
Abstract
The global epidemic of prediabetes and diabetes has led to a corresponding epidemic of complications of these disorders. The most prevalent complication is neuropathy, of which distal symmetric polyneuropathy (for the purpose of this Primer, referred to as diabetic neuropathy) is very common. Diabetic neuropathy is a loss of sensory function beginning distally in the lower extremities that is also characterized by pain and substantial morbidity. Over time, at least 50% of individuals with diabetes develop diabetic neuropathy. Glucose control effectively halts the progression of diabetic neuropathy in patients with type 1 diabetes mellitus, but the effects are more modest in those with type 2 diabetes mellitus. These findings have led to new efforts to understand the aetiology of diabetic neuropathy, along with new 2017 recommendations on approaches to prevent and treat this disorder that are specific for each type of diabetes. In parallel, new guidelines for the treatment of painful diabetic neuropathy using distinct classes of drugs, with an emphasis on avoiding opioid use, have been issued. Although our understanding of the complexities of diabetic neuropathy has substantially evolved over the past decade, the distinct mechanisms underlying neuropathy in type 1 and type 2 diabetes remains unknown. Future discoveries on disease pathogenesis will be crucial to successfully address all aspects of diabetic neuropathy, from prevention to treatment.
Similar content being viewed by others
Introduction
The International Diabetes Federation estimates that 425 million people worldwide have diabetes1, making it the largest global epidemic of the 21st century2. 115 million people in China, 73 million in India and 30 million in the United States have diabetes3. These numbers are dwarfed by the number of individuals with prediabetes, which is estimated to be 388 million in China4, 133 million in India5 and 85 million in the United States6. 12% of global health expenditure, or $727 billion, is directed towards diabetes and its complications, and similar to the number of individuals with diabetes, this number continues to increase at an unsustainable rate1.
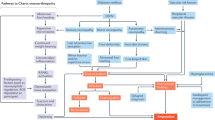
Among the complications of diabetes, a group of clinical syndromes caused by damage to the peripheral and autonomic nervous systems are by far the most prevalent. Generally referred to as different forms of neuropathy, these syndromes are caused by diffuse and focal nervous system damage and occur in up to half of all individuals with diabetes7. The most common form of diabetic neuropathy — distal symmetric polyneuropathy — is the focus of this Primer, and as such will be referred to as diabetic neuropathy throughout. Distal symmetric polyneuropathy manifests with a ‘stocking and glove’ distribution, whereby the hands and lower limbs are commonly affected. Other diffuse neuropathies secondary to diabetes can occur (Fig. 1) and include the constellation of autonomic neuropathies, such as cardiac autonomic neuropathy, gastrointestinal dysmotility and diabetic cystopathy and impotence (Box 1). Focal neuropathies, although less common, include dysfunction of individual peripheral nerves leading to isolated mononeuropathies, or less commonly to nerve roots leading to radiculopathy or polyradiculopathy (Fig. 1).

Several different patterns of neuropathy can present in individuals with diabetes. Of these, the most common is distal symmetric polyneuropathy (DSP). Examples of patterns of neuropathy are DSP, small-fibre-predominant neuropathy or treatment-induced neuropathy (part a); radiculoplexopathy or radiculopathy (part b); mononeuropathy (part c); and autonomic neuropathy or treatment-induced neuropathy (part d). Small-fibre-predominant neuropathy has the same distribution as DSP, although the neurological examination and results from nerve conduction velocity studies are different. Diabetic radiculoplexopathy or radiculopathy can respond to immunotherapy and usually improves with time, unlike other types of nerve injury in individuals with diabetes. Treatment-induced neuropathy is under-recognized, is caused by overaggressive glycaemic control and can present in multiple forms (parts a and d). Adapted by permission from BMJ Publishing Group Limited. BMJ Peltier, A., Goutman, S. A. & Callaghan, B. C. 348, (2014)230.
This Primer reviews the current knowledge on the epidemiology and pathogenesis of diabetic neuropathy and the optimal approaches for diagnosis and screening. Treatment approaches are outlined and are personalized for patients with different types of diabetes and for those with and without associated pain. We close with a call to action. The global epidemic of diabetes and its most common complication, neuropathy, requires a public health mandate to address modifiable risk factors with growing urgency. Without successful intervention, it is estimated that of the expected 9.7 billion individuals living in 2050, one-third will have diabetes and half of those will have neuropathy8. The cost to the individual in terms of both physical and mental function, and to society in terms of productivity, is staggering.
Epidemiology
Diabetic neuropathy is a highly prevalent condition that substantially affects patients by increasing falls, causing pain and reducing quality of life (QOL)9. The annual costs of diabetic neuropathy and its complications are more than $10 billion in the United States10.
Several studies have assessed the prevalence and/or incidence of neuropathy, although the definition of neuropathy used is different in each study. Two population-based studies using door-to-door screening reported prevalence estimates of 1%–4% for neuropathy, with 40–55% of these cases secondary to diabetes11,12. Similarly, in another study13, the cause of neuropathy was attributed to diabetes in over half of cases after diagnostic work-up by a neurologist. In the Netherlands, the incidence of neuropathy increases dramatically with age14, from <50 cases per 100,000 person-years in those <50 years of age to ~300 per 100,000 person-years in those >75 years of age, with diabetes accounting for 32% of all cases.
In addition to these studies evaluating the incidence and prevalence of diabetic neuropathy in the entire population, many epidemiological studies are confined to patients with either type 1 diabetes mellitus (T1DM) or T2DM. The incidence of neuropathy is higher in individuals with T2DM (6,100 per 100,000 person-years) than in those with T1DM (2,800 per 100,000 person-years)9,15,16,17. By contrast, the prevalence of neuropathy is similar in those with T2DM (8–51%18,19,20) to those with T1DM (11–50%20,21,22). Importantly, the prevalence is even higher when asymptomatic neuropathy is included, with 45% of patients with T2DM and 54% of those with T1DM developing neuropathy20. The higher incidence of neuropathy in patients with T2DM, with a similar prevalence in those with T2DM or T1DM, is probably secondary to multiple factors, including differences in age of onset of diabetes and differences in the underlying pathophysiology.
The prevalence of diabetic neuropathy also changes with disease duration. Indeed, the prevalence of diabetic neuropathy increased from 8% to 42% in patients with T2DM when patients were monitored for 10 years19. In the Danish Addition study, patients with newly diagnosed screen-detected T2DM23 had a prevalence of diabetic neuropathy of 13% at study entry, with a cumulative incidence of 10% over the 13-year follow-up period in a cohort with very mild T2DM that adhered to good metabolic control. On the other hand, in a large cohort of patients with more-advanced T2DM and confirmed coronary artery disease participating in the BARI 2D trial, 50% had confirmed diabetic neuropathy at baseline17, and the 4-year cumulative incidence of diabetic neuropathy was 66–72% in those without neuropathy at baseline17. Given how common neuropathy is in individuals with diabetes, effective diagnostic, screening and prevention strategies are of paramount importance.
Risk factors
The duration of diabetes and haemoglobin A1c (HbA1c) levels (a measurement of glycated haemoglobin as a surrogate for average daily glucose levels) are major predictors of diabetic neuropathy22. These two predictors commonly associate with other metabolic factors that are correlated with diabetic neuropathy, particularly in T2DM, such as insulin resistance and hypertension. Obesity is common in patients with neuropathy in population-based studies in multiple countries, including the United States, Denmark, China and the Netherlands23,24,25,26,27,28. Independent of HbA1c levels, the number of metabolic syndrome components, such as hypertriglyceridaemia, hypertension, abdominal obesity and low high-density lipoprotein (HDL) levels, is consistently associated with diabetic neuropathy in patients with T2DM24,25 and in selected T1DM cohorts29. Other independent risk factors for the development of diabetic neuropathy include smoking, alcohol abuse, increased height and older age30.
Several genes are linked to diabetic neuropathy, but only ACE (encoding angiotensin-converting enzyme) and MTHFR (encoding methylenetetrahydrofolate reductase) polymorphisms have been studied in multiple populations including large cohorts. Much more research is needed to better understand the role of genetics in the development of diabetic neuropathy, and several studies of existing cohorts are currently underway31,32.
Mechanisms/pathophysiology
Diabetic neuropathy is a unique neurodegenerative disorder of the peripheral nervous system that preferentially targets sensory axons, autonomic axons and later, to a lesser extent, motor axons. How diabetes mellitus targets sensory neurons remains debated. Progressive diabetic neuropathy involves retraction and ‘dying back’ of terminal sensory axons in the periphery, with relative preservation of the perikarya (cell bodies). Its ‘stocking and glove’ pattern of involvement reflects damage to the longest sensory axons first with, for example, loss of distal leg epidermal axons preceding loss in more proximal limbs; for this reason, diabetic neuropathy is considered a length-dependent neuropathy.
Substantial experimental evidence supports the notion that the entire neuron, from the perikaryon to the terminal, is targeted by diabetes. However, whether damage first targets peripheral axons and their associated Schwann cells or the neuron perikarya that reside in the dorsal root ganglia (DRG) and act to support the axons are debated (Fig. 2).

Sensory neurons relay sensory information from their nerve terminals (which are located throughout the periphery) to the dorsal horn of the spinal cord. The cell bodies of these sensory neurons are located in the dorsal root ganglia (DRG). Conversely, the cell bodies of motor neurons reside in the spinal cord ventral horn and transmit information from here to the periphery. Thin and unmyelinated sensory axons (C fibres or small fibres) are grouped together by non-myelinating Schwann cells into Remak bundles and represent a large portion of neurons of the peripheral nervous system. By comparison, other sensory axons are myelinated by associated Schwann cells, which have an important role in preserving axonal function. The precise order of cellular injury (whether, for example, damage to Schwann cells or axons occurs before damage to neuronal cell bodies) in diabetes is currently unknown. These changes include alterations in Schwann cell–axon transport, alterations in protein expression in the DRG, demyelination and degeneration. GAP43, growth-associated protein 43; HSP, heat shock protein; PARP, poly(ADP-ribose) polymerase. Adapted with permission from ref.37, Elsevier.
Although diabetic neuropathy is not considered primarily a demyelinating neuropathy, Schwann cells are targeted by chronic hyperglycaemia, and more severe cases of diabetic neuropathy in patients include features of demyelination33,34,35. Given the close and intimate mutual support between axons and Schwann cells, Schwann cell damage might lead to several alterations in the axon. For example, Schwann cells have a fundamental role in regulating the cytoskeletal properties of axons, including the position of proteins at the nodes of Ranvier and axon trafficking parameters36. Failure by Schwann cells to support axons might involve inadequate provision of cytoskeletal support, trophic factors or failure of Schwann cell–axon ribosome transfer that supports intra-axonal mRNA translation within distal axons37. In mice, Schwann cells contain ribosome-filled vesicles that, when transferred to desomatized axons, can control axonal protein synthesis38. In settings of axonal damage and stress, this transfer of ribosomes may place increased importance on axon–Schwann cell interactions38,39.
Whether diabetes promotes intrinsic programmes within axons that facilitate axonal degeneration is unclear. Studies of Wallerian degeneration have identified intracellular signalling pathways that actively induce axonal degeneration, and mononucleotide adenylyltransferase (NMNAT; also known as NMN/NaMN adenylyltransferase) seems to be a key regulator of this pathway. However, whether these pathways are activated in diabetes is not yet clear40.
Changes in axons, especially distal terminals, are associated with changes in the neuronal perikarya. Indeed, sensory neurons within the DRG alter their phenotype in chronic experimental diabetes, which might be critical in how they support distal axon branches. For example, in chronic T1DM in rats, there is progressive loss of synthesis and export of neurofilament polymers, which are essential structural scaffolds of the axon. Reduced mRNA expression encoding neurofilament has been proposed to underlie this loss of neurofilament polymers41. Preclinical studies in diabetic rodents also associate endoplasmic reticulum stress with diabetes-mediated peripheral nerve damage42 that would affect nerve function. Similarly, in vitro and in vivo experiments in rodent models have demonstrated that hyperglycaemia alters the function of key plasticity molecules, such as growth-associated protein 43 (GAP43; also known as neuromodulin) and β-tubulin, and the expression patterns of heat shock proteins (HSPs)43,44 and poly(ADP-ribose) polymerase (PARP)45,46 in the DRG. Although the mechanisms of injury remain under investigation, data suggest that dysfunction in these pathways promotes abnormal protein processing, oxidative damage and mitochondrial dysfunction, leading to loss of peripheral nerve function37. In support of this theory, modulating specific molecules in these pathways can lead to improvement in nerve function. For example, regulating HSP90 activity improved nerve conduction velocity (NCV) and responses to thermal and mechanical stimuli (both of which are clinically relevant end points), most likely by improving mitochondrial function44.
More recent array studies have also demonstrated a range of both mRNA and microRNA alterations in DRG sensory neurons exposed to chronic diabetes47,48,49,50,51. Indeed, upregulation of pathways involved in inflammation, bioenergetics and lipid processing have been reported in arrays of sciatic nerves from preclinical models of T1DM and T2DM52,53. In addition, one study comparing gene expression patterns in peripheral nerves from mouse models of diabetic neuropathy with nerves from patients with T1DM and T2DM revealed multiple highly conserved pathways involved in adipogenesis, lipid synthesis and inflammation54.
Other specific changes in the DRG and nerve function can be linked to diabetic neuropathy, including altered spliceosome function, changes in expression of survival motor neuron protein and upregulation of GW-bodies (sites of mRNA processing)55. Analysis of rodent models with longstanding diabetes has been essential to model chronic human disease. DRG have reductions in local blood flow, but whether this contributes to neuronal damage or follows lower oxygen demand is unclear56.
Hyperglycaemia and hyperlipidaemia
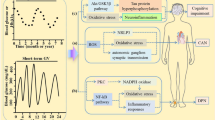
How the peripheral nervous system uses substrates for energy, especially in diabetes, is necessary to understand the pathogenesis of diabetic neuropathy. In Schwann cells, DRG neurons and axons, both glucose and fatty acids produce NADH and FADH2 via glycolysis and the tricarboxylic acid cycle (glucose) and β-oxidation (fatty acids). When long-chain fatty acids are transported into Schwann cells to undergo β-oxidation, each β-oxidation cycle forms one molecule of acetyl-CoA, which is transported to the tricarboxylic acid cycle for NADH and FADH2 formation. However, during substrate overload, such as in diabetes, the transport system becomes saturated, and acetyl-CoA molecules are converted to acylcarnitines. The accumulation of acylcarnitines is toxic to both Schwann cells and DRG neurons, adding to the ongoing nervous system injury in diabetic neuropathy. Accumulated acylcarnitines are released from Schwann cells and can induce axonal degeneration, which has been proposed to involve mitochondrial dysfunction and a maladaptive integrated stress response in Schwann cells57.
NADH and FADH2 are shuttled in the mitochondria through Complexes I–IV to produce ATP through oxidative phosphorylation. A byproduct of oxidative phosphorylation is the production of low levels of reactive oxygen species (ROS) that are easily neutralized by innate cellular antioxidants, such as superoxide dismutase, glutathione and catalase58,59,60,61,62,63. However, during excess substrate load, such as in diabetes, oxidative phosphorylation fails, leading to loss of ATP production and increased ROS levels, which subsequently leads to mitochondrial failure and metabolic and oxidative damage of Schwann cells and DRG neurons64,65,66. Dysfunctional mitochondria produce insufficient energy and lose the ability to normally traffic down axons, further promoting axonal disruption and injury67.
Increased glucose levels leads to glucose metabolism via the polyol and hexosamine pathways, resulting in increased ROS and inflammation, respectively, largely owing to mitochondrial injury37, which contributes to ongoing nervous system dysfunction. Increased glucose levels lead to the glycation of numerous structural and functional proteins to produce advanced glycation end-products (AGEs). AGEs result in altered or loss of protein function and interact with AGE-specific receptor (RAGE) to modify gene expression and intracellular signalling and promote the release of pro-inflammatory molecules and free radicals68. In parallel, the excessive free fatty acids catabolized by β-oxidation in response to hyperlipidaemia can injure the peripheral nervous system, particularly Schwann cells69, through ROS generation and systemic and local inflammation via macrophage activation with subsequent cytokine and chemokine production70 (Fig. 3).

Hyperglycaemia and dyslipidaemia, together with altered insulin signalling, lead to several pathological alterations in neurons, glia and vascular cells that can lead to nerve dysfunction and ultimately, neuropathy, including DNA damage, endoplasmic reticulum stress, mitochondrial dysfunction, neurodegeneration and loss of neurotrophic signalling, and can trigger macrophage activation. The importance of these pathways in the development of neuropathy varies with cell type, disease profile and time, as distinct cell types are more or less susceptible to injury depending on the metabolic impairments. AGE, advanced glycation end-product; FFAs, free fatty acids; Glucosamine-6-P, glucosamine 6-phosphate; LDL, low-density lipoprotein; LOX1, oxidized LDL receptor 1; RAGE, AGE-specific receptor; ROS, reactive oxygen species; TLR4, Toll-like receptor 4; UDP-GlcNAc, uridine diphosphate N-acetylglucosamine.
Other lipids adversely affect the peripheral nervous system in diabetic neuropathy. Oxidation of cholesterol to oxysterols in neurons mediates tissue injury58,71, whereas plasma lipoproteins, particularly low-density lipoproteins (LDLs), are oxidized by ROS and bind oxidized LDL receptor 1 (LOX1) (ref.72), Toll-like receptor 4 (TLR4)73 and RAGE74. Binding of oxidized LDLs to these receptors activates a series of signalling cascades, including activation of caspase 3 and nuclear DNA degradation74, that mediate additional inflammation and ROS accumulation, with continued and progressive nerve injury72,75,76 (Fig. 3).
Microvascular contributions
Although many studies show no change in blood flow associated with the development of diabetic neuropathy, deficiencies in the blood supply to peripheral nerves is considered a possible additional pathological mechanism of diabetic neuropathy (reviewed in ref.77). Microcirculatory dysfunction is strongly associated with peripheral nerve dysfunction, and a cycle of poor microcirculation leading to additional nerve damage has been proposed. Increases in endoneurial capillary density are present in patients with diabetes compared with healthy individuals, suggesting that capillary density may respond to diabetes-induced nerve ischaemia78. Blood vessels develop thickening of their basement membrane that correlates with nerve damage in patients79. Moreover, poor vasodilation of epineurial arterioles has been reported in diabetic rats, and this change appears before decreases in NCV80. In preclinical models, reduced endoneurial blood flow could be improved by treatment with vasodilators. Finally, diabetes has been reported to decrease mediators of blood vessel formation, including insulin growth factors, vascular endothelial growth factor (VEGF), nerve growth factor (NGF) and angiopoietins. This view is supported by preclinical studies in which administration of VEGF in diabetic rats increased NCV and vasa nervora (small arteries that supply peripheral nerves) density81. Together, these findings suggest that addressing microvascular problems in diabetes should be considered as an adjunct therapy.
Impaired insulin signalling
As structural similarities between NGF and insulin were first recognized, evidence of direct neuronal actions of insulin have emerged82. Initial work demonstrated that insulin acts as a growth factor for cultured adult sensory neurons, leading to increasing neurite outgrowth83. Subsequent studies demonstrated the expression of insulin receptors by sensory neurons in DRGs and axons, particularly at nodes of Ranvier84,85, and the reversal of features of experimental diabetic neuropathy with intrathecally or intranasally delivered insulin independent of glucose levels48,84. Insulin administered near the nerve, or in the plantar skin where it accesses dermal axons, also repairs abnormalities of diabetes in experimental animal models86,87.
Despite these findings, correcting hyperglycaemia with insulin has little effect on diabetic neuropathy in patients with T2DM, in whom the disorder correlates more strongly with components of the metabolic syndrome. By contrast, normoglycaemia achieved with insulin treatment provides a substantial therapeutic benefit for those with T1DM and diabetic neuropathy. This conundrum might be, in part, due to the development of insulin resistance in neurons in those with T2DM, which is not unlike the resistance developed in fat, muscle and adipose tissue88,89. Indeed, mice with T2DM have systemic resistance to insulin therapy, and controlling glucose with insulin has little effect on diabetic neuropathy90. Altered phosphorylation of insulin-receptor substrate 2, part of the downstream insulin transduction pathway, seems important for the development of insulin resistance91.
Mechanisms of pain
Neuropathic pain is defined as pain caused by a lesion or disease of the somatosensory nervous system. Approximately 30–50% of patients with diabetic neuropathy develop neuropathic pain92, which most commonly takes the form of spontaneous (that is, stimulus-independent) burning pain of the feet. Patients can also report other positive sensory symptoms, such as brush-evoked allodynia (when a normally non-noxious stimulus evokes pain) and paresthesias. These positive sensory symptoms are often accompanied by sensory loss, and patients will comment on the paradox that their feet are continuously painful yet insensate to touch. Why only some patients with diabetic neuropathy develop neuropathic pain whereas others do not remains unclear, although this likely reflects a complex interplay of vulnerabilities, including genetic factors, the somatosensory circuitry and psychological factors in the face of stressors, such as the metabolic dysfunction of diabetes and neuropathy severity93.
Risk factors for painful diabetic neuropathy
Most studies assessing the risk factors for neuropathic pain in diabetic neuropathy are cross-sectional rather than prospective, use univariate rather than multivariate analysis and do not always specify the comparator (ideally the comparison should be painful versus painless diabetic neuropathy). However, despite these caveats, a number of interesting factors have emerged that have recently been summarized94. Consistent with risk factors for many neuropathic pain disorders, female sex is a risk factor for painful diabetic neuropathy95. In addition, although neuropathic pain can be observed at all severities of neuropathy, a higher prevalence of pain has been consistently reported in patients with more severe neuropathy as defined by clinical scoring scales and sensory loss upon quantitative sensory testing75,95,96. Several metabolic factors are associated with painful diabetic neuropathy compared with painless diabetic neuropathy, including poor glycaemic control96, impaired renal function95 and high body mass index (BMI)75. These factors might be associated with neuropathy progression, but some could be associated with the hyperexcitability of sensory neurons and the development of pain.
Hyperexcitability of sensory neurons
Injured sensory neurons, such as in diabetic neuropathy, develop hyperexcitability and can generate action potentials in the absence of a stimulus (spontaneous activity) and develop an altered stimulus–response function97,98,99 (Fig. 4). This aberrant activity is crucial for the maintenance of neuropathic pain, even in patients with longstanding pain. Indeed, in one study, a targeted local anaesthetic block to the nerves innervating the lower limb in patients with neuropathic pain in the feet led to the resolution of ongoing pain and evoked pain for the duration of the nerve block100. This finding suggests that primary afferent hyperexcitability is a critical pathophysiological driver of pain, and we need to both understand its basis and develop therapeutics to target it.

a | Several alterations to peripheral and central neurons contribute to the pathophysiology of painful diabetic neuropathy. Ion channels at the terminals of nociceptors can undergo glycation through the addition of methylglyoxal to form advanced glycation end-products (AGEs), which can contribute to gain of function of these channels and neuronal hyperexcitability. Changes at the perikaryon include increased expression of voltage-gated sodium channels, such as Nav1.8, which can lead to hyperexcitability. In myelinated axons, the expression of shaker-type potassium (Kv) channels is reduced, which can also contribute to hyperexcitability. Hyperexcitability of neurons leads to increased stimulus responses and ectopic neuronal activity, leading to excessive nociceptive input to the spinal cord. In the spinal cord, microglia become activated and further enhance excitability within the dorsal horn. b | Several ascending pathways are involved in pain perception and the psychological changes associated with pain, for example, the spinothalamic pathway (1), which is involved in pain perception, and the spinoreticular tract. In addition, ascending pathways that travel via the parabrachial nucleus (2) to the hypothalamus and amygdala (3) are involved in autonomic function, fear and anxiety. Descending pathways inhibit or facilitate the transmission of nociceptive information at the spinal level (4). Changes induced by peripheral neuropathy are shown in boxes. Adapted with permission from ref.37, Elsevier, and from American Diabetes Association, Diabetes Care, American Diabetes Association (2013)231. Copyright and all rights reserved. Material from this publication has been used with the permission of American Diabetes Association.
The expression, trafficking and phosphorylation status of ion channels within sensory neurons are critical determinants of excitability101. Nociceptors (sensory neurons that detect tissue injury) express specialized voltage-gated sodium channels (VGSCs) to detect injurious stimuli102. Multiple α-subunits of VGSCs exist, of which Nav1.7, Nav1.8 and Nav1.9 are enriched in nociceptors. Nav1.7 and Nav1.9 are important as threshold channels, setting the excitability of the terminal and amplifying the sensory signal, whereas Nav1.8 is important for the upstroke of the action potential in nociceptors103. Hyperpolarization-activated cyclic nucleotide-gated channels also act to regulate neuronal excitability and are important for repetitive firing, whereas potassium channels act as important breaks on excitability.
Genetic variants in ion channels as well as alterations in their expression, trafficking and post-translational modifications have been implicated in the pathogenesis of neuropathic pain. For example, painful diabetic neuropathy is associated with increased expression of Nav1.8 in sensory neurons; in rodent models of painful diabetic neuropathy, this increase reduces conduction failure in C fibre nociceptors, resulting in increased impulse conduction to the central nervous system (CNS), which promotes neuropathic pain104. Furthermore, diabetes results in increased expression of reactive metabolites such as methylglyoxal, which can modify cellular proteins and alter their function. In one study, patients with painful diabetic neuropathy had higher levels of methylglyoxal than patients with painless diabetic neuropathy105, although this finding was not replicated in a subsequent study106. Methyglyoxal can post-translationally modify Nav1.8, resulting in Nav1.8 gain of function and sensory neuron hyperexcitability, which can contribute to pain in rodent models of diabetic neuropathy105. In addition, methylglyoxal might modify and enhance the activity of the non-selective cation channel TRPA1, leading to sensory neuron hyperexcitability in preclinical models107. The shaker-type potassium channels attenuate axonal excitability, particularly in the context of injury108; however, their expression at the juxtaparanodes of myelinated axons is reduced in animal models of diabetes and in patient-derived samples109, suggesting that this adaptive mechanism to suppress hyperexcitability is lost in painful diabetic neuropathy.
Gain-of-function variants in SCN9A (encoding Nav1.7) are present in a number of pain disorders, such as erythromelalgia and small-fibre neuropathy110, therefore this gene was a natural candidate to test in painful diabetic neuropathy. Rare variants in SCN9A were more common in patients with painful diabetic neuropathy than in those with painless diabetic neuropathy in two studies111,112, although this finding was not replicated in one study of painful neuropathy owing to multiple aetiologies including diabetes113. A number of the rare SCN9A variants resulted in gain of function of Nav1.7, owing to impaired inactivation, following expression in HEK cells112. This finding is an example of a gene–environment interaction in that patients with these variants did not report pain before the onset of diabetes. A recent trial of a small-molecule selective Nav1.7 blocker in painful diabetic neuropathy was negative114; however, the study did not stratify patients according to the presence of SCN9A variants. Selective blockers of Nav1.7 have been used in other neuropathic pain disorders with some success, such as trigeminal neuralgia, therefore further trials in diabetic neuropathy are expected115.
T-Type Ca2+ channels have also been implicated in painful diabetic neuropathy through the regulation of the subthreshold excitability of nociceptors. For example, CaV3.2 activity is enhanced in diabetes through the glycosylation of extracellular arginine residues resulting in DRG neuron hyperexcitability116. Reducing this calcium current via gene silencing of CaV3.2 reduces pain-related hypersensitivity in animal models of diabetes117.
Local factors within the DRG perikarya might also contribute to sensory neuron dysfunction and pain development. For example, mice that were fed a high-fat diet and underwent exercise had lower levels of pro-inflammatory cytokines, such as TNF and IL-1β, as well as reduced mechanical hypersensitivity, compared with sedentary mice fed a high-fat diet118. These cytokines are capable of directly sensitizing sensory neurons.
Dysfunction within the CNS
In diabetic neuropathy, enhanced input from spontaneously active nociceptors increases synaptic transmission within the spinal cord, further amplifying nociceptive signalling in a process termed central sensitization119. This process occurs as a consequence of spatial and temporal summation of nociceptive inputs, such that neurons in the spinal cord dorsal horn have an enhanced response to the same nociceptive input. In animal models of diabetic neuropathy, spinal neurons have hyperexcitability to peripheral stimuli, which is associated with altered shape (increased length and spine head diameter), increased density and redistribution of dendritic spines120. Changes in glial cells are also apparent in diabetic neuropathy. Microglia (the resident immune cells of the CNS) transform to a pro-inflammatory phenotype in diabetic neuropathy, although the mechanism by which this occurs is currently unknown. These cells can release factors, such as brain-derived neurotrophic factor (BDNF), that amplify nociceptive synaptic signalling within the spinal cord121 and contribute to mechanical pain-related hypersensitivity in animal models of painful diabetic neuropathy122. The role of astrocytes is less clear, as some studies demonstrated activation of astrocytes in models of painful diabetic neuropathy123 whereas others have not124.
The processing of nociceptive information within the dorsal horn of the spinal cord is regulated by a descending pain modulatory system that is mediated by projections from the brainstem, which can either inhibit or facilitate transmission of nociceptive information at the spinal level125 (Fig. 4). The balance between such facilitation and inhibition has been suggested to have a role in the development of neuropathic pain. Data from both animal models and patients suggest that spinal disinhibition can contribute to the development of neuropathic pain in diabetic neuropathy126. Functional MRI studies suggest that facilitation via the ventrolateral periaqueductal grey is increased in patients with painful diabetic neuropathy compared with those with painless diabetic neuropathy127. Conditioned pain modulation can be used as an experimental measure to test the integrity of this descending pain modulatory system; in this process, a painful conditioning stimulus is applied to one body site and will usually reduce pain evoked by a stimulus applied to a different body site. Conditioned pain modulation is impaired in some patients with painful diabetic neuropathy128.
Changes have also been noted in higher brain centres, including reduced connectivity between the thalamus and cortex129 and structural changes, such as a greater reduction in thalamic grey matter volume, in patients with painful diabetic neuropathy compared with those with painless diabetic neuropathy130. Such changes could partly reflect deafferentation due to damage of the somatosensory pathways, and the reduced connectivity between the thalamus and cortex could represent reduced thalamic feedback and lead to aberrant pain processing. In concert with these changes, painful diabetic neuropathy is associated with important psychological changes and comorbidities, including increased anxiety, depression and impaired sleep131. Recent preclinical studies emphasize the importance of chronic neuropathic pain in the aetiology of anxiety132. Further prospective clinical studies will be needed to understand the directionality of the relationship between pain and these psychological factors.
Diagnosis, screening and prevention
Diagnosis
Diabetic neuropathy is the presence of symptoms and/or signs of peripheral nerve dysfunction in patients with diabetes after other aetiologies have been excluded9. Typically, the presence of more symptoms or signs of nerve dysfunction confers higher certainty about the diagnosis9, although abnormalities in lower-limb NCV and sensory and motor nerve amplitudes assessed in nerve conduction studies (NCS) provide even further evidence. For the vast majority of patients, the diagnosis of diabetic neuropathy is based solely on the history and examination and no additional testing is needed9,133. Objective confirmatory testing is most commonly used in the research setting or as part of the diagnostic work-up of patients with atypical clinical presentations.
The symptoms of diabetic neuropathy are numbness, tingling, pain and weakness and unsteadiness, starting distally (at the toes) and spreading proximally and then to the upper limb digits when the lower-limb symptoms reach the knees. Patients often have predominantly small-fibre neuropathy early in the course of diabetic neuropathy or when diagnosed with prediabetes134, and have distal painful symptoms of burning, lancinating, freezing pain that are greater at rest. Large-fibre injury usually occurs later in the disease course, but this is not always the case.
Clinical findings of diabetic neuropathy are a loss of sensation to pinprick, temperature (mostly cold), vibration and proprioception in a ‘stocking and glove’ distribution. These sensory modalities are tested initially by the application of the sensory stimulus to a region where normal responses are expected, such as the forehead. Following this, the stimulus is applied to the great toe and then moved proximally up the limb to the level where the sensation is felt to be normal. Pinprick sensation is tested using a sharp object, such as a safety pin, that is discarded after each patient, whereas temperature is tested using a cool material, such as a metallic object. Vibration is tested by application of a vibrating tuning fork to the bony prominence at the dorsum of the great toe and then determining when the vibration stops, and proprioception is examined by small movements of the distal interphalangeal joint of the great toe. Pinprick and temperature sensations are mediated via small nerve fibres, whereas vibration sensation and proprioception are mediated by large nerve fibres.
Loss of ankle reflexes occurs early in diabetic neuropathy; thus, initial examination should include reflex testing. Later, weakness of small foot muscles and dorsiflexors is observed. Although many patients notice symptomatic weakness, major weakness on examination is only observed in later stages of advanced diabetic neuropathy. Early neurological dysfunction in the upper limbs should raise suspicion of a mononeuropathy or an alternative diagnosis.
The symptoms and clinical signs of diabetic neuropathy can be combined in scales, such as in the Toronto Clinical Neuropathy Score135, the modified Toronto Clinical Neuropathy Score136 or the Michigan Diabetic Neuropathy Score137, which have defined cut-off values for the presence of neuropathy. Other scales include signs only or a combination of signs and ancillary tests.
As previously mentioned, in research settings, a diagnosis of confirmed diabetic neuropathy commonly requires abnormality of objective tests, usually changes in NCS (Fig. 5), or validated measures of small nerve fibres if an NCS is normal138 as NCS do not assess small-fibre function. NCS are performed with surface stimulating and recording techniques that test motor and sensory nerve fibres in the upper and lower limbs. Changes in NCS in patients with diabetic neuropathy include decreased amplitudes, decreased conduction velocities and prolonged F responses139. Changes in the amplitude of motor nerve fibres typically follow changes in the amplitude of sensory nerve fibres, and lower-limb changes precede upper-limb changes as diabetic neuropathy is a length-dependent process.

Abnormal sural nerve recording from a patient with diabetic neuropathy showing a decreased sural sensory nerve action potential amplitude (normal >6 µV) and slow sural sensory nerve conduction velocity (normal >39 m s–1; panel a). Intraepidermal nerve fibres (arrows) and branched fibres (arrowhead) in a skin biopsy sample from a healthy individual (panel b) and from a patient with small-fibre neuropathy (panel c). A sural nerve biopsy sample exhibiting evidence of axonal loss of small-diameter and large-diameter nerves in diabetic neuropathy (panel d). Image (×20 magnification) of an Epon-embedded, 0.5 µm thick section stained with toluidine blue (panel d). AMP, amplitude; CV, conduction velocity; Dist, distance; Lat, latency; NCS, nerve conduction studies; Rec, recording; Stim, stimulating. Parts b and c adapted from ref.232, Springer Nature Limited.
NCS are normal in patients with primarily small-fibre neuropathy, and these patients typically also have an almost normal clinical examination140. The gold standard for the diagnosis of small-fibre neuropathy is measurement of intraepidermal nerve fibre density (IENFD) by skin punch biopsy141,142, but this invasive approach is rarely necessary in routine diagnosis and is primarily used for research purposes. Other confirmatory tests of small nerve fibre damage that are most commonly used for research purposes include quantitative sensory thermal thresholds for reduced cooling detection thresholds or elevated heat thresholds143, laser Doppler flare imaging studies144 and corneal confocal microscopy to measure nerve fibre length in Bowman’s layer of the cornea, which is reduced in diabetic neuropathy145. However, the validity of these tests is not as well defined as for NCS, and these tests have no clear role in routine clinical diagnosis146.
If a patient with numbness, tingling, pain and/or weakness presents with atypical features, such as acute or subacute presentation of neuropathy, non-length dependence, motor predominance and/or asymmetry of neuropathic signs and/or symptoms, a neurological consult and additional testing should be prompted30. Additional testing depends on clinical presentation but typically includes measuring serum vitamin B12 levels, thyroid function tests, serum protein electrophoresis with immunofixation and markers of autoimmune disorders. Cerebrospinal fluid examination using lumbar puncture to assess protein levels, genetic testing and MRI of nerve roots and peripheral nerves is frequently required for the correct diagnosis in atypical clinical presentations. Rarely, sural or radial nerve biopsy is necessary.
Differential diagnosis
Assessing other aetiologies for which the clinical presentation can mimic that of diabetic neuropathy is essential during diagnosis of diabetic neuropathy133. A complete history and examination are most helpful to assess other potential aetiologies of neuropathy, such as alcohol abuse (alcoholic neuropathy), genetic neuropathies, neoplasia, medication-induced neuropathy (medications such as chemotherapy and HIV treatments) and amyloidosis. Laboratory assessment includes measurement of vitamin B12 levels (methylmalonic acid with or without homocysteine can provide additional information) to evaluate for vitamin B12 deficiency, particularly in patients receiving metformin147, thyroid function tests and serum immunoelectrophoresis with immunofixation to evaluate for a monoclonal gammopathy. More genetic forms of polyneuropathy are being discovered, but the role of routine genetic testing is unclear unless the patient has the phenotype of genetic polyneuropathy148. As new treatments become available for disorders such as familial transthyretin amyloidosis, and as more information about the phenotypes of genetic neuropathies becomes available, it might be advisable to consider genetic testing earlier rather than later in the course of polyneuropathy149,150. However, determining which patients benefit from genetic testing needs further study.
Screening
Screening for diabetic neuropathy using a recommended evidence-based screening algorithm is advised for all patients with diabetes9. Current position statements from the American Diabetes Association (ADA) and guidelines from the Canadian Diabetes Association recommend screening for diabetic neuropathy at diagnosis and annually for patients with T2DM and 5 years after diagnosis and then annually for patients with T1DM9,151.
The tests for screening need to be rapid, reliable and simple, and advocating for anything other than very simple test paradigms will lead to a lack of screening. Several simple sensory tests can be carried out to detect diabetic neuropathy152,153, for example, the 10 g monofilament test can be used to predict incident diabetic neuropathy154. The value of this monofilament is that higher insensitivities predict a high risk of foot ulceration; thus, the practitioner needs to use only a single tool for screening for diabetic neuropathy and to assess risk of foot ulceration155. Vibration testing with a 128 Hz tuning fork (timed or number of times felt) has similar discriminating abilities to the monofilament test and is also quick and easy to perform156. Assessment of deep tendon reflexes has good test characteristics, although not quite as high as monofilament or vibration testing156,157. Other screening methods, such as the Michigan Neuropathy Screening Instrument, that use a questionnaire and simple examination have also been validated and are useful for screening and assessing the severity of neuropathy137,158.
Prevention
The consistent feature between T1DM and T2DM is hyperglycaemia; therefore, treatment of hyperglycaemia logically would be the best preventive treatment for diabetic neuropathy. However, although enhanced glycaemic control effectively reduced the incidence of diabetic neuropathy in patients with T1DM, the effect was much smaller, or in some studies absent, in patients with T2DM in one Cochrane systematic review159. Indeed, the difference in patients with T2DM did not reach statistical significance in either the meta-analysis or in individual studies. The T1DM meta-analysis was dominated primarily by the Diabetes Control and Complications Trial (DCCT), which accounted for 1,186 of the 1,228 patients in the meta-analysis16 and demonstrated an annualized risk difference of –1.84 (95% CI –2.56 to –1.11) in favour of enhanced glycaemic control160. The T2DM meta-analysis was dominated primarily by the ACCORD and VADT studies, which accounted for 6,568 of the 6,669 patients in the meta-analysis161,162 and reported an annualized risk difference of –0.58 (95% CI –1.17 to 0.01) in favour of enhanced glucose control, although this value did not reach statistical significance159. Since the publication of this systematic review, another study reported no difference in the prevalence of diabetic neuropathy in patients with screen-detected T2DM who received routine care compared with those who received intensive treatment (encompassing goal-directed glycaemia and cholesterol and blood-pressure management)163. Importantly, the two groups had little to no differences in glycaemic and other metabolic measurements. Taken together, current data indicate that enhanced glucose control has a large effect on the prevention of diabetic neuropathy in patients with T1DM, whereas the effect in T2DM is much less, although it is likely still important.
Exercise is emerging as a promising prevention strategy in diabetic neuropathy. One study demonstrated increased distal leg IENFD by 1.5 fibres mm–1 in patients with diabetes (without neuropathy) who received a weekly structured and supervised exercise programme, but IENFD was unchanged in patients who received lifestyle counselling (–0.1 fibres mm–1; P = 0.03)164. This study indicates the potential for exercise to prevent nerve injury and even promote nerve regeneration, although the study was not randomized and the effect on patient-oriented neuropathy outcomes is still not clear. Currently, routine exercise is recommended to all patients with diabetes, but no firm recommendations can be made pertaining to the role of exercise and the prevention of neuropathy. Additional studies investigating the effect of exercise on neuropathy outcomes in patients with established diabetic neuropathy are discussed below in the Management section.
Only one study has focused on neuropathy outcomes after bariatric surgery, although this study did not focus on prevention. This prospective cohort study demonstrated significant improvements in neuropathy outcome measures 6 months after Roux-en-Y gastric bypass surgery in patients with T2DM and preoperative diabetic neuropathy165. However, of note, no control group was provided and outcome measures were not masked to the intervention. Despite the results from this study, whether bariatric surgery can prevent neuropathy is still unclear. No studies to date have focused on medical or pharmacological weight loss and the prevention of neuropathy. Accordingly, future studies need to determine the role of bariatric surgery and medical weight loss in the prevention of diabetic neuropathy, including whether there is a difference between the two approaches.
Management
The current approaches to management of diabetic neuropathy focus on improving glycaemic control (mainly in patients with T1DM), lifestyle modifications (mainly in patients with T2DM) and management of neuropathic pain. The optimal therapeutic approach for patients with T2DM includes lifestyle interventions, specifically diet and exercise, coupled with optimal lipid and blood pressure control. Glycaemic control with a HbA1c goal of <6 increases mortality in patients with T2DM166 and has little effect on diabetic neuropathy, therefore it is not recommended as standard of care162,166,167. Rather, good glycaemic control as part of a more holistic, personalized approach to the treatment of T2DM is the optimal choice. Many therapeutic interventions have failed; however, several promising therapies are in ongoing clinical trials.
Improved glycaemic control
As previously mentioned, improved glycaemic control plays a role in preventing the onset and progression of diabetic neuropathy in patients with T1DM. The landmark trial with the most robust data supporting this is the DCCT/Epidemiology of Diabetes Interventions and Complications (EDIC); although diabetic neuropathy was uncommon at the start of DCCT/EDIC, intensive glucose control significantly delayed its development and progression over time16, and similar improvements in neuropathy outcomes with intensive insulin treatment were reported by two other smaller European cohorts168,169.
As previously discussed, large meta-analyses have demonstrated little to no effect of glucose control on diabetic neuropathy in patients with T2DM. However, some studies support the idea that glucose control remains important. For example, a relatively small trial of Japanese patients with early T2DM and diabetic neuropathy demonstrated improvement in several measures of diabetic neuropathy, including NCS, with intensive insulin treatment170. Most recently, data from two cohorts with uncontrolled T2DM and neuropathy at baseline demonstrated improvement in several measures of large-fibre and small-fibre neuropathy with improvement in HbA1c to near-normal levels after 2 years171. In addition, because factors other than hyperglycaemia, including metabolic factors such as dyslipidaemia or other components of the metabolic syndrome, insulin resistance and chronic inflammation, are involved in the pathophysiology of diabetic neuropathy, particularly in patients with T2DM, specific classes of glucose-lowering agents that target these factors are emerging as potentially effective in delaying progression of neuropathy17.
Diet and lifestyle interventions
Three uncontrolled studies and one small randomized study have shown the potential for exercise to improve neuropathy outcomes in patients with established neuropathy172,173,174,175. One study evaluated the effect of a lifestyle intervention consisting of individualized diet and exercise for 12 months in 32 patients with neuropathy caused by impaired glucose tolerance175. Although the BMI of participants decreased by only an average of 1.1 kg m–2, IENFD levels in the proximal thigh significantly increased by 1.4 fibres mm–1 and significantly correlated with decreased neuropathic pain, indicating that the IENFD increase is likely clinically relevant to patients.
A second study evaluated the benefit of 4 months of a lifestyle intervention of 30–90 min of supervised exercise twice weekly, with the addition of home exercise, in 36 patients with diabetes and/or metabolic syndrome174. Dietary counselling was provided only twice during this study, and the BMI decreased by an average of only 0.11 kg m–2. Following the exercise intervention, the cutaneous nerve regenerative capacity (measured by IENFD) increased from 0.051 to 0.072 fibres mm–1 per day (P = 0.002), and, notably, those with improvements in more components of the metabolic syndrome had a greater increase in cutaneous nerve regenerative capacity (P < 0.012)174.
The third study demonstrated improved intraepidermal nerve fibre branching at the proximal thigh after 10 weeks of aerobic and strengthening exercise (0.11 branch nodes per fibre; P = 0.008), despite no change in BMI, in 17 patients with diabetic neuropathy173. Furthermore, neuropathic symptoms, including pain, were significantly reduced. IENFD at the proximal thigh also improved, although this result did not meet statistical significance (1.68 fibres mm–1; P = 0.09).
Finally, a small randomized trial that included a mixture of patients with T1DM and T2DM demonstrated improvements in both groups in some NCS parameters and vibration perception thresholds after 4 years of an aerobic exercise regimen172. BMI changed little over the study duration, and IENFD was not measured. Limitations of this study include the lack of designated primary and secondary outcomes, no blinded outcome assessments, inclusion of individuals with T1DM and T2DM, unequal randomization of the study population and no patient-oriented outcomes.
These studies show the promise of exercise regimens to improve IENFD without significant weight loss, but importantly only one study included a control group. Overall, promising data for exercise to prevent and/or improve diabetic neuropathy exist, but well-designed future studies are needed to firmly establish this as an effective intervention.
Disease-modifying therapies
Several therapies have been designed to target the pathogenesis of diabetic neuropathy. These studies have revealed some evidence of efficacy of these therapies, but unequivocal evidence of benefit from phase III studies is currently lacking.
α-Lipoic acid has been shown to improve symptoms in diabetic neuropathy. The multicentre, randomized, double-blind, placebo-controlled ALADIN III trial demonstrated significant improvement in the Neuropathy Impairment Score (NIS) in patients who received α-lipoic acid but no significant improvement in the Total Symptom Score (TSS)176. In addition, in the SYDNEY2 trial, 181 patients with diabetic neuropathy received once-daily oral doses of 600 mg, 1,200 mg or 1,800 mg α-lipoic acid or placebo for 5 weeks177. The primary outcome measure was the change from baseline of the TSS, which decreased by 51% in the 600 mg group, 48% in the 1,200 mg group and 52% in the 1,800 mg group compared with 32% in the placebo group (P < 0.05 versus placebo); on the basis of these results, a dose of 600 mg once daily seems to provide the optimum risk-to-benefit ratio177. The largest and longest trial, Nathan I178, a multicentre, randomized, double-blind, parallel-group trial, evaluated the efficacy and safety of 600 mg α-lipoic acid over 4 years in patients with mild-to-moderate diabetic neuropathy. No significant difference in the primary end point, a composite score (NIS-Lower Limbs (NIS-LL) and seven neurophysiological tests), was observed between α-lipoic acid treatment and placebo. However, clinical subscores assessing components of neuropathy, such as distal muscle weakness alone, or a combination of weakness, reflexes and sensory changes in the arms and legs or legs alone, showed some marginal improvement from baseline in the intervention group. In addition, more patients showed a clinical improvement and fewer showed progression of the NIS (P = 0.013) and NIS-LL (P = 0.025) with α-lipoic acid than with placebo. One potential reason that could have contributed to the lack of difference in the primary end point was that NCS and quantitative sensory testing results did not significantly worsen in the placebo group as originally predicted. Global assessment of treatment tolerability did not differ between the groups. Despite these caveats, when the primary end point of a clinical trial is not met, the US FDA does not support the use of the study drug.
Benfotiamine administration has been shown to increase the levels of intracellular thiamine and reduces AGEs that induce experimental diabetic neuropathy179,180. In a short 6-week, double-blind, placebo-controlled, phase III trial181, a borderline difference in the primary outcome parameter, the Neuropathy Symptom Score, was observed between the benfotiamine and placebo groups in the per-protocol population but not in the intention-to-treat population (P = 0.055). However, no significant differences in changes in peripheral nerve function or soluble inflammatory biomarkers were observed in a longer, 24-month trial assessing high-dose benfotiamine compared with placebo182.
In a small, single-arm, open-label trial of seal oil v-3 polyunsaturated fatty acid (PUFA) supplementation (consisting of eicosapentaenoic acid, docosapentaenoic acid and docosahexaenoic acid) for 1 year in patients with T1DM and diabetic neuropathy, a 29% improvement in the primary end point, the corneal nerve fibre length, was observed, although there were no changes in other measures of neuropathy183. Confirmation of these observations is required in a larger trial.
Aldose reductase inhibitors should be effective in the treatment of diabetic neuropathy owing to the proposed involvement of the sorbitol pathway in the pathophysiology of this disorder. Since 1980, numerous clinical studies with aldose reductase inhibitors have been performed but have failed to meet regulatory requirements to be marketed in the United States. However, epalrestat is marketed in Japan for the treatment of diabetic neuropathy. Although several uncontrolled trials have been carried out, one controlled trial demonstrated a small but significant change from baseline in motor NCV in patients with diabetic neuropathy and a median motor NCV ≥40 m s–1 and HbA1c ≤9% with epalrestat compared with control184. Secondary end points such as the median nerve minimum F-wave latency, the vibration perception threshold and subjective symptoms were statistically significant between groups; however, significance was not reached for the cardiovascular autonomic nerve function variables.
Pain management
The consensus from multiple guidelines and systematic reviews is that calcium channel a2δ ligands, serotonin and noradrenaline reuptake inhibitors (SNRIs) and tricyclic antidepressants (TCAs) have the best evidence to support their use in the treatment of diabetic neuropathic pain185,186,187,188,189. However, comparative effectiveness studies to inform the best choice of medications are lacking. Given the similar evidence between these classes of medications, cost and severity or frequency of adverse effects should be important considerations190. A detailed approach for pain management is provided in Fig. 6 (ref.9), and evidence and recommendations for treatment are discussed below.

First-line and second-line treatments for painful diabetic neuropathy include several drug classes, such as anticonvulsants (gabapentin or pregabalin), serotonin and noradrenaline reuptake inhibitors (SNRIs; duloxetine or venlafaxine) and tricyclic antidepressants (amitriptyline, nortriptyline, desipramine or imipramine). Opioids should be avoided owing to their serious adverse effects and association with addiction.
Anticonvulsants
Of the anticonvulsants, the a2δ ligands gabapentin and pregabalin are effective for painful diabetic neuropathy. Gabapentin has been effective in most9,187,188, but not all9,187,191, trials of painful diabetic neuropathy. Given its pharmacokinetic profile, gabapentin requires gradual titration9,192,193. Pregabalin has a linear and dose-proportional absorption in the therapeutic dose range. The majority of studies of pregabalin show efficacy in painful diabetic neuropathy, with at least 30–50% improvement in pain9,192,193, although not all trials have been positive187,188,194,195, particularly in those with advanced refractory patients9,196. Some studies have suggested a dose response, with a weaker therapeutic effect at lower doses of pregabalin192. The adverse effects of both medications can include confusion and dizziness, are more severe in older patients197 and can be attenuated by lower starting doses and more gradual titration. Gabapentin is currently less expensive, an important implication for patients as out-of-pocket costs continue to rise, especially for those in high-deductible health plans in the United States190.
SNRIs
Duloxetine is a selective SNRI with demonstrated efficacy for the treatment of painful diabetic neuropathy in several multicentre randomized trials9,198,199. Duloxetine treatment can also improve neuropathy-related QOL200,201. Venlafaxine is another SNRI that can be effective in the treatment of pain in diabetic neuropathy202,203. SNRIs are associated with a range of adverse effects that can be more severe than those observed with gabapentin and pregabalin, such as dizziness, fatigue, nausea and insomnia.
TCAs
Amitriptyline is the most frequently used TCA and has demonstrated efficacy in painful diabetic neuropathy in small randomized, blinded, placebo-controlled clinical trials204,205. Nortriptyline and desipramine have fewer adverse effects than amitriptyline and imipramine and might be safer in older adults206,207. However, there are fewer and smaller randomized controlled trials indicating efficacy of nortriptyline and desipramine206,207,208.
Opioid and atypical opioid analgesics
Although there is evidence of efficacy of opioids for pain relief, these drugs are associated with a high risk of addiction and safety concerns; thus, the most recent ADA position statement does not recommend opioid use as first-line or second-line therapies for treating neuropathic pain associated with diabetic neuropathy9. An American Academy of Neurology (AAN) position statement and Centers for Disease Control and Prevention (CDC) guideline have both urged great caution in using opioids for chronic, non-cancer pain, including neuropathic pain from diabetic neuropathy209,210.
Tapentadol has demonstrated efficacy in painful diabetic neuropathy in two phase III trials211,212, although a systematic review and meta-analysis by the Special Interest Group on Neuropathic Pain (NeuPSIG) found that the evidence of tapentadol efficacy in reducing neuropathic pain was inconclusive187. Tramadol has a similar mode of action to tapentadol213. Two large studies have demonstrated the efficacy of tramadol for painful diabetic neuropathy214,215, and the effect might be long-lasting216. Oxycodone improved pain scores in two single-centre trials in patients with painful diabetic neuropathy; however, one trial had a small sample size217,218. Although tapentadol and tramadol have evidence supporting their efficacy in diabetic neuropathic pain, whether the risk-to-benefit ratio of these drugs is different from that of stronger opioids deserves further study. The role, if any, of other opioids is unclear, but these drugs should likely be avoided in most if not all patients given the mounting evidence of harm. Numerous serious adverse effects and cautions are associated with opioid analgesics, but the most concerning are prescription abuse, addiction and increased mortality219.
Quality of life
As diabetes is a chronic condition that requires lifelong medications, monitoring and adherence to dietary advice, the majority of patients experience issues with their physical and mental well-being. ‘Diabetes distress’ is a term used to describe the hidden emotional burden of diabetes220. QOL further decreases if the patient with diabetes develops diabetic complications or comorbidities, such as retinopathy, nephropathy and neuropathy. The development of neuropathic foot ulcers can lead to substantial reductions in QOL owing to the prolonged immobilization required to heal the ulcers.
Diabetic neuropathy causes significant impairment in QOL. Indeed, the QOL of patients with diabetic neuropathy is lower than in patients without neuropathy, and this difference started years before and continued for years after their neuropathy diagnosis221. Moreover, painful diabetic neuropathy has a particularly strong effect on QOL222. A high prevalence of pain due to diabetic neuropathy, with substantial sleep impairment and comorbid mood disorder, was reported in a study in India conducted by the INdINeP study group223. In addition, pain was associated with an adverse effect on employment status and reduced productivity, which in turn creates a negative effect on the economy.
Several psychometric tools are available to assess the effect of both diabetes and its complications on the lives of patients as well as the effect of medical interventions. These include the Diabetes Quality of Life (DQOL) measure, the Diabetes-Specific Quality of Life Scale (DSQOLS), the Appraisal of Diabetes Scale, the ATT-39, the Questionnaire on Stress in Patients with Diabetes-Revised, the Type 2 Diabetes Symptom Checklist, the Problem Areas in Diabetes Scale (PAID-1) and the Audit of Diabetes-Dependent Quality of Life (ADDQoL). The utility of these scales was recently reviewed224. The Nottingham Health Profile (NHP) is useful to assess the QOL in patients with diabetic neuropathic pain and involves six domains (energy, sleep, pain, physical mobility, emotional reaction and social isolation)225. Other health-related QOL instruments are used, for example, the 36-item Short Form Health Survey (SF-36)226.
It is well established that successful treatment of diabetic neuropathic pain improves QOL, evaluated using the quality assessment tools listed above. Indeed, QOL is now a routine measurement in all studies assessing the efficacy of therapeutic interventions for diabetic neuropathy and diabetic neuropathic pain. QOL is also an important consideration for individuals with severe end-stage diabetic neuropathy. Daily intensive psychological counselling rendered before and after foot amputation secondary to diabetic neuropathy reduces the sudden psychological trauma associated with amputation and significantly improves psychological well-being and QOL among amputees227.
One new way to improve the QOL in people with painful diabetic neuropathy is to use cognitive behavioural therapy (CBT). CBT can help reduce pain intensity and improve physical function. Indeed, CBT had a beneficial effect on mixed chronic pain and QOL in one recent study228. Ten weekly 90-min group CBT sessions involving motivational reinforcement and training aimed at reducing pain intensity and depression had a positive enduring benefit on a patient’s QOL for up to 6 months.
Outlook
Our understanding of diabetic neuropathy continues to advance, although at a rate slower than needed to meet the impending health-care crisis. Preclinical and large, well-conducted clinical trials have changed our practice parameters and have led to a more personalized approach to the treatment of diabetic neuropathy.
Advances in our understanding of the clinical presentation and optimal therapeutic management of diabetic neuropathy form the foundation for the current paradigm shift in the preclinical research space. Previously, preclinical studies focused on cell culture and animal models of glucose metabolism alone, and between 1980 and the present, findings from these studies were translated into 70 clinical trials, with a focus on ten different aldose reductase inhibitors, all of which failed. Although the knowledge gap in the field remains large as the previous nerve-centric focus on glucose alone did not move the field forward, the future is promising. Preclinical research is moving towards understanding global whole-nerve metabolism, nutrient overload and the sharing of energy between Schwann cells and axons in T1DM and T2DM. Progress is being made as fundamental questions are being asked by basic scientists, such as whether there is metabolic reprogramming of the peripheral nervous system during diabetes, the separate and combined roles of excessive glucose and lipids on nerve bioenergetics and the role of insulin and insulin resistance in the peripheral nervous system. Additional ongoing questions include whether axoglial sharing of energy and/or transfer of toxic byproducts occur during diabetes, how metabolic perturbations of diabetes regulate mitochondrial function in both the neurons and axons and, ultimately, how our understanding of basic peripheral nervous system metabolism and bioenergetics during diabetes will translate to the changes in neuronal and axonal structure and function that define diabetic neuropathy. Understanding these aspects of global metabolism and energy use by the peripheral nervous system is our only chance of developing meaningful therapies for diabetic neuropathy.
There is a clear call for action in the field of diabetic neuropathy. As the pandemic of diabetes and obesity continues to escalate, effective therapies to prevent and treat diabetic neuropathy are needed now. Unfortunately, large pharmaceutical companies have reduced research and clinical trials in diabetic neuropathy owing to our lack of basic understanding of this disease. This change has occurred despite the growing burden of this disease. The societal costs of diabetic neuropathy are outnumbered only by the individual costs to each patient, including pain, inability to work, poor QOL, multiple hospitalizations for ulcers and eventual amputations. Although diabetic neuropathy is the strongest predictor of mortality in T2DM, it remains the only microvascular complication of diabetes without a specific treatment. Accumulating clinical and preclinical research in the next decade can and will change this scenario.
References
International Diabetes Federation. IDF Diabetes Atlas - 8th edition: key messages. IDF https://diabetesatlas.org/key-messages.html (2019).
Tabish, S. A. Is diabetes becoming the biggest epidemic of the twenty-first century? Int. J. Health Sci. (Qassim) 1, V–VIII (2007).
World Health Organization. Diabetes. WHO https://www.who.int/news-room/fact-sheets/detail/diabetes (2018).
Wang, L. et al. Prevalence and ethnic pattern of diabetes and prediabetes in China in 2013. JAMA 317, 2515–2523 (2017).
Anjana, R. M. et al. Prevalence of diabetes and prediabetes in 15 states of India: results from the ICMR-INDIAB population-based cross-sectional study. Lancet Diabetes Endocrinol. 5, 585–596 (2017).
Centers for Disease Control and Prevention. Prediabetes: your chance to prevent type 2 diabetes. CDC https://www.cdc.gov/diabetes/basics/prediabetes.html (updated 21 Jun 2018).
Callaghan, B. C., Price, R. S., Chen, K. S. & Feldman, E. L. The importance of rare subtypes in diagnosis and treatment of peripheral neuropathy: a review. JAMA Neurol. 72, 1510–1518 (2015).
Boyle, J. P., Thompson, T. J., Gregg, E. W., Barker, L. E. & Williamson, D. F. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul. Health Metr. 8, 29 (2010).
Pop-Busui, R. et al. Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care 40, 136–154 (2017). This article contains the most recent recommendations from the ADA for the prevention, screening, diagnosis, management and treatment of diabetic neuropathy, as well as recommended research and clinical trial neuropathy end points.
Gordois, A., Scuffham, P., Shearer, A., Oglesby, A. & Tobian, J. A. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 26, 1790–1795 (2003).
Italian General Practitioner Study Group (IGPSG). Chronic symmetric symptomatic polyneuropathy in the elderly: a field screening investigation in two Italian regions. I. Prevalence and general characteristics of the sample. Neurology 45, 1832–1836 (1995).
Bharucha, N. E., Bharucha, A. E. & Bharucha, E. P. Prevalence of peripheral neuropathy in the Parsi community of Bombay. Neurology 41, 1315–1317 (1991).
Callaghan, B. C. et al. Role of neurologists and diagnostic tests on the management of distal symmetric polyneuropathy. JAMA Neurol. 71, 1143–1149 (2014).
Visser, N. A., Notermans, N. C., Linssen, R. S., van den Berg, L. H. & Vrancken, A. F. Incidence of polyneuropathy in Utrecht, the Netherlands. Neurology 84, 259–264 (2015).
Ang, L., Jaiswal, M., Martin, C. & Pop-Busui, R. Glucose control and diabetic neuropathy: lessons from recent large clinical trials. Curr. Diab. Rep. 14, 528–0528 (2014).
Martin, C. L., Albers, J. W. & Pop-Busui, R. Neuropathy and related findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care 37, 31–38 (2014).
Pop-Busui, R. et al. Impact of glycemic control strategies on the progression of diabetic peripheral neuropathy in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) Cohort. Diabetes Care 36, 3208–3215 (2013).
Franklin, G. M., Kahn, L. B., Baxter, J., Marshall, J. A. & Hamman, R. F. Sensory neuropathy in non-insulin-dependent diabetes mellitus. The San Luis Valley Diabetes study. Am. J. Epidemiol. 131, 633–643 (1990).
Partanen, J. et al. Natural history of peripheral neuropathy in patients with non-insulin-dependent diabetes mellitus. N. Engl. J. Med. 333, 89–94 (1995).
Dyck, P. J. et al. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population-based cohort: the Rochester Diabetic Neuropathy Study. Neurology 43, 817–824 (1993).
Boulton, A. J., Knight, G., Drury, J. & Ward, J. D. The prevalence of symptomatic, diabetic neuropathy in an insulin-treated population. Diabetes Care 8, 125–128 (1985).
Tesfaye, S. et al. Vascular risk factors and diabetic neuropathy. N. Engl. J. Med. 352, 341–350 (2005).
Andersen, S. T. et al. Risk factors for incident diabetic polyneuropathy in a cohort with screen-detected type 2 diabetes followed for 13 years: ADDITION-Denmark. Diabetes Care 41, 1068–1075 (2018).
Callaghan, B. C. et al. Diabetes and obesity are the main metabolic drivers of peripheral neuropathy. Ann. Clin. Transl Neurol. 5, 397–405 (2018).
Callaghan, B. C. et al. Metabolic syndrome components are associated with symptomatic polyneuropathy independent of glycemic status. Diabetes Care 39, 801–807 (2016). This study demonstrates a link between the number of metabolic syndrome components and neuropathy prevalence that is independent of glycaemic status.
Callaghan, B. C. et al. Association between metabolic syndrome components and polyneuropathy in an obese population. JAMA Neurol. 73, 1468–1476 (2016).
Hanewinckel, R. et al. Metabolic syndrome is related to polyneuropathy and impaired peripheral nerve function: a prospective population-based cohort study. J. Neurol. Neurosurg. Psychiatry 87, 1336–1342 (2016).
Lu, B. et al. Determination of peripheral neuropathy prevalence and associated factors in Chinese subjects with diabetes and pre-diabetes - ShangHai Diabetic neuRopathy Epidemiology and Molecular Genetics Study (SH-DREAMS). PLOS ONE 8, e61053 (2013).
Tesfaye, S. & Selvarajah, D. The Eurodiab study: what has this taught us about diabetic peripheral neuropathy? Curr. Diab. Rep. 9, 432–434 (2009).
Callaghan, B. C., Price, R. S. & Feldman, E. L. Distal symmetric polyneuropathy: a review. JAMA 314, 2172–2181 (2015).
Prabodha, L. B. L., Sirisena, N. D. & Dissanayake, V. H. W. Susceptible and prognostic genetic factors associated with diabetic peripheral neuropathy: a comprehensive literature review. Int. J. Endocrinol. 2018, 8641942 (2018).
Politi, C. et al. Recent advances in exploring the genetic susceptibility to diabetic neuropathy. Diabetes Res. Clin. Pract. 120, 198–208 (2016).
Dunnigan, S. K. et al. Conduction slowing in diabetic sensorimotor polyneuropathy. Diabetes Care 36, 3684–3690 (2013).
Gumy, L. F., Bampton, E. T. & Tolkovsky, A. M. Hyperglycaemia inhibits Schwann cell proliferation and migration and restricts regeneration of axons and Schwann cells from adult murine DRG. Mol. Cell Neurosci. 37, 298–311 (2008).
Mizisin, A. P., Shelton, G. D., Wagner, S., Rusbridge, C. & Powell, H. C. Myelin splitting, Schwann cell injury and demyelination in feline diabetic neuropathy. Acta Neuropathol. 95, 171–174 (1998).
Pan, S. & Chan, J. R. Regulation and dysregulation of axon infrastructure by myelinating glia. J. Cell Biol. 216, 3903–3916 (2017).
Feldman, E. L., Nave, K. A., Jensen, T. S. & Bennett, D. L. H. New horizons in diabetic neuropathy: mechanisms, bioenergetics, and pain. Neuron 93, 1296–1313 (2017). This article provides a detailed review of advances in our understanding of the pathways underlying peripheral nerve injury and pain in diabetic neuropathy, including systems biology insights and ideas related to bioenergetic crosstalk and the axon–Schwann cell relationship.
Court, F. A., Hendriks, W. T., MacGillavry, H. D., Alvarez, J. & van Minnen, J. Schwann cell to axon transfer of ribosomes: toward a novel understanding of the role of glia in the nervous system. J. Neurosci. 28, 11024–11029 (2008).
Willis, D. E. & Twiss, J. L. The evolving roles of axonally synthesized proteins in regeneration. Curr. Opin. Neurobiol. 16, 111–118 (2006).
Cashman, C. R. & Hoke, A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci. Lett. 596, 33–50 (2015).
Scott, J. N., Clark, A. W. & Zochodne, D. W. Neurofilament and tubulin gene expression in progressive experimental diabetes: failure of synthesis and export by sensory neurons. Brain 122, 2109–2118 (1999).
Lupachyk, S., Watcho, P., Stavniichuk, R., Shevalye, H. & Obrosova, I. G. Endoplasmic reticulum stress plays a key role in the pathogenesis of diabetic peripheral neuropathy. Diabetes 62, 944–952 (2013).
Ma, J., Pan, P., Anyika, M., Blagg, B. S. & Dobrowsky, R. T. Modulating molecular chaperones improves mitochondrial bioenergetics and decreases the inflammatory transcriptome in diabetic sensory neurons. ACS Chem. Neurosci. 6, 1637–1648 (2015).
Urban, M. J. et al. Modulating molecular chaperones improves sensory fiber recovery and mitochondrial function in diabetic peripheral neuropathy. Exp. Neurol. 235, 388–396 (2012).
Ilnytska, O. et al. Poly(ADP-ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes 55, 1686–1694 (2006).
Lupachyk, S., Shevalye, H., Maksimchyk, Y., Drel, V. R. & Obrosova, I. G. PARP inhibition alleviates diabetes-induced systemic oxidative stress and neural tissue 4-hydroxynonenal adduct accumulation: correlation with peripheral nerve function. Free Radic. Biol. Med. 50, 1400–1409 (2011).
Cheng, C. et al. Evidence for epigenetic regulation of gene expression and function in chronic experimental diabetic neuropathy. J. Neuropathol. Exp. Neurol. 74, 804–817 (2015).
Toth, C., Brussee, V. & Zochodne, D. W. Remote neurotrophic support of epidermal nerve fibres in experimental diabetes. Diabetologia 49, 1081–1088 (2006).
Fernyhough, P., Diemel, L. T., Brewster, W. J. & Tomlinson, D. R. Altered neurotrophin mRNA levels in peripheral nerve and skeletal muscle of experimentally diabetic rats. J. Neurochem. 64, 1231–1237 (1995).
Fernyhough, P., Diemel, L. T. & Tomlinson, D. R. Target tissue production and axonal transport of neurotrophin-3 are reduced in streptozotocin-diabetic rats. Diabetologia 41, 300–306 (1998).
Delcroix, J. D., Michael, G. J., Priestley, J. V., Tomlinson, D. R. & Fernyhough, P. Effect of nerve growth factor treatment on p75NTR gene expression in lumbar dorsal root ganglia of streptozocin-induced diabetic rats. Diabetes 47, 1779–1785 (1998).
Hur, J. et al. The metabolic syndrome and microvascular complications in a murine model of type 2 diabetes. Diabetes 64, 3294–3304 (2015).
Hur, J. et al. Transcriptional networks of murine diabetic peripheral neuropathy and nephropathy: common and distinct gene expression patterns. Diabetologia 59, 1297–1306 (2016).
McGregor, B. A. et al. Conserved transcriptional signatures in human and murine diabetic peripheral neuropathy. Sci. Rep. 8, 17678 (2018).
Kobayashi, M. et al. Diabetic polyneuropathy, sensory neurons, nuclear structure and spliceosome alterations: a role for CWC22. Dis. Model. Mech. 10, 215–224 (2017).
Zochodne, D. W. & Ho, L. T. The influence of sulindac on experimental streptozotocin-induced diabetic neuropathy. Can. J. Neurol. Sci. 21, 194–202 (1994).
Viader, A. et al. Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron 77, 886–898 (2013).
Vincent, A. M., Callaghan, B. C., Smith, A. L. & Feldman, E. L. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat. Rev. Neurol. 7, 573–583 (2011).
Vincent, A. M., Kato, K., McLean, L. L., Soules, M. E. & Feldman, E. L. Sensory neurons and schwann cells respond to oxidative stress by increasing antioxidant defense mechanisms. Antioxid. Redox Signal 11, 425–438 (2009).
Russell, J. W. et al. Oxidative injury and neuropathy in diabetes and impaired glucose tolerance. Neurobiol. Dis. 30, 420–429 (2008).
Vincent, A. M., Edwards, J. L., Sadidi, M. & Feldman, E. L. The antioxidant response as a drug target in diabetic neuropathy. Curr. Drug Targets 9, 94–100 (2008).
Vincent, A. M., Calabek, B., Roberts, L. & Feldman, E. L. Biology of diabetic neuropathy. Handb. Clin. Neurol. 115, 591–606 (2013).
Vincent, A. M., Russell, J. W., Low, P. & Feldman, E. L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 25, 612–628 (2004).
Fernyhough, P. Mitochondrial dysfunction in diabetic neuropathy: a series of unfortunate metabolic events. Curr. Diab. Rep. 15, 89 (2015).
Fernyhough, P. & McGavock, J. Mechanisms of disease: mitochondrial dysfunction in sensory neuropathy and other complications in diabetes. Handb. Clin. Neurol. 126, 353–377 (2014).
Chowdhury, S. K., Smith, D. R. & Fernyhough, P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol. Dis. 51, 56–65 (2013).
Rumora, A. E. et al. Dyslipidemia impairs mitochondrial trafficking and function in sensory neurons. FASEB J. 32, 195–207 (2018).
Singh, V. P., Bali, A., Singh, N. & Jaggi, A. S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 18, 1–14 (2014).
Padilla, A., Descorbeth, M., Almeyda, A. L., Payne, K. & De Leon, M. Hyperglycemia magnifies Schwann cell dysfunction and cell death triggered by PA-induced lipotoxicity. Brain Res. 1370, 64–79 (2011).
Legrand-Poels, S. et al. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem. Pharmacol. 92, 131–141 (2014).
Jang, E. R. & Lee, C. S. 7-Ketocholesterol induces apoptosis in differentiated PC12 cells via reactive oxygen species-dependent activation of NF-kappaB and Akt pathways. Neurochem. Int. 58, 52–59 (2011).
Vincent, A. M. et al. Dyslipidemia-induced neuropathy in mice: the role of oxLDL/LOX-1. Diabetes 58, 2376–2385 (2009).
Nowicki, M. et al. Oxidized low-density lipoprotein (oxLDL)-induced cell death in dorsal root ganglion cell cultures depends not on the lectin-like oxLDL receptor-1 but on the toll-like receptor-4. J. Neurosci. Res. 88, 403–412 (2010).
Vincent, A. M. et al. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology 148, 548–558 (2007).
Keller, J. N., Hanni, K.B. & Markesbery, W. R. Oxidized low-density lipoprotein induces neuronal death: implications for calcium, reactive oxygen species, and caspases. J. Neurochem. 72, 2601–2609 (1999).
Cotter, M. A. & Cameron, N. E. Effect of the NAD(P)H oxidase inhibitor, apocynin, on peripheral nerve perfusion and function in diabetic rats. Life Sci. 73, 1813–1824 (2003).
Kim, H., Kim, J. J. & Yoon, Y. S. Emerging therapy for diabetic neuropathy: cell therapy targeting vessels and nerves. Endocr. Metab. Immune Disord. Drug Targets 12, 168–178 (2012).
Thrainsdottir, S. et al. Endoneurial capillary abnormalities presage deterioration of glucose tolerance and accompany peripheral neuropathy in man. Diabetes 52, 2615–2622 (2003).
Nowicki, M., Kosacka, J., Serke, H., Bluher, M. & Spanel-Borowski, K. Altered sciatic nerve fiber morphology and endoneural microvessels in mouse models relevant for obesity, peripheral diabetic polyneuropathy, and the metabolic syndrome. J. Neurosci. Res. 90, 122–131 (2012).
Coppey, L. J. et al. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes 50, 1927–1937 (2001).
Schratzberger, P. et al. Reversal of experimental diabetic neuropathy by VEGF gene transfer. J. Clin. Invest. 107, 1083–1092 (2001).
Frazier, W. A., Angeletti, R. H. & Bradshaw, R. A. Nerve growth factor and insulin. Science 176, 482–488 (1972).
Fernyhough, P., Willars, G. B., Lindsay, R. M. & Tomlinson, D. R. Insulin and insulin-like growth factor I enhance regeneration in cultured adult rat sensory neurones. Brain Res. 607, 117–124 (1993).
Brussee, V., Cunningham, F. A. & Zochodne, D. W. Direct insulin signaling of neurons reverses diabetic neuropathy. Diabetes 53, 1824–1830 (2004).
Sugimoto, K., Murakawa, Y., Zhang, W., Xu, G. & Sima, A. A. Insulin receptor in rat peripheral nerve: its localization and alternatively spliced isoforms. Diabetes Metab. Res. Rev. 16, 354–363 (2000).
Guo, G., Kan, M., Martinez, J. A. & Zochodne, D. W. Local insulin and the rapid regrowth of diabetic epidermal axons. Neurobiol. Dis. 43, 414–421 (2011).
Singhal, A., Cheng, C., Sun, H. & Zochodne, D. W. Near nerve local insulin prevents conduction slowing in experimental diabetes. Brain Res. 763, 209–214 (1997).
Kim, B., McLean, L. L., Philip, S. S. & Feldman, E. L. Hyperinsulinemia induces insulin resistance in dorsal root ganglion neurons. Endocrinology 152, 3638–3647 (2011).
Singh, B. et al. Resistance to trophic neurite outgrowth of sensory neurons exposed to insulin. J. Neurochem. 121, 263–276 (2012).
Grote, C. W. et al. Peripheral nervous system insulin resistance in ob/ob mice. Acta Neuropathol. Commun. 1, 15 (2013).
Grote, C. W., Morris, J. K., Ryals, J. M., Geiger, P. C. & Wright, D. E. Insulin receptor substrate 2 expression and involvement in neuronal insulin resistance in diabetic neuropathy. Exp. Diabetes Res. 2011, 212571 (2011).
Abbott, C. A., Malik, R. A., van Ross, E. R., Kulkarni, J. & Boulton, A. J. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care 34, 2220–2224 (2011).
von Hehn, C. A., Baron, R. & Woolf, C. J. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 73, 638–652 (2012).
Hebert, H. L., Veluchamy, A., Torrance, N. & Smith, B. H. Risk factors for neuropathic pain in diabetes mellitus. Pain 158, 560–568 (2017).
Raputova, J. et al. Sensory phenotype and risk factors for painful diabetic neuropathy: a cross-sectional observational study. Pain 158, 2340–2353 (2017).
Themistocleous, A. C. et al. The Pain in Neuropathy Study (PiNS): a cross-sectional observational study determining the somatosensory phenotype of painful and painless diabetic neuropathy. Pain 157, 1132–1145 (2016).
Suzuki, Y., Sato, J., Kawanishi, M. & Mizumura, K. Lowered response threshold and increased responsiveness to mechanical stimulation of cutaneous nociceptive fibers in streptozotocin-diabetic rat skin in vitro — correlates of mechanical allodynia and hyperalgesia observed in the early stage of diabetes. Neurosci. Res. 43, 171–178 (2002).
Garcia-Perez, E. et al. Behavioural, morphological and electrophysiological assessment of the effects of type 2 diabetes mellitus on large and small nerve fibres in Zucker diabetic fatty, Zucker lean and Wistar rats. Eur. J. Pain 22, 1457–1472 (2018).
Orstavik, K. et al. Abnormal function of C-fibers in patients with diabetic neuropathy. J. Neurosci. 26, 11287–11294 (2006).
Haroutounian, S. et al. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 155, 1272–1279 (2014).
Bennett, D. L. & Woods, C. G. Painful and painless channelopathies. Lancet Neurol. 13, 587–599 (2014). This review presents insights into pain mechanisms, diagnosis and treatment that have emanated from studies of heritable pain disorders.
Dubin, A. E. & Patapoutian, A. Nociceptors: the sensors of the pain pathway. J. Clin. Invest. 120, 3760–3772 (2010).
Blair, N. T. & Bean, B. P. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci. 22, 10277–10290 (2002).
Sun, W. et al. Reduced conduction failure of the main axon of polymodal nociceptive C-fibres contributes to painful diabetic neuropathy in rats. Brain 135, 359–375 (2012).
Bierhaus, A. et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 18, 926–933 (2012).
Hansen, C. S. et al. The role of serum methylglyoxal on diabetic peripheral and cardiovascular autonomic neuropathy: the ADDITION Denmark study. Diabet. Med. 32, 778–785 (2015).
Andersson, D. A. et al. Methylglyoxal evokes pain by stimulating TRPA1. PLOS ONE 8, e77986 (2013).
Calvo, M. et al. Altered potassium channel distribution and composition in myelinated axons suppresses hyperexcitability following injury. eLife 5, e12661 (2016).
Zenker, J. et al. Altered distribution of juxtaparanodal kv1.2 subunits mediates peripheral nerve hyperexcitability in type 2 diabetes mellitus. J. Neurosci. 32, 7493–7498 (2012).
Dib-Hajj, S. D., Yang, Y., Black, J. A. & Waxman, S. G. The NaV1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62 (2013).
Li, Q. S. et al. SCN9A variants may be implicated in neuropathic pain associated with diabetic peripheral neuropathy and pain severity. Clin. J. Pain 31, 976–982 (2015).
Blesneac, I. et al. Rare Nav1.7 variants associated with painful diabetic peripheral neuropathy. Pain 159, 469–480 (2017).
Wadhawan, S. et al. NaV channel variants in patients with painful and nonpainful peripheral neuropathy. Neurol. Genet. 3, e207 (2017).
McDonnell, A. et al. Efficacy of the Nav1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain 159, 1465–1476 (2018).
Zakrzewska, J. M. et al. Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: a double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. 16, 291–300 (2017).
Orestes, P. et al. Reversal of neuropathic pain in diabetes by targeting glycosylation of CaV3.2T-type calcium channels. Diabetes 62, 3828–3838 (2013).
Messinger, R. B. et al. In vivo silencing of the Ca(V)3.2T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain 145, 184–195 (2009).
Cooper, M. A. et al. Modulation of diet-induced mechanical allodynia by metabolic parameters and inflammation. J. Peripher. Nerv. Syst. 22, 39–46 (2017).
Woolf, C. J. Central sensitization: implications for the diagnosis and treatment of pain. Pain 152, S2–15 (2011).
Tan, A. M. et al. Maladaptive dendritic spine remodeling contributes to diabetic neuropathic pain. J. Neurosci. 32, 6795–6807 (2012).
Salter, M. W. & Beggs, S. Sublime microglia: expanding roles for the guardians of the CNS. Cell 158, 15–24 (2014).
Tsuda, M., Ueno, H., Kataoka, A., Tozaki-Saitoh, H. & Inoue, K. Activation of dorsal horn microglia contributes to diabetes-induced tactile allodynia via extracellular signal-regulated protein kinase signaling. Glia 56, 378–386 (2008).
Liao, Y. H. et al. Spinal astrocytic activation contributes to mechanical allodynia in a mouse model of type 2 diabetes. Brain Res. 1368, 324–335 (2011).
Wodarski, R., Clark, A. K., Grist, J., Marchand, F. & Malcangio, M. Gabapentin reverses microglial activation in the spinal cord of streptozotocin-induced diabetic rats. Eur. J. Pain 13, 807–811 (2009).
West, S. J., Bannister, K., Dickenson, A. H. & Bennett, D. L. Circuitry and plasticity of the dorsal horn — toward a better understanding of neuropathic pain. Neuroscience 300, 254–275 (2015).
Marshall, A. G. et al. Spinal disinhibition in experimental and clinical painful diabetic neuropathy. Diabetes 66, 1380–1390 (2017).
Segerdahl, A. R., Themistocleous, A. C., Fido, D., Bennett, D. L. & Tracey, I. A brain-based pain facilitation mechanism contributes to painful diabetic polyneuropathy. Brain 141, 357–364 (2018).
Yarnitsky, D., Granot, M., Nahman-Averbuch, H., Khamaisi, M. & Granovsky, Y. Conditioned pain modulation predicts duloxetine efficacy in painful diabetic neuropathy. Pain 153, 1193–1198 (2012).
Cauda, F. et al. Low-frequency BOLD fluctuations demonstrate altered thalamocortical connectivity in diabetic neuropathic pain. BMC Neurosci. 10, 138 (2009).
Selvarajah, D. et al. Magnetic resonance neuroimaging study of brain structural differences in diabetic peripheral neuropathy. Diabetes Care 37, 1681–1688 (2014).
Vileikyte, L. & Gonzalez, J. S. Recognition and management of psychosocial issues in diabetic neuropathy. Handb. Clin. Neurol. 126, 195–209 (2014).
Sieberg, C. B. et al. Neuropathic pain drives anxiety behavior in mice, results consistent with anxiety levels in diabetic neuropathy patients. Pain Rep. 3, e651 (2018).
Tesfaye, S. et al. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 33, 2285–2293 (2010). This update presents the results of the October 2009 Toronto Diabetic Neuropathy Expert Group discussion on the classification, definition, diagnostic criteria and treatments of diabetic peripheral, autonomic and painful neuropathies.
Divisova, S. et al. Prediabetes/early diabetes-associated neuropathy predominantly involves sensory small fibres. J. Peripher. Nerv. Syst. 17, 341–350 (2012).
Bril, V. & Perkins, B. A. Validation of the Toronto Clinical Scoring System for diabetic polyneuropathy. Diabetes Care 25, 2048–2052 (2002).
Bril, V., Tomioka, S., Buchanan, R. A. & Perkins, B. A., the mTCNS Study Group. Reliability and validity of the modified Toronto Clinical Neuropathy Score in diabetic sensorimotor polyneuropathy. Diabet. Med. 26, 240–246 (2009).
Feldman, E. L. et al. A practical two-step quantitative clinical and electrophysiological assessment for the diagnosis and staging of diabetic neuropathy. Diabetes Care 17, 1281–1289 (1994).
Dyck, P. J. et al. Diabetic polyneuropathies: update on research definition, diagnostic criteria and estimation of severity. Diabetes Metab. Res. Rev. 27, 620–628 (2011).
Weisman, A. et al. Identification and prediction of diabetic sensorimotor polyneuropathy using individual and simple combinations of nerve conduction study parameters. PLOS ONE 8, e58783 (2013).
Singleton, J. R. et al. The Utah Early Neuropathy Scale: a sensitive clinical scale for early sensory predominant neuropathy. J. Peripher. Nerv. Syst. 13, 218–227 (2008).
Andersson, C., Guttorp, P. & Sarkka, A. Discovering early diabetic neuropathy from epidermal nerve fiber patterns. Stat. Med. 35, 4427–4442 (2016).
Devigili, G. et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 131, 1912–1925 (2008).
Jensen, T. S., Bach, F. W., Kastrup, J., Dejgaard, A. & Brennum, J. Vibratory and thermal thresholds in diabetics with and without clinical neuropathy. Acta Neurol. Scand. 84, 326–333 (1991).
Krishnan, S. T. & Rayman, G. The LDIflare: a novel test of C-fiber function demonstrates early neuropathy in type 2 diabetes. Diabetes Care 27, 2930–2935 (2004).
Sivaskandarajah, G. A. et al. Structure-function relationship between corneal nerves and conventional small-fiber tests in type 1 diabetes. Diabetes Care 36, 2748–2755 (2013).
Breiner, A., Lovblom, L. E., Perkins, B. A. & Bril, V. Does the prevailing hypothesis that small-fiber dysfunction precedes large-fiber dysfunction apply to type 1 diabetic patients? Diabetes Care 37, 1418–1424 (2014).
Yang, W., Cai, X. L., Wu, H. & Ji, L. Associations between metformin use and vitamin B12 level, anemia and neuropathy in patients with diabetes: a meta-analysis. J. Diabetes. https://doi.org/10.1111/1753-0407.12900 (2019).
England, J. D. et al. Practice parameter: the evaluation of distal symmetric polyneuropathy: the role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. PM R. 1, 5–13 (2009).
Adams, D. et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 11–21 (2018).
Benson, M. D. et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 22–31 (2018).
Diabetes Canada Clinical Practice Guidelines Expert Committee, Bril, V., Breiner, A., Perkins, B. A. & Zochodne, D. Neuropathy. Can. J. Diabetes 42 (Suppl. 1), S217–S221 (2018).
Olaleye, D., Perkins, B. A. & Bril, V. Evaluation of three screening tests and a risk assessment model for diagnosing peripheral neuropathy in the diabetes clinic. Diabetes Res. Clin. Pract. 54, 115–128 (2001).
Perkins, B. A., Olaleye, D., Zinman, B. & Bril, V. Simple screening tests for peripheral neuropathy in the diabetes clinic. Diabetes Care 24, 250–256 (2001).
Perkins, B. A. et al. Prediction of incident diabetic neuropathy using the monofilament examination: a 4-year prospective study. Diabetes Care 33, 1549–1554 (2010).
Boulton, A. J. et al. Comprehensive foot examination and risk assessment: a report of the task force of the foot care interest group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care 31, 1679–1685 (2008).
Kanji, J. N., Anglin, R. E., Hunt, D. L. & Panju, A. Does this patient with diabetes have large-fiber peripheral neuropathy? JAMA 303, 1526–1532 (2010).
Beghi, E., Treviso, M., Ferri, P. & Di Mascio, R. Diagnosis of diabetic polyneuropathy. Correlation between clinical and instrumental findings and assessment of simple diagnostic criteria. Ital. J. Neurol. Sci. 9, 577–582 (1988).
Herman, W. H. et al. Use of the Michigan Neuropathy Screening Instrument as a measure of distal symmetrical peripheral neuropathy in type 1 diabetes: results from the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications. Diabet Med. 29, 937–944 (2012).
Callaghan, B. C., Little, A. A., Feldman, E. L. & Hughes, R. A. Enhanced glucose control for preventing and treating diabetic neuropathy. Cochrane Database Syst. Rev. 6, CD007543 (2012). This analysis of clinical studies evaluating the impact of glycaemic control on neuropathy outcomes in T1DM and T2DM reveals that enhanced glucose control significantly attenuates neuropathy development in T1DM but not in T2DM.
The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 329, 977–986 (1993).
Duckworth, W. et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 360, 129–139 (2009).
Ismail-Beigi, F. et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 376, 419–430 (2010).
Charles, M. et al. Prevalence of neuropathy and peripheral arterial disease and the impact of treatment in people with screen-detected type 2 diabetes: the ADDITION-Denmark study. Diabetes Care 34, 2244–2249 (2011).
Singleton, J. R. et al. Exercise increases cutaneous nerve density in diabetic patients without neuropathy. Ann. Clin. Transl Neurol. 1, 844–849 (2014).
Muller-Stich, B. P. et al. Gastric bypass leads to improvement of diabetic neuropathy independent of glucose normalization—results of a prospective cohort study (DiaSurg 1 study). Ann. Surg. 258, 760–765 (2013).
Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 358, 2545–2559 (2008).
Qaseem, A. et al. Hemoglobin A1c targets for glycemic control with pharmacologic therapy for nonpregnant adults with type 2 diabetes mellitus: a guidance statement update from the American College of Physicians. Ann. Intern. Med. 168, 569–576 (2018).
Ziegler, D., Behler, M., Schroers-Teuber, M. & Roden, M. Near-normoglycaemia and development of neuropathy: a 24-year prospective study from diagnosis of type 1 diabetes. BMJ Open 5, e006559 (2015).
Dahl-Jorgensen, K. et al. Effect of near normoglycaemia for two years on progression of early diabetic retinopathy, nephropathy, and neuropathy: the Oslo study. Br. Med. J. (Clin. Res. Ed) 293, 1195–1199 (1986).
Ohkubo, Y. et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res. Clin. Pract. 28, 103–117 (1995).
Ishibashi, F., Taniguchi, M., Kosaka, A., Uetake, H. & Tavakoli, M. Improvement in neuropathy outcomes with normalizing HbA1c in patients with type 2 diabetes. Diabetes Care 42, 110–118 (2018).
Balducci, S. et al. Exercise training can modify the natural history of diabetic peripheral neuropathy. J. Diabetes Complicat. 20, 216–223 (2006).
Kluding, P. M. et al. The effect of exercise on neuropathic symptoms, nerve function, and cutaneous innervation in people with diabetic peripheral neuropathy. J. Diabetes Complicat. 26, 424–429 (2012).
Singleton, J. R., Marcus, R. L., Lessard, M. K., Jackson, J. E. & Smith, A. G. Supervised exercise improves cutaneous reinnervation capacity in metabolic syndrome patients. Ann. Neurol. 77, 146–153 (2015).
Smith, A. G. et al. Lifestyle intervention for pre-diabetic neuropathy. Diabetes Care 29, 1294–1299 (2006).
Ziegler, D. et al. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: a 7-month multicenter randomized controlled trial (ALADIN III Study). ALADIN III Study Group. Alpha-lipoic acid in diabetic neuropathy. Diabetes Care 22, 1296–1301 (1999).
Ziegler, D. et al. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care 29, 2365–2370 (2006).
Ziegler, D. et al. Efficacy and safety of antioxidant treatment with alpha-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes Care 34, 2054–2060 (2011). This multicentre, randomized, double-blind, parallel-group trial of α-lipoic acid in 460 individuals with diabetes and neuropathy does not meet the primary composite end point. There is a beneficial effect in the α-lipoic-acid-treated cohort on secondary end points, including the NIS.
Balakumar, P., Rohilla, A., Krishan, P., Solairaj, P. & Thangathirupathi, A. The multifaceted therapeutic potential of benfotiamine. Pharmacol. Res. 61, 482–488 (2010).
Zilliox, L. & Russell, J. W. Treatment of diabetic sensory polyneuropathy. Curr. Treat. Options Neurol. 13, 143–159 (2011).
Stracke, H., Gaus, W., Achenbach, U., Federlin, K. & Bretzel, R. G. Benfotiamine in diabetic polyneuropathy (BENDIP): results of a randomised, double blind, placebo-controlled clinical study. Exp. Clin. Endocrinol. Diabetes 116, 600–605 (2008).
Fraser, D. A. et al. The effects of long-term oral benfotiamine supplementation on peripheral nerve function and inflammatory markers in patients with type 1 diabetes: a 24-month, double-blind, randomized, placebo-controlled trial. Diabetes Care 35, 1095–1097 (2012).
Lewis, E. J. H. et al. Effect of omega-3 supplementation on neuropathy in type 1 diabetes: a 12-month pilot trial. Neurology 88, 2294–2301 (2017).
Hotta, N. et al. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: the 3-year, multicenter, comparative Aldose Reductase Inhibitor-Diabetes Complications Trial. Diabetes Care 29, 1538–1544 (2006).
Attal, N. et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur. J. Neurol. 17, 1113–e88 (2010).
Bril, V. et al. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology 76, 1758–1765 (2011). This article presents an evidenced-based guideline for the treatment of painful diabetic neuropathy.
Finnerup, N. B. et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 14, 162–173 (2015). This systematic review and meta-analysis provides support for a revision to the NeuPSIG recommendations for the treatment of neuropathic pain.
Griebeler, M. L. et al. Pharmacologic interventions for painful diabetic neuropathy: An umbrella systematic review and comparative effectiveness network meta-analysis. Ann. Intern. Med. 161, 639–649 (2014).
Waldfogel, J. M. et al. Pharmacotherapy for diabetic peripheral neuropathy pain and quality of life: a systematic review. Neurology 88, 1958–1967 (2017).
Callaghan, B. C. & Feldman, E. L. Painful diabetic neuropathy: many similarly effective therapies with widely dissimilar costs. Ann. Intern. Med. 161, 674–675 (2014).
Backonja, M. & Glanzman, R. L. Gabapentin dosing for neuropathic pain: evidence from randomized, placebo-controlled clinical trials. Clin. Ther. 25, 81–104 (2003).
Freeman, R., Durso-Decruz, E. & Emir, B. Efficacy, safety, and tolerability of pregabalin treatment for painful diabetic peripheral neuropathy: findings from seven randomized, controlled trials across a range of doses. Diabetes Care 31, 1448–1454 (2008).
Moore, R. A., Straube, S., Wiffen, P. J., Derry, S. & McQuay, H. J. Pregabalin for acute and chronic pain in adults. Cochrane Database Syst. Rev. 8, CD007076 (2009).
Ziegler, D., Duan, W. R., An, G., Thomas, J. W. & Nothaft, W. A randomized double-blind, placebo-, and active-controlled study of T-type calcium channel blocker ABT-639 in patients with diabetic peripheral neuropathic pain. Pain 156, 2013–2020 (2015).
Quilici, S. et al. Meta-analysis of duloxetine versus pregabalin and gabapentin in the treatment of diabetic peripheral neuropathic pain. BMC Neurol. 9, 6 (2009).
Raskin, P. et al. Pregabalin in patients with inadequately treated painful diabetic peripheral neuropathy: a randomized withdrawal trial. Clin. J. Pain 30, 379–390 (2014).
Dworkin, R. H., Jensen, M. P., Gammaitoni, A. R., Olaleye, D. O. & Galer, B. S. Symptom profiles differ in patients with neuropathic versus non-neuropathic pain. J. Pain 8, 118–126 (2007).
Goldstein, D. J., Lu, Y., Detke, M. J., Lee, T. C. & Iyengar, S. Duloxetine versus placebo in patients with painful diabetic neuropathy. Pain 116, 109–118 (2005).
Tesfaye, S. et al. Duloxetine and pregabalin: High-dose monotherapy or their combination? The “COMBO-DN study” - a multinational, randomized, double-blind, parallel-group study in patients with diabetic peripheral neuropathic pain. Pain 154, 2616–2625 (2013).
Wernicke, J. F. et al. A randomized controlled trial of duloxetine in diabetic peripheral neuropathic pain. Neurology 67, 1411–1420 (2006).
Zilliox, L. & Russell, J. W. Maintaining efficacy in the treatment of diabetic peripheral neuropathic pain: role of duloxetine. Diabetes Metab. Syndr. Obes. 3, 7–17 (2010).
Rowbotham, M. C., Goli, V., Kunz, N. R. & Lei, D. Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo-controlled study. Pain 110, 697–706 (2004).
Sindrup, S. H., Bach, F. W., Madsen, C., Gram, L. F. & Jensen, T. S. Venlafaxine versus imipramine in painful polyneuropathy: a randomized, controlled trial. Neurology 60, 1284–1289 (2003).
Boyle, J. et al. Randomized, placebo-controlled comparison of amitriptyline, duloxetine, and pregabalin in patients with chronic diabetic peripheral neuropathic pain: impact on pain, polysomnographic sleep, daytime functioning, and quality of life. Diabetes Care 35, 2451–2458 (2012).
Max, M. B. et al. Amitriptyline relieves diabetic neuropathy pain in patients with normal or depressed mood. Neurology 37, 589–596 (1987).
Max, M. B. et al. Efficacy of desipramine in painful diabetic neuropathy: a placebo-controlled trial. Pain 45, 3–9 (1991).
Max, M. B. et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N. Engl. J. Med. 326, 1250–1256 (1992).
Derry, S., Wiffen, P. J., Aldington, D. & Moore, R. A. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst. Rev. 1, CD011209 (2015).
Dowell, D., Haegerich, T. M. & Chou, R. CDC guideline for prescribing opioids for chronic pain—United States, 2016. JAMA 315, 1624–1645 (2016).
Franklin, G. M. Opioids for chronic noncancer pain: a position paper of the American Academy of Neurology. Neurology 83, 1277–1284 (2014). This article reviews the safety and efficacy evidence, state and federal policies and recommendations for practising neurologists regarding safe and effective opioid use in chronic pain conditions.
Schwartz, S. et al. Safety and efficacy of tapentadol ER in patients with painful diabetic peripheral neuropathy: results of a randomized-withdrawal, placebo-controlled trial. Curr. Med. Res. Opin. 27, 151–162 (2011).
Vinik, A. I. et al. A randomized withdrawal, placebo-controlled study evaluating the efficacy and tolerability of tapentadol extended release in patients with chronic painful diabetic peripheral neuropathy. Diabetes Care 37, 2302–2309 (2014).
Raffa, R. B. et al. Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an ‘atypical’ opioid analgesic. J. Pharmacol. Exp. Ther. 260, 275–285 (1992).
Harati, Y. et al. Double-blind randomized trial of tramadol for the treatment of the pain of diabetic neuropathy. Neurology 50, 1842–1846 (1998).
Freeman, R. et al. Randomized study of tramadol/acetaminophen versus placebo in painful diabetic peripheral neuropathy. Curr. Med. Res. Opin. 23, 147–161 (2007).
Harati, Y. et al. Maintenance of the long-term effectiveness of tramadol in treatment of the pain of diabetic neuropathy. J. Diabetes Complicat. 14, 65–70 (2000).
Gimbel, J. S., Richards, P. & Portenoy, R. K. Controlled-release oxycodone for pain in diabetic neuropathy: a randomized controlled trial. Neurology 60, 927–934 (2003).
Watson, C. P., Moulin, D., Watt-Watson, J., Gordon, A. & Eisenhoffer, J. Controlled-release oxycodone relieves neuropathic pain: a randomized controlled trial in painful diabetic neuropathy. Pain 105, 71–78 (2003).
Jalal, H. et al. Changing dynamics of the drug overdose epidemic in the United States from 1979 through 2016. Science 361, eaau1184 (2018).
Fisher, L., Hessler, D. M., Polonsky, W. H. & Mullan, J. When is diabetes distress clinically meaningful?: establishing cut points for the Diabetes Distress Scale. Diabetes Care 35, 259–264 (2012).
Callaghan, B. et al. Longitudinal patient-oriented outcomes in neuropathy: Importance of early detection and falls. Neurology 85, 71–79 (2015).
Van Acker, K. et al. Prevalence and impact on quality of life of peripheral neuropathy with or without neuropathic pain in type 1 and type 2 diabetic patients attending hospital outpatients clinics. Diabetes Metab. 35, 206–213 (2009).
Ind, I. S. G. Burden of neuropathic pain in Indian patients attending urban, specialty clinics: results from a cross sectional study. Pain Pract. 8, 362–378 (2008).
Trikkalinou, A., Papazafiropoulou, A. K. & Melidonis, A. Type 2 diabetes and quality of life. World J. Diabetes 8, 120–129 (2017).
Benbow, S. J., Wallymahmed, M. E. & MacFarlane, I. A. Diabetic peripheral neuropathy and quality of life. QJM 91, 733–737 (1998).
Meyer-Rosberg, K. et al. Peripheral neuropathic pain — a multidimensional burden for patients. Eur. J. Pain 5, 379–389 (2001).
Amalraj, M. J., Anitha Rani, A. & Viswanathan, V. A study on positive impact of intensive psychological counseling on psychological well-being of type 2 diabetic patients undergoing amputation. Int. J. Psychol. Couns. 9, 10–16 (2017).
Thorn, B. E. et al. Literacy-adapted cognitive behavioral therapy versus education for chronic pain at low-income clinics: a randomized controlled trial. Ann. Intern. Med. 168, 471–480 (2018).
Freeman, R. Diabetic autonomic neuropathy. Handb. Clin. Neurol. 126, 63–79 (2014).
Peltier, A., Goutman, S. A. & Callaghan, B. C. Painful diabetic neuropathy. BMJ 348, g1799 (2014).
Tesfaye, S., Boulton, A. J. M. & Dickenson, A. H. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care 36, 2456–2465 (2013).
Lauria, G. & Devigili, G. Skin biopsy as a diagnostic tool in peripheral neuropathy. Nat. Clin. Pract. Neurol. 3, 546–557 (2007).
Acknowledgements
E.L.F. acknowledges support from the NIH (R24DK082841 and R01D107956) and the NovoNordisk Foundation (NNF14OC0011633). B.C.C. acknowledges support from the NIH (K23NS079417 and R01DK115687) and a VA Clinical Science Research and Development (CSRD) Merit (CX001504). R.P.B. acknowledges support from the NIH (R01D107956). D.W.Z. acknowledges support from the Canadian Institutes of Health Research (RN192747-298730) and Diabetes Canada (RN271389-OG-3-15-5025-DZ). D.E.W. acknowledges support from the NIH (R01NS0433314-14). D.L.B. acknowledges support from the NovoNordisk Foundation (NNF14OC0011633) and the Wellcome Trust (102645/Z/13/Z, 202747/Z/16/Z) and is a member of the DOLORisk Consortium funded by the European Commission Horizon 2020 (ID633491). J.W.R. acknowledges support from the NIH (R01DK107007), the US Department of Veterans Affairs (101RX001030), the Diabetes Action Research and Education Foundation and the Baltimore Geriatric Research Education and Clinical Center (GRECC). The authors thank S. Sakowski Jacoby for manuscript preparation and editorial assistance.
Author information
Authors and Affiliations
Contributions
Introduction (E.L.F.); Epidemiology (B.C.C. and E.L.F.); Diagnosis, screening and prevention (B.C.C. and V.B.); Mechanisms/pathophysiology of diabetic neuropathy (D.W.Z., D.E.W. and E.L.F.); Mechanisms/pathophysiology of pain (D.L.B.); Management (R.P.-B., J.W.R. and E.L.F.); Quality of life (V.V.); Outlook (E.L.F.); Overview of the Primer (E.L.F.).
Corresponding author
Ethics declarations
Competing interests
B.C.C. consults for a Patient-Centered Outcomes Research Institute (PCORI) grant, the Immune Tolerance Network and DynaMed and performs medical legal consultations. D.L.B. has undertaken consultancy work on behalf of Oxford Innovation for Abide, Biogen, GSK, Lilly, Mitsubishi Tanabe, Mundipharma, Teva and Theranexus. All other authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Feldman, E.L., Callaghan, B.C., Pop-Busui, R. et al. Diabetic neuropathy. Nat Rev Dis Primers 5, 41 (2019). https://doi.org/10.1038/s41572-019-0092-1
Accepted:
Published:
DOI: https://doi.org/10.1038/s41572-019-0092-1
This article is cited by
-
Potential applications of artificial intelligence in image analysis in cornea diseases: a review
Eye and Vision (2024)
-
Magnetization transfer ratio of the sciatic nerve differs between patients in type 1 and type 2 diabetes
European Radiology Experimental (2024)
-
Chitosan as a promising materials for the construction of nanocarriers for diabetic retinopathy: an updated review
Journal of Biological Engineering (2024)
-
Association of interleukin-36α and interleukin-38 with type 2 diabetes mellitus and diabetic neuropathy
The Egyptian Journal of Internal Medicine (2024)
-
Diabetic peripheral neuropathy among adult type 2 diabetes patients in Adama, Ethiopia: health facility-based study
Scientific Reports (2024)
