
Abstract
Small molecules have extensive untapped potential to benefit society, but access to this potential is too often restricted by limitations inherent to the highly customized approach that is currently used to synthesize this class of chemical matter. An alternative ‘building block approach’ — that is, generalized iterative assembly of interchangeable parts — has now proved to be a highly efficient and flexible method of constructing things ranging from skyscrapers and macromolecules to artificial intelligence algorithms. The structural redundancy found in many small molecules suggests that they possess a similar capacity for generalized building block-based construction. It is also encouraging that many customized iterative synthesis methods have been developed that already improve access to specific classes of small molecules. There has also been substantial recent progress towards the iterative assembly of many different types of small molecules, including complex natural products, pharmaceuticals, biological probes and materials, using common building blocks and coupling chemistry. Collectively, these advances suggest that a generalized building block approach for small-molecule synthesis may be within reach.
Similar content being viewed by others
Introduction
Small molecules can serve as powerful tools for improving society, with wide-ranging applications in medicine, science and technology. They make the world a more enjoyable place to see, touch, taste and smell, by acting as popular colourants, lotions, flavourings and perfumes. In fact, there is unlikely to be a household on the planet that is not positively affected by this class of chemical matter on a daily basis. Small molecules also possess substantial untapped potential to perform many frontier functions1, including modulating protein–protein interactions2, allosterically modifying protein function3, acting as prostheses on the molecular scale4,5, serving as next-generation biological probes6–9, enabling miniaturized diagnostics10, transducing energy11–16, emitting light17, initiating self-healing18, acting as molecular magnets19, and enabling next-generation computing20 and superconducting21,22. However, mainly owing to limitations in synthesis, much of this functional potential remains untapped. Eliminating this synthesis bottleneck thus represents both a major challenge and an extraordinary opportunity for the field of chemistry.
Currently, most small molecules are synthesized using customized approaches. For each target, a unique set of starting materials and a specialized sequence of different chemical reactions are developed de novo and then extensively optimized. This approach is a useful strategy for the large-scale production of a particular target. But it is also a laborious bottleneck for the discovery and optimization of new function, which depend on rapid access to many different chemical compounds. Although it is now widely considered possible to synthesize any physically accessible small molecule using this customized approach, both the design and execution phases of this process are time intensive, challenging to automate and inherently restricted to specialists.
In many other disciplines that share the challenge of assembling complex structures to access new functions, the development of a more generalized strategy that involves the iterative assembly of interchangeable building blocks has been transformative. Early examples can be found in Samuel Bentham's pioneering use of interchangeable parts to facilitate the rapid repair of wooden pulley blocks23, and Honoré Blanc's24 and Eli Whitney's25 modularization of musket production. Henry Ford dramatically increased the efficiency of this approach with the development of the assembly line, which revolutionized human transportation by making automobiles a household commodity26. The building block approach is now recognized in a remarkably wide range of different areas, including architecture27, computers28, space stations29, robotics30, college curricula31, music32, smart technology apps28 and artificial intelligence algorithms33. The advantages of building block-based construction for efficiency, flexibility and scalability are well-documented and widely appreciated34. Perhaps even more exciting is the capacity of this approach to inspire and enable innovation. This is evidenced by the explosion of applications for 3D printing35–38 and the joyful creativity that is unleashed in a child when they are handed their first bucket of Lego bricks.
During the latter half of the 20th century, iterative building block assembly was extended to the molecular scale, yielding automated synthesizers that now provide on-demand access to peptides39 and oligonucleotides40. The corresponding impact has been substantial. To highlight just a few examples, automatically synthesized oligonucleotide probes that correspond to every gene in the human genome printed on a glass slide helped to usher in the era of genomics41,42. Countless peptide- and oligonucleotide-based drug candidates were rapidly tested and optimized, yielding entirely new classes of therapeutics43,44. Total synthesis of genes, proteins and even complete genomes became possible, launching the field of synthetic biology45–47. Substantial recent progress in the automated iterative synthesis of oligosaccharides has also led to important advances in vaccine development48,49.
Extending the building block method to small-molecule synthesis brings a unique set of challenges. The remarkable structural complexity and diversity of small molecules will require a greater number of building blocks, more versatile assembly reactions and new strategies for the generalized purification of intermediates. Encouragingly, many different iterative building block-based synthesis approaches have already increased access to particular molecules and regions of chemical space. Heading towards a more general platform, iteration of metal-mediated cross-coupling reactions has provided increasingly broad access to a diverse range of small molecules, and such a process has even been fully automated. Many challenges remain, but as progress in this direction enables access to previously untapped small-molecule functions, the impetus for finding solutions is expected to continue to grow.
Inherent modularity of small molecules
Small molecules are highly structurally diverse, which makes the development of a generalized building block approach for this class of chemical matter especially challenging. However, many types of small molecule are inherently modular, suggesting that such structures and their accompanying functions should be accessible using a generalized modular synthesis approach. Most natural products, which represent the source or inspiration for more than 50% of all human therapeutics50,51, are derived from only a few major biosynthetic pathways that each involve the iterative assembly of a small number of discrete molecular building blocks (Fig. 1a). For example, polyketides are biosynthesized from malonyl-CoA and methyl malonyl-CoA, polyterpenes from isopentenyl pyrophosphate and dimethylallylpyrophosphate, fatty acids primarily from malonyl-CoA, non-ribosomal peptides from amino acids and polyphenylpropanoids from phenylpyruvic acid.

a | Although it results in a huge diversity of structures, the biosynthesis of natural products relies on the iterative assembly of a few building blocks. b | More than 75% of all known polyene natural product chemical space can be covered using only 12 bifunctional halo N-methyliminodiacetic acid (MIDA) boronate building blocks52. c | Common heteroaryl motifs found in pharmaceuticals56. d | Modularity in organic materials.
Even highly complex molecular structures from natural products can usually be traced back to these same modular pathways. Although the biosynthesis of many natural products also involves rearrangements, oxidations and cyclizations, there is still evidence that this inherent modularity translates to the final products. A recent analysis revealed that more than 75% of all polyene motifs found in natural products can be prepared using only twelve building blocks and one coupling reaction52 (Fig. 1b). Increasing evidence further suggests that natural product chemical space is bounded53, enabling the consideration of generalized approaches for studying the complete natural productome54. An expanded effort is now under way to find the minimal number of building blocks required to access most of the natural product chemical space55.
Even many non-natural small molecules, which lack such biosynthetic constraints, still contain a remarkable degree of structural redundancy. For example, a 2014 analysis of 1,086 Food and Drug Administration (FDA)-approved small-molecule drugs revealed many recurring heterocyclic building blocks (Fig. 1c), including piperidine (72 drugs), pyridine (62 drugs), piperazine (59 drugs), cephem (41 drugs), pyrrolidine (37 drugs) and thiazole (30 drugs)56. Seventeen additional heterocycles are found in at least ten drugs. This modularity suggests that much of the chemical space relevant to synthesizing pharmaceuticals should also be accessible from a defined set of building blocks. Material components also display a high degree of modularity. Despite performing a wide range of different functions, they are often composed of common repeating substructural motifs, such as oligoarenes, oligothiophenes and polystyrenes (Fig. 1d). Collectively, this inherent modularity suggests that a wide range of different molecular functions should be accessible by simply assembling building blocks that come from a finite set of common substructural motifs.
Customized iterative synthesis
Iterative synthesis of small molecules from building blocks has already improved access to a diverse range of molecular structures. In each case, building blocks are consecutively added to a growing molecule by repeating a series of chemical transformations. The two main distinguishing characteristics of different iterative synthesis methods are the type of bond used to connect the building blocks and the reactions used to form that bond; both of these characteristics can be optimized for a given target or area of chemical space. Representative methods of iterative synthesis are summarized in Fig. 2 and Fig. 3 (natural products), and Fig. 4 (unnatural molecules), with building blocks highlighted in different colours. The schemes highlight the iterative section of often more complex syntheses (detailed reaction schemes are provided with corresponding figure numbering in Supplementary Fig. S1).

a | Aldohexose synthesis via iterative Sharpless asymmetric epoxidation (AE) with L-mannose synthesis shown as an example. A repeated series of nine synthetic steps is seen between one allylic alcohol and the next57,58. b | The Crimmins asymmetric aldol method has been used in the formal synthesis of 6-deoxyerythronolide B65. c | Iterative allylboration was used in the preparation of an intermediate in the proposed structure of passifloricin A69. d | (+)-Roxaticin via iterative C–C bond-forming transfer hydrogenation71. e | (−)-Borrelidin via iterative Myers alkylation78,79. f | Phthioceranic acid via iterative conjugate addition80. g | Iterative synthesis of polydeoxypropionates via stereospecific displacement of triflates82. Detailed reaction schemes are included in Supplementary Fig. S1. TBS, tert-butyldimethylsilyl; TIPS, triisopropylsilyl; PMP, para-methoxyphenyl; TBDPS, tert-butyldiphenylsilyl.

a | The iterative homologation of boronic esters has been applied to the synthesis of (+)-kalkitoxin85. b | A key intermediate in the convergent synthesis of coenzyme Q10 was prepared by iterative palladium-catalysed couplings of alkylzinc reagents93. c | Iterative Horner–Wadsworth–Emmons olefinations were applied to the synthesis of a key polyene fragment of amphotericin B94,95. d | Synthesis of goniocin through iterative THF ring formation96. e | The ABCDEF ring system of yessotoxin and adriatoxin has been prepared using an iterative oxiranyl anion strategy97. f | Structure of natural product halichondrin B showing the four key substructures. Eribulin is an anticancer drug developed as a result of structure–activity relationship studies on the natural product. Detailed reaction schemes are included in Supplementary Fig. S2. TMS, trimethylsilyl; TBDPS, tert-butyldiphenylsilyl; TES, triethylsilyl; TIB, 2,4,6-triisopropyl benzoyl.

a | Iterative arene homologation105. b | Iterative aryne cycloadditions have been used in the synthesis of polyacenes109. c | Phenylacetylene oligomers110,111 and dendrimers113–115 have been prepared by iterative Sonogashira-type reactions. d | Extended iptycene structures have been prepared by iterative Diels–Alder–like cycloadditions116. e | Selective iterative cross-coupling of pre-installed boronate esters has been used to access polyarylated structures117. Detailed reaction schemes are included in Supplementary Fig. S3. TMS, trimethylsilyl; DMAP; 4-dimethylaminopyridine; LAH, lithium aluminium hydride.
These examples reflect some of the most efficient routes for accessing particular regions of chemical space and, in some cases, they have led to important discoveries. However, the assembly chemistries can impose some practical limitations, such as imperfect stereoselectivity in bond formations or harsh reaction conditions. The synthesis of more complex targets can require many transformations to complete one iteration cycle, lowering the overall yield and efficiency of the process. These methods also differ by how much structural variation is allowed in the building blocks, which is ultimately what determines the scope of molecules that can be made using an iterative method. Although these customized platforms all use different types of building blocks and assembly reactions and thus are limited with respect to their generality, they have substantially increased synthetic access to specific types of small-molecule structure.
Iterative approaches to naturally occurring molecules. Seminal work by Masamume and Sharpless58 established that an iterative multistep cycle could be applied to access all eight l-hexoses. Figure 2a shows the sequence applied to the synthesis of l-mannose as a representative example. The cycle begins with the asymmetric epoxidation of an allylic alcohol. In basic medium, the product rearranges to the more thermodynamically favourable terminal epoxide (Payne rearrangement), allowing for nucleophilic attack by sodium thiophenolate. A sequence of acetal protection of the nascent 1,2-diol followed by oxidation to the sulfoxide enables a Pummerer rearrangement to a gem-acetoxysulfide. Subsequent hydrolysis through reductive (DIBAL) or basic (K2CO3/MeOH) conditions enables selective retention or inversion, respectively, of the C2 stereogenic centre in the formation of the corresponding aldehydes. Finally, Wittig olefination and aldehyde reduction produces allylic alcohols for further iteration. Changing the conditions of the two stereocontrolling steps, Sharpless asymmetric epoxidation and gem-acetoxysulfide reduction, enables access to all eight L-hexoses. The versatility of this strategy has inspired the development of further methods for the de novo enantioselective synthesis of sugars57,58, and many iterative methods have similarly been developed to access various different classes of small molecule.
The modular nature of polypropionates has inspired several customized iterative synthesis methods to access even highly stereochemically complex products. In early examples by Evans and Paterson, substrate-controlled diastereoselective aldol reactions followed by stereodivergent ketone reductions provided efficient access to a library of stereotetrad motifs59–61. Auxiliary cleavage and the regeneration of aldehyde intermediates enable both of these processes to be iterated. More recently, Crimmins developed a variant of the Evans oxazolidinone methodology62 that uses N-acylthiazolidinethiones as chiral auxiliaries to facilitate a more readily iterated aldol approach63,64 (Fig. 2b). After a diastereoselective aldol reaction, cleavage of the chiral auxiliary directly generates an aldehyde, priming the substrate for further homologation. Notably, the same chiral building block can grant access to either stereoisomer of the syn aldol product depending on whether the additive is TiCl4 or (−)-sparteine. During the synthesis of 6-deoxyerythronolide B, Crimmins iterated this sequence five times, setting 10 of the 11 stereocentres using a single type of reaction65 (Supplementary Fig. S1b).
A related polyketide motif, the 1,3-polyol unit, can also be prepared using several different types of iterative chemistry. Brown developed an iterative allylboration reaction using chiral boranes to carry out highly enantioselective reactions with aldehydes66–68. The olefin motif of the resulting homoallylic alcohols can be oxidatively cleaved to reveal a new aldehyde for further allylation. This approach was used to generate the 1,3-polyol fragment in the synthesis of passifloricin A69 (Fig. 2c).
Recently, Krische and co-workers pioneered a highly efficient C–C bond-forming transfer hydrogenation strategy involving the in situ generation of an aldehyde and an organometallic nucleophile70. Application of this iterative strategy in a two-directional manner expedited the construction of the key polyol portion of (+)-roxaticin71 (Fig. 2d). This methodology has proved to be highly versatile and readily scalable, enabling practical gram-scale access to stereochemically complex building blocks for a wide range of highly complex polyketide natural products72–74. This efficiency has further enabled the practical synthesis and testing of bryostatin derivatives, shedding new light on the relationship between potency and biological activity75.
Polydeoxypropionates are an important subclass of the polyketide family, but the absence of functional handles has rendered their stereoselective synthesis challenging. Myers has developed a robust iterative alkylation protocol using stoichiometric ephedrine-based auxiliaries that provides access to all possible stereochemical variants76,77, and Theodorakis has applied this methodology to the total synthesis of (−)-borrelidin78,79 (Fig. 2e).
Minnaard and Feringa80 have recently developed an alternative iterative three-step protocol to access syn deoxypropionate motifs based on catalytic asymmetric conjugate additions (Fig. 2f). Starting from an α,β-unsaturated thioester, a highly enantioselective 1,4-addition of MeMgBr in the presence of a chiral copper catalyst sets the first methyl-bearing stereogenic centre. A reduction and Wittig olefination sequence generates a new α,β-unsaturated thioester for further 1,4-addition reactions. Repetition of these three steps allows seven methyl stereocentres to be installed with excellent levels of stereoselectivity, enabling the first total synthesis of phthioceranic acid80, as well as the first total synthesis of sulfolipid-1 (Ref. 81).
Breit has also developed an iterative zinc-catalysed sp3–sp3 coupling method for the synthesis of deoxypropionates82 (Fig. 2g). Treatment of an alkyl Grignard with ZnCl2 generates a triorganozincate species (R3ZnMgCl), which displaces a secondary triflate to generate a new C–C bond with inversion of configuration. Reduction of the ester followed by conversion to a primary alkyl chloride enables further iteration. The lack of reactivity with alkyllithium species suggests that magnesium coordination to the triflate may play a role in Lewis acid activation of the electrophile. Using this iterative method, Breit was able to access a library of different diastereomers of trideoxypropionates, all in >99% diastereomeric excess. The modular nature of polydeoxypropionates has also inspired the development of several other iterative synthesis strategies83,84.
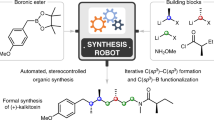
Aggarwal's versatile approach for the iterative synthesis of various stereochemically complex Csp3-rich motifs is demonstrated with his route to (+)-kalkitoxin85 (Fig. 3a). This approach leverages the stereospecificity of the 1,2-metallate rearrangement of boronate complexes86 to install both stereochemistry and functionality through iterative chain extension of boronic esters. In a one-pot procedure, a boronic ester was subjected to a series of six homologations, installing three methylene spacer units and three methyl-bearing stereocentres derived from the requisite enantiomerically pure lithiated benzoates87. Amination followed by amide formation furnished the core of (+)-kalkitoxin in an overall 52% yield. The same approach has been used to synthesize baulamycin A88, tatanan A89, fluorohexestrol90 and C30 botryococcene91, among many other targets92. More broadly, the versatility of this homologation method, which tolerates diverse structural variation in its building blocks, opens the door for divergent synthesis. This can be seen in Aggarwal's assembly-line production method of hydrocarbons with tailored shapes87.
Polyisoprenoids are attractive targets for iterative synthesis due to the numerous 1,5-trisubstituted olefin motifs spanning their structures. Negishi93 has developed an iterative and convergent synthesis for these motifs and applied it to the synthesis of coenzyme Q10 (Fig. 3b). A one-pot iterative cycle begins with the formation of a primary alkylzinc iodide followed by a chemoselective cross-coupling with a diiodo building block. Two further iterations followed by coupling with a diene-containing building block installs five of the trisubstituted olefins of coenzyme Q10. To enable convergent synthesis, the trimethylsilyl (TMS)-protected alkyne also serves as an attachment point. TMS deprotection and subsequent carbometalation–iodination generates a new vinyl iodide that undergoes a strategic and convergent cross-coupling with an earlier homologue. A final round of deprotection, hydrozirconation-iodination and cross-coupling gives coenzyme Q10 in only 11 steps. Only part of the iterative sequence is shown in the figure, but a detailed reaction scheme is available in Supplementary Fig. S2b (Fig. 3b).
Long polyene chains are another modular structure found in natural products. However, these motifs are often sensitive to many common reagents (for example, protic and Lewis acids) and conditions (for example, light and oxygen), rendering their synthesis challenging. In a landmark total synthesis of the complex polyene macrolide amphotericin B (AmB), Nicolaou used an iterative Horner–Wadsworth–Emmons (HWE) strategy to complete the all-trans-polyene motif. Starting from an aldehyde, the first triene unit was installed using a diene-containing phosphonate94 (Fig. 3c). Subsequent conversion of the terminal ethyl ester into an aldehyde set the stage for a second homologation using the phosphonate building block. Deprotection and redox modification furnished a hexaenal, which was esterified with a highly functionalized carboxylic acid containing a phosphonate to generate the open-chain molecule. An intramolecular HWE reaction initiated by K2CO3/18-crown-6 formed the desired cyclic heptaene, completing the carbocyclic core of AmB95 (Supplementary Fig. S2c).
Developing iterative routes to complex, polycyclic molecules represents a substantial challenge, but provided that common repeating units and assembly methods can be established, iterative synthesis can both be practical and expeditious. For example, Uenishi and co-workers developed an iterative protocol for the stereoselective synthesis of linked tetrahydrofuran (THF) rings96 (Fig. 3d). The iterative sequence commences with the formation of a homoallylic alcohol through Grignard addition to an aldehyde or epoxide, followed by cross-metathesis with a stereodefined allylic alcohol. Pd(II)-mediated ring closure forms the THF ring and, finally, ozonolysis of the resultant olefin forms an aldehyde to allow further iteration. Synthesis of either trans-threo-trans or trans-threo-cis THF rings is simply a case of exchanging the allylic alcohol building blocks during the metathesis stage of the iterative cycle.
Mori and co-workers leveraged the modularity inherent in structurally complex polycyclic ether natural products to enable their iterative construction97 (Fig. 3e). In this approach, diastereomerically pure oxiranyl anions were utilized to displace primary tosylates, and the resulting epoxy sulfone products were then subjected to an acid-catalysed 6-endo cyclization. A five-step sequence then generated a new triflate for further iteration. Six repetitions of this protocol led to the efficient construction of the ABCDEF-ring fragments of yessotoxin and adriatoxin, with the stereochemistry of the cyclic ethers introduced through the selection of the appropriate oxirane building blocks. This iterative oxiranyl anion strategy has further been used for the synthesis of hemibrevetoxin B98, gambierol99 and even gymnocin-A with 14 contiguous fused rings100. Other iterative synthesis strategies have also been developed for polycyclic eithers, including iterative ring closing metathesis–hydroboration, iterative reductive cyclizations, and iterative oxonium ylide formation-[2,3]-shift processes101.
Kishi's important synthesis of halichondrin B101 used iterative Nozaki–Hiyama–Kishi (NHK) reactions interspaced by tailoring steps to build cyclic ether moieties in an iterative fashion (Fig. 3f). This strategy allowed highly complex building blocks to be efficiently assembled in an iterative manner. Other family members of the halichondrin family could also be accessed by incorporating variant building blocks into a similar synthetic sequence102. This efficient and modular synthesis enabled Kishi and co-workers to perform a structure–activity relationship study and discover that the macrocyclic portion of halichondrin B was almost as equipotent as an anticancer agent as the natural product itself. A close variant of the synthesis was successfully scaled up by chemists at Eisai to produce enough of this fragment, dubbed eribulin, to enable its clinical study. Significant benefits were observed for patients with metastatic breast cancer and liposarcoma, leading to the FDA approval of eribulin for the treatment of these cancers in 2010 and 2016, respectively103,104.
Iterative approaches to oligomeric small molecules. Beyond natural products, customized iterative assembly methods have also enabled the synthesis of many other types of inherently oligomeric materials (Fig. 4). For example, polyaromatic hydrocarbons have numerous applications in solar cells and light-emitting diodes but can often prove challenging to prepare when site-specific functionalization is required. Kwon and co-workers have developed an efficient strategy for the iterative synthesis of polyaromatic hydrocarbons through the union of a 1,2-dialdehyde and ethyl allenoate105 (Fig. 4a). Oxidative modification of the annulated product furnishes a 2,3-dialdehyde that is primed for another annulation reaction. This iterative cycle rapidly generates 2,3-substituted anthracene and tetracene structures.
The synthesis of larger acenes is complicated by their higher reactivity, but these structures are highly sought after for their applications in organic electronic materials. For example, pentacene is currently the best available organic p-type semiconductor, but larger members could be even more useful106. Excellent syntheses of octacene and nonacene derivatives have been carried out by Echavarren107,108, and Bettinger and co-workers have developed a building block-based approach using iterative Diels–Alder reactions109 (Fig. 4b). Combination of 5,6,7,8-tetramethylenebicyclo[2.2.2]oct-2-ene and an aryne dienophile, generated by the treatment of 1,2-dibromobenzene with n-BuLi, led to the formation of a cycloadduct with a terminating diene moiety. This was treated with an aryne generated from 1,2,4,5-tetrabromobenzene, resulting in a product terminating in another dibromide moiety. An additional iteration of aryne formation and cycloaddition generated a stable precursor to octacene. After a sequence of aromatization and oxidation reactions, exposure to low-wavelength UV light generated octacene for functional studies.
Although most iterative synthesis methods are based on the linear assembly of building blocks, Moore and co-workers have developed a convergent iterative synthesis of phenylacetylene oligomers (Fig. 4c; left) using Sonogashira coupling110,111. Moore's work highlights a crucial advance that enables the application of iterative synthesis in a convergent fashion, the ability to orthogonally protect and deprotect each of the two different functional groups required for building block assembly (Supplementary Fig. S3c). Here, a bifunctional building block can be selectively activated in two different ways: a dialkyltriazene can be converted to an iodide or a TMS protecting group can be removed to reveal a reactive terminal alkyne. These two differently activated building blocks can then be assembled via Sonagashira coupling to form an advanced intermediate containing a dialkyltriazene and a protected alkyne at opposite termini. Repeating this process of using advanced intermediates as building blocks enables exponential molecular growth. In such a manner, it is possible to generate repeating tetramers, octamers and longer oligomers with precise control of the sequence of building blocks. Additionally, building blocks with other functional groups can also be incorporated to prepare diverse oligomeric products with a range of important functions112.
Moore has also developed a convergent approach to iterative synthesis for the assembly of large dendrimers113–115 (Fig. 4c; right). Compared with the strategy for making oligomers, this dendrimer method is different in two respects. In this case, the building blocks are activated in a single direction, by unmasking a reactive terminal alkyne for Sonogashira coupling through the removal of a TMS protecting group. However, exponential molecule growth can still be achieved via double Sonogashira coupling onto a trifunctional monomer containing two bromines. Four iterations of TMS deprotection and double Sonogashira coupling enabled the rapid construction of a monodendron containing 31 building blocks113. This double Sonogashira approach is capable of quickly making large molecules, but generating unsymmetrical targets represents an additional challenge. Such a limitation is not problematic for dendrimer synthesis, and these examples illustrate that convergent iterative synthesis can be most versatile when it has two separate masking and deprotection strategies for orthogonal functional groups.
Iterative synthesis methods have also been developed for molecules with defined 3D architectures. Iptycenes are of interest in materials science and supramolecular chemistry due to their structural rigidity and three-dimensionality. Swager and co-workers devised an iterative solid-state synthesis of extended iptycenes in which a Diels–Alder reaction between anthracene and 1,4-anthraquinone was followed by rearomatization to form a new anthracene unit for further iteration (Fig. 4d). This cycle was then repeated to form longer iptycenes in a modular fashion116.
Many methods have recently been developed to enable boronic esters to be chemoselectively functionalized. For example, Crudden discovered and harnessed such selectivity to facilitate a different approach to iterative synthesis (Fig. 4e). Instead of generating new positions for appending building blocks during each iteration cycle, all of the attachment points were pre-installed in advance. In a one-pot procedure, a Csp2–Csp2 coupling of an aryl boronic ester was followed by group-selective cross-coupling of a Csp3 primary boronic ester. Simple filtration through silica gel enabled a Crudden Csp3 cross-coupling of the remaining secondary boronic ester in the presence of Ag2O, creating a defined polyarylated structure117.
Iterative synthesis has also been applied to create mechanically interlocked molecules. In Goldup's iterative synthesis of oligo[n]rotaxanes, copper-templating via a bipyridyl-containing macrocycle is harnessed to control a three-component click reaction118 (Supplementary Fig. S3g). The product rotaxane has a terminus containing a triisopropyl (TIPS)-protected alkyne. Cleavage of the silyl group and iteration of this reaction allow for the incorporation of different macrocyclic moieties119.
Each of these examples represents an important advance towards efficient and flexible synthetic access to specific types of small-molecule motifs. In most cases, the iterative syntheses proceed in a linear way, but there are also multiple strategies for convergent iterative methods (Fig. 4c) that enable even more efficient molecule growth. There is a trade-off: the more convergent an iterative synthesis method, the more limited its scope of targets. By contrast, divergent approaches to iterative synthesis have the greatest potential to accelerate the discovery of new function, but may require more steps. It is evident from several divergent iterative synthesis methods60,77,82,87,120 that the greater the potential for structural variation in the building blocks, the greater the scope of possible products. This suggests that highly generalized coupling chemistry may provide an opportunity for more generalized iterative synthesis approaches to be achieved.
A general platform for iterative synthesis
A more general iterative platform for small-molecule synthesis requires a common type of assembly chemistry that can form a wide range of different types of C–C and C–X bonds while being tolerant of many functional groups. Although many methods fall short of the high bar required to enable generalized synthesis, metal-mediated cross-coupling represents an exceptionally attractive candidate. The Suzuki–Miyaura and Buchwald–Hartwig couplings, in particular, can use non-toxic and shelf-stable building blocks, are highly efficient and stereospecific, and can proceed under mild reaction conditions with high levels of functional group tolerance. Moreover, the scope of both C–C and C–X bonds that can be formed using such methodology is already very broad and continues to expand, and the versatility of these types of coupling has placed them among the most widely used reactions in both academic and industrial synthesis groups. Most recently, this scope has been extended to include a wide range of Csp3 and Xsp3 coupling partners121–123, even including stereospecific Csp3 cross-couplings of stereochemically defined chiral non-racemic building blocks, a concept that was pioneered by Crudden124–134. These new methods, combined with the anticipated major additional advances in the area of sp3 cross-coupling over the next few decades, suggest that iterative cross-coupling (ICC) could represent a generalizable approach for building block-based small-molecule synthesis.
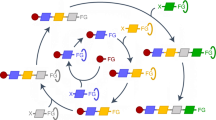
In such a platform, the complex problem of molecular construction can be simplified into simply making and coupling building blocks. In theory, all the required functional groups, oxidation states and stereochemistry can be pre-installed into such building blocks and then faithfully translated into the growing target structure using only mild and stereospecific cross-coupling reactions. Achieving this goal requires compatible bifunctional building blocks that can be iteratively assembled in a precisely controlled manner. This in turn requires the development of methods for reversibly attenuating the reactivity of such building blocks towards metal-mediated coupling110,111 (Fig. 5a).

a | Reversible attenuation of organometallic reactivity enables iterative coupling between dual-functionalized building blocks. b | Silane activation by a proximal hydroxyl group enables iterative Hiyama couplings in the synthesis of oligofluorene-type structures135. c | Iterative Suzuki couplings by reversible protection of the boronic acid functionality with 1,8-diaminonaphthalene (dan)136. d | Iterative Suzuki couplings by reversible protection with N-methyliminodiacetic acid (MIDA)169. THF, tetrahydrofuran; DMF, N,N-dimethylformamide; RT, room temperature.
Several approaches to achieve such reactivity attenuation have leveraged the sensitivity of Lewis acidic organometallic coupling partners in different ways. Hiyama has devised a strategy for reversibly attenuating the reactivity of arylsilanes using specially designed organo[(2-hydroxymethyl)phenyl]dimethylsilanes (Fig. 5b). Under normal cross-coupling conditions, these arylsilanes are ‘switched off’ and unreactive towards transmetalation, but upon deprotection of a strategically positioned neighbouring alcohol, the resulting intramolecular O–Si coordination activates the silane and promotes cross-coupling. Using this method of building block assembly in an iterative fashion, Hiyama completed the synthesis of highly conjugated linear oligoarenylsilanes135.
Suginome reported an alternative method for switching off organometallic coupling partners (Fig. 5c). Complexing boronic acids with the 1,8-diaminonaphthalene (DAN) group decreases the Lewis acidity of the p-orbital of the sp2-hybridized boron atom via electron donation from neighbouring lone pairs on planar nitrogen atoms. The resulting BDAN compounds are stable to both anhydrous and aqueous biphasic cross-coupling conditions, but exposure to strong aqueous HCl or H2SO4 removes the DAN group and releases the corresponding boronic acid. This iterative building block-based method has been applied to the synthesis of oligoarenes136 and oligo(phenylenevinylene)s137.
Work by our group identified that complexation with the trivalent ligand N-methyliminodiacetic acid (MIDA) can alternatively attenuate boronic acid reactivity by rehybridizing the boron atom from sp2 to sp3 (Ref. 138) (Fig. 5d). As described below, MIDA boronates139 represent a very promising platform for generalized building block-based molecular synthesis, and the collective efforts of many different research groups have continued to expand the scope of this approach. Continued advances in these and other methods to reversibly attenuate the reactivity of organometallic building blocks are set to continue to enable generalized synthesis of small molecules and the discovery of new molecular functions.
MIDA boronates have many advantageous physical and chemical features. They are readily purified by silica gel chromatography and/or recrystallization and are usually indefinitely stable on the benchtop as free-flowing crystalline solids138. Moreover, many alkylboronic, vinylboronic and arylboronic acids can be directly converted to their MIDA boronate counterparts in quantitative yields under mild conditions. To drive the MIDA complexation to completion, water can be removed simply via toluene azeotrope with a Dean–Stark trap139 or by the addition of a drying agent such as magnesium sulfate. MIDA boronates are inert to anhydrous cross-coupling reactions, but they can be quickly deprotected under mild aqueous basic conditions. Deprotection can proceed through two different mechanisms: either under a strong aqueous base in which a rate-limiting attack of hydroxide occurs on the carbonyl carbon, or under weakly basic or neutral aqueous conditions in which a slower rate-limiting attack of water occurs on the B–N bond140. This mechanistic divergence is consistent with extensive reaction kinetics, kinetic isotope effects, 18O labelling and computational studies. In situ slow release of MIDA boronates has been advantageous for many reactions, including couplings of unstable heteroarylboronates141,142, polymerization reactions143,144, asymmetric methodologies145, the synthesis of organic photovoltaics146 and a one-pot homologation of boronic acids147.
The durability of MIDA boronates to a wide range of different reagents and reaction conditions further facilitates the synthesis of otherwise challenging to access boronate building blocks from simple boron-containing starting materials (Box 1; Supplementary Fig. S4). For example, many standard oxidations, reductions and protecting group manipulations are well-tolerated148–154. Numerous other common synthetic transformations leave MIDA boronates intact, including aldol reactions148, carbonyl olefination reactions148, Mitsunobu reactions148,155, electrophilic substitution reactions152,156, hydroborations and hydrostannylations154, Diels–Alder cycloadditions154,157 and cyclopropanations150. A variety of transition metal-catalysed reactions are also well-tolerated, including Heck reactions150,158, Grubbs alkene metathesis150, Sonagashira couplings154 and Suzuki cross-couplings138.
MIDA boronates are, however, not without their limitations in terms of scope and application. A large number of Suzuki–Miyaura cross-coupling reactions require the use of an aqueous base, which causes the hydrolysis of MIDA boronates, and Buchwald–Hartwig aminations can involve the use of strong bases that are incompatible with the acidic protons on the backbone of the MIDA ligand. As many Csp3 cross-coupling methods involve aqueous basic reaction conditions, these important limitations currently prevent the use of these reactions in MIDA boronate-based ICC.
Only a few of the many recent advances in MIDA boronate-based building block synthesis from many different research groups are highlighted in this Review. By harnessing the ability of MIDA to rehybridize boron from sp2 to sp3, Zard used vinyl MIDA boronates as acceptors for xanthate-derived radicals159. The resulting α-boryl radicals are sufficiently destabilized to ensure that the reaction is irreversible. Making use of similar vinyl MIDA boronate substrates, Wang149 developed a new oxidative difunctionalization reaction. After treating with SIBX along with either a fluorinating or a trifluoromethylating reagent, the resulting α-boryl ketones can be converted to highly functionalized furans bearing MIDA boronates. A variety of other heterocycle-forming reactions have also been developed using MIDA boronate-containing substrates. Making use of amphoteric molecules containing MIDA boronates and electrophilic ketones or aldehydes, Yudin developed syntheses of a wide range of otherwise challenging to access borylated building blocks, including pyridazines and pyrroles160, imidazoles151, thiazoles152, imidazo[1,2-a]pyridines161 and many other motifs162. Watson163 has also developed a synthesis of 2-borylated indoles and benzofurans involving the use of a palladium-catalysed cascade reaction with ethynyl BMIDA and 2-iodoanilines. Similarly, borylated indolenes and benzofurans have been synthesized through gold-catalysed intramolecular cyclizations onto alkynes164. These reactions are expanding the collection of MIDA boronate building blocks available for small-molecule construction, and hundreds of MIDA boronates are already commercially available.
The protection of boron with the MIDA ligand has also enabled previously inaccessible borylated molecules to be created such as α-boryl adehydes162,164, which without MIDA complexation would rearrange to O-boryl enolates. Yudin and our group have explored applications of this unique class of molecules by exploiting the divergent reactivity of the aldehyde and MIDA boronate motifs165,164. Yudin has extensively expanded this platform to enable the efficient synthesis of a variety of heterocycles and functionalized boronic esters and even the formation of complex tertiary organoboronates through Tsuji–Trost allylations166.
Beyond the synthesis of simple achiral and racemic building blocks, chiral non-racemic MIDA ligand variants can direct diastereoselective epoxidation of vinyl boronates to generate stereoisomerically pure oxyranyl boronates167. Furthermore, installation of enantiopure chiral MIDA ligands onto racemic boronic acids generates diastereomers that can be separated via column chromatography to ultimately yield enantiomerically pure boronic acids168.
ICC with MIDA boronates. Leveraging these many advantageous features, the ICC of MIDA boronates has now been applied by many research groups to the synthesis of a wide range of structurally diverse small molecules138,148,167,169–182 (Fig. 6, Supplementary Fig. S5) This rapidly growing list includes natural products from every major biosynthetic pathway, pharmaceuticals, biological probes and materials components. Moreover, these completed syntheses include linear molecules for which the capacity for iterative building block-based assembly is more apparent, as well as an increasing number of highly complex macrocyclic and polycyclic frameworks for which the inherent modularity is less obvious. Moreover, many of these syntheses took advantage of common off-the-shelf MIDA boronate building blocks, hundreds of which are now commercially available. Collectively, this rapidly growing collection of successful applications of ICC for making a wide range of complex small molecules suggests that the opportunity to drive functional discovery with this type of chemical matter will be increasingly accessible.

a | A generic scheme for iterative cross-coupling. b | Molecules with a linear polyene core represent the largest group of molecules that have so far been prepared via an N-methyliminodiacetic acid (MIDA) boronate-derived iterative synthetic route52,169,172,177,191. Linear precursors synthesized via an MIDA boronate route have been converted into the polycyclic core structures of several natural products180,182. c | Polyarene natural products and drugs are obvious targets of such an iterative Suzuki-type method138,176,179,182,184. d | Polyene substructures of larger molecules, such as amphotericin B173, filipin III174, elansolid B1 (Ref. 178), myxalamide A175 and crocacin B148, have also been prepared using the MIDA-boronate method. Detailed molecular structures are included in Supplementary Fig. S5.
A few illustrative examples are highlighted in detail in Fig. 7. Ratanhine was the first natural product to be synthesized by ICC138 (Fig. 7a). Notably, the mild nature of the deprotection and coupling reactions preserved even hydrolytically sensitive functional groups such as esters. The stability of MIDA also assisted in several additional ways. The benzofuran building block, which in its boronic acid form is prone to decomposition, proved to be bench-stable under air. Even couplings that required elevated temperatures of 80 °C tolerated the MIDA boronate and proceeded in high yield.

A generic scheme for iterative cross-coupling is shown in part a. Detailed reaction schemes are shown for the synthesis of ratanhine138 (part b), a histamine H3 antagonist184 (part c) and peridinin171 (part d). Detailed reaction schemes are included in Supplementary Fig. S6. MIDA, N-methyliminodiacetic acid; TBS, tert-butyldimethylsilyl; THF, tetrahydrofuran; TMS, trimethylsilyl. C and D represent coupling and deprotection steps respectively.
Kobayashi developed an efficient ICC-based synthesis to the natural product myxalamide A175 (Supplementary Fig. S6b). A key challenge in this synthesis was the construction of the central trans-trans-cis-trans tetraene, as such motifs are difficult to install in a stereocontrolled fashion. The capacity of olefinic stereochemical elements pre-installed in shelf-stable bifunctional MIDA boronate building blocks to be faithfully translated into growing targets by using mild and stereospecific cross-coupling methods simplifies this type of complex problem169,170,183. In this specific case, a cis-bifunctional vinyl MIDA boronate building block was iteratively cross-coupled to unite the two key fragments of the polyene core of myxalamide A in a stereospecific fashion. Moreover, no protecting groups were required, as the cross-coupling reactions proceeded in the presence of two free alcohols.
The same concept of leveraging stereospecific cross-couplings to transfer pre-installed stereochemistry from pre-fabricated building blocks into products has also been extended to stereogenic Csp3 centres. During the synthesis of a glucagon receptor inhibitor, the presence of several Csp3 carbons offered strategic disconnection points (Supplementary Fig. S6c). The first two building blocks were assembled by a Csp3 coupling between an aryl iodide and a primary alkylzinc reagent167. In the second coupling reaction, an enantiomerically enriched benzylic boronate was then subjected to Crudden's stereoretentive Csp3 coupling to yield the targeted chiral pharmaceutical candidate in a very simple manner124.
Another powerful reaction in the synthesis of pharmaceutical compounds is C–N bond formation; however, strong bases are sometimes required for less reactive amines and, as described above, these conditions can be incompatible with MIDA boronates. Hamann and co-workers pioneered a strategy that selectively protects MIDA boronates in situ by enolization with Li(N(SiMe3)2) (LiHMDS) at low temperatures184. This more robust Li-MIDA enolate enabled an efficient one-pot ICC synthesis of the histamine H3 agonist through iterative C–N and then C–C coupling reactions (Fig. 7b). This Li-MIDA enolate strategy may have other applications beyond C–N coupling where use of a strong base causes loss or cleavage of the MIDA protecting group.
Even some highly complex small molecules have been prepared via ICC. For example, the natural product peridinin (Fig. 7c) is a norcarotenoid that contains several complex functional groups and stereochemical elements, including a butenolide, an epoxide and an allene motif. Because of the mild and stereospecific nature of MIDA-boronate ICC, all of these functional groups and stereochemical elements were pre-installed in the four complex building blocks and faithfully translated into the final product, yielding the first fully stereocontrolled synthesis of peridinin171. Each MIDA boronate intermediate, including the final heptaenyl MIDA boronate, proved stable to chromatography and storage. Although the boronic acid in the final coupling reaction proved unstable to isolation, an in situ deprotection of the MIDA boronate allowed the coupling to proceed both in high yield and with complete stereoretention.
Polycyclic molecules present an especially formidable challenge for building block-based construction. However, the biosynthesis of complex, Csp3-rich polycyclic natural products often involves an actionable two-part strategy: the assembly of building blocks into a linear precursor and then a cyclization reaction to transform this linear molecule into a complex (poly)cyclic skeleton185. In theory, ICC could be used in a similar linear-to-cyclized approach. This was confirmed by the synthesis of the pentacyclic core of secodaphnane natural products182 (Fig. 8a). Two cycles of Csp3 coupling were initially used to construct a linear precursor, which was then subjected to a bioinspired cyclization cascade involving amine condensation and intramolecular Diels–Alder and Prins cyclizations186.

a | Alinear tetra ene intermediate was prepared using the N-methyliminodiacetic acid (MIDA) boronate method. Alkene reduction, ester reduction and deprotection leads to a linear primary alcohol. Swern oxidation followed by condensation with methylamine leads to construction of the secodaphnane core182. b | Iterative MIDA-boronate couplings form a key part of a strategy to produce a modified version of amphotericin B173. C and D represent coupling and deprotection steps respectively. Detailed reaction schemes are included in Supplementary Fig. S6. TBS, tert-butyldimethylsilylation; TIPS, triisopropylsilyl.
An exceptionally efficient linear-to-cyclized approach was also used by Vosburg to make highly complex polycyclic natural product frameworks, including the ethyl ester of the tetracyclic natural product cryptobeilic acid D179 (Supplementary Fig. S6g). Vosburg and co-workers used all commercially available MIDA boronate building blocks to stereospecifically construct the complex trans-cis-cis-trans-tetraene linear precursor. This tetraene was primed for an 8π/6π electrocyclization cascade to generate a fused 4–6 ring system, which ultimately led to the completion of cryptobeilic acid D180.
This linear-to-cyclized strategy has also enabled biological studies of clinically relevant natural products. AmB has served for more than 50 years as a last line of defence against invasive and drug-resistant fungal infections, but it has dose-limiting side effects. With the goal of gaining a better understanding of the mechanism of toxicity of AmB, an AmB derivative lacking a single hydroxyl group was synthesized via ICC followed by macrocyclization187 (Fig. 8b). This new compound, C35deOAmB, lacked the ability to form ion channels but retained its toxicity to fungal pathogens, overturning decades of prior thinking about the primary mechanism of action of this clinically vital but unfortunately highly toxic natural product187. This discovery helped to build a strong foundation for ongoing efforts to rationally optimize the therapeutic index of AmB188,189. It also enabled the ion channel-forming capacity of this natural product to be rationally separated from its cell-killing effects. This, in turn, facilitated the development of small molecules that replace missing protein ion channels and thereby restore physiology, akin to acting as prostheses on the molecular scale4,5.
MIDA boronates have also been used independently of iteration in a wide range of other small-molecule synthesis applications143,144,146,153,184,190–197.
Automation of small-molecule synthesis
The automation of small-molecule synthesis has the potential to dramatically improve the efficiency with which new molecular functions can be discovered and optimized. It also represents an actionable path for bringing the power of making small molecules to non-specialists. One strategy for automating small-molecule synthesis involves translating current customized synthesis approaches into an automated format198–201. Although this approach has the advantage of utilizing known manual solutions, it necessitates the automation of many different types of chemical reaction, each of which requires different reagents, conditions, optimization protocols, reaction vessels and/or purification protocols.
A major advantage of ICC is that, once the necessary building blocks are in hand, it only requires the same two reactions — deprotection and coupling — to complete the molecular assembly process. This presents the strategic opportunity for collective efforts to be deployed to extensively optimize these two reactions and, ultimately, to reach highly generalized conditions, as was previously achieved for iterative peptide202 and oligonucleotide synthesis203, and is now being increasingly realized for oligosaccharides204.
It is also notable that every intermediate in a MIDA boronate-based ICC sequence contains the same common MIDA boronate functional group. It was recognized that this could potentially serve as a handle for generalized purification. In this vein, MIDA boronates were discovered to possess an unusual binary affinity for silica gel with certain pairs of eluents182 (Fig. 9a). Regardless of size, functional group content or polarity, MIDA boronates show minimal mobility when eluting with MeOH:Et2O, but simply switching the solvent to THF causes rapid elution. This was the key to developing a generalized ‘catch-and-release’ purification platform. A crude reaction is first loaded onto a silica gel plug and washed with 1.5% MeOH in Et2O to remove any excess reagents, catalysts or other impurities. Then, by simply switching the solvent to THF, the purified MIDA boronate is rapidly released. This methodology was readily converted into a general automated purification module.

a | The binary affinities of N-methyliminodiacetic acid (MIDA) boronates for silica gel when eluting with diethyl ether and methanol (top) and tetrahydrofuran (THF; bottom) enable catch and release purification. b | Design of a machine for automated iterative cross-coupling using modules for deprotection, coupling and purification. c | Automated synthesis of ratanhine and 19 derivatives; the target structure can be broken down into four substructures. Combinations of these haloboronate building blocks allow the automated synthesis of many different structural derivatives. d | Off-machine cyclization can be used to convert linear precursors prepared by automated iterative cross-coupling into complex polycyclic structures.
This catch-and-release purification module was then combined with a deprotection module and a cross-coupling module to create a machine capable of carrying out each step of ICC in a fully automated fashion182. This synthesis machine (Fig. 9b) proved capable of making natural products from every major biosynthetic pathway (crocacin C, β-parinaric acid, all-trans-retinal and ratanhine) and many natural product derivatives. In addition, materials components (oligophenylene and oligothiophene), pharmaceuticals (PDE472 and BRAF kinase inhibitors), and biological probes (BTP2) were also made in the same fully automated fashion. In each case, all the functional groups, oxidation states and stereochemical information were pre-installed into the corresponding building blocks and then faithfully translated to the products via automated stereospecific couplings.
Another advantage of building block-based synthesis is the ability to rapidly prepare many different structural derivatives of a targeted compound. For the natural product ratanhine, the automated synthesizer was capable of combining four sets of variant building blocks in all possible combinations to make 19 unique structural derivatives of the parent natural product (Fig. 9c). All 20 of these syntheses were completed without any customized optimization of the conditions for deprotection or coupling.
It is not immediately obvious that the same building block-based approach can be used to create complex polycyclic scaffolds consisting of an intricate network of Csp3-linkages. However, by integrating the abovementioned biomimetic ‘linear-to-cyclized’ strategy (Fig. 9d), even Csp3-rich cyclic and polycyclic natural products can be accessed using automated synthesis. For the macrocyclic natural product citreofuran, the fully automated assembly of building blocks into a linear precursor set the stage for an atropdiastereoselective Mitsunobu cyclization. In the case of oblongolide, the stereochemical information encoded in the building blocks was faithfully translated through the automated assembly of the linear precursor and then used to direct the subsequent diastereoselective polycyclization. A steroid-like core was made through an automatically assembled linear precursor followed by a catalyst-promoted enantioselective and diastereoselective cation-π cyclization. The same first two building blocks were also used to achieve the automated synthesis of the linear precursor for the pentacyclic secodaphnane natural product core. This strategy of combining the automated assembly of prefabricated molecular building blocks with biomimetic cyclization reactions has substantial potential to remove the synthetic bottleneck to accessing many other complex, biologically relevant natural products. Thus, although many important challenges remain, the scope of the now fully automated MIDA boronate-based ICC is already substantial and rapidly expanding.
Summary and prospectus
Over the past several centuries, the strategy of the iterative assembly of building blocks has repeatedly accelerated the discovery of new functions on many scales. The recent development of automated synthesis platforms for oligopeptides and oligonucleotides, and increasingly for oligosaccharides, removed the synthetic barrier and led to countless discoveries and applications of these classes of chemical matter. Small molecules are different, and the development of a highly general building block synthesis for this type of chemical matter will require solving a unique set of challenges. That said, increasing evidence suggests that this is a solvable problem.
Achieving this goal will require very efficient and versatile ways of both making and coupling a wide range of building blocks that correspond to some of the most common substructural motifs found in small molecules. For specific classes of small molecules, customized iterative strategies have already demonstrated some of this potential and have led to important discoveries. A more general platform for small-molecule synthesis now represents a highly attractive and accessible target. As an important step in this direction, ICC with MIDA boronates has already enabled the synthesis of many different types of small molecules in a fully automated fashion.
Continued progress towards this goal will depend on the development of advanced chemical methodologies. The past several decades have seen tremendous progress in the expansion of cross-coupling chemistry, even including stereocontrolled Csp3 coupling methods. Another important opportunity involves the integration of customized iterative approaches into more generalized synthesis platforms, as some of these customized approaches represent especially efficient routes to specific areas of chemical space. These advances will enable generalized synthesis strategies to make an increasingly broad range of different small molecules, and the resulting discoveries of new molecular functions will inspire further efforts to improve the versatility of generalized synthesis.
It is worth noting that customized synthesis retains many practical advantages in the scale-up of routes to particular target compounds, where a premium is placed on step-count, atom economy and overall efficiency. Thus, removing the synthesis bottleneck on the discovery scale will create many more opportunities for chemists to find the best ways of making exceptionally functional molecules practically accessible at scale.
Given the extraordinary untapped potential that small molecules have in terms of helping to solve some of the most important problems facing society, it is exciting to consider the prospect of generalized and automated synthesis platforms bringing the power of synthesis to non-specialists. There are many great ideas out there just waiting to be harnessed.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
How to cite this article
Lehmann, J. W., Blair, D. J. & Burke, M. D. Towards the generalized iterative synthesis of small molecules. Nat. Rev. Chem. 2, 0115 (2018).
Change history
07 March 2018
In the version of this Article originally published, the author affiliations were incorrect. This error has now been corrected.
References
Wender, P. A. & Miller, B. L. Synthesis at the molecular frontier. Nature 460, 197–201 (2009).
Villar, E. A. et al. How proteins bind macrocycles. Nat. Chem. Biol. 10, 723–731 (2014).
Du, J., Lu, W., Wu, S., Cheng, Y. & Gouaux, E. Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 526, 224–229 (2015).
Cioffi, A. G., Hou, J., Grillo, A. S., Diaz, K. A. & Burke, M. D. Restored physiology in protein-deficient yeast by a small molecule channel. J. Am. Chem. Soc. 137, 10096–10099 (2015).
Grillo, A. S. et al. Restored iron transport by a small molecule promotes absorption and hemoglobinization in animals. Science 356, 608–616 (2017).
Kocaoglu, O. & Carlson, E. E. Progress and prospects for small-molecule probes of bacterial imaging. Nat. Chem. Biol. 12, 472–478 (2016).
Chan, J., Dodani, S. C. & Chang, C. J. Reaction-based small-molecule fluorescent probes for chemoselective bioimaging. Nat. Chem. 4, 973–984 (2012).
Weber, J., Beard, P. C. & Bohndiek, S. E. Contrast agents for molecular photoacoustic imaging. Nat. Methods 13, 639–650 (2016).
Li, H., Zhang, P., Smaga, L. P., Hoffman, R. A. & Chan, J. Photoacoustic probes for ratiometric imaging of copper(II) J. Am. Chem. Soc. 137, 15628–15631 (2015).
Schumacher, S., Sartorius, D., Ehrentreich-Forster, E. & Bier, F. F. Miniaturization for point-of-care analysis: platform technology for almost every biomedical assay. eJIFCC 23, 70–75 (2012).
Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075–10166 (2016).
Kiser, P. D., Golczak, M. & Palczewski, K. Chemistry of the retinoid (visual) cycle. Chem. Rev. 114, 194–232 (2014).
Mart, R. J. & Allemann, R. K. Azobenzene photocontrol of peptides and proteins. Chem. Commun. 52, 12262–12277 (2016).
Koumura, N., Zijlstra, R. W., van Delden, R. A., Harada, N. & Feringa, B. L. Light-driven monodirectional molecular rotor. Nature 401, 152–155 (1999).
Hickenboth, C. R. et al. Biasing reaction pathways with mechanical force. Nature 446, 423–427 (2007).
Davis, D. A. et al. Force-induced activation of covalent bonds in mechanoresponsive polymeric materials. Nature 459, 68–72 (2009).
Aizawa, N. et al. Solution-processed multilayer small-molecule light-emitting devices with high-efficiency white-light emission. Nat. Commun. 5, 5756 (2014).
Wilson, G. O. et al. Evaluation of ruthenium catalysts for ring-opening metathesis polymerization-based self-healing applications. Chem. Mater. 20, 3288–3297 (2007).
Thiele, S. et al. Electrically driven nuclear spin resonance in single-molecule magnets. Science 344, 1135–1138 (2014).
Heinrich, B. W., Braun, L., Pascual, J. I. & Franke, K. J. Protection of excited spin states by a superconducting energy gap. Nat. Phys. 9, 765–768 (2013).
Valade, L., Caro, D. d., Faulmann, C. & Jacob, K. TTF[Ni(dmit)2]2: From single-crystals to thin layers, nanowires, and nanoparticles. Coord. Chem. Rev. 308, 433–444 (2016).
Cui, H. et al. A single-component molecular superconductor. J. Am. Chem. Soc. 136, 7619–7622 (2014).
Cooper, C. C. The Portsmouth system of manufacture. Technol. Cult. 25, 185–225 (1984).
Alder, K. Innovation and amnesia: engineering rationality and the fate of interchangeable parts manufacturing in France Technol. Cult. 38, 273–311 (1997).
Rattenbury, R. C. A Legacy in Arms: American Firearm Manufacture, Design, and Artistry, 1800–1900 (University of Oklahoma Press, 2014).
Hounshell, D. From the American System to Mass Production, 1800-1932: The Development of Manufacturing Technology in the United States (Johns Hopkins University Press, 1985).
Ali, M. M. & Moon, K. S. Structural developments in tall buildings: current trends and future prospects. Architect. Sci. Rev. 50, 205–223 (2007).
Telamarthi, K., Aman, M. S. & Abdelgawad, A. An application-driven modular IoT architecture. Wireless Commun. Mobile Comput. 2017, 1350929 (2017).
Kitmacher, G. H. Design of the Space Station Habitable Modules (American Institute of Aeronautics and Astronautics, 2002).
Meng, Y., Johnson, K., Simms, B. & Conforth, M. in 2008 IEEE/RSJ International Conference on Intelligent Robots and Systems 3725–3730 (Nice, France, 2008).
Garud, R., Kumaraswamy, A. & Langlois, R. Managing in the Modular Age: Architectures, Networks, and Organizations (Blackwell, 2002).
Peretz, I. & Coltheart, M. Modularity of music processing. Nat. Neurosci. 6, 688–691 (2003).
d’Orazio, F. Introducing modules: artificial intelligence on demand on Pulsar. Pulsarhttps://www.pulsarplatform.com/blog/2016/introducing-modules-artificial-intelligence-on-demand-on-pulsar/ (2016).
Baldwin, C. Y. & Clark, K. B. Managing in an age of modularity. Harvard Business Review 75, 84–93 (1997).
Schubert, C., van Langeveld, M. C. & Donoso, L. A. Innovations in 3D printing: a 3D overview from optics to organs. Br. J. Ophthalmol. 98, 159–161 (2014).
MacDonald, E. et al. 3D printing for the rapid prototyping of structural electronics. IEEE Access 2, 234–242 (2014).
Ventola, C. L. Medical applications for 3D printing: current and projected uses. P T 39, 704–711 (2014).
Gross, B. C., Erkal, J. L., Lockwood, S. Y., Chen, C. & Spence, D. M. Evaluation of 3D printing and its potential impact on biotechnology and the chemical sciences. Anal. Chem. 86, 3240–3253 (2014).
Merrified, R. B. Automated synthesis of peptides. Science 150, 178–185 (1965).
Caruthers, M. H. Gene synthesis machines: DNA chemistry and its uses. Science 230, 281–285 (1985).
Trevino, V., Falciani, F. & Barrera-Saldana, H. A. DNA microarrays: a powerful genomic tool for biomedical and clinical research. Mol. Med. 13, 527–541 (2007).
Lenoir, T. & Giannella, E. The emergence and diffusion of DNA microarray technology. J. Biomed. Discov. Collabor. 1, 11 (2006).
Fosgerau, K. & Hoffmann, T. Peptide therapeutics: current status and future directions. Drug Discov. Today 20, 122–128 (2015).
Khvorova, A. & Watts, J. K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 35, 238–248 (2017).
Gibson, D. G. et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–56 (2010).
Khorana, H. G. Total synthesis of a gene. Science 203, 614–625 (1979).
Kent, S. B. Total chemical synthesis of proteins. Chem. Soc. Rev. 38, 338–351 (2009).
Seeberger, P. H. & Werz, D. B. Automated synthesis of oligosaccharides as a basis for drug discovery. Nat. Rev. Drug Discov. 4, 751–763 (2005).
Plante, O. J., Palmacci, E. R. & Seeberger, P. H. Automated solid-phase synthesis of oligosaccharides. Science 291, 1523–1527 (2001).
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661 (2016).
Rodrigues, T., Reker, D., Schneider, P. & Schneider, G. Counting on natural products for drug design. Nat. Chem. 8, 531–541 (2016).
Woerly, E. M., Roy, J. & Burke, M. D. Synthesis of most polyene natural product motifs using just 12 building blocks and one coupling reaction. Nat. Chem. 6, 484–491 (2014).
Pye, C. R., Bertin, M. J., Lokey, R. S. Gerwick, W. H. & Linington, R. G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl Acad. Sci. USA 114, 5601–5606 (2017).
Palazzolo, A. M. E., Simons, C. L. W. & Burke, M. D. The natural productome. Proc. Natl Acad. Sci. USA 114, 5564–5566 (2017).
Service, R. F. Billion-dollar project would synthesize hundreds of thousands of molecules in search of new medicines. Sciencehttp://www.sciencemag.org/news/2017/04/billion-dollar-project-would-synthesize-hundreds-thousands-molecules-search-new (2017).
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U. S. FDA approved pharmaceuticals. J. Med. Chem. 57, 10257–10274 (2014).
Frihed, T. G., Bols, M. & Pedersen, C. M. Synthesis of L-hexoses. Chem. Rev. 115, 3615–3676 (2015).
Ko, S. Y. et al. Total synthesis of the L-hexoses. Science 220, 949–951 (1983).
Paterson, I. & Scott, J. P. Polyketide library synthesis: iterative assembly of extended polypropionates using (R )- and (S)-l-(benzyloxy)-2-methylpentan-3-one. Tetrahedron Lett. 38, 7441–7444 (1997).
Paterson, I., Donghi, M. & Gerlach, K. A. Combinatorial approach to polyketide-type libraries by iterative asymmetric aldol reactions performed on solid support. Angew. Chem. Int. Ed. 39, 3315–3319 (2000).
Evans, D. A., Clark, J. S., Metternich, R., Novack, V. J. & Sheppard, G. S. Diastereoselective aldol reactions using.beta.-keto imide derived enolates. A versatile approach to the assemblage of polypropionate systems. J. Am. Chem. Soc. 112, 866–868 (1990).
Evans, D. A., Nelson, J. V., Vogel, E. & Taber, T. R. Stereoselective aldol condensations via boron enolates. J. Am. Chem. Soc. 103, 3099–3111 (1981).
Crimmins, M. T. & Chaudhary, K. Titanium enolates of thiazolidinethione chiral auxiliaries: versatile tools for asymmetric aldol additions. Org. Lett. 2, 775–777 (2000).
Crimmins, M. T., King, B. W., Tabet, E. A. & Chaudhary, K. Asymmetric aldol additions: use of titanium tetrachloride and (-)-sparteine for the soft enolization of N-acyl oxazolidinones, oxazolidinethiones, and thiazolidinethiones. J. Org. Chem. 66, 894–902 (2001).
Crimmins, M. T. & Slade, D. J. Formal synthesis of 6-deoxyerythronolide B. Org. Lett. 8, 2191–2194 (2006).
Brown, H. C., Bhat, K. S. & Randad, R. S. β-Allyldiisopinocampheylborane: a remarkable reagent for the diastereoselective allylboration of.alpha.-substituted chiral aldehydes. J. Org. Chem. 52, 319–320 (1987).
Brown, H. C. & Bhat, K. S. Enantiomeric Z− and E-crotyldiisopinocampheylboranes. Synthesis in high optical purity of all four possible stereoisomers of β-methylhomoallyl alcohols. J. Am. Chem. Soc. 108, 293–294 (1986).
Brown, H. C. & Bhat, K. S. Chiral synthesis via organoboranes. 7. Diastereoselective and enantioselective synthesis of erythro- and threo-.beta.-methylhomoallyl alcohols via enantiomeric (Z )- and (E)-crotylboranes. J. Am. Chem. Soc. 108, 5919–5923 (1986).
Garcia-Fortanet, J., Murga, J., Carda, M. & Marco, J. A. On the structure of passifloricin A: asymmetric synthesis of the delta-lactones of (2Z, 5S,7R,9S,11S)- and (2Z,5R,7R,9S,11S)tetrahydroxyhexacos-2-enoic acid. Org. Lett. 5, 1447–1449 (2003).
Dechert-Schmitt, A. M., Schmitt, D. C., Gao, X., Itoh, T. & Krische, M. J. Polyketide construction via hydrohydroxyalkylation and related alcohol C–H functionalizations: reinventing the chemistry of carbonyl addition. Nat. Prod. Rep. 31, 504–513 (2014).
Han, S. B., Hassan, A., Kim, I. S. & Krische, M. J. Total synthesis of (+)-roxaticin via C–C bond forming transfer hydrogenation: a departure from stoichiometric chiral reagents, auxiliaries, and premetalated nucleophiles in polyketide construction. J. Am. Chem. Soc. 132, 15559–15561 (2010).
Shin, I., Hong, S. & Krische, M. J. Total synthesis of swinholide A: an exposition in hydrogen-mediated C–C Bond formation. J. Am. Chem. Soc. 138, 14246–14249 (2016).
Gao, X., Woo, S. K. & Krische, M. J. Total synthesis of 6-deoxyerythronolide B via C–C bond-forming transfer hydrogenation. J. Am. Chem. Soc. 135, 4223–4226 (2013).
Feng, J., Kasun, Z. A. & Krische, M. J. Enantioselective alcohol C–H functionalization for polyketide construction: unlocking redox-economy and site-selectivity for ideal chemical synthesis. J. Am. Chem. Soc. 138, 5467–5478 (2016).
Ketcham, J. M. et al. Evaluation of chromane-based bryostatin analogues prepared via hydrogen-mediated C–C bond formation: potency does not confer bryostatin-like biology. J. Am. Chem. Soc. 138, 13415–13423 (2016).
Myers, A. G., Yang, B. H., Chen, H. & Gleason, J. L. Use of pseudoephedrine as a practical chiral auxiliary for asymmetric synthesis. J. Am. Chem. Soc. 116, 9361–9362 (1994).
Myers, A. G., Yang, B. H., Chen, H. & Kopecky, D. J. Asymmetric synthesis of 1,3-dialkyl-substituted carbon chains of any stereochemical configuration by an iterable process. Synlett 1997, 457–459 (1997).
Vong, B. G., Abraham, S., Xiang, A. X. & Theodorakis, E. A. Synthetic studies on borrelidin: enantioselective synthesis of the C1–C12 fragment. Org. Lett. 5, 1617–1620 (2003).
Vong, B. G., Kim, S. H., Abraham, S. & Theodorakis, E. A. Stereoselective total synthesis of (-)-borrelidin. Angew. Chem. Int. Ed. 43, 3947–3951 (2004).
ter Horst, B., Feringa, B. L. & Minnaard, A. J. Catalytic asymmetric synthesis of phthioceranic acid, a heptamethyl-branched acid from Mycobacterium tuberculosis. Org. Lett. 9, 3013–3015 (2007).
Geerdink, D. & Minnaard, A. J. Total synthesis of sulfolipid-1. Chem. Commun. 50, 2286–2288 (2014).
Brand, G. J., Studte, C. & Breit, B. Iterative synthesis of (oligo)deoxypropionates via zinc-catalyzed enantiospecific sp3-sp3 cross-coupling. Org. Lett. 11, 4668–4670 (2009).
ter Horst, B., Feringa, B. L. & Minnaard, A. J. Iterative strategies for the synthesis of deoxypropionates. Chem. Commun. 46, 2535–2547 (2010).
Schmid, F., Baro, A. & Laschat, S. Strategies for the synthesis of deoxypropionates. Synthesis 49, 237–251 (2017).
Balieu, S. et al. Toward ideality: the synthesis of (+)-kalkitoxin and (+)-hydroxyphthioceranic acid by assembly-line synthesis. J. Am. Chem. Soc. 137, 4398–4403 (2015).
Thomas, S. P., French, R. M., Jheengut, V. & Aggarwal, V. K. Homologation and alkylation of boronic esters and boranes by 1,2-metallate rearrangement of boronate complexes. Chem. Rec. 9, 24–39 (2009).
Burns, M. et al. Assembly-line synthesis of organic molecules with tailored shapes. Nature 513, 183–188 (2014).
Wu, J. et al. Synergy of synthesis, computation and NMR reveals correct baulamycin structures. Nature 547, 436–440 (2017).
Noble, A., Roesner, S. & Aggarwal, V. K. Short enantioselective total synthesis of tatanan a and 3-epi-tatanan a using assembly-line synthesis. Angew. Chem. Int. Ed. 55, 15920–15924 (2016).
Roesner, S., Blair, D. J. & Aggarwal, V. K. Enantioselective installation of adjacent tertiary benzylic stereocentres using lithiation–borylation–protodeboronation methodology. Application to the synthesis of bifluranol and fluorohexestrol. Chem. Sci. 6, 3718–3723 (2015).
Pulis, A. P. & Aggarwal, V. K. Synthesis of enantioenriched tertiary boronic esters from secondary allylic carbamates. Application to the synthesis of C30 botryococcene. J. Am. Chem. Soc. 134, 7570–7574 (2012).
Leonori, D. & Aggarwal, V. K. Lithiation-borylation methodology and its application in synthesis. Acc. Chem. Res. 47, 3174–3183 (2014).
Negishi, E., Liou, S. Y., Xu, C. & Huo, S. A novel, highly selective, and general methodology for the synthesis of 1,5-diene-containing oligoisoprenoids of all possible geometrical combinations exemplified by an iterative and convergent synthesis of coenzyme Q(10). Org. Lett. 4, 261–264 (2002).
Nicolaou, K. C., Chakraborty, T. K., Daines, R. A. & Simpkins, N. S. Retrosynthetic and Synthetic Chemistry on amphotericin b. synthesis of C(1)-C(20) and C(21)-C(38) fragments and construction of the 38-membered macrocycle. J. Chem. Soc., Chem. Commun. 413–416 (1986).
Nicolaou, K. C., Daines, R. A., Chakraborty, T. K. & Ogawa, Y. Total synthesis of amphoteronolide B and amphotericin B. 2. Total synthesis of amphoteronolide B. J. Am. Chem. Soc. 110, 4685–4696 (1988).
Suzuki, A. et al. Construction of iterative tetrahydrofuran ring units and total synthesis of (+)-goniocin. Org. Lett. 18, 2248–2251 (2016).
Mori, Y., Nogami, K., Hayashi, H. & Noyori, R. Sulfonyl-stabilized oxiranyllithium-based approach to polycyclic ethers. Convergent synthesis of the ABCDEF-ring system of yessotoxin and adriatoxin. J. Org. Chem. 68, 9050–9060 (2003).
Mori, Y., Yaegashi, K. & Furukawa, H. Formal total synthesis of hemibrevetoxin B by an oxiranyl anion strategy. J. Org. Chem. 63, 6200–6209 (1998).
Furuta, H., Hasegawa, Y., Hase, M. & Mori, Y. Total synthesis of gambierol by using oxiranyl anions. Chem. Eur. J. 16, 7586–7595 (2010).
Sakai, T. et al. Total synthesis of gymnocin-A. J. Am. Chem. Soc. 137, 14513–14516 (2015).
Marmsater, F. P. & West, F. G. New efficient iterative approaches to polycyclic ethers. Chem. Eur. J. 8, 4346–4353 (2002).
Yamamoto, A., Ueda, A., Bremond, P., Tiseni, P. S. & Kishi, Y. Total synthesis of halichondrin C. J. Am. Chem. Soc. 134, 893–896 (2012).
Ledford, H. Complex synthesis yields breast-cancer therapy. Nature 468, 608–609 (2010).
Cortes, J. et al. Eribulin monotherapy versus treatment of physician's choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet 377, 914–923 (2011).
Zhang, K., Cai, L., Jiang, X., Garcia-Garibay, M. A. & Kwon, O. Phosphine-mediated iterative arene homologation using allenes. J. Am. Chem. Soc. 137, 11258–11261 (2015).
Anthony, J. E. The larger acenes: versatile organic semiconductors. Angew. Chem. Int. Ed. 47, 452–483 (2008).
Zuzak, R. et al. Nonacene generated by on-surface dehydrogenation. ACS Nano 11, 9321–9329 (2017).
Dorel, R. & Echavarren, A. M. Strategies for the synthesis of higher acenes. Eur. J. Org. Chem. 2017, 14–24 (2017).
Tonshoff, C. & Bettinger, H. F. Photogeneration of octacene and nonacene. Angew. Chem. Int. Ed. 49, 4125–4128 (2010).
Zhang, J., Moore, J. S., Xu, Z. & Aguirre, R. A. Nanoarchitectures. 1. Controlled synthesis phenylacetylene sequences. J. Am. Chem. Soc. 114, 2273–2274 (1992).
Moore, J. S. Shape-persistent molecular architectures of nanoscale dimension. Acc. Chem. Res. 30, 402–413 (1997).
Zhang, J., Pesak, D. J., Ludwick, J. L. & Moore, J. S. Geometrically-controlled and site-specifically-functionalized phenylacetylene macrocycles. J. Am. Chem. Soc. 116, 4227–4239 (1993).
Bharathi, P., Patel, U., Kawaguchi, T., Pesak, D. J. & Moore, J. S. Improvements in the synthesis of phenylacetylene monodendrons including a solid-phase convergent method. Macromolecules 28, 5955–5963 (1995).
Xu, Z., Kahr, M., Walker, K. L., Wilkins, C. L. & Moore, J. S. Phenylacetylene dendrimers by the divergent, convergent, and double-stage convergent methods. J. Am. Chem. Soc. 116, 4537–4550 (1994).
Xu, Z. & Moore, J. S. Rapid construction of large-size phenylacetylene dendrimers up to 12.5 nanometers in molecular diameter. Angew. Chem. Int. Ed. Engl. 32, 1354–1357 (1993).
Zhao, Y., Rocha, S. V. & Swager, T. M. Mechanochemical synthesis of extended iptycenes. J. Am. Chem. Soc. 138, 13834–13837 (2016).
Crudden, C. M. et al. Iterative protecting group-free cross-coupling leading to chiral multiply arylated structures. Nat. Commun. 7, 11065 (2016).
Denis, M. & Goldup, S. M. The active template approach to interlocked molecules. Nat. Rev. Chem. 1, 0061 (2017).
Lewis, J. E., Winn, J., Cera, L. & Goldup, S. M. Iterative synthesis of oligo[n]rotaxanes in excellent yield. J. Am. Chem. Soc. 138, 16329–16336 (2016).
Bredenkamp, A., Wegener, M., Hummel, S., Haring, A. P. & Kirsch, S. F. Versatile process for the stereodiverse construction of 1,3-polyols: iterative chain elongation with chiral building blocks. Chem. Commun. 52, 1875–1878 (2016).
Johansson Seechurn, C. C., Kitching, M. O., Colacot, T. J. & Snieckus, V. Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 51, 5062–5085 (2012).
Hartwig, J. F. Organotransition Metal Chemistry: From Bonding to Catalysis (University Science Books, 2010).
Martin, R. & Buchwald, S. L. Palladium-catalyzed Suzuki-Miyaura cross-coupling reactions employing dialkylbiaryl phosphine ligands. Acc. Chem. Res. 41, 1461–1473 (2008).
Imao, D., Glasspoole, B. W., Laberge, V. S. & Crudden, C. M. Cross coupling reactions of chiral secondary organoboronic esters with retention of configuration. J. Am. Chem. Soc. 131, 5024–5025 (2009).
Matthew, S. C., Glasspoole, B. W., Eisenberger, P. & Crudden, C. M. Synthesis of enantiomerically enriched triarylmethanes by enantiospecific Suzuki–Miyaura cross-coupling reactions. J. Am. Chem. Soc. 136, 5828–5831 (2014).
Sandrock, D. L., Jean-Gerard, L., Chen, C. Y., Dreher, S. D. & Molander, G. A. Stereospecific cross-coupling of secondary alkyl beta-trifluoroboratoamides. J. Am. Chem. Soc. 132, 17108–17110 (2010).
Molander, G. A. & Wisniewski, S. R. Stereospecific cross-coupling of secondary organotrifluoroborates: potassium 1-(benzyloxy)alkyltrifluoroborates. J. Am. Chem. Soc. 134, 16856–16868 (2012).
Ohmura, T., Awano, T. & Suginome, M. Stereospecific Suzuki-Miyaura coupling of chiral alpha-(acylamino)benzylboronic esters with inversion of configuration. J. Am. Chem. Soc. 132, 13191–13193 (2010).
Awano, T., Ohmura, T. & Suginome, M. Inversion or retention? Effects of acidic additives on the stereochemical course in enantiospecific Suzuki-Miyaura coupling of alpha-(acetylamino)benzylboronic esters. J. Am. Chem. Soc. 133, 20738–20741 (2011).
Lee, J. C., McDonald, R. & Hall, D. G. Enantioselective preparation and chemoselective cross-coupling of 1,1-diboron compounds. Nat. Chem. 3, 894–899 (2011).
Li, L., Wang, C. Y., Huang, R. & Biscoe, M. R. Stereoretentive Pd-catalysed Stille cross-coupling reactions of secondary alkyl azastannatranes and aryl halides. Nat. Chem. 5, 607–612 (2013).
Li, L., Zhao, S., Joshi-Pangu, A., Diane, M. & Biscoe, M. R. Stereospecific Pd-catalyzed cross-coupling reactions of secondary alkylboron nucleophiles and aryl chlorides. J. Am. Chem. Soc. 136, 14027–14030 (2014).
Wang, C. Y., Ralph, G., Derosa, J. & Biscoe, M. R. Stereospecific palladium-catalyzed acylation of enantioenriched alkylcarbastannatranes: a general alternative to asymmetric enolate reactions. Angew. Chem. Int. Ed. 56, 856–860 (2017).
Hoang, G. L. & Takacs, J. M. Enantioselective gamma-borylation of unsaturated amides and stereoretentive Suzuki-Miyaura cross-coupling. Chem. Sci. 8, 4511–4516 (2017).
Nakao, Y., Chen, J., Tanaka, M. & Hiyama, T. A silicon-based approach to oligoarenes by iterative cross-coupling reactions of halogenated organo[(2-hydroxymethyl)phenyl]dimethylsilanes. J. Am. Chem. Soc. 129, 11694–11695 (2007).
Noguchi, H., Hojo, K. & Suginome, M. Boron-masking strategy for the selective synthesis of oligoarenes via iterative Suzuki-Miyaura coupling. J. Am. Chem. Soc. 129, 758–759 (2007).
Iwadate, N. & Suginome, M. Synthesis of B-protected β-styrylboronic acids via iridium-catalyzed hydroboration of alkynes with 1,8-naphthalenediaminatoborane leading to iterative synthesis of oligo(phenylenevinylene)s. Org. Lett. 11, 1899–1902 (2009).
Gillis, E. P. & Burke, M. D. A simple and modular strategy for small molecule synthesis: iterative Suzuki-Miyaura coupling of B-protected haloboronic acid building blocks. J. Am. Chem. Soc. 129, 6716–6717 (2007).
Mancilla, T. & Contreras, R. New bicyclic organylboronic esters derived from iminodiacetic acids. J. Organomet. Chem. 307, 1–6 (1986).
Gonzalez, J. A. et al. MIDA boronates are hydrolysed fast and slow by two different mechanisms. Nat. Chem. 8, 1067–1075 (2016).
Knapp, D. M., Gillis, E. P. & Burke, M. D. A general solution for unstable boronic acids: slow-release cross-coupling from air-stable MIDA boronates. J. Am. Chem. Soc. 131, 6961–6963 (2009).
Dick, G. R., Woerly, E. M. & Burke, M. D. A general solution for the 2-pyridyl problem. Angew. Chem. Int. Ed. 51, 2667–2672 (2012).
Carrillo, J. A., Ingleson, M. J. & Turner, M. L. Thienyl MIDA boronate esters as highly effective monomers for Suzuki–Miyaura polymerization reactions. Macromolecules 48, 979–986 (2015).
Carrillo, J. A., Turner, M. L. & Ingleson, M. J. A. General protocol for the polycondensation of thienyl N-methyliminodiacetic acid boronate esters to form high molecular weight copolymers. J. Am. Chem. Soc. 138, 13361–13368 (2016).
Brak, K. & Ellman, J. A. Asymmetric Rh(I)-catalyzed addition of MIDA boronates to N-tert-butanesulfinyl aldimines: development and comparison to trifluoroborates. J. Org. Chem. 75, 3147–3150 (2010).
Davy, N. C. et al. Contorted hexabenzocoronenes with extended heterocyclic moieties improve visible-light absorption and performance in organic solar cells. Chem. Mater. 28, 673–681 (2016).
Muir, C. W., Vantourout, J. C., Isidro-Llobet, A., Macdonald, S. J. & Watson, A. J. One-pot homologation of boronic acids: a platform for diversity-oriented synthesis. Org. Lett. 17, 6030–6033 (2015).
Gillis, E. P. & Burke, M. D. Multistep synthesis of complex boronic acids from simple MIDA boronates. J. Am. Chem. Soc. 130, 14084–14085 (2008).
Lv, W. X. et al. Oxidative difunctionalization of alkenyl MIDA boronates: a versatile platform for halogenated and trifluoromethylated alpha-boryl ketones. Angew. Chem. Int. Ed. 55, 10069–10073 (2016).
Uno, B. E., Gillis, E. P. & Burke, M. D. Vinyl MIDA boronate: a readily accessible and highly versatile building block for small molecule synthesis. Tetrahedron 65, 3130–3138 (2009).
Lee, C. F. et al. Oxalyl boronates enable modular synthesis of bioactive imidazoles. Angew. Chem. Int. Ed. 56, 6264–6267 (2017).
St Denis, J. D. et al. Boron-containing enamine and enamide linchpins in the synthesis of nitrogen heterocycles. J. Am. Chem. Soc. 136, 17669–17673 (2014).
Zajdlik, A. et al. alpha-Boryl isocyanides enable facile preparation of bioactive boropeptides. Angew. Chem. Int. Ed. 52, 8411–8415 (2013).
Struble, J. R., Lee, S. J. & Burke, M. D. Ethynyl MIDA boronate: a readily accessible and highly versatile building block for small molecule synthesis. Tetrahedron 66, 4710–4718 (2010).
Adachi, S. et al. Facile synthesis of borofragments and their evaluation in activity-based protein profiling. Chem. Commun. 51, 3608–3611 (2015).
Close, A. J., Kemmitt, P., Mark Roe, S. & Spencer, J. Regioselective routes to orthogonally-substituted aromatic MIDA boronates. Org. Biomol. Chem. 14, 6751–6756 (2016).
Eberlin, L., Carboni, B. & Whiting, A. Regioisomeric and substituent effects upon the outcome of the reaction of 1-borodienes with nitrosoarene compounds. J. Org. Chem. 80, 6574–6583 (2015).
Cornil, J. et al. Heck coupling using a vinyliodo-MIDA boronate: an efficient and modular access to polyene frameworks. Org. Lett. 17, 948–951 (2015).
Quiclet-Sire, B. & Zard, S. Z. Radical instability in aid of efficiency: a powerful route to highly functional MIDA boronates. J. Am. Chem. Soc. 137, 6762–6765 (2015).
Trinchera, P., Corless, V. B. & Yudin, A. K. Synthesis of previously inaccessible borylated heterocycle motifs using novel boron-containing amphoteric molecules. Angew. Chem. Int. Ed. 54, 9038–9041 (2015).
Adachi, S. et al. Condensation-driven assembly of boron-containing bis(heteroaryl) motifs using a linchpin approach. Org. Lett. 17, 5594–5597 (2015).
Denis, J. D. S., He, Z. & Yudin, A. K. Amphoteric α-boryl aldehyde linchpins in the synthesis of heterocycles. ACS Catal. 5, 5373–5379 (2015).
Seath, C. P., Wilson, K. L., Campbell, A., Mowat, J. M. & Watson, A. J. Synthesis of 2-BMIDA 6,5-bicyclic heterocycles by Cu(i)/Pd(0)/Cu(ii) cascade catalysis of 2-iodoaniline/phenols. Chem. Commun. 52, 8703–8706 (2016).
Chan, J. M., Amarante, G. W. & Toste, F. D. Tandem cycloisomerization/suzuki coupling of arylethynyl MIDA boronates. Tetrahedron 67, 4306–4312 (2011).
He, Z. & Yudin, A. K. Amphoteric α-boryl aldehydes. J. Am. Chem. Soc. 133, 13770–13773 (2011).
St Denis, J. D., He, Z. & Yudin, A. K. Chemoselective palladium-catalyzed α-allylation of α-boryl aldehydes. Org. Biomol. Chem. 10, 7900–7902 (2012).
Li, J. & Burke, M. D. Pinene-derived iminodiacetic acid (PIDA): a powerful ligand for stereoselective synthesis and iterative cross-coupling of C(sp3) boronate building blocks. J. Am. Chem. Soc. 133, 13774–13777 (2011).
Yoon, J. M., Lee, C. Y., Jo, Y. I. & Cheon, C. H. Synthesis of optically pure 3,3′-disubstituted-1,1′-bi-6-methoxy-2-phenol (BIPhOL) derivatives via diastereomeric resolution. J. Org. Chem. 81, 8464–8469 (2016).
Lee, S. J., Gray, K. C., Paek, J. S. & Burke, M. D. Simple, efficient, and modular syntheses of polyene natural products via iterative cross-coupling. J. Am. Chem. Soc. 130, 466–468 (2008).
Lee, S. J., Anderson, T. M. & Burke, M. D. A simple and general platform for generating stereochemically complex polyene frameworks by iterative cross-coupling. Angew. Chem. Int. Ed. 49, 8860–8863 (2010).
Woerly, E. M., Cherney, A. H., Davis, E. K. & Burke, M. D. Stereoretentive Suzuki-Miyaura coupling of haloallenes enables fully stereocontrolled access to (-)-peridinin. J. Am. Chem. Soc. 132, 6941–6943 (2010).
Fujii, S., Chang, S. Y. & Burke, M. D. Total synthesis of synechoxanthin through iterative cross-coupling. Angew. Chem. Int. Ed. 50, 7862–7864 (2011).
Gray, K. C. et al. Amphotericin primarily kills yeast by simply binding ergosterol. Proc. Natl Acad. Sci. USA 109, 2234–2239 (2012).
Brun, E., Bellosta, V. & Cossy, J. Synthesis of the acyclic carbon skeleton of filipin III. J. Org. Chem. 81, 8206–8221 (2016).
Fujita, K., Matsui, R., Suzuki, T. & Kobayashi, S. Concise total synthesis of (-)-myxalamide A. Angew. Chem. Int. Ed. 51, 7271–7274 (2012).
Dennis, E. G., Jeffrey, D. W., Johnston, M. R., Perkins, M. V. & Smith, P. A. Procyanidin oligomers. A new method for 4–8 interflavan bond formation using C8-boronic acids and iterative oligomer synthesis through a boron-protection strategy. Tetrahedron 68, 340–348 (2012).
Mohamed, Y. M. A. & Hansen, T. V. Synthesis of methyl (5Z, 8Z,10E,12E,14Z)-eicosapentaenoate. Tetrahedron Lett. 52, 1057–1059 (2011).
Weber, A., Dehn, R., Schlager, N., Dieter, B. & Kirschning, A. Total synthesis of the antibiotic elansolid B1. Org. Lett. 16, 568–571 (2014).
Davoren, J. E. et al. Discovery of the potent and selective M1 PAM-agonist N-[(3R,4S)-3-hydroxytetrahydro-2H-pyran-4-yl]-5-methyl-4-[4-(1,3-thiazol-4-yl)benzyl]pyridine-2-carboxamide (PF-06767832): evaluation of efficacy and cholinergic side effects. J. Med. Chem. 59, 6313–6328 (2016).
Go, E. B., Wetzler, S. P., Kim, L. J., Chang, A. Y. & Vosburg, D. A. Concise, diastereoconvergent synthesis of endriandric-type tetracycles by iterative cross-coupling. Tetrahedron 72, 3790–3794 (2016).
Nishioka, Y. et al. Stereocontrolled synthesis of paracentrone. Synlett 28, 327–332 (2017).
Li, J. et al. Synthesis of many different types of organic small molecules using one automated process. Science 347, 1221–1226 (2015).
Woerly, E. M., Struble, J. R., Palyam, N., O’Hara, S. P. & Burke, M. D. (Z)-(2-bromovinyl)-MIDA boronate: a readily accessible and highly versatile building block for small molecule synthesis. Tetrahedron 67, 4333–4343 (2011).
Grob, J. E. et al. One-pot C-N/C-C cross-coupling of methyliminodiacetic acid boronyl arenes enabled by protective enolization. Org. Lett. 14, 5578–5581 (2012).
Yoder, R. A. & Johnston, J. N. A case study in biomimetic total synthesis: polyolefin carbocyclizations to terpenes and steroids. Chem. Rev. 105, 4730–4756 (2005).
Heathcock, C. H., Piettre, S., Ruggeri, R. B., Ragan, J. A. & Kath, J. C. Daphniphyllum alkaloids. 12. A proposed biosynthesis of the pentacylic skeleton. proto-Daphniphylline. J. Org. Chem. 57, 2554–2566 (1992).
Monk, B. C. & Goffeau, A. Outwitting multidrug resistance to antifungals. Science 321, 367–369 (2008).
Wilcock, B. C., Endo, M. M., Uno, B. E. & Burke, M. D. C2’-OH of amphotericin B plays an important role in binding the primary sterol of human cells but not yeast cells. J. Am. Chem. Soc. 135, 8488–8491 (2013).
Davis, S. A. et al. Nontoxic antimicrobials that evade drug resistance. Nat. Chem. Biol. 11, 481–487 (2015).
Brak, K. & Ellman, J. A. Total synthesis of (-)-aurantioclavine. Org. Lett. 12, 2004–2007 (2010).
Igarashi, Y., Aoki, K., Nishimura, H., Morishita, I. & Usui, K. Total synthesis of hydroxy-α- and hydroxy-β-sanshool using Suzuki-Miyaura coupling. Chem. Pharm. Bull. 60, 1088–1091 (2012).
Lindsay, A. C. & Sperry, J. Extending the utility of the bartoli indolization: synthesis of marinoquinolines C and E. Synlett 24, 461–464 (2013).
Coluccini, C. et al. Quaterpyridine ligands for panchromatic Ru(II) dye sensitizers. J. Org. Chem. 77, 7945–7956 (2012).
Ronga, L. et al. Design, synthesis and biological evaluation of novel 4-alkapolyenylpyrrolo[1,2-a]quinoxalines as antileishmanial agents—part III. Eur. J. Med. Chem. 81, 378–393 (2014).
Llona-Minguez, S. et al. Discovery of the first potent and selective inhibitors of human dCTP pyrophosphatase 1. J. Med. Chem. 59, 1140–1148 (2016).
Hornum, M., Kumar, P., Podsiadly, P. & Nielsen, P. Increasing the stability of DNA:RNA duplexes by introducing stacking phenyl-substituted pyrazole, furan, and triazole moieties in the major groove. J. Org. Chem. 80, 9592–9602 (2015).
Webster, A. M. & Cobb, S. L. Synthesis of biaryl-linked cyclic peptoides. Tetrahedron Lett. 58, 1010–1014 (2017).
Godfrey, A. G., Masquelin, T. & Hemmerle, H. A remote-controlled adaptive medchem lab: an innovative approach to enable drug discovery in the 21st Century. Drug Discov. Today 18, 795–802 (2013).
Pastre, J. C., Browne, D. L. & Ley, S. V. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 42, 8849–8869 (2013).
Adamo, A. et al. On-demand continuous-flow production of pharmaceuticals in a compact, reconfigurable system. Science 352, 61–67 (2016).
Peplow, M. Organic synthesis: the robo-chemist. Nature 512, 20–22 (2014).
Made, V., Els-Heindl, S. & Beck-Sickinger, A. G. Automated solid-phase peptide synthesis to obtain therapeutic peptides. Beilstein J. Org. Chem. 10, 1197–1212 (2014).
Iyer, R. P. & Beaucage, S. L. in Comprehensive Natural Products Chemistry Vol. 7 (ed. Kool, E. T. ) 105–152 (Elsevier, Amsterdam, 1999).
Hahm, H. S. et al. Automated glycan assembly using the Glyconeer 2.1 synthesizer. Proc. Natl Acad. Sci. USA 114, e3385–e3389 (2017).
Acknowledgements
J.W.L. is funded by NSF grant NSF CHE 15–66071, D.J.B. is supported by the Damon Runyon Cancer Research Foundation (Damon Runyon) postdoctoral fellowship and M.D.B. is supported by the US Department of Health & Human Services NIH grant R35GM118185.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to the preparation of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The University of Illinois has filed patent applications on MIDA boronate chemistry and the automated synthesis platform described herein. These inventions have been licensed to REVOLUTION Medicines, a company for which M.D.B. is a founder.
Supplementary information
Supplementary information S1 (figure) | S2 (figure) | S3 (figure) | S4 (figure) | S5 (figure) | S6 (figure)
Supplementary Information (PDF 497 kb)
Rights and permissions
About this article

Cite this article
Lehmann, J., Blair, D. & Burke, M. Towards the generalized iterative synthesis of small molecules. Nat Rev Chem 2, 0115 (2018). https://doi.org/10.1038/s41570-018-0115
Published:
DOI: https://doi.org/10.1038/s41570-018-0115
This article is cited by
-
Iterative SuFEx approach for sequence-regulated oligosulfates and its extension to periodic copolymers
Nature Communications (2024)
-
Stepwise on-demand functionalization of multihydrosilanes enabled by a hydrogen-atom-transfer photocatalyst based on eosin Y
Nature Chemistry (2023)
-
Rational design of N-heterocyclic compound classes via regenerative cyclization of diamines
Nature Communications (2023)
-
Iterative synthesis of 1,3-polyboronic esters with high stereocontrol and application to the synthesis of bahamaolide A
Nature Chemistry (2023)
-
A computer algorithm to discover iterative sequences of organic reactions
Nature Synthesis (2022)
