
Abstract
The lungs are the primary target of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, with severe hypoxia being the cause of death in the most critical cases. Coronavirus disease 2019 (COVID-19) is extremely heterogeneous in terms of severity, clinical phenotype and, importantly, global distribution. Although the majority of affected patients recover from the acute infection, many continue to suffer from late sequelae affecting various organs, including the lungs. The role of the pulmonary vascular system during the acute and chronic stages of COVID-19 has not been adequately studied. A thorough understanding of the origins and dynamic behaviour of the SARS-CoV-2 virus and the potential causes of heterogeneity in COVID-19 is essential for anticipating and treating the disease, in both the acute and the chronic stages, including the development of chronic pulmonary hypertension. Both COVID-19 and chronic pulmonary hypertension have assumed global dimensions, with potential complex interactions. In this Review, we present an update on the origins and behaviour of the SARS-CoV-2 virus and discuss the potential causes of the heterogeneity of COVID-19. In addition, we summarize the pathobiology of COVID-19, with an emphasis on the role of the pulmonary vasculature, both in the acute stage and in terms of the potential for developing chronic pulmonary hypertension. We hope that the information presented in this Review will help in the development of strategies for the prevention and treatment of the continuing COVID-19 pandemic.
Key points
-
A thorough understanding of the dynamic behaviour of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is essential to understanding its heterogeneous effects on the pulmonary vasculature in patients with coronavirus disease 2019 (COVID-19).
-
The severity and clinical phenotype of COVID-19 are influenced by host factors, including socioeconomic factors and genetics.
-
Silent hypoxia is a major and independent cause of lung damage in COVID-19; the use of modern imaging techniques is proving to be very valuable in identifying silent hypoxia.
-
The pulmonary vascular system has a major role in the pathobiology of COVID-19.
-
Both COVID-19 and chronic pulmonary hypertension are global diseases with a complex interaction.
Similar content being viewed by others
Introduction
Following the initial reports from China in December 2019 of atypical pneumonia caused by a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)1, and confirmation of human-to-human transmission2, coronavirus disease 2019 (COVID-19) rapidly progressed to a global pandemic. As of September 2021, there have been >217,558,771 reported cases and >4,517,240 deaths associated with COVID-19 across all inhabited continents3, with heterogeneous incidence, severity and phenotype. The lungs seem to be the primary target of infection, with major involvement of the pulmonary vasculature. However, the exact pathobiology of the pulmonary vascular effects of COVID-19, in both the acute and the chronic stages, are poorly understood.
In this Review, we describe the possible virus-related and host-related factors that could be responsible for the heterogeneity of COVID-19. In addition, we discuss in detail the pathobiology of the pulmonary vascular effects of SARS-CoV-2 infection and their clinical implications for chronic pulmonary hypertension. A thorough understanding of these changes should inform the evolving global pre-emptive strategies for dealing with the COVID-19 pandemic and its sequelae.
The SARS-CoV-2 virus: an update
Studying the origins and dynamic behaviour of the SARS-CoV-2 virus is essential to understanding the pathobiology of the infection and in developing strategies to manage it. SARS-CoV-2 is the seventh coronavirus known to infect humans. Four coronaviruses are endemic in the population: these viruses typically cause a mild respiratory tract infection in the winter months, although the disease can be severe in neonates, elderly individuals and those with predisposing conditions. The highly pathogenic coronaviruses severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) each caused outbreaks in humans, with a case fatality rate of 9% and 40%, respectively4. However, these two viruses do not transmit readily between humans and, therefore, containment of the outbreaks was relatively simple. By contrast, SARS-CoV-2 is intermediate in virulence between the endemic coronaviruses and the highly pathogenic viruses but its high human-to-human transmissibility has led to the current human pandemic.
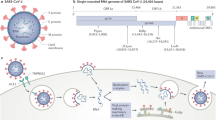
Like the other coronaviruses, SARS-CoV-2 has a non-segmented, positive-sense RNA genome4 (Fig. 1a,b). The structure of the SARS-CoV-2 virus particle (virion) is shown in Fig. 1c,d. The virion is enclosed in a lipid envelope, derived from the host cell. Projecting from the virion envelope is the spike (S) protein, producing the familiar crown-like appearance that gives the coronavirus its name. The host protease furin cleaves the S protein into two moieties: S1 and S2. S1 binds to the angiotensin-converting enzyme 2 (ACE2) receptor on the cell surface, which thereby serves as the cellular entry point for the virus5. S2 fuses the virion envelope with the cell membrane, resulting in infection of the cell (Fig. 1e). A second cell-surface protein, neuropilin 1, acts as a cofactor for SARS-CoV-2 infection by binding the furin-cleaved S1 protein6,7. The S protein is an important target of both the antibody response8 and the T cell immune response9 (Fig. 1f) and the majority of the existing vaccines use the S protein as the immunogen.

a | Genetic structure of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and other representative human coronaviruses. The genome of each virus is ~30 kb long. The replicase is encoded in two long open reading frames (ORF1a and ORF1b). The genetic composition of the respective viruses differs chiefly in the 3′ third of the genome. SARS-CoV-2 has 79% nucleotide sequence identity with severe acute respiratory syndrome coronavirus (SARS-CoV), the virus that caused the SARS epidemic in 2003. Four coronaviruses circulate seasonally in the human population, usually causing mild upper respiratory tract infection: the representative virus Human coronavirus-229E (HCoV-229E) is depicted. Three coronaviruses are highly pathogenic in humans: SARS-CoV and the Middle East respiratory syndrome coronavirus (MERS-CoV) cause infections with a fatality rate substantially greater than that of SARS-CoV-2; however, of these three viruses, SARS-CoV-2 transmits most efficiently between humans. b | Phylogenetic tree depicting the relatedness between SARS-CoV (from the 2003 SARS epidemic), SARS-CoV-2, and coronaviruses isolated from bats and civets. SARS-CoV-2 has ~97% nucleotide identity with the bat coronavirus RaTG13. c | Electron micrograph of a SARS-CoV-2 particle, covered in spike (S) proteins. d | Structure of a SARS-CoV-2 particle, showing the structural proteins of the virus. The particle has a diameter of ~125 nm. The trimerized S protein, which is the receptor-binding protein, gives the virion its crown-like appearance. The membrane (M) protein is the most abundant structural protein in the virus and seems to give the virus its shape. The third protein in the viral lipid envelope is the envelope (E) protein, which has ion channel activity and facilitates the assembly and release of the virion from the infected cell. The nucleoprotein (N) binds to the single-stranded RNA genome and regulates its structure and replication. The non-structural proteins are not illustrated. e | Structure of the S protein monomer. The surface that binds angiotensin-converting enzyme 2 (ACE2), the cellular receptor for the virus, is shown in green. Marked in yellow are the positions of three key mutations present in the SARS-CoV-2 virus variant B.1.351, which was first identified in South Africa. The N501Y mutation, which has evolved independently in different geographical locations, including the variant detected first in the UK (B.1.1.7), increases the affinity of binding of the (trimerized) S protein to the ACE2 receptor, resulting in greater transmissibility of the virus. Further mutations that increase the transmissibility of the virus have been identified in other variants of concern (Table 1). f | Immune responses to SARS-CoV-2 infection include the generation of interferons, natural killer cells, antibodies, and CD4+ and CD8+ T cells. The efficacy of the host immune response in the first week of infection is an important determinant of the risk of developing coronavirus disease 2019. SARS-CoV-2 encodes proteins that suppress the interferon response, resulting in efficient viral replication. UTR, untranslated region. Part a adapted with permission from refs4,229, Elsevier and Springer Nature Ltd, respectively. Part b published with permission from M. Escalera Zamudio (University of Oxford, UK). Part c © National Infection Service/Science Photo Library.
SARS-CoV-2 shares up to 97% nucleotide sequence identity with coronaviruses of bats (Fig. 1b) and, like the other highly pathogenic coronaviruses SARS-CoV and MERS-CoV, is likely to have originated in bats10,11 and spread to humans either directly or via an unidentified intermediate mammalian host10,12.
Coronaviruses mutate less quickly than other RNA-based viruses because they express a ‘proof-reading’ 3′–5′ exonuclease, which increases the fidelity of replication of the virus. Nonetheless, mutants do arise by nucleotide misincorporation and recombination and by the action of the host innate immune response13. On average, any two circulating SARS-CoV-2 isolates differ by about 10 nucleotides14. Each isolate that differs in its genome sequence from other identified viruses can be called a ‘variant’, and the total number of variants of SARS-CoV-2 is therefore very large — many thousands have been identified. However, the great majority of these sequence variants do not substantially alter either the infectiousness of the virus or the severity of the disease that it causes.
The resulting sequence diversity between isolates of SARS-CoV-2 has important practical and theoretical implications. First, sequence diversity makes it possible to trace the origin and spread of the infection both at the population level and in local outbreaks, thereby providing valuable information for contact tracing, epidemiology and public health. For example, sequence analysis showed that there were >1,170 importations of the virus into the UK in February to March 2020, mostly from southern Europe, with <1% from China15. Second, the rapid sequence variation allowed SARS-CoV-2 to evolve quickly, raising the question of whether newly emerging variants of the virus might differ from the currently circulating strains either in virulence (pathogenicity) or transmissibility. Weak correlations have indeed been observed between some 11 sequence variants of the virus and the case fatality rate in patients with COVID-19 (ref.16) but the mechanisms involved are not yet known. The rapid spread of a variant in the spike protein (D614G) might result from increased transmissibility or infectiousness of this variant17,18 and does not seem to confer a difference in virulence (Fig. 1b).
Third, the question arises whether a mutation in the viral RNA might render the vaccines ineffective. At present, this outcome is improbable, because the vaccines that are now being given use the full-length S protein as the immunogen; given that the S protein encodes several antibody epitopes and several T cell epitopes, a small number of mutations are unlikely to abrogate the immune response to the vaccine. However, direct evidence is needed to verify the effectiveness of existing vaccines against newly emerging variants. Additionally, monoclonal antibodies are used to treat some patients with severe COVID-19. Given that monoclonal antibodies are specific to a single epitope, often in the receptor-binding domain of SARS-CoV-2, even a point mutation might confer resistance to a monoclonal antibody.
A small minority of the variant sequences that arise confer substantial changes in the behaviour of the virus, chiefly to increase its transmissibility. These variants, known as variants of concern, include the viruses designated Alpha19, Beta20 and Delta21 (Table 1), are substantially more infectious and spread more rapidly in the population than the preceding variants and, in some cases, might cause more severe disease. These variants of concern typically contain certain point mutations in the receptor-binding domain of the S protein of SARS-CoV-2 (notably N501Y, E484K and K417N) (Fig. 1e; Table 1), with two major consequences. First, the mutations increase the binding affinity of the S protein to its receptor (ACE2) and thereby increase the efficiency of infection. Secondly, the mutation can diminish the recognition of the spike protein by some antibodies — that is, immune escape. However, this escape is only partial22,23, and current evidence indicates that the existing vaccines provide substantial protection against the variants of concern24,25,26,27, although the incidence of mild-to-moderate disease might be higher28. The variant designated Delta (Table 1), first isolated in India in October 2020, similarly contains two mutations (E484Q and L452R) that seem to confer the greater infectiousness and transmissibility of this variant. Whether the Delta variant causes more severe disease than other currently circulating variants remains uncertain29. In addition, the immunity resulting either from previous infection with another strain of SARS-CoV-2 or from vaccination with existing vaccines seems to give an important degree of protection against serious disease29. The Iota variant (Table 1), first detected in New York, USA, in November 2020, carries the E484K mutation and a further mutation at amino acid 477 (S477N). In the future, modification of vaccines might be necessary, perhaps to contain more than one variant sequence of the S protein, in order to provide efficient protection against new variants that differ substantially from the currently circulating strains.
Heterogeneity of COVID-19 disease
Severity
COVID-19 is mild in the majority of infected individuals and is therefore associated with a substantial rate of asymptomatic and undocumented infection30. However, 4–5% of people develop more severe disease that requires hospitalization, thought to be related to hyperinflammation and/or a failure to resolve the initial infection. Although pneumonitis with hypoxia is a major feature, severe COVID-19 has multisystem manifestations, including systemic thrombosis, cardiac injury, renal failure and hepatic dysfunction. In the initial China-wide outbreak of COVID-19, the overall case fatality rate was 2.3%, rising to 49% in those with critical disease (defined as respiratory failure, septic shock, and/or multiple organ dysfunction or failure)31. However, mortality among hospitalized patients with COVID-19 has fallen to 20–25% with advances in management and the introduction of disease-modifying drugs such as dexamethasone32.
Determinants of severity
Current data suggest that the severity and outcome of COVID-19 are determined mainly by host factors rather than by viral genetic variation33 (Box 1). Although the latter might affect transmissibility17,18, large-scale, population-based data suggest that factors that predispose patients with COVID-19 to death include increasing age (>60 years), male sex, socioeconomic deprivation, pre-existing comorbidities (notably cardiovascular disease and diabetes mellitus) and a non-white ethnic background34. These data represent the combined effect of factors at all points in the disease trajectory, from initial infection to the development of severe disease and life-threatening complications. However, different risk factors might have an effect at different stages of the disease. For example, analyses based on hospitalized patients with COVID-19 confirm that people with pre-existing cardiovascular comorbidities have higher mortality, but the presence of diabetes had only a minor independent effect in this study35. Predicting outcomes from the phenotype and severity of clinical presentation can be challenging36,37, but can be aided by expanding the use of clinical, haemodynamic, imaging, biochemical, immunological and genetic biomarkers38,39,40,41,42,43,44.
Geographical distribution
The global epidemiology of COVID-19 is striking in its heterogeneity. Particularly high case and death rates (per 100,000 population) were observed in Europe, the Americas and parts of the Middle East during 2020 but relatively low rates were observed across much of Africa3 (Fig. 2). Countries such as India initially showed a low number of deaths relative to reported cases45. Although some of this disparity might reflect differences in the extent of testing or the intensity of suppression strategies (such as lockdowns), many other factors are likely to be involved. Differences in population age structure and the rates of prevalent comorbidities might be important. The adaptive46 and trained innate47,48,49,50 immune systems have also been implicated in these differences. In addition, higher vitamin D levels in sunnier climates were speculated to be protective51. However, 2021 has seen a reversal of some of these trends, with substantial increases in the number of cases reported in many African countries, South and Central America, and Asia. India has seen the highest case numbers of any country since the beginning of the pandemic52. At the time of writing, probable contributors to this resurgence seem to include the spread of new SARS-CoV-2 variants, public behaviour and government policies53,54.

a | As of 24 August 2021, a total of 212,357,898 cases of coronavirus disease 2019 (COVID-19) have been confirmed. b | As of 24 August 2021, a total of 4,439,843 deaths have been reported to the WHO. Data from ref.3.
Ethnicity and socioeconomic factors
Numerous reports from multi-ethnic regions in the UK and the USA suggest disproportionately higher mortality from COVID-19 in non-white groups than in the majority white population34,55. Children of non-white ethnicity are also over-represented among those admitted to hospital with COVID-19-associated inflammatory syndromes56. The relationship between ethnicity and the severity of COVID-19 is likely to be complex and might involve many factors, including worse socioeconomic deprivation in minority communities, larger and intergenerational households, a higher risk of occupational exposure, poorer access to health care and a higher burden of comorbidities. UK data suggest that individuals of South Asian ethnicity have worse outcomes than other minority groups for reasons that are unclear57; genetic factors58 and/or an amplified response to cardiovascular risk factors59 might contribute (see next section). However, the effect of socioeconomic factors might be more nuanced in developing countries. A striking example is provided by a study in Mumbai, India, in which slum dwellers had a higher rate of infection but much lower death rates than non-slum residents (0.076% versus 0.263%), indicating a higher rate of infection and/or transmission but potentially less severe disease and higher levels of antibodies to SARS-CoV-2 (ref.60), possibly as a result of a more robust trained innate immunity47,48,49,50.
Host genetics
Host genetic background can influence the severity of the initial effects of COVID-19 in several ways61,62,63,64. Rare or moderately rare host genetic variants can confer a dominant trait with regard to COVID-19 infection65. This situation is exemplified by the familial incidence of severe disease reported in two families of Dutch and African ancestries with monogenic, rare, putative loss-of-function variants of TLR7 on the X chromosome66. Other studies reported monogenic inborn errors of interferon immunity in patients with life-threatening COVID-19 (refs67,68). The influence of monogenic disorders in different populations needs to be studied further.
In addition, genome-wide association studies have shown that common variants in multiple loci can induce additive effects that weaken defence mechanisms and increase COVID-19 incidence and severity65,69,70,71. Some of these studies might explain the heterogeneous global incidence described above58,72. A study from 2021 combined multiomics and machine learning approaches to decode the effects of sequence variation identified by genome-wide association studies73. The researchers showed a strong association with COVID-19 at the 3p21.31 locus and identified rs17713054 as a probable causative variant that interacts with LZTFL1, one of the regulators of epithelial-to-mesenchymal transformation in pulmonary epithelial cells73. To date, the genes associated with COVID-19 host susceptibility can be categorized into the following groups according to biological function: innate and adaptive immune system (CCR9, CXCR6, TLR7, XCR1 and mixed evidence for HLA genes)66,69,74,75,76,77, blood group (blood group A and blood group O)69, cell-entry proteins (ACE2, ACE1 (an analogue of ACE2), TMPRSS2 (encoding a cell receptor) and FURIN (encoding a host protease))75,78,79,80,81,82,83,84,85,86,87,88,89,90,91 and epithelial-to-mesenchymal transformation231. Studying the overlap between susceptibility genes for COVID-19 and those for other vascular diseases such as diabetes, atherosclerosis and pulmonary hypertension can further our understanding of the fundamental mechanisms of severity and the development of strategies to manage related pathways.
Gene-regulatory mechanisms, such as DNA methylation, histone modifications and RNA-based mechanisms, might affect viral entry, replication, transcription and protein maturation92,93,94. Epigenetic regulation of genes encoding cell-entry proteins such as ACE2 (refs87,94,95,96), genes encoding proteins with innate immune functions involved in vaccine-induced increased immunity97 and NETosis98, and genes encoding proteins with adaptive immune functions such as interferons99 might explain the increased severity of COVID-19 in some patients. Given that epigenetic alterations are reversible, they are attractive drug targets and, therefore, research is now focusing on deciphering the epigenetic landscape of COVID-19 for the development and/or repurposing of drugs92,93,100.
Pulmonary vascular pathobiology in COVID-19
The major cause of death in patients with COVID-19 is hypoxic respiratory failure due to a combination of parenchymal and vascular changes, similar to those encountered in acute respiratory distress syndrome (ARDS) due to other causes101. However, several studies have suggested possibly specific features in ARDS caused by COVID-19 (refs38,102). In addition to lung parenchymal changes, such as diffuse alveolar damage with hyaline membranes and acute fibrinous and organizing pneumonia, important vascular changes have been observed in patients with COVID-19.
ARDS caused by COVID-19 versus other causes
Unlike in ‘typical’ ARDS, lung mechanics in patients with COVID-19 are preserved regardless of the severity of hypoxaemia38. This preservation was thought to be due to the presence of large intrapulmonary shunts and, importantly, the loss of regulation of lung perfusion by hypoxia, as suggested by the presence of hyperperfusion in areas with markedly reduced ventilation (gasless tissue)38,103. These changes could have important therapeutic implications for the management of these patients, such as using the lowest positive end-expiratory pressure to prevent further lung damage38.
Functional and structural changes in the arterial wall
Vascular changes affect the entire pulmonary vascular tree, from large-calibre vessels104 to capillaries38,39,40,41,42,104,105. In patients with severe COVID-19, enlargement of the large-calibre vessels proximal to the opacities on pulmonary angiograms was observed during the acute phase of the disease39,40 (Fig. 3a,c). In a study from the USA using CT pulmonary angiography, 41 out of 48 patients with COVID-19 had dilated lobar and subsegmental vessels within parenchymal radiological opacities39. In a study from the UK also using CT pulmonary angiography, 21 out of 33 patients with COVID-19 and at least two assessable lobes with no evidence of acute pulmonary emboli had dilated peripheral vessels, with 100% of the patients showing perfusion defects on dual-energy CT, suggesting microvascular dysfunction41 (Fig. 3b,d).

Pulmonary angiograms and dual-energy CT (DECT) scans in two patients with coronavirus disease 2019 (COVID-19) pneumonia without evidence of pulmonary emboli. a | Peripheral ground-glass opacities are present in the lower lobes (black arrowheads), and central ground-glass opacities are noted in the right middle and left lower lobes (white arrows). b | The corresponding DECT image shows a peripheral perfusion defect with a halo of increased perfusion in the left lower lobe (black arrows). Areas of increased perfusion corresponding to the central ground-glass opacities are also present (white arrowheads). c | Areas of peripheral ground-glass opacity are present in the posterior lungs (black arrowheads), and central ground-glass opacities with enlarged vessels are present in the upper lobes (white arrows). d | The corresponding DECT image shows peripheral areas of decreased perfusion with surrounding halos of increased perfusion (black arrows). Reproduced with permission from ref.40, Elsevier.
Cellular changes in the arterial wall
All components of the vascular wall from the lumen to the perivascular regions can be affected and contribute to the overall pathobiology of COVID-19 (refs104,106,107,108,109,110). The lumen can contain fresh microthrombi as well as chronic thrombotic lesions with acute or chronic inflammatory cells and fibroblasts, displaying varying degrees of organization (Fig. 4). However, the frequency of the microthrombi in patients with COVID-19 is strikingly varied in different case series. This observation is exemplified in one study in which fibrin thrombi were observed in only one out of eight patients with COVID-19 studied (perhaps because this patient cohort was recruited from a long-term care facility, with a high baseline prevalence of anticoagulation therapy)108. By contrast, fibrin thrombi were found in the alveolar capillaries in a series of seven patients who died from COVID-19 (ref.104). The true incidence could be in between, as suggested by a study in which microthrombi were found in five out of 14 patients with COVID-19 (ref.107).

Haematoxylin and eosin (H&E) staining and immunostaining images showing microvasculature changes in patients with severe coronavirus disease 2019 (COVID-19). a | Arteriole filled with neutrophils that are in part adherent to the endothelium. b,c | Pulmonary microvessels (either arterioles or venules) displaying perivascular lymphocytic infiltrate. d | Immunostaining with anti-CD3 showing the vessel displayed in part c. e | Anti-CD4 staining on a serial section of the same vessel. f | Anti-CD8 staining on another serial section of the same vessel. g | Lymphocytic endothelialitis or venulitis with transmural infiltrate involving the intima; note the immediate vicinity of lymphocytes (dark blue, round nuclei) and endothelial cells (arrows). This inflammatory pattern is not frequently encountered and seems also to involve post-capillary vessels, as shown. h | Elastic-type artery (>500 μm in diameter) containing a wall-adherent, organized thrombotic lesion with endothelium-lined, cushion-like intimal fibrosis protruding into the vascular lumen.
Changes in the morphology and function of endothelial cells are a characteristic feature of COVID-19 (refs104,106,111,112) (Fig. 5a). Changes in morphology include disruption of intercellular junctions, cell swelling and a loss of contact with the basal membrane107. These changes are commonly associated with the induction of inflammatory and pro-coagulant responses111,113. Another finding in pulmonary endothelial cells of patients with COVID-19 is the presence of higher levels of intussusceptive proliferation than in the pulmonary endothelium of patients with ARDS caused by infection with influenza A virus subtype H1N1 (ref.104) (Fig. 5a).

a | Mechanisms of endothelial injury in coronavirus disease 2019 (COVID-19). On the left is a histological image showing endothelial swelling (arrows). The cartoon on the right shows the mode of entry of severe acute respiratory syndrome coronavirus (SARS-CoV-2) via the angiotensin-converting enzyme 2 (ACE2) receptor, which induces endothelial apoptosis. To the right are scanning electron micrographs of microvascular corrosion casts from the thin-walled alveolar plexus of a healthy lung (upper left panel) and the substantial architectural distortion seen in lungs injured by COVID-19 (upper right panel). The loss of a clearly visible vessel hierarchy in the alveolar plexus is the result of new blood vessel formation by intussusceptive angiogenesis. The lower left panel shows the intussusceptive pillar localizations (arrowheads) at higher magnification. The lower right panel is a transmission electron micrograph showing ultrastructural features of endothelial cell destruction and SARS-CoV-2 visible within the cell membrane (arrowheads). Red cells (RCs) are labelled. b | Contribution of pericytes to vascular injury. The cartoon shows the different types of pericyte located along a vessel, from the arteriole to the venule. After infection with SARS-CoV-2, pericytes detach and undergo apoptosis, leading to increased microvascular permeability and cytokine storm. The electron microscopy images show the intimate association between pericytes (P) and endothelial cells in human lung capillaries. The pericytes are located within the basement membrane (B) and extend long, thin pericyte processes (PPs) that establish contacts with the adjacent endothelial cell. Also labelled are the alveolus (A), endoplasmic reticulum (er), endothelium (E) and cytoplasmic filament (F). The lower panel is a higher magnification of a section of the upper panel. c | Smooth muscle cells (SMCs). A histological image and cartoon showing de-differentiation and proliferation of SMCs in COVID-19, resulting in increased medial wall thickness and muscularization. d | Fibroblasts. A histological image and cartoon showing fibroblast proliferation and deposition of fibrin and extracellular matrix in COVID-19, resulting in adventitial thickening and parenchymal lung fibrosis (arrows). Micrographs in part a adapted with permission from ref.104, Massachusetts Medical Society. Cartoon of types of pericyte in part b adapted from ref.230, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). Micrographs in part b adapted with permission from ref.122, Wiley.
Pericytes are mural cells present in the abluminal surface of blood vessels and have a pivotal role in blood vessel maturation and stability114. Studies performed using pericytes isolated from the lungs of patients with pulmonary hypertension as well as from a transgenic mouse model of pulmonary hypertension demonstrate a dynamic contribution of pericytes to blood vessel remodelling in pulmonary hypertension115,116. ACE2, together with cathepsin B and cathepsin L, is highly expressed in microvascular pericytes in the heart, making them targets for SARS-CoV-2 infection117. In addition, a single-cell and spatial atlas of autopsy tissues from patients with COVID-19 showed that pericytes from the lungs are positive for viral RNA, indicating that SARS-CoV-2 had entered these pericytes118. Interestingly, post-mortem histological analysis of lung biopsy samples from patients with COVID-19 revealed pericyte loss or detachment from the pulmonary vasculature, which might lead to decreased ACE2 expression in the vascular microenvironment119,120 (Fig. 5b). Similar findings in post-mortem studies have been reported by others121. The loss of pericytes in these patients is associated with a reduction in ACE2 receptors and could shift the humoral balance so that generation of angiotensin II by ACE would increase locally, leading to vessel constriction. Furthermore, pericyte loss or detachment would damage the endothelial barrier, leading to increased microvascular permeability, and would facilitate the entry of pro-inflammatory cells, which promote the ‘cytokine storm’ (Fig. 5b). Although doubts exist about the presence of pericytes on the capillaries of lung alveoli, a systematic electron microscopy study showed that pericytes were present in human lungs as well as in the lungs of dogs, guinea pigs and rats, whereas they could not be found in the lungs of smaller mammals122.
The media in SARS-CoV-2 infection shows varying degrees of pulmonary arterial smooth muscle cell proliferation and de-differentiation123 (Fig. 5c). These changes might be due to either direct or indirect effects of the SARS-CoV-2 virus. Patients who died from COVID-19 had increased pulmonary arterial medial wall thickness and reduced lumen area compared with pulmonary arteries from patients who died from influenza A virus subtype H1N1 infection124,125. Unpublished observations (S.S.P. and P.D.) in several autopsies and in three explanted lungs from patients with COVID-19 who underwent lung transplantation showed moderate muscular remodelling of the arterial wall (Fig. 5c). A single-cell, spatial landscape study of lung pathology in SARS-CoV-2 infection using high-parameter imaging mass cytometry showed a thickened medial layer with an increased number of cells positive for α-smooth muscle actin125. Patients with COVID-19 also have striking anomalies in the distribution of blood volume within the pulmonary vascular tree, similar to that in patients with pulmonary hypertension126. Of note, patients with COVID-19 have a substantial reduction in the pulmonary blood volume contained in blood vessels with a cross-sectional area of <5 mm2 and a substantial increase in the pulmonary blood volume contained in vessels with a cross-sectional area of 5–10 mm2 or >10 mm2 (refs126,127,128). All these changes could predispose patients to chronic pulmonary hypertension in the future123.
The adventitia and perivascular region have been shown to have a major regulatory effect on the function of the vascular wall129,130. Interestingly, in specimens from patients with COVID-19 who underwent lung transplantation, we have observed massive and uniform interstitial fibrosis of the lung parenchyma involving the pulmonary vasculature, including the adventitia (S.S.P. and P.D., unpublished observations). Moreover, we have noted the contribution of the pulmonary arterial adventitia to the fibrotic process, which suggests that the interstitial fibrosis observed in various patients with COVID-19 might be caused by diffuse pathological remodelling of fibroblasts or myofibroblasts in response to SARS-CoV-2 infection (Fig. 5d). Perivascular inflammation is a common finding in patients with advanced COVID-19 (ref.104). This finding is in accordance with unpublished observations (S.S.P. and P.D.) in patients with COVID-19 who died at the end of the first or during the second wave of infection, all of whom received anticoagulant therapy. These patients all showed intravascular neutrophil aggregations (Fig. 4a) and moderate perivascular lymphocytic infiltrates (Fig. 4b,c) at the microvascular level (arterioles and/or venules <100 μm in diameter). Phenotyping of these infiltrates demonstrated a clear T cell predominance, with the presence of both CD4+ and CD8+ cells (Fig. 4d–f). Only some patients with abundant interstitial and microvascular inflammation showed a transmural inflammatory cell infiltrate involving the intima, which led to variable endothelialitis (Fig. 4g,h).
Mechanisms of vascular lung injury
Following entry of the SARS-CoV-2 virus into different cells131,132, the exact mechanisms of injury remain controversial44,111,112. Several mechanisms have been suggested, including the induction of apoptosis or autophagy133, and involve several pathways.
Virus–cell interaction
With regard to SARS-CoV-2 virus entry into pulmonary vascular cells, most reports indicate that endothelial cells are a primary target of the virus118. However, this proposal has been questioned for two main reasons: the size of SARS-CoV-2 viral particles shown in the electron microscopic images of earlier studies are four times larger than that of the expected virion size120 and the lack of detectable expression of two important receptors (ACE2 and TMPRSS2)112 in endothelial cells through which SARS-CoV-2 gains entry. ACE2 receptors have been shown to be essential for the entry of SARS-CoV-2 into cells, and soluble recombinant ACE2 can inhibit viral infection in animal models and tissue-engineered organoids134. In addition, in vitro infection studies using endothelial cells also provided no evidence that the virus could directly infect these cells112. Therefore, whether SARS-CoV-2-induced lung endothelialitis is caused by direct viral infection of endothelial cells or by indirect perivascular inflammation and complement activation is uncertain. Understanding how SARS-CoV-2 causes endothelial dysfunction and pulmonary vascular abnormalities is of paramount importance.
An alternative route is that the SARS-CoV-2 virus enters smooth muscle cells and/or fibroblasts. The human ACE2 receptor is widely expressed in arterial smooth muscle cells across multiple organs, including the lungs131. Lung fibroblasts express both ACE2 and TMPRSS2 (ref.132), and data from single-cell sequencing studies showed high levels of ACE2 expression in pericytes117,122,135, suggesting that the alteration of adventitial fibroblasts and pericytes might be linked to direct or indirect SARS-CoV-2 infection122. Furthermore, a study has demonstrated that hypoxia upregulates the expression of ACE2 in human and rat pulmonary arterial smooth muscle cells136, which lends support to the theory that SARS-CoV-2-induced hypoxia might further exacerbate the disease. As in pulmonary hypertension, sex hormones and age can also influence ACE2 expression (levels are lower in women137 and increase with age138), which might explain the observed sex-specific differences in COVID-19 (ref.137). The SARS-CoV-2 spike protein alone, without additional viral components, is enough to elicit aberrant growth signalling (increased MEK phosphorylation) in human cultured pulmonary arterial smooth muscle cells124. MEK phosphorylation is associated with de-differentiation, nuclear activation and proliferation of pulmonary arterial smooth muscle cells in pulmonary hypertension139. More detailed studies to define the exact route of entry of SARS-CoV-2 into the arterial wall as well as the long-term effects of SARS-CoV-2 infection on the pulmonary microvasculature are urgently needed.
Pathways involved in vascular lung injury
Several pathways (hypoxia, inflammation and complement activation) have been proposed as possible drivers of pulmonary vascular injury mediated by SARS-CoV-2 infection.
Hypoxia
Hypoxia pathways have important roles in health and disease as highlighted by the recipients of the Nobel Prize in Physiology or Medicine in 2019 (refs140,141,142). In addition, hypoxia has been shown to have an important role in virus–host interactions, including in COVID-19 (refs143,144). Silent hypoxaemia, despite an apparent lack of dyspnoea, is observed in patients with COVID-19 (Fig. 6). Several reports revealed that the prevalence of silent hypoxia in patients with COVID-19 ranges from 20% to 40%145,146. However, the pathophysiology of silent hypoxaemia in COVID-19 lung injury and its influence on the pulmonary vasculature is still poorly understood. Hypoxia itself is known to have substantial deleterious effects on the cells of the lungs, in particular endothelial cells lining the pulmonary arteries and capillaries147,148, caused by various mediators, including activation of the NF-κB transcription factor149,150. Moreover, hypoxia can result in the overexpression of ACE2 receptors, thereby increasing the risk of damage through SARS-CoV-2 infection136. In addition, hypoxia can contribute to the mechanisms of cytokine storm142 and complement activation151.

a | Radiographic grading of lung fibrosis in a patient with coronavirus disease 2019 (COVID-19) and severe pulmonary fibrosis. (1) Chest high-resolution CT at discharge from hospital. (2) Extent of lesions marked in red using artificial intelligence (AI) at discharge from hospital. (3) Chest high-resolution CT at 30 days after discharge from hospital. (4) Extent of lesions marked in red using AI at 30 days after discharge from hospital, indicating a substantial reduction in lung fibrosis. b | Mechanisms of acute and chronic COVID-19-related pulmonary hypertension. Part a adapted from ref.179, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). Part b adapted from ref.183 (Springer Nature Ltd).
Inflammation
Several inflammatory pathways are involved in the severe vascular injury associated with COVID-19 (refs104,152,153,154) in the pulmonary and extrapulmonary vasculature, affecting both the macrovasculature and the microvasculature104 (Fig. 7). The underlying molecular mechanisms might include direct endothelial cell infection by SARS-CoV-2, which subsequently leads to endothelial damage and a shift to a pro-coagulant state104; cytokine storm (a sudden and large increase in the circulating levels of classical pro-inflammatory cytokines, such as IL-1β, IL-6 and tumour necrosis factor, which can induce inflammation of the alveolar space and immunothrombosis of the adjoining pulmonary vasculature155); or a type 3 hypersensitivity reaction as reported in some patients with COVID-19, which leads to a pro-coagulant environment in the vasculature156. Coagulation is closely linked to endothelial inflammation signalling; inflammatory moieties on the endothelium increase leukocyte infiltration and alter coagulation control, promoting a pro-coagulant status157. Together, these inflammatory and immunological changes have a major role in COVID-19-induced pulmonary vascular pathobiology and chronic pulmonary hypertension158,159.

Various inflammatory signalling pathways become dysregulated in patients with coronavirus disease 2019 (COVID-19). The figure shows the receptor-mediated transduction pathways from receptors to second messengers and ultimately to the nucleus and/or mitochondria. Targeting these pathways using selective anti-inflammatory drugs (pink boxes) might be therapeutically beneficial in patients with COVID-19. AAK1, AP2 associated protein kinase 1; ADAM17, a disintegrin and metalloproteinase 17; AP1, activator protein 1; ARB, angiotensin II receptor blocker; AT2R, angiotensin II receptor; COX2, cyclooxygenase 2; GHR, growth hormone receptor; ICAM, intercellular adhesion molecule; IFNγ, interferon-γ; JAK, Janus kinase; IκB, inhibitor of NF-κB; mTOR, mechanistic target of rapamycin; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase; MyD88, myeloid differentiation primary response protein MyD88; NF-κB, nuclear factor-κB; NSAID, non-steroidal anti-inflammatory drug; PI3K, phosphoinositol 3-kinase; PPAR, peroxisome proliferative activator receptor; RAF, RAF proto-oncogene serine/threonine-protein kinase; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; STAT, signal transducer and activator of transcription; sTNF, soluble tumour necrosis factor; TAK1, nuclear receptor subfamily 2 group C member 2; TGFβ, transforming growth factor-β; TNF, tumour necrosis factor; TLR, Toll-like receptor; TRADD, TNF receptor type 1-association DEATH domain protein; TRAF, TNF receptor-associated factor; TZD, thiazolidinedione; VCAM, vascular cell adhesion molecule. Adapted with permission from ref.152, Elsevier.
Complement
Several studies have shown that the complement system is activated in an uncontrolled manner in response to SARS-CoV-2 infection44,104,160,161. Strong immunohistochemical signals for many different complement components were detected in the lung tissue of patients with COVID-19, and complement deposition is also observed in the damaged lung microvasculature44,162. Complement components have also been found to be deposited in type 1 and type 2 alveolar epithelial cells as well as in inflammatory cells and their exudates (hyaline membranes) in the alveolar spaces filled with necrotic debris163. This deposition in turn triggers the activation of the membrane attack complex, causing microvascular endothelial injury and activation of the clotting pathways. High levels of d-dimer have also been measured in samples in which complement activation has been documented, which might in part explain COVID-19-associated coagulopathies160.
Obliterating endothelialitis was associated with an accumulation of complement component 5a receptor 1 (C5aR1)-positive macrophages around the arteries and in the thrombus164 (Fig. 8a,b). C5a can also promote the secretion of CCL2, leading indirectly to the recruitment of inflammatory cells. The levels of soluble C5a increased proportionately to the severity of COVID-19, and high levels of C5aR1 expression were found in blood and pulmonary myeloid cells, which supports a role for the C5a–C5aR1 axis in the pathophysiology of ARDS164. Complement activation in plasma from patients with COVID-19 was greater than in patients with influenza-associated lung injury165. The alternative pathway is activated in patients with COVID-19 and is associated with worse outcomes. Higher complement levels identified those patients at increased risk of worse outcomes165. These observations support the hypothesis that complement activation and C5a production lead to the chemoattraction and activation of myeloid cells in the lungs and contribute to the excessive release of inflammatory cytokines in SARS-CoV-2-infected lungs164 (Fig. 8c).

a | Haematoxylin and eosin staining of obliterating endothelialitis lesions in the lung of a representative patient with coronavirus disease 2019 (COVID-19). b | Representative multiplexed immunohistochemical staining of complement component 5a receptor 1 (C5aR1; green), CD68 (red) and CD163 (orange) showing that obliterating endothelialitis is associated with C5aR1+ macrophages surrounding the arteries and endothrombus (white dashed line). c | Hypothetical pathway for complement-mediated inflammation of the pulmonary alveolus in COVID-19. (1) Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) attaches to an angiotensin-converting enzyme 2 (ACE2) receptor on a type II alveolar epithelial cell (AEC-II), and the cell undergoes apoptosis. (2) Complement activation is initiated upon recognition of viral glycans by lectins (such as collectin 11 and ficolin 1, which are secreted by AEC-II) complexed with mannose-binding lectin-associated serine proteases (MASPs), including MASP2. Direct binding of MASP2 to the nucleoprotein of SARS-CoV-2 has also been suggested to initiate lectin pathway activation161. (3) Complement deposition and membrane attack complex (MAC)–C5b-9 formation on AECs cause inflammasome activation and cell damage. (4) Release of complement C5a increases vascular permeability and recruitment/activation of polymorphonuclear leukocytes (PMNs) and monocytes to the alveolus. (5) Monocytes differentiated into inflammatory macrophages overproduce pro-inflammatory cytokines in response to C3a and C5a stimulation. (6) Endothelial cell (EC) activation by C5a and MAC predisposes to thrombus formation, which is further increased through mannose-binding lectin recognition of viral particles in the vascular compartment, leading to the cleavage of thrombin and fibrinogen by MASPs. Drugs that can target complement activation are shown in the pink boxes. DAMP, damage-associated molecular pattern; mAB, monoclonal antibody. Parts a and b adapted from ref.164, Springer Nature Ltd. Part c adapted from ref.231, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Complement component C3 exacerbates disease in SARS-CoV-associated ARDS166. A study in C3-deficient mice showed that they were protected against SARS-CoV lung injury despite a high viral load and that fewer neutrophils and inflammatory monocytes were present in their lungs166. This finding has raised interest in using anti-complement regimens for the treatment of COVID-19. C3 activation is upstream of most anaphylatoxin signalling (C3a, C5a) and the formation of membrane attack complex–C5b-9. C3 is implicated as an initial effector mechanism that can exacerbate injury. These observations have led to the initiation of several small clinical trials using various anti-complement strategies. Complement C3 inhibitor AMY101 was safely and successfully used to treat a severe case of ARDS in a patient with COVID-19-related pneumonia167. In another study, four patients with COVID-19 were treated with the C5-blocking monoclonal antibody eculizumab, which reduced the levels of inflammatory biomarkers168. All patients successfully recovered (mean duration of disease 12.8 days). However, these four patients were not severely ill and had only moderately elevated levels of C-reactive protein, which nevertheless decreased with eculizumab treatment from 14.6 mg/dl to 3.5 mg/dl. In a third study, treatment with the monoclonal antibody LFG316 blocked C5 activity and complement activation for ≥4 days169. Four out of five patients with severe COVID-19 showed a sustained improvement in their clinical state, which persisted beyond the duration of C5 blockade169. These results suggest that transient blockade of C5 is sufficient to interrupt the hyperinflammatory cycle in COVID-19 and allow recovery. The findings also suggest that prolonged complement blockade might not only be unnecessary for patient benefit but might actually be harmful by increasing the risk of infection.
Therefore, accumulating evidence supports the idea that interrupting activated complement signalling might be beneficial in patients with severe COVID-19. However, to date, the number of patients tested has been small and, importantly, no randomized trials have been performed. Much work needs to be done to understand how and when to target the complement cascade to ameliorate SARS-CoV-2-related lung disease.
Chronic sequelae of COVID-19
Following the acute phase, many patients with COVID-19 continue to have symptoms due to persistent damage in several organs, including the lung vasculature170,171, and the sequelae of this damage such as chronic lung fibrosis and possibly chronic pulmonary hypertension170. Given that the COVID-19 pandemic started only in 2019, no long-term data similar to those reported in SARS and MERS or in ARDS due to other causes172,173 are so far available. However, several specific pathological changes during the acute phase could be used to predict chronic changes.
Lung fibrosis during or after acute COVID-19
Lung fibrosis is increasingly being recognized as one of the important sequelae of COVID-19 (refs174,175,176,177,178). The exact incidence depends on the radiographic criteria used for diagnosis. Using artificial intelligence with the development of a lung fibrosis score, researchers analysed a series of 284 patients discharged from hospital after COVID-19 and reported lung fibrosis in 100% of patients who had had severe disease and in 78.9% of those who had had moderate disease179. Lung fibrosis is attributable to chronic vascular ischaemia as well as parenchymal inflammation179. Probable risk factors for developing this complication include air pollution, smoking, exposure to toxins, infections and other factors that cause lung injury176. Certain radiological features of the lung opacities, age, IL-6 levels and other markers of the severity of inflammation have been shown to be independent risk factors for the development of lung fibrosis179 (Fig. 6a). The rate of progression and the influence of lung fibrosis after COVID-19 on long-term prognosis are still unknown.
Chronic pulmonary hypertension after COVID-19
The pathobiology of the pulmonary vasculature in patients with COVID-19 shares many features with that of the pulmonary vasculature in patients with pulmonary hypertension159,180,181,182,183. Medial hypertrophy and smooth muscle cell proliferation, essential features of pulmonary hypertension, have been reported in acute COVID-19 (ref.123) (Fig. 5c). In addition, the reported intravascular clotting in the lung vessels of patients with COVID-19 during the acute phase39,41,170,184,185,186,187 could predispose these individuals to chronic thromboembolic pulmonary hypertension171,185 in the future.
Furthermore, identified genetic drivers of chronic pulmonary hypertension188,189,190 are similar to the putative genetic factors that influence the severity of COVID-19 (described above). These similarities could have important implications for the incidence and severity of chronic pulmonary hypertension following SARS-CoV-2 infection and therefore need to be studied further.
The prevalence of pulmonary hypertension during the acute phase of COVID-19 is fairly high. In 200 consecutive patients with COVID-19 disease who were admitted to non-intensive care units in Milan, Italy, the prevalence of pulmonary hypertension (defined as a systolic pulmonary artery pressure >35 mmHg) and right ventricular dysfunction was 12.0% and 14.5%, respectively191. Another study from Sweden published in 2021 reported that 26 out of 67 patients (39%) with severe COVID-19 had acute pulmonary hypertension192. Taken together, the available evidence suggests that the pulmonary vascular pathology during the acute stage of COVID-19 can predispose these patients to develop chronic pulmonary hypertension.
Interaction between COVID-19 and pulmonary hypertension: a global perspective
Pulmonary hypertension is increasingly considered to be a global disease193,194,195. Similarly, COVID-19 has affected all continents, including remote areas. The two diseases interact at both ‘individual’ and global levels.
The ‘individual’ level
Evidence on the effect of pulmonary hypertension PAH on the severity and outcomes of COVID-19 is conflicting196,197. An observational study from a large pulmonary hypertension centre in New York City, USA, showed that pre-existing chronic pulmonary hypertension has major adverse effects on both the clinical course and mortality associated with COVID-19 (ref.198). By contrast, another study on data from 32 pulmonary hypertension centres in the USA reported a paucity of hospitalized patients with both pulmonary hypertension and COVID-19, and those who developed COVID-19 had mild symptoms and tended to recover quickly199. In a large, international survey from 47 pulmonary hypertension centres, including patients with pulmonary arterial hypertension or chronic thromboembolic pulmonary hypertension, the findings were mixed in terms of the number of reported cases per centre in different countries (ranging from 0 to 19) and the number of deaths (ranging from 0 to 6)200, and we agree with Farha and Heresi201 that the interaction between COVID-19 and chronic pulmonary hypertension at the individual level requires further intensive investigation.
The global level
Globally, pulmonary hypertension and COVID-19 interact in different ways according to prevailing conditions. This interaction is exemplified by populations living at high altitudes202,203, those with endemic HIV infection, tuberculosis or polyparasitism204,205,206,207,208,209,210, and those with high levels of illicit drug use211,212,213.
Nearly 140 million people live in high-altitude locations worldwide in a hypoxic environment with an increased prevalence (5–18%) of pulmonary hypertension214,215. As discussed above, hypoxia has a major and varied role in the pathophysiology of COVID-19 (refs103,202,216,217,218) and might therefore influence the phenotype and outcome of COVID-19 disease in these high-altitude locations. However, evidence on the incidence and progression of COVID-19 at high altitudes is incomplete and contradictory202,219,220,221, with one study reporting that high altitude was protective222.
The presence of endemic infections and polyparasitism has been identified as an important driver of pulmonary hypertension at the global level193,194,223. Many infectious diseases, in particular schistosomiasis, HIV infection and, potentially, tuberculosis, can induce pulmonary vascular diseases207,208,209,210 by altering the Zinnate and adaptive immune response to COVID-19 (ref.224) and by inducing herd immunity225.
Pulmonary hypertension due to toxins and the use of illicit drugs is a major problem of global dimensions226. The effect of these substances on COVID-19 infection is the subject of ongoing studies227,228. For example, in a retrospective study of the electronic health records of 73,099,850 patients in the USA, patients with substance use disorder had an increased risk of COVID-19 (adjusted OR 8.699, P <10–30)227.
Taken together, the emerging picture of an intense interaction between COVID-19 and chronic pulmonary hypertension at the global level is anticipated to have an important effect on the future of both diseases.
Conclusions
In this Review, we have presented a detailed description of the pathobiology of the pulmonary vasculature in COVID-19 and its potential interactions with pulmonary hypertension and other sequelae. These observations could be of value in developing novel strategies to deal with the ongoing COVID-19 global pandemic.
References
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Chan, J. F. W. et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395, 514–523 (2020).
World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard https://covid19.who.int/ (2021).
Wang, Y., Grunewald, M. & Perlman, S. Coronaviruses: an updated overview of their replication and pathogenesis. Methods Mol. Biol. 2203, 1–29 (2020).
Gheblawi, M. et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ. Res. 126, 1456–1474 (2020).
Cantuti-Castelvetri, L. et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370, 856–860 (2020).
Daly, J. L. et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 370, 861–865 (2020).
Robbiani, D. F. et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 584, 437–442 (2020).
Grifoni, A. et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 181, 1489–1501 (2020).
Boni, M. F. et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 5, 1408–1417 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Holmes, E. C. et al. The origins of SARS-CoV-2: a critical review. Cell 184, 4848–4856 (2021).
Kosuge, M., Furusawa-Nishii, E., Ito, K., Saito, Y. & Ogasawara, K. Point mutation bias in SARS-CoV-2 variants results in increased ability to stimulate inflammatory responses. Sci. Rep. 10, 17766 (2020).
van Dorp, L. et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 83, 104351 (2020).
du Plessis, L. et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 371, 708–712 (2021).
Gussow, A. B. et al. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. PNAS 117, 15193–15199 (2020).
Daniloski, Z., Guo, X. & Sanjana, N. E. The D614G mutation in SARS-CoV-2 spike increases transduction of multiple human cell types. eLife 10, e65365 (2021).
Korber, B. et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182, 812–827 (2020).
Rambaut, A. et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. ARTIC Network https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (2020).
ECDC. Risk related to spread of new SARS-CoV-2 variants of concern in the EU/EEA. European Centre for Disease Prevention and Control. https://www.ecdc.europa.eu/en/publications-data/covid-19-risk-assessment-spread-new-sars-cov-2-variants-eueea (2020).
Cherian, S. et al. SARS-CoV-2 spike mutations, L452R, T478K, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. Microorganisms 9, 1542 (2021).
Zhou, H. et al. B.1.526 SARS-CoV-2 variants identified in New York City are neutralized by vaccine-elicited and therapeutic monoclonal antibodies. mBio 12, e0138621 (2021).
West, A. P. et al. Detection and characterization of the SARS-CoV-2 lineage B.1.526 in New York. Nat. Commun. 12, 4886 (2021).
Emary, K. R. W. et al. Efficacy of ChAdOx1 nCoV-19 (AZD1222) vaccine against SARS-CoV-2 variant of concern 202012/01 (B.1.1.7): an exploratory analysis of a randomised controlled trial. Lancet 397, 1351–1362 (2021).
Shinde, V. et al. Efficacy of NVX-CoV2373 Covid-19 vaccine against the B.1.351 variant. N. Engl. J. Med. 384, 1899–1909 (2021).
Bernal, J. L. et al. Effectiveness of the Pfizer-BioNTech and Oxford-AstraZeneca vaccines on COVID-19 related symptoms, hospital admissions, and mortality in older adults in England: test negative case-control study. BMJ 373, n1088 (2021).
Edara, V. V. et al. Infection- and vaccine-induced antibody binding and neutralization of the B.1.351 SARS-CoV-2 variant. Cell Host Microbe 29, 516–521 (2021).
Madhi, S. A. et al. Efficacy of the ChAdOx1 nCoV-19 COVID-19 vaccine against the B.1.351 variant. N. Engl. J. Med. 384, 1885–1898 (2021).
Yadav, P. D. et al. Neutralization of variant under investigation B.1.617 with sera of BBV152 vaccines. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciab411 (2021).
Li, R. et al. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science 368, 489–493 (2020).
Wu, Z. & McGoogan, J. M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 323, 1239–1242 (2020).
Wiersinga, W. J., Rhodes, A., Cheng, A. C., Peacock, S. J. & Prescott, H. C. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA 324, 782–793 (2020).
Zhang, X. et al. Viral and host factors related to the clinical outcome of COVID-19. Nature 583, 437–440 (2020).
Williamson, E. J. et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 584, 430–436 (2020).
Docherty, A. B. et al. Features of 20 133 UK patients in hospital with COVID-19 using the ISARIC WHO clinical characterisation protocol: prospective observational cohort study. BMJ 369, m1985 (2020).
Carr, E. et al. Evaluation and improvement of the national early warning score (NEWS2) for COVID-19: a multi-hospital study. BMC Med. 19, 23 (2021).
Knight, S. R. et al. Risk stratification of patients admitted to hospital with COVID-19 using the ISARIC WHO clinical characterisation protocol: development and validation of the 4C mortality score. BMJ 370, m3339 (2020).
Gattinoni, L. et al. COVID-19 does not lead to a “typical” acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 201, 1299–1300 (2020).
Lang, M. et al. Pulmonary vascular manifestations of COVID-19 pneumonia. Radiol. Cardiothorac. Imaging 2, e200277 (2020).
Lang, M. et al. Hypoxaemia related to COVID-19: vascular and perfusion abnormalities on dual-energy CT. Lancet Infect. Dis. 20, 1365–1366 (2020).
Patel, B. V. et al. Pulmonary angiopathy in severe COVID-19: physiologic, imaging, and hematologic observations. Am. J. Respir. Crit. Care Med. 202, 690–699 (2020).
Ridge, C. A. et al. Dual-energy CT pulmonary angiography quantifies vasculopathy in severe COVID-19 pneumonia. Radiol. Cardiothorac. Imaging 2, e200428 (2020).
Caravita, S. et al. Haemodynamic characteristics of COVID-19 patients with acute respiratory distress syndrome requiring mechanical ventilation. An invasive assessment using right heart catheterization. Eur. J. Heart Fail. 22, 2228–2237 (2020).
Stenmark, K. R. et al. Mechanisms of SARS-CoV-2-induced lung vascular disease: potential role of complement. Pulm. Circ. 11, 204589402110157 (2021).
Jain, V. K., Iyengar, K., Vaish, A. & Vaishya, R. Differential mortality in COVID-19 patients from India and western countries. Diabetes Metab. Syndr. Clin. Res. Rev. 14, 1037–1041 (2020).
Sette, A. & Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 184, 861–880 (2021).
Netea, M. G. et al. Trained immunity: a program of innate immune memory in health and disease. Science 352, aaf1098 (2016).
Netea, M. G. et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 20, 375–388 (2020).
Fu, W. et al. Reconcile the debate over protective effects of BCG vaccine against COVID-19. Sci. Rep. 11, 8356 (2021).
O’Neill, L. A. J. & Netea, M. G. BCG-induced trained immunity: can it offer protection against COVID-19? Nat. Rev. Immunol. 20, 335–337 (2020).
Meltzer, D. O. et al. Association of vitamin D levels, race/ethnicity, and clinical characteristics with COVID-19 test results. JAMA Netw. Open 4, e214117 (2021).
World Health Organization. Weekly Epidemiological Update on COVID-19 - 4 May 2021 https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---4-may-2021 (2021).
Welch, S. B. et al. The interplay between policy and COVID-19 outbreaks in south Asia: longitudinal trend analysis of surveillance data. JMIR Public Health Surveill. 7, e24251 (2021).
Kumar, G. et al. Clinical profile of hospitalized COVID-19 patients in first & second wave of the pandemic: insights from an Indian registry based observational study. Indian J. Med. Res. 153, 619–628 (2021).
Karaca-Mandic, P., Georgiou, A. & Sen, S. Assessment of COVID-19 hospitalizations by race/ethnicity in 12 states. JAMA Intern. Med. 181, 131–134 (2021).
Davies, P. et al. Intensive care admissions of children with paediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2 (PIMS-TS) in the UK: a multicentre observational study. Lancet Child. Adolesc. Health 4, 669–677 (2020).
Nafilyan, V. et al. Ethnic differences in COVID-19 mortality during the first two waves of the coronavirus pandemic: a nationwide cohort study of 29 million adults in England. Eur. J. Epidemiol. 36, 605–617 (2021).
Zeberg, H. & Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587, 610–612 (2020).
Patel, A. P., Wang, M., Kartoun, U., Ng, K. & Khera, A. V. Quantifying and understanding the higher risk of atherosclerotic cardiovascular disease among South Asian individuals: results from the UK Biobank prospective cohort study. Circulation 144, 410–422 (2021).
Malani, A. et al. Seroprevalence of SARS-CoV-2 in slums versus non-slums in Mumbai, India. Lancet Glob. Health 9, e110–e111 (2021).
Carter-Timofte, M. E. et al. Deciphering the role of host genetics in susceptibility to severe COVID-19. Front. Immunol. 11, 1606 (2020).
Ovsyannikova, I. G., Haralambieva, I. H., Crooke, S. N., Poland, G. A. & Kennedy, R. B. The role of host genetics in the immune response to SARS-CoV-2 and COVID-19 susceptibility and severity. Immunol. Rev. 296, 205–219 (2020).
Thierry, A. R. Host/genetic factors associated with COVID-19 call for precision medicine. Precis. Clin. Med. 3, 228–234 (2020).
Williams, F. M. et al. Self-reported symptoms of COVID-19, including symptoms most predictive of SARS-CoV-2 infection, are heritable. Twin Res. Hum. Genet. 23, 316–321 (2020).
Darbeheshti, F. & Rezaei, N. Genetic predisposition models to COVID-19 infection. Med. Hypotheses 142, 109818 (2020).
Van Der Made, C. I. et al. Presence of genetic variants among young men with severe COVID-19. JAMA 324, 663–673 (2020).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020).
Zhang, S. Y. et al. Severe COVID-19 in the young and healthy: monogenic inborn errors of immunity? Nat. Rev. Immunol. 20, 455–456 (2020).
Severe Covid-19 GWAS Group. Genomewide association study of severe COVID-19 with respiratory failure. N. Engl. J. Med. 383, 1522–1534 (2020).
The COVID-19 Host Genetics Initiative. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28, 715–718 (2020).
Pairo-Castineira, E. et al. Genetic mechanisms of critical illness in COVID-19. Nature 591, 92–98 (2021).
Public Health England. COVID-19: review of disparities in risks and outcomes https://www.gov.uk/government/publications/covid-19-review-of-disparities-in-risks-and-outcomes (2020).
Downes, D. J. et al. Identification of LZTFL1 as a candidate effector gene at a COVID-19 risk locus. Nat. Genet. 53, 1606–1615 (2021).
Tal, Y., Adini, A., Eran, A. & Adini, I. Racial disparity in COVID-19 mortality rates - a plausible explanation. Clin. Immunol. 217, 108481 (2020).
Jacob, C. O. On the genetics and immunopathogenesis of COVID-19. Clin. Immunol. 220, 108591 (2020).
Nguyen, A. et al. Human leukocyte antigen susceptibility map for severe acute respiratory syndrome coronavirus 2. J. Virol. 94, e00510–e00520 (2020).
Warren, R. L. & Birol, I. HLA predictions from the bronchoalveolar lavage fluid and blood samples of eight COVID-19 patients at the pandemic onset. Bioinformatics 36, 5271–5273 (2020).
Stawiski, E. et al. Human ACE2 receptor polymorphisms predict SARS-CoV-2 susceptibility. Commun. Biol. 4, 475 (2021).
Yamamoto, N. et al. SARS-CoV-2 infections and COVID-19 mortalities strongly correlate with ACE1 I/D genotype. Gene 758, 144944 (2020).
Saab, Y. B., Gard, P. R. & Overall, A. D. J. The geographic distribution of the ACE II genotype: a novel finding. Genet. Res. 89, 259–267 (2007).
Asselta, R., Paraboschi, E. M., Mantovani, A. & Duga, S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. Aging 12, 10087–10098 (2020).
Russo, R., Andolfo, I., Lasorsa, V. A., Iolascon, A. & Capasso, M. Genetic analysis of the coronavirus SARS-CoV-2 host protease TMPRSS2 in different populations. Front. Genet. 11, 872 (2020).
Dobrindt, K. et al. Common genetic variation in humans impacts in vitro susceptibility to SARS-CoV-2 infection. Stem Cell Rep. 16, 505–518 (2021).
Benetti, E. et al. ACE2 gene variants may underlie interindividual variability and susceptibility to COVID-19 in the Italian population. Eur. J. Hum. Genet. 28, 1602–1614 (2020).
Li, Q., Cao, Z. & Rahman, P. Genetic variability of human angiotensin-converting enzyme 2 (hACE2) among various ethnic populations. Mol. Genet. Genom. Med. 8, e1344 (2020).
Fujikura, K. & Uesaka, K. Genetic variations in the human severe acute respiratory syndrome coronavirus receptor ACE2 and serine protease TMPRSS2. J. Clin. Pathol. 74, 307–313 (2021).
Pinto, B. G. G. et al. ACE2 expression is increased in the lungs of patients with comorbidities associated with severe COVID-19. J. Infect. Dis. 222, 556–563 (2020).
Li, M. Y., Li, L., Zhang, Y. & Wang, X. S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 9, 45 (2020).
Cao, Y. et al. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 6, 11 (2020).
Chen, J. et al. Individual variation of the SARS-CoV-2 receptor ACE2 gene expression and regulation. Aging Cell 19, e13168 (2020).
Delanghe, J. R., Speeckaert, M. M. & De Buyzere, M. L. The host’s angiotensin-converting enzyme polymorphism may explain epidemiological findings in COVID-19 infections. Clin. Chim. Acta 505, 192–193 (2020).
Atlante, S. et al. The epigenetic implication in coronavirus infection and therapy. Clin. Epigenet. 12, 156 (2020).
El Baba, R. & Herbein, G. Management of epigenomic networks entailed in coronavirus infections and COVID-19. Clin. Epigenet. 12, 118 (2020).
Pruimboom, L. Methylation pathways and SARS-CoV-2 lung infiltration and cell membrane-virus fusion are both subject to epigenetics. Front. Cell. Infect. Microbiol. 10, 290 (2020).
Chlamydas, S., Papavassiliou, A. G. & Piperi, C. Epigenetic mechanisms regulating COVID-19 infection. Epigenetics 16, 263–270 (2021).
Corley, M. J. & Ndhlovu, L. C. DNA methylation analysis of the COVID-19 host cell receptor, angiotensin I converting enzyme 2 gene (ACE2) in the respiratory system reveal age and gender differences. Preprints https://doi.org/10.20944/preprints202003.0295.v1 (2020).
Mantovani, A. & Netea, M. G. Trained innate immunity, epigenetics, and COVID-19. N. Engl. J. Med. 383, 1078–1080 (2020).
Barnes, B. J. et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J. Exp. Med. 217, e20200652 (2020).
Sawalha, A. H., Zhao, M., Coit, P. & Lu, Q. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin. Immunol. 215, 108410 (2020).
Crimi, E., Benincasa, G., Figueroa-Marrero, N., Galdiero, M. & Napoli, C. Epigenetic susceptibility to severe respiratory viral infections and its therapeutic implications: a narrative review. Br. J. Anaesth. 125, 1002–1017 (2020).
Ma, X. et al. Characteristics of 1738 patients with coronavirus disease 2019 (COVID-19) in Wuhan, China. Disaster Med. Public Health Prep. https://doi.org/10.1017/dmp.2021.129 (2021).
Fan, E. et al. COVID-19-associated acute respiratory distress syndrome: is a different approach to management warranted? Lancet Respir. Med. 8, 816–821 (2020).
Archer, S. L., Sharp, W. W. & Weir, E. K. Differentiating COVID-19 pneumonia from acute respiratory distress syndrome and high altitude pulmonary edema: therapeutic implications. Circulation 142, 101–104 (2020).
Ackermann, M. et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 383, 120–128 (2020).
Borczuk, A. C. et al. COVID-19 pulmonary pathology: a multi-institutional autopsy cohort from Italy and New York City. Mod. Pathol. 33, 2156–2168 (2020).
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418 (2020).
Bradley, B. T. et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington state: a case series. Lancet 396, 320–332 (2020).
Martines, R. B. et al. Pathology and pathogenesis of SARS-CoV-2 associated with fatal coronavirus disease, United States. Emerg. Infect. Dis. 26, 2005–2015 (2020).
Konopka, K. E. et al. Diffuse alveolar damage (DAD) resulting from coronavirus disease 2019 infection is morphologically indistinguishable from other causes of DAD. Histopathology 77, 570–578 (2020).
Bussani, R. et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. eBioMedicine 61, 103104 (2020).
Conde, J. N., Schutt, W. R., Gorbunova, E. E. & Mackow, E. R. Recombinant ACE2 expression is required for SARS-CoV-2 to infect primary human endothelial cells and induce inflammatory and procoagulative responses. mBio 11, e03185–20 (2020).
McCracken, I. R. et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation 143, 865–868 (2021).
Aid, M. et al. Vascular disease and thrombosis in SARS-CoV-2-infected rhesus macaques. Cell 183, 1354–1366 (2020).
Díaz-Flores, L. et al. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol. Histopathol. 24, 909–969 (2009).
Bordenave, J. et al. Lineage tracing reveals the dynamic contribution of pericytes to the blood vessel remodeling in pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 40, 766–782 (2020).
Yuan, K. et al. Loss of endothelium-derived Wnt5a is associated with reduced pericyte recruitment and small vessel loss in pulmonary arterial hypertension. Circulation 139, 1710–1724 (2019).
Chen, L., Li, X., Chen, M., Feng, Y. & Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 116, 1097–1100 (2020).
Delorey, T. M. et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 595, 107–113 (2021).
Burel-Vandenbos, F., Cardot-Leccia, N. & Passeron, T. Apoptosis and pericyte loss in alveolar capillaries in COVID-19 infection: choice of markers matters. Author’s reply. Intensive Care Med. 46, 1967–1968 (2020).
Som, A., Lang, M. & Little, B. Pulmonary vascular pathology in COVID-19. N. Engl. J. Med. 383, 887 (2020).
Cardot-Leccia, N., Hubiche, T., Dellamonica, J., Burel-Vandenbos, F. & Passeron, T. Pericyte alteration sheds light on micro-vasculopathy in COVID-19 infection. Intensive Care Med. 46, 1777–1778 (2020).
Yuan, K. et al. Lung pericytes in pulmonary vascular physiology and pathophysiology. Compr. Physiol. 11, 2227–2247 (2021).
Suzuki, Y. J., Nikolaienko, S. I., Shults, N. V. & Gychka, S. G. COVID-19 patients may become predisposed to pulmonary arterial hypertension. Med. Hypotheses 147, 110483 (2021).
Suzuki, Y. J. et al. SARS-CoV-2 spike protein-mediated cell signaling in lung vascular cells. Vasc. Pharmacol. 137, 106823 (2021).
Rendeiro, A. F. et al. The spatial landscape of lung pathology during COVID-19 progression. Nature 593, 564–569 (2021).
Lins, M. et al. Assessment of small pulmonary blood vessels in COVID-19 patients using HRCT. Acad. Radiol. 27, 1449–1455 (2020).
Morris, M. F. et al. Altered pulmonary blood volume distribution as a biomarker for predicting outcomes in COVID-19 disease. Eur. Respir. J. 58, 2004133 (2021).
Thillai, M. et al. Functional respiratory imaging identifies redistribution of pulmonary blood flow in patients with COVID-19. Thorax 76, 182–184 (2021).
Stenmark, K. R., Bouchey, D., Nemenoff, R., Dempsey, E. C. & Das, M. Hypoxia-induced pulmonary vascular remodeling: contribution of the adventitial fibroblasts. Physiol. Res. 49, 503–517 (2000).
Stenmark, K. R., Davie, N., Frid, M., Gerasimovskaya, E. & Das, M. Role of the adventitia in pulmonary vascular remodeling. Physiology 21, 134–145 (2006).
Hamming, I. et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203, 631–637 (2004).
Muus, C. et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med. 27, 546–559 (2021).
Randhawa, P. K., Scanlon, K., Rappaport, J. & Gupta, M. K. Modulation of autophagy by SARS-CoV-2: a potential threat for cardiovascular system. Front. Physiol. 11, 1560 (2020).
Monteil, V. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181, 905–913 (2020).
Ziegler, C. G. K. et al. SARS-CoV-2 Receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181, 1016–1035 (2020).
Zhang, R. et al. Role of HIF-1α in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L631–L640 (2009).
Kalidhindi, R. S. R. et al. Sex steroids skew ACE2 expression in human airway: a contributing factor to sex differences in COVID-19? Am. J. Physiol. Lung Cell. Mol. Physiol. 319, L843–L847 (2020).
Baker, S. A., Kwok, S., Berry, G. J. & Montine, T. J. Angiotensin-converting enzyme 2 (ACE2) expression increases with age in patients requiring mechanical ventilation. PLoS One 16, e0247060 (2021).
Xing, J. et al. TRPM7 channel inhibition exacerbates pulmonary arterial hypertension through MEK/ERK pathway. Aging 11, 4050–4065 (2019).
The Nobel Prize. The Nobel Prize in Physiology or Medicine 2019 https://www.nobelprize.org/prizes/medicine/2019/advanced-information/ (2019).
Richalet, J.-P. The invention of hypoxia. J. Appl. Physiol. 130, 1573–1582 (2021).
Jahani, M., Dokaneheifard, S. & Mansouri, K. Hypoxia: a key feature of COVID-19 launching activation of HIF-1 and cytokine storm. J. Inflamm. 17, 33 (2020).
Gan, E. S. et al. Dengue virus induces PCSK9 expression to alter antiviral responses and disease outcomes. J. Clin. Invest. 130, 5223–5234 (2020).
Huang, R., Huestis, M., Gan, E. S., Ooi, E. E. & Ohh, M. Hypoxia and viral infectious diseases. JCI Insight 6, e147190 (2021).
Tobin, M. J., Laghi, F. & Jubran, A. Why COVID-19 silent hypoxemia is baffling to physicians. Am. J. Respir. Crit. Care Med. 202, 356–360 (2020).
Swenson, K. E., Ruoss, S. J. & Swenson, E. R. The pathophysiology and dangers of silent hypoxemia in COVID-19 lung injury. Ann. Am. Thorac. Soc. 18, 1098–1105 (2021).
Stenmark, K. R., Tuder, R. M. & El Kasmi, K.C. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxic pulmonary hypertension. J. Appl. Physiol. 119, 1164–1172 (2015).
Pugliese, S. C. et al. A time- and compartment-specific activation of lung macrophages in hypoxic pulmonary hypertension. J. Immunol. 198, 4802–4812 (2017).
Taylor, C. T. & Colgan, S. P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 17, 774–785 (2017).
Marchetti, M. COVID-19-driven endothelial damage: complement, HIF-1, and ABL2 are potential pathways of damage and targets for cure. Ann. Hematol. 99, 1701–1707 (2020).
Ward, P. A., Fattahi, F. & Bosmann, M. New insights into molecular mechanisms of immune complex-induced injury in lung. Front. Immunol. 7, 86 (2016).
Yarmohammadi, A., Yarmohammadi, M., Fakhri, S. & Khan, H. Targeting pivotal inflammatory pathways in COVID-19: a mechanistic review. Eur. J. Pharmacol. 890, 173620 (2021).
Song, C. Y., Xu, J., He, J. Q. & Lu, Y. Q. Immune dysfunction following COVID-19, especially in severe patients. Sci. Rep. 10, 15838 (2020).
Peng, Y. et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 21, 1336–1345 (2020).
Levi, M. & Coppens, M. Vascular mechanisms and manifestations of COVID-19. Lancet Respir. Med. 9, 551–553 (2021).
Roncati, L. et al. Type 3 hypersensitivity in COVID-19 vasculitis. Clin. Immunol. 217, 108487 (2020).
Van Hinsbergh, V. W. M. Endothelium-role in regulation of coagulation and inflammation. Semin. Immunopathol. 34, 93–106 (2012).
Rabinovitch, M., Guignabert, C., Humbert, M. & Nicolls, M. R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 115, 165–175 (2014).
Humbert, M. et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur. Respir. J. 53, 1801887 (2019).
Magro, C. et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl. Res. 220, 1–13 (2020).
Gao, T. et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv https://doi.org/10.1101/2020.03.29.20041962 (2020).
Noris, M., Benigni, A. & Remuzzi, G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 98, 314–322 (2020).
Page, E. M. & Ariëns, R. A. S. Mechanisms of thrombosis and cardiovascular complications in COVID-19. Thromb. Res. 200, 1–8 (2021).
Carvelli, J. et al. Association of COVID-19 inflammation with activation of the C5a–C5aR1 axis. Nature 588, 146–150 (2020).
Ma, L. et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci. Immunol. 6, eabh2259 (2021).
Gralinski, L. E. et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio 9, e01753–18 (2018).
Mastaglio, S. et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin. Immunol. 215, 108450 (2020).
Diurno, F. et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur. Rev. Med. Pharmacol. Sci. 24, 4040–4047 (2020).
Zelek, W. M. et al. Complement inhibition with the C5 blocker LFG316 in severe COVID-19. Am. J. Respir. Crit. Care Med. 202, 1304–1308 (2020).
Nalbandian, A. et al. Post-acute COVID-19 syndrome. Nat. Med. 27, 601–615 (2021).
Greenhalgh, T., Knight, M., A’Court, C., Buxton, M. & Husain, L. Management of post-acute COVID-19 in primary care. Brit. Med. J. 370, m3026 (2020).
Herridge, M. S. et al. Functional disability 5 years after acute respiratory distress syndrome. N. Engl. J. Med. 364, 1293–1304 (2011).
Ahmed, H. et al. Long-term clinical outcomes in survivors of severe acute respiratory syndrome and Middle East respiratory syndrome coronavirus outbreaks after hospitalisation or ICU admission: a systematic review and meta-analysis. J. Rehabil. Med. 52, jrm00063 (2020).
Ojo, A. S., Balogun, S. A., Williams, O. T. & Ojo, O. S. Pulmonary fibrosis in COVID-19 survivors: predictive factors and risk reduction strategies. Pulm. Med. 2020, 6175964 (2020).
Lechowicz, K. et al. COVID-19: the potential treatment of pulmonary fibrosis associated with SARS-CoV-2 infection. J. Clin. Med. 9, 1917 (2020).
Yim, J., Lim, H. H. & Kwon, Y. COVID-19 and pulmonary fibrosis: therapeutics in clinical trials, repurposing, and potential development. Arch. Pharm. Res. 44, 499–513 (2021).
Ambardar, S. R. et al. Post-COVID-19 pulmonary fibrosis: novel sequelae of the current pandemic. J. Clin. Med. 10, 2452 (2021).
Ali, R. M. M. & Ghonimy, M. B. I. Post-COVID-19 pneumonia lung fibrosis: a worrisome sequelae in surviving patients. Egypt J. Radiol. Nucl. Med. 52, 101 (2021).
Zou, J. N. et al. The characteristics and evolution of pulmonary fibrosis in COVID-19 patients as assessed by AI-assisted chest HRCT. PLoS One 16, e0248957 (2021).
Tuder, R. M. et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 62, D4–D12 (2013).
Butrous, G. & Mathie, A. Infection in pulmonary vascular diseases: would another consortium really be the way to go? Glob. Cardiol. Sci. Pract. 2019, 1 (2019).
Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Invest. 118, 2372–2379 (2008).
Malczyk, M. et al. NADPH oxidases - do they play a role in TRPC regulation under hypoxia? Pflug. Arch. Eur. J. Physiol. 468, 23–41 (2016).
Pepke-Zaba, J. et al. Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation 124, 1973–1981 (2011).
Lang, I. M. & Jais, X. Chronic thromboembolic pulmonary hypertension. Prog. Respr. Res. 41, 226–236 (2021).
Engelen, M. M. et al. Incidence of venous thromboembolism in patients discharged after COVID-19 hospitalisation [abstract LB/CO01/3]. Res. Pract. Thromb. Haemost. 47, 362–371 (2020).
Leonard-Lorant, I. et al. Acute pulmonary embolism in COVID-19 patients on CT angiography and relationship to D-dimer levels. Radiology 296, E189–E191 (2020).
Morrell, N. W. et al. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 53, 1801899 (2019).
Southgate, L., Machado, R. D., Gräf, S. & Morrell, N. W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 17, 85–95 (2020).
Swietlik, E. M., Gräf, S. & Morrell, N. W. The role of genomics and genetics in pulmonary arterial hypertension. Glob. Cardiol. Sci. Pract. 2020, e202013 (2020).
Pagnesi, M. et al. Pulmonary hypertension and right ventricular involvement in hospitalised patients with COVID-19. Heart 106, 1324–1331 (2020).
Norderfeldt, J. et al. Acute pulmonary hypertension and short-term outcomes in severe COVID-19 patients needing intensive care. Acta Anaesthesiol. Scand. 65, 761–769 (2021).
Butrous, G. Pulmonary hypertension: from an orphan disease to a global epidemic. Glob. Cardiol. Sci. Pract. 2020, e202005 (2020).
Rich, S., Haworth, S. G., Hassoun, P. M. & Yacoub, M. H. Pulmonary hypertension: the unaddressed global health burden. Lancet Respir. Med. 6, 577–579 (2018).
Emmons-Bell, S. et al. Prevalence, incidence and survival of pulmonary arterial hypertension: a systematic review for the Global Burden of Disease 2020 study. Pulm. Circ. (in the press).
Ryan, J. T., de Jesus Perez, V. A. & Ryan, J. J. Health disparities in pulmonary arterial hypertension and the impact of the COVID-19 pandemic. Adv. Pulm. Hypertens. 20, 6–15 (2021).
Ryan, J. J. The impact of COVID-19 on pulmonary hypertension. American College of Cardiology https://www.acc.org/latest-in-cardiology/articles/2020/08/13/08/35/the-impact-of-covid-19-on-pulmonary-hypertension (2020).
Sulica, R. et al. COVID-19 in pulmonary artery hypertension (PAH) patients: observations from a large PAH center in New York City. Diagnostics 11, 128 (2021).
Horn, E. M., Chakinala, M., Oudiz, R., Joseloff, E. & Rosenzweig, E. B. Could pulmonary arterial hypertension patients be at a lower risk from severe COVID-19? Pulm. Circ. 10, 2045894020922799 (2020).
Belge, C. et al. COVID-19 in pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: a reference centre survey. ERJ Open Res. 6, 00520–02020 (2020).
Farha, S. & Heresi, G. A. COVID-19 and pulmonary arterial hypertension: early data and many questions. Ann. Am. Thorac. Soc. 17, 1528–1530 (2020).
Pun, M., Turner, R., Strapazzon, G., Brugger, H. & Swenson, E. R. Lower incidence of COVID-19 at high altitude: facts and confounders. High Alt. Med. Biol. 21, 217–222 (2020).
Thomson, T. M. et al. Potential protective effect from COVID-19 conferred by altitude: a longitudinal analysis in Peru during full lockdown. High Alt. Med. Biol. 22, 209–224 (2021).
Hopman, J., Allegranzi, B. & Mehtar, S. Managing COVID-19 in low- and middle-income countries. JAMA 323, 1549–1550 (2020).
Boulle, A. et al. Risk factors for coronavirus disease 2019 (COVID-19) death in a population cohort study from the Western Cape province, South Africa. Clin. Infect. Dis. 73, e2005-e2015 (2021).
Brown, L. B., Spinelli, M. A. & Gandhi, M. The interplay between HIV and COVID-19: summary of the data and responses to date. Curr. Opin. HIV AIDS 16, 63–73 (2021).
Butrous, G. The global challenge of pulmonary vascular diseases and its forgotten impact in the developing world. Adv. Pulm. Hypertens. 11, 117–118 (2012).
Butrous, G. Human immunodeficiency virus-associated pulmonary arterial hypertension considerations for pulmonary vascular diseases in the developing world. Circulation 131, 1361–1370 (2015).
Butrous, G., Ghofrani, H. A. & Grimminger, F. Pulmonary vascular disease in the developing world. Circulation 118, 1758–1766 (2008).
Butrous, G. Schistosome infection and its effect on pulmonary circulation. Glob. Cardiol. Sci. Pract. 2019, 5 (2019).
Alexander, G. C., Stoller, K. B., Haffajee, R. L. & Saloner, B. An epidemic in the midst of a pandemic: opioid use disorder and COVID-19. Ann. Intern. Med. 173, 57–58 (2020).
Columb, D., Hussain, R. & O’Gara, C. Addiction psychiatry and COVID-19: impact on patients and service provision. Ir. J. Psychol. Med. 37, 164–168 (2020).
Sharma, Gupta, P., Pal, A. & Parmar, A. Opioid use disorder and COVID-19 in India: waiting for an epidemic within the pandemic? Indian J. Soc. Psychiatry 37, 117 (2021).
Maggiorini, M. & Léon-Velarde, F. High-altitude pulmonary hypertension: a pathophysiological entity to different diseases. Eur. Respir. J. 22, 1019–1025 (2003).
Mirrakhimov, A. E. & Strohl, K. P. High-altitude pulmonary hypertension: an update on disease pathogenesis and management. Open Cardiovasc. Med. J. 10, 19–27 (2016).
Luks, A. M. & Swenson, E. R. COVID-19 lung injury and high altitude pulmonary edema: a false equation with dangerous implications. Ann. Am. Thorac. Soc. 17, 918–921 (2020).
Pujhari, S., Paul, S., Ahluwalia, J. & Rasgon, J. L. Clotting disorder in severe acute respiratory syndrome coronavirus 2. Rev. Med. Virol. 31, e2177 (2020).
Kotwal, J., Apte, C. V., Kotwal, A., Mukherjee, B. & Jayaram, J. High altitude: a hypercoagulable state: results of a prospective cohort study. Thromb. Res. 120, 391–397 (2007).
Perez-Castilla, J. High altitude and COVID-19: a relationship difficult to assess. Arch. Bronconeumol. 57, 68–70 (2021).
Millet, G. P., Debevec, T., Brocherie, F., Burtscher, M. & Burtscher, J. Altitude and COVID-19: friend or foe? A narrative review. Physiol. Rep. 8, e14615 (2021).
Gavriatopoulou, M. et al. Organ-specific manifestations of COVID-19 infection. Clin. Exp. Med. 20, 493–506 (2020).
Accinelli, R. A. & Leon-Abarca, J. A. At high altitude COVID-19 is less frequent: the experience of Peru. Arch. Bronconeumol. 56, 760–761 (2020).
Hoeper, M. M. et al. A global view of pulmonary hypertension. Lancet Respir. Med. 4, 306–322 (2016).
Chacon, N., Chacin-Bonilla, L. & Cesar, I. M. Implications of helminth immunomodulation on COVID-19 co-infections. Life Res. 4, 26 (2021).
Randolph, H. E. & Barreiro, L. B. Herd immunity: understanding COVID-19. Immunity 52, 737–741 (2020).
Ramirez, R. L. III et al. Drug- and toxin-induced pulmonary arterial hypertension: current state of the literature. Glob. Cardiol. Sci. Pract. https://doi.org/10.21542/gcsp.2019.19 (2020).
Wang, Q. Q., Kaelber, D. C., Xu, R. & Volkow, N. D. COVID-19 risk and outcomes in patients with substance use disorders: analyses from electronic health records in the United States. Mol. Psychiatry 26, 30–39 (2021).
Becker, W. C. & Fiellin, D. A. When epidemics collide: coronavirus disease 2019 (COVID-19) and the opioid crisis. Ann. Intern. Med. 173, 59–60 (2020).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921.e10 (2020).
Hartmann, D. A. et al. Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics 2, 041402 (2015).
Polycarpou, A. et al. Rationale for targeting complement in COVID-19. EMBO Mol. Med. 12, e1264 (2020).
Acknowledgements
We are grateful to the Infection and Pulmonary Vascular Diseases (iPVD) Consortium for their work on the global nature of chronic pulmonary hypertension.
Author information
Authors and Affiliations
Contributions
G.B. and M.H.Y. conceptualized the content of the manuscript. All the authors researched data for the article, discussed its content and wrote the manuscript. S.H., Y.A. and M.H.Y. reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Cardiology thanks Mary Beasley and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Halawa, S., Pullamsetti, S.S., Bangham, C.R.M. et al. Potential long-term effects of SARS-CoV-2 infection on the pulmonary vasculature: a global perspective. Nat Rev Cardiol 19, 314–331 (2022). https://doi.org/10.1038/s41569-021-00640-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-021-00640-2
This article is cited by
-
Blood and marrow transplant within 4 weeks of SARS-CoV-2 infection is associated with increased risk of mortality: a National COVID Cohort Collaborative (N3C) Study
Bone Marrow Transplantation (2024)
-
Damage to endothelial barriers and its contribution to long COVID
Angiogenesis (2024)
-
SARS-CoV-2 spike protein receptor-binding domain perturbates intracellular calcium homeostasis and impairs pulmonary vascular endothelial cells
Signal Transduction and Targeted Therapy (2023)
-
Unsupervised discovery of tissue architecture in multiplexed imaging
Nature Methods (2022)
