statement. HPB 17, 669–680 (2015).
Weber, S. M. et al. Intrahepatic cholangiocarcinoma: resectability, recurrence pattern, and outcomes. J. Am. Coll. Surg. 193, 384–391 (2001).
Spolverato, G. et al. The impact of surgical margin status on long-term outcome after resection for intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 22, 4020–4028 (2015).
Buettner, S. et al. Survival after resection of multiple tumor foci of intrahepatic cholangiocarcinoma. J. Gastrointest. Surg. 23, 2239–2246 (2019).
Lamarca, A. et al. Liver metastases of intrahepatic cholangiocarcinoma: implications for an updated staging system. Hepatology 73, 2311–2325 (2021).
Kim, Y. et al. Surgical management of intrahepatic cholangiocarcinoma: defining an optimal prognostic lymph node stratification schema. Ann. Surg. Oncol. 22, 2772–2778 (2015).
Kizy, S. et al. Surgical resection of lymph node positive intrahepatic cholangiocarcinoma may not improve survival. HPB 21, 235–241 (2019).
Sapisochin, G., de Sevilla, E. F., Echeverri, J. & Charco, R. Management of “very early” hepatocellular carcinoma on cirrhotic patients. World J. Hepatol. 6, 766–775 (2014).
Sapisochin, G. et al. Liver transplantation for “very early” intrahepatic cholangiocarcinoma: international retrospective study supporting a prospective assessment. Hepatology 64, 1178–1188 (2016).
Lunsford, K. E. et al. Liver transplantation for locally advanced intrahepatic cholangiocarcinoma treated with neoadjuvant therapy: a prospective case-series. Lancet Gastroenterol. Hepatol. 3, 337–348 (2018).
Hyder, O. et al. Intra-arterial therapy for advanced intrahepatic cholangiocarcinoma: a multi-institutional analysis. Ann. Surg. Oncol. 20, 3779–3786 (2013).
Cercek, A. et al. Assessment of hepatic arterial infusion of floxuridine in combination with systemic gemcitabine and oxaliplatin in patients with unresectable intrahepatic cholangiocarcinoma: a phase 2 clinical trial. JAMA Oncol. 6, 60–67 (2020).
Hong, T. S. et al. Multi-institutional phase II study of high-dose hypofractionated proton beam therapy in patients with localized, unresectable hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Clin. Oncol. 34, 460–468 (2016).
Bird, N. et al. Role of staging laparoscopy in the stratification of patients with perihilar cholangiocarcinoma. Br. J. Surg. 104, 418–425 (2017).
Nuzzo, G. et al. Improvement in perioperative and long-term outcome after surgical treatment of hilar cholangiocarcinoma: results of an Italian multicenter analysis of 440 patients. Arch. Surg. 147, 26–34 (2012).
Nagino, M. et al. Evolution of surgical treatment for perihilar cholangiocarcinoma: a single-center 34-year review of 574 consecutive resections. Ann. Surg. 258, 129–140 (2013).
Abbas, S. & Sandroussi, C. Systematic review and meta-analysis of the role of vascular resection in the treatment of hilar cholangiocarcinoma. HPB 15, 492–503 (2013).
de Jong, M. C. et al. The impact of portal vein resection on outcomes for hilar cholangiocarcinoma: a multi-institutional analysis of 305 cases. Cancer 118, 4737–4747 (2012).
Ebata, T. et al. Surgical resection for Bismuth type IV perihilar cholangiocarcinoma. Br. J. Surg. 105, 829–838 (2018).
van Vugt, J. L. A. et al. The prognostic value of portal vein and hepatic artery involvement in patients with perihilar cholangiocarcinoma. HPB 20, 83–92 (2018).
Dickson, P. V. & Behrman, S. W. Distal cholangiocarcinoma. Surg. Clin. North. Am. 94, 325–342 (2014).
Rea, D. J. et al. Liver transplantation with neoadjuvant chemoradiation is more effective than resection for hilar cholangiocarcinoma. Ann. Surg. 242, 451–458 (2005). discussion 458-461.
Sudan, D. et al. Radiochemotherapy and transplantation allow long-term survival for nonresectable hilar cholangiocarcinoma. Am. J. Transpl. 2, 774–779 (2002).
Darwish Murad, S. et al. Predictors of pretransplant dropout and posttransplant recurrence in patients with perihilar cholangiocarcinoma. Hepatology 56, 972–981 (2012).
Valle, J. et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 362, 1273–1281 (2010). This study established gemcitabine and cisplatin as the first-line systemic therapy for cholangiocarcinoma.
Lamarca, A. et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a phase 3, open-label, randomised, controlled trial. Lancet Oncol. 22, 690–701 (2021).
Morizane, C. et al. Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: the FUGA-BT (JCOG1113) randomized phase III clinical trial. Ann. Oncol. 30, 1950–1958 (2019).
Phelip, J. M. et al. Modified FOLFIRINOX versus CISGEM as first-line chemotherapy for advanced biliary tract cancer: results of AMEBICA PRODIGE 38 randomized phase II trial [abstract 52P]. Ann. Oncol. 31, S260–S261 (2020).
Shroff, R. T. et al. Gemcitabine, cisplatin, and nab-paclitaxel for the treatment of advanced biliary tract cancers: a phase 2 clinical trial. JAMA Oncol. 5, 824–830 (2019).
Lamarca, A. et al. Advanced intrahepatic cholangiocarcinoma: post hoc analysis of the ABC-01, -02, and -03 clinical trials. J. Natl Cancer Inst. 112, 200–210 (2020).
Abou-Alfa, G. K. et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 21, 796–807 (2020).
Abou-Alfa, G. K. et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 21, 671–684 (2020). Pemigatinib was the first targeted therapy to receive FDA approval in cholangiocarcinoma.
Javle, M. et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J. Clin. Oncol. 36, 276–282 (2018).
Rizvi, S. & Gores, G. J. Emerging molecular therapeutic targets for cholangiocarcinoma. J. Hepatol. 67, 632–644 (2017).
Goeppert, B. et al. BRAF V600E-specific immunohistochemistry reveals low mutation rates in biliary tract cancer and restriction to intrahepatic cholangiocarcinoma. Mod. Pathol. 27, 1028–1034 (2014).
Javle, M. et al. Biliary cancer: utility of next-generation sequencing for clinical management. Cancer 122, 3838–3847 (2016).
Subbiah, V. et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol. 21, 1234–1243 (2020).
Piha-Paul, S. A. et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: results from the KEYNOTE-158 and KEYNOTE-028 studies. Int. J. Cancer 147, 2190–2198 (2020).
Asaoka, Y., Ijichi, H. & Koike, K. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 373, 1979 (2015).
Kim, R. D. et al. A phase 2 multi-institutional study of nivolumab for patients with advanced refractory biliary tract cancer. JAMA Oncol. 6, 888–894 (2020).
Klein, O. et al. Evaluation of combination nivolumab and ipilimumab immunotherapy in patients with advanced biliary tract cancers: subgroup analysis of a phase 2 nonrandomized clinical trial. JAMA Oncol. 6, 1405–1409 (2020).
Finn, R. S. et al. KEYNOTE-966: A randomized, double-blind, placebo-controlled, phase 3 study of pembrolizumab in combination with gemcitabine and cisplatin for the treatment of advanced biliary tract carcinoma [abstract CT283]. Cancer Res. 80 (Suppl. 16), CT283 (2020).
Nathan, H. et al. Trends in survival after surgery for cholangiocarcinoma: a 30-year population-based SEER database analysis. J. Gastrointest. Surg. 11, 1488–1496; discussion 1496–1487 (2007).
Wang, Y. et al. Prognostic nomogram for intrahepatic cholangiocarcinoma after partial hepatectomy. J. Clin. Oncol. 31, 1188–1195 (2013).
Edeline, J. et al. Gemcitabine and oxaliplatin chemotherapy or surveillance in resected biliary tract cancer (PRODIGE 12-ACCORD 18-UNICANCER GI): a randomized phase III study. J. Clin. Oncol. 37, 658–667 (2019).
Ebata, T. et al. Randomized clinical trial of adjuvant gemcitabine chemotherapy versus observation in resected bile duct cancer. Br. J. Surg. 105, 192–202 (2018).
Primrose, J. N. et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): a randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 20, 663–673 (2019).
Shroff, R. T. et al. Adjuvant therapy for resected biliary tract cancer: ASCO clinical practice guideline. J. Clin. Oncol. 37, 1015–1027 (2019).
Fitzmaurice, C., Seiler, C. M., Buchler, M. W. & Diener, M. K. Survival, mortality and quality of life after pylorus-preserving or classical Whipple operation. A systematic review with meta-analysis. Chirurg 81, 454–471 (2010).
Dasgupta, D. et al. Quality of life after liver resection for hepatobiliary malignancies. Br. J. Surg. 95, 845–854 (2008).
Loeuillard, E., Fischbach, S. R., Gores, G. J. & Rizvi, S. Animal models of cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 982–992 (2019).
Lavu, S. et al. Effect of statins on the risk of extrahepatic cholangiocarcinoma. Hepatology 72, 1298–1309 (2020).
Fabris, L., Cadamuro, M., Cagnin, S., Strazzabosco, M. & Gores, G. J. Liver matrix in benign and malignant biliary tract disease. Semin. Liver Dis. 40, 282–297 (2020).
Abou-Alfa, G. K., Pandya, S. S. & Zhu, A. X. Ivosidenib for advanced IDH1-mutant cholangiocarcinoma – Authors’ reply. Lancet Oncol. 21, e371 (2020).
Lamarca, A. et al. Molecular profiling in daily clinical practice: practicalities in advanced cholangiocarcinoma and other biliary tract cancers. J. Clin. Med. 9, 2854 (2020).
Smout, M. J. et al. Infection with the carcinogenic human liver fluke, Opisthorchis viverrini. Mol. BioSyst. 7, 1367–1375 (2011).
Sirica, A. E., Strazzabosco, M. & Cadamuro, M. Intrahepatic cholangiocarcinoma: morpho-molecular pathology, tumor reactive microenvironment, and malignant progression. Adv. Cancer Res. 149, 321–387 (2021).
Chuchuen, O. et al. Rapid label-free analysis of Opisthorchis viverrini eggs in fecal specimens using confocal Raman spectroscopy. PLoS ONE 14, e0226762 (2019).
Suwannatrai, A., Saichua, P. & Haswell, M. Epidemiology of Opisthorchis viverrini infection. Adv. Parasitol. 101, 41–67 (2018).
Diemert, D. J., Bottazzi, M. E., Plieskatt, J., Hotez, P. J. & Bethony, J. M. Lessons along the critical path: developing vaccines against human helminths. Trends Parasitol. 34, 747–758 (2018).
McManus, D. P. Recent progress in the development of liver fluke and blood fluke vaccines. Vaccines (Basel) 8, 553 (2020).
Sun, H. et al. Bacillus subtilis spore with surface display of paramyosin from Clonorchis sinensis potentializes a promising oral vaccine candidate. Parasit. Vectors 11, 156 (2018).
Wang, X. et al. Surface display of Clonorchis sinensis enolase on Bacillus subtilis spores potentializes an oral vaccine candidate. Vaccine 32, 1338–1345 (2014).
Mekonnen, G. G., Pearson, M., Loukas, A. & Sotillo, J. Extracellular vesicles from parasitic helminths and their potential utility as vaccines. Expert Rev. Vaccines 17, 197–205 (2018).
Phumrattanaprapin, W. et al. Orally administered Bacillus spores expressing an extracellular vesicle-derived tetraspanin protect hamsters against challenge infection with carcinogenic human liver fluke. J. Infect. Dis. 223, 1445–1455 (2021).
Acknowledgements
G.J.G. is supported by the SPORE grant CA210964. P.J.B. and A.L. receive grant support (R01CA164719) from the National Institute of Health. S.A.K. is grateful for support from the UK National Institute for Health Research (NIHR) Biomedical Facilities at Imperial College London. B.T.T. receives support from The National Medical Research Council (grant MOH-000248). S.I.I. receives support from the National Cancer Institute (1K08CA236874) and the Mayo Foundation. A.L. receives support from National Health and Medical Research Council Senior Principal Research Fellowship 1117504. S.W. receives support from National Science and Technology Development Agency (NSTDA), Thailand, and the e-ASIA JRP.
Author information
Authors and Affiliations
Contributions
Introduction (G.J.G.); Epidemiology (P.J.B. and S.A.K.); Mechanisms/pathophysiology (B.T.T., A.L., S.W. and A.E.S.); Diagnosis, screening and prevention (S.I.I., P.J.B. and G.J.G.); Management (S.I.I.); Quality of life (M.B.); Outlook (G.J.G.); Overview of Primer (G.J.G.).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Disease Primers thanks B. Koerkamp; D.-Y. Oh, who co-reviewed with J. Yoon; R. T. Shroff, who co-reviewed with M. Savani; and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Brindley, P.J., Bachini, M., Ilyas, S.I. et al. Cholangiocarcinoma. Nat Rev Dis Primers 7, 65 (2021). https://doi.org/10.1038/s41572-021-00300-2
Accepted:
Published:
DOI: https://doi.org/10.1038/s41572-021-00300-2
This article is cited by
-
Development and validation of a clinical prediction model for the risk of distal metastasis in intrahepatic cholangiocarcinoma: a real-world study
BMC Gastroenterology (2024)
-
Identification of FOXP1 as a favorable prognostic biomarker and tumor suppressor in intrahepatic cholangiocarcinoma
BMC Cancer (2024)
-
Tumor burden score and carcinoembryonic antigen predict outcomes in patients with intrahepatic cholangiocarcinoma following liver resection: a multi‑institutional analysis
BMC Cancer (2024)
-
Predictive value of HTS grade in patients with intrahepatic cholangiocarcinoma undergoing radical resection: a multicenter study from China
World Journal of Surgical Oncology (2024)
-
RPL35A promotes the progression of cholangiocarcinoma by mediating HSPA8 ubiquitination
Biology Direct (2024)
