
Abstract
Mechanical stress from haemodynamic perturbations or interventional manipulation of epicardial coronary atherosclerotic plaques with inflammatory destabilization can release particulate debris, thrombotic material and soluble substances into the coronary circulation. The physical material obstructs the coronary microcirculation, whereas the soluble substances induce endothelial dysfunction and facilitate vasoconstriction. Coronary microvascular obstruction and dysfunction result in patchy microinfarcts accompanied by an inflammatory reaction, both of which contribute to progressive myocardial contractile dysfunction. In clinical studies, the benefit of protection devices to retrieve atherothrombotic debris during percutaneous coronary interventions has been modest, and the treatment of microembolization has mostly relied on antiplatelet and vasodilator agents. The past 25 years have witnessed a relative proportional increase in non-ST-segment elevation myocardial infarction in the presentation of acute coronary syndromes. An associated increase in the incidence of plaque erosion rather than rupture has also been recognized as a key mechanism in the past decade. We propose that coronary microembolization is a decisive link between plaque erosion at the culprit lesion and the manifestation of non-ST-segment elevation myocardial infarction. In this Review, we characterize the features and mechanisms of coronary microembolization and discuss the clinical trials of drugs and devices for prevention and treatment.
Key points
-
Coronary microembolization results from spontaneous or interventional erosion or rupture of an epicardial coronary atherosclerotic plaque.
-
Atherothrombotic plaque debris causes physical obstruction of coronary microvessels and results in microinfarcts with an inflammatory response.
-
The surviving myocardium around the patchy microinfarcts has reduced contractile function as a result of inflammatory signal transduction leading to myofibrillar oxidation.
-
Atherosclerotic plaque erosion or rupture releases soluble substances that induce coronary microvascular endothelial dysfunction, vasoconstriction, platelet aggregation and coagulation.
-
Routine use of interventional protection devices does not improve patient outcomes but these devices are indicated in cases of high atherothrombotic plaque burden.
-
Plaque stabilization with statins and canakinumab prevents coronary microembolization, whereas platelet inhibitors, vasodilators and interleukin antagonists attenuate coronary microvascular impairment.
Similar content being viewed by others

Introduction
Interest in coronary microembolization and its sequelae has fluctuated over time. In the 1980s, several investigators (notably Davies and Falk), performed autopsy studies in patients with coronary artery disease who had died suddenly. Together with the rupture or fissure of epicardial coronary atherosclerotic plaques1,2,3, obstruction of the coronary microcirculation by microemboli was frequently identified. These microemboli predominantly consisted of platelet aggregates, fibrin, hyalin and atherosclerotic plaque material, including cholesterol1,2,3,4,5,6. In the same decade, the development of platelet aggregation in the stenotic coronary arteries of dogs was characterized by Folts and Willerson. They identified endothelial dysfunction involving serotonin, thrombin and thromboxane A2 as mediators and causing cyclic flow variations with embolization of platelet aggregates into the microcirculation7,8,9,10.
With the increasing use of elective and primary percutaneous coronary intervention (PCI), interest in coronary microembolization was renewed in the early 2000s prompted by the question of what happened to plaque material that was released during PCI11,12. The use of protection devices during PCI enabled particulate plaque debris, thrombotic material and soluble factors to be retrieved from native and graft coronary vessels13. However, the clinical benefit of protection devices was modest at best, and interest in coronary microembolization understandably subsided.
During the past decade, several investigators (notably Libby) have emphasized an increasing incidence of plaque erosion rather than rupture as the key underlying event of acute coronary syndromes, particularly as a consequence of increasing statin use14,15,16,17. The increasing incidence of plaque erosion has been associated with a quantitative shift from ST-segment elevation myocardial infarction (STEMI) to non-ST-segment elevation myocardial infarction (NSTEMI) over the past 25 years15,16,18. Coronary microemboli are more frequently found at autopsy in patients with plaque erosion who have died suddenly than in those with plaque rupture19. The fact that not all patients with NSTEMI need routine intervention but do require intensified antithrombotic treatment20,21 places the spotlight on the obstruction and dysfunction of the coronary microcirculation. Indeed, the treatment of coronary microembolization is likely to be more important in the outcome of patients with acute coronary syndromes than during elective PCI. In this Review, we characterize the features and mechanisms of coronary microembolization and discuss clinical trials of drugs and devices for prevention and treatment.
Experimental coronary microembolization
Animal models
Intracoronary injection of inert particles (such as talcum, lycopodium spores or plastic microspheres of various diameters) in anaesthetized dogs has been used since the early twentieth century to study the effects of a fairly standardized, partial coronary obstruction on cardiac rhythm and contractile function, including cardiogenic shock22,23,24,25,26,27,28,29. Intracoronary injection of inert polystyrene microspheres is the current standard method of inducing a regional acute coronary syndrome30,31 and, with repetitive microembolization, heart failure32,33 in animal models (for example, dogs34,35,36, pigs37,38,39,40, sheep41, mice42 and rats43). The advantage of using polystyrene microspheres is that the diameter (usually around 40 µm) and the number of injected spheres can be standardized and adjusted to coronary inflow. The disadvantage of this method is the inert nature of these microspheres36, which cause physical obstruction of the coronary microcirculation but have no prothrombotic, pro-inflammatory or vasoconstrictor potential.
An alternative but somewhat less standardized approach is the intracoronary infusion of exogenously (ex vivo) generated homologous or autologous thrombotic material that, although not inert, still does not include atherosclerotic debris and soluble factors released from an atherosclerotic culprit lesion. This method has been used in rats44 and pigs45.
A model of severe epicardial coronary stenosis with endothelial dysfunction, developed by Folts et al.7, causes the spontaneous formation of platelet aggregates in the stenotic segment with progressive inflow reduction, followed by sudden restoration of blood flow when the platelet aggregate is dislodged from the stenotic segment and embolized into the coronary microcirculation. This model has been used in dogs8,9,46 and pigs47 and is characterized by cyclic coronary flow variations, which enable prothrombotic and vasoconstrictor factors, such as thrombin8, serotonin and thromboxane A2 (ref.9), to be studied.
Blood flow and cardiac function
In anaesthetized dogs, intracoronary injection of inert microparticles results in an immediate decrease in coronary blood flow24,48, followed by reactive hyperaemia28,35,37,48 and then a return to normal blood flow24,48 (Fig. 1). Regional myocardial contractile function, as measured by sonomicrometry, in the perfusion territory of the embolized coronary artery decreases immediately, then partially recovers over minutes but does not return to baseline function48. The response of global left ventricular (LV) function depends on the number and size of embolizing particles and the size of the affected coronary perfusion territory. The response can range from transient and subtle LV dysfunction48 to protracted and severe cardiogenic shock49. With repetitive coronary microembolization, baseline coronary blood flow can still be normal or even above normal but coronary reserve in response to adenosine is markedly reduced48. Both the slightly increased baseline flow (reflecting reactive hyperaemia) and, more importantly, the reduced maximal blood flow secondary to physical obstruction of the coronary microvasculature contribute to reduced coronary reserve48. The reduction in regional contractile function with repetitive coronary microembolization is cumulative (Fig. 1), and repetitive coronary microembolization has become a standard method to induce LV dysfunction and heart failure32,33. The dissociation between profound contractile dysfunction and normal or even slightly elevated coronary blood flow (that is, a perfusion–contraction mismatch pattern), is a salient feature of microembolized myocardium36,50.

The arrows along the top indicate infusion of 30,000 polystyrene microspheres of 42 µm in diameter. Coronary blood flow (red) is immediately reduced after infusion, recovers quickly and displays some reactive hyperaemia (exceeds baseline flow). Systolic myocardial wall thickening (blue) is also immediately reduced and recovers quickly but not back to baseline. After four infusions of microspheres, coronary blood flow is normal but systolic wall thickening has a cumulative deficit. Adapted with permission from ref.48, APS.
Morphological sequelae
Coronary arterioles are functional end arteries, and their physical obstruction causes microinfarction (Fig. 2). The size of the microinfarcted area corresponds to the site of vascular obstruction which, in turn, is determined by the diameter of the embolizing particles34. On haematoxylin and eosin staining, the microinfarction from coronary microembolization appears as typical necrosis in dogs34,36, pigs51, mice42 and rats43,44. There is also evidence for apoptosis in the microinfarcted area shown by terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL)30,38,52,53,54,55 and increased expression of caspase 3, caspase 9 and Bax 2 (also known as apoptosis regulator BAX)52,53,55,56. However, the contribution of apoptosis to cell death from microinfarction is minor compared with that from necrosis. The microinfarct resulting from physical obstruction of a microvessel by inert microspheres is characterized by a profound inflammatory response leading to pyroptotic cell death57,58. Morphologically, oedema and neutrophil and macrophage infiltration occur around the microinfarct site in dogs36,59, pigs51,60, mice42 and rats43. The inflammatory response is also characterized by the increased expression of cytokines, such as tumour necrosis factor (TNF) in macrophages51 and cardiomyocytes61, as well as of interleukins43,56 and inducible nitric oxide synthase (iNOS)56 at the mRNA and protein levels. In rats, the activation of extracellular signal-regulated kinase 1 (ERK1) and ERK2 seems to be involved in initiating the inflammatory response62. Inflammation around microinfarcts in pigs is associated with increased insulin-like growth factor 1 (IGF1) mRNA expression in infiltrating monocytes63, which is a potential start signal for an angiogenic response and for the promotion of collateral growth, as seen in a long-term model of coronary microembolization in dogs64. CT and MRI in pig models of coronary microembolization demonstrated not only a decrease in regional and global contractile function but also patchy perfusion defects, oedema and microinfarcts60,65,66,67.

a | Haematoxylin and eosin-stained microinfarct with surrounding infiltration (enlarged insert) distal to an embolizing microsphere. b | Phase-contrast microscopy of the microinfarct for improved demarcation. c | Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining of a few apoptotic cardiomyocytes within a microinfarct (phalloidin staining). d | In situ hybridization of tumour necrosis factor mRNA in the viable myocardium surrounding a microinfarct. Parts a and b adapted with permission from ref.36, APS. Parts c and d adapted with permission from ref.61, Elsevier.
Signal transduction of contractile dysfunction
Regional contractile function in microembolized myocardium follows a typical time course, with a decrease immediately after microembolization, followed by a partial recovery and then a subsequent deterioration over several hours, with a nadir at 6–12 h after the procedure36,38,61,68,69. A nadir in global LV function is also seen at around 12 h after microembolization43. The magnitude of contractile dysfunction exceeds that of acute ischaemia, reflecting the contribution of loss of viable contractile cardiomyocytes from microinfarction and, more importantly, contractile impairment in viable myocardium from the effect of negative inotropic inflammatory mediators (Fig. 3). With the intracoronary injection of microspheres in anaesthetized pigs, the total surface area of microperfusion defects was more closely correlated with LV contractile dysfunction than with the total volume of microperfusion defects on electron-beam CT39. This finding supports the notion that interaction with the surrounding myocardium and inflammation are more important for contractile dysfunction than the loss of viable tissue39.

a | Rupture or erosion of an atherosclerotic plaque in an epicardial coronary artery releases particulate atherothrombotic debris and soluble substances into the coronary microcirculation. Debris causes physical obstruction of coronary microvessels, whereas soluble substances cause endothelial dysfunction, vasoconstriction and platelet aggregation. b | Microinfarcts occur in the myocardium accompanied by an inflammatory reaction, which impairs contractile function in adjacent surviving cardiomyocytes through signal transduction involving nitric oxide (NO), tumour necrosis factor (TNF), sphingosine, reactive oxygen species (ROS) and, ultimately, myofibrillar oxidation. Ab, antibody; l-NAME, NG-nitro-l-arginine methyl ester; NOE, N-oleoylethanolamine.
The increased expression of TNF in macrophages and cardiomyocytes is central among the inflammatory mediators to exert negative inotropic effects on the microembolized myocardium43,51,61. Therefore, antibodies to TNF abrogate the progressive contractile dysfunction between 1 and 8 h after microembolization in anaesthetized dogs61. In anaesthetized pigs, nitric oxide is formed upstream of increased TNF expression and sphingosine is synthesized downstream of increased TNF expression38. As such, inhibition of nitric oxide formation by NG-nitro-l-arginine methyl ester (l-NAME) or inhibition of sphingosine formation by N-oleoylethanolamine (NOE) abrogates progressive contractile dysfunction between 1 and 8 h after microembolization38. The ultimate effect of such inflammatory signal transduction is apparently the increased formation of reactive oxygen species (ROS), oxidative modification of contractile myofibrils69 and reduced Ca2+ responsiveness of the contractile machinery70. The causal role of ROS is supported by the prevention of myofibrillar oxidation and progressive contractile dysfunction by high-dose vitamin C69. In chronically instrumented dogs, regional contractile dysfunction has its nadir 4–12 h after coronary microembolization but recovers almost completely over 1 week68. Recovery is accelerated by intravenous methylprednisolone, further supporting the causal role of inflammation in the observed regional contractile dysfunction68. Although the causal role of inflammatory mediators in contractile dysfunction from coronary microembolization is obvious, we do not currently know the exact biochemical, cellular and subcellular source of each participating mediator nor the full sequence of signalling steps.
Several drugs attenuate contractile dysfunction but where and how they interact with signal transduction is unclear. Some of these drugs, including metoprolol52, nobiletin54, resveratrol55, breviscapine71, puerarin72, nicorandil73 and glycyrrhizin56 reduce markers of apoptosis (reduced caspase 3 activation and expression of p53 and Bax, reduced TUNEL staining) and activate classical cardioprotection pathways74 such as phosphoinositide 3-kinase (PI3K), protein kinase B (Akt) and glycogen synthase kinase 3β (GSK3β)54,71,72. Some of these drugs, including pyrrolidine dithiocarbamate44, glycyrrhizin56 and breviscapine71, also attenuate the expression of inflammatory cytokines44,56,71 or ROS54. In chronically instrumented pigs with coronary microembolization, long-term treatment with the angiotensin II receptor antagonist irbesartan improved both perfusion and contractile dysfunction75. Pretreatment with trimetazidine in anaesthetized pigs with coronary microembolization attenuated apoptosis (reduced activation of caspase 3 and caspase 9, reduced TUNEL staining) and global LV dysfunction (as shown on echocardiography) 12 h after coronary microembolization53.
Cardioprotection
Cardioprotection in its strictest sense refers to the reduction of infarct size that results from sustained and severe myocardial ischaemia and subsequent reperfusion by mechanical or pharmacological interventions74. Whereas microembolization induces patchy microinfarcts, the increased expression of TNF not only causes progressive contractile dysfunction61 but is also cardioprotective. In anaesthetized pigs, TNF expression was increased 6 h after coronary microembolization, and the infarct that resulted from 90 min of severe coronary hypoperfusion and 2 h of reperfusion was then reduced in size; this cardioprotection was abrogated by TNF antibodies76. While coronary microembolization per se apparently exerts cardioprotection under specific circumstances, it does not interfere with cardioprotection produced by ischaemic preconditioning or postconditioning. Conversely, in anaesthetized pigs, the immediate increase in coronary venous adenosine concentration associated with coronary microembolization did not result in infarct size reduction with the 90 min of severe myocardial ischaemia and 120 min of reperfusion that followed 10 min after microembolization77. In addition, coronary microembolization did not interfere with cardioprotection from a true ischaemic preconditioning protocol (one 10-min cycle of severe ischaemia and 15 min of reperfusion), which was associated with a substantial increase in interstitial adenosine before 90 min of sustained ischaemia followed by 120 min of reperfusion, although coronary microembolization slightly increased infarct size78. These findings contrast with the delayed cardioprotection associated with increased TNF expression described above76. Collectively, these data suggest that coronary microembolization per se might induce some delayed protection but neither contributes to nor interferes with cardioprotection from pre-infarction angina. Almost the same is true for ischaemic postconditioning. The microinfarcts from coronary microembolization at early reperfusion following 90 min of severe myocardial ischaemia increased infarct size after 2 h of reperfusion in anaesthetized pigs79. However, microembolization did not prevent infarct size reduction by an ischaemic postconditioning protocol (six cycles of 20 s of coronary occlusion and 20 s of reperfusion)79. Therefore, coronary microembolization, which is possibly associated with manipulation of the culprit lesion when applying an ischaemic postconditioning protocol, does not abrogate protection from postconditioning. Cardioprotective conditioning procedures are therefore still effective and could blunt deleterious infarct expansion from coronary microembolization.
Coronary no-reflow
Coronary no-reflow is the manifestation of coronary microvascular injury from sustained and severe myocardial ischaemia and reperfusion80,81,82,83. No-reflow results from a number of pathomechanisms, including interstitial, endothelial and cardiomyocyte oedema, impaired vasomotion, leukocyte adherence and infiltration, stasis of blood elements, and capillary destruction and rupture83. No-reflow can occur after sustained coronary occlusion and reperfusion in a previous virgin coronary vascular territory; therefore, it does not require physical obstruction by atherothrombotic material or the action of soluble factors released from upstream atherosclerotic lesions. Nevertheless, there are typical leukocyte–platelet aggregates84,85,86,87 and erythrocyte aggregates88 that obstruct the capillaries in the myocardium with no-reflow. Additionally, in a realistic clinical scenario of acute plaque rupture or erosion, there will be physical obstruction by atherothrombotic debris and microvascular dysfunction from prothrombotic, pro-inflammatory and vasoconstrictor soluble substances31,82,83. Coronary microembolization with sodium laurate in rats induced microthrombi together with endothelial and cardiomyocyte injury, which were all attenuated by pretreatment with prostaglandin E1 (ref.89).
Heart failure
Progressive LV dysfunction following coronary microembolization with glass beads (400–600 µm diameter) in chronically instrumented dogs was first used as a preclinical heart failure model by Franciosa et al.32. Sabbah et al. then refined this dog model by using polystyrene microspheres (70–110 µm diameter) and a protocol of sequential microembolizations to establish stable heart failure33 characterized by patchy myocardial fibrosis and LV hypertrophy33, apoptosis (demonstrated by DNA fragmentation and electron microscopy)90 and neurohumoural activation33,91,92. With more limited coronary microembolization, heart failure (neurohumoural activation and volume expansion) with preserved ejection fraction could also be established92. This model with characteristic features of LV dysfunction and neurohumoural activation was repeated in sheep93. Sabbah et al. used coronary microembolization-induced heart failure in dogs to study the effects of several drugs. Metoprolol not only improved LV function91,94 but also attenuated apoptosis95 and endoplasmic reticulum stress94 and increased the expression of excitation–contraction coupling proteins94. Metoprolol and enalapril also prevented progressive LV dilatation in this model91. In a similar model of heart failure, in which microspheres of 45 µm diameter were used to induce coronary embolization in pigs, intracoronary cardiosphere-derived cell infusion reduced infarct size and collagen volume, increased myocyte number and capillary density, and improved regional and global LV function96.
Animal experiments on coronary microembolization have strengths and limitations (Box 1). Collectively, however, these models are imperfect surrogates for the human clinical situation of acute or chronic coronary syndromes, with or without PCI.
Clinical coronary microembolization
The clinical presentation of coronary microembolization is unspecific, ranging from lack of symptoms and retrospective detection by chance to symptoms of an acute coronary syndrome. Coronary microembolization occurs spontaneously and as a result of cardiac, mostly coronary, interventions. Spontaneous coronary microembolization, typically symptomatic as an acute coronary syndrome, can be ascertained only at autopsy after sudden cardiac death3,4,5,6,97. Increases in levels of biomarkers, such as creatine kinase or troponin (particularly with high-sensitivity assays), above the normal range reflect myocardial injury but are unspecific and fairly common in the general population98. Such elevations in biomarkers become more frequent with increasing age and with the presence of other risk factors for atherosclerosis and are indicative of poor prognosis98,99. The true contribution of coronary microembolization to an increase in biomarker levels cannot be distinguished from other causes, such as alternative forms of myocardial ischaemia, myocarditis, stroke, trauma and excessive exercise. Coronary microembolization is therefore more unequivocally identified in cardiac (notably coronary) interventions. Massive and often disastrous coronary embolization can arise from aortic or mitral valve endocarditis or iatrogenic valve interventions, atrial fibrillation or peripheral thrombosis through a patent foramen ovale. However, a coronary embolus is rarely the cause of an acute coronary syndrome and is seen in no more than 3% of patients undergoing catheterization for acute myocardial infarction (MI)100.
In the early reperfusion era, 80% of patients with acute MI who had undergone balloon angioplasty or thrombolysis and died within 3 weeks of the procedure had coronary emboli at autopsy101. The emboli consisted of atherothrombotic material within the primary infarction, extending the infarction, inducing new infarction or involving non-infarcted myocardium101. Microvascular obstruction/no-reflow is frequently seen on MRI in patients with reperfused acute STEMI, and microvascular obstruction is a determinant of patient prognosis, independent from and in addition to infarct size102,103,104. However, MRI visualizes microvascular obstruction only in the infarcted tissue and not in the non-infarcted area at risk. MRI is usually performed within 1 week of reperfusion, and microvascular obstruction is typically identified at this point. However, the duration of microvascular obstruction and its distinction from scarring or angiogenesis on later MRI scans is unclear. Importantly, the contribution of coronary microembolization compared with other causes of microvascular obstruction, such as endothelial and interstitial oedema, vasoconstriction, leukocyte adherence, capillary destruction or haemorrhage, is impossible to quantify83,105. We will therefore address only the clinical scenarios for which clear evidence for coronary microembolization exists.
To date, the recognition of coronary microembolization in patients with spontaneous acute coronary syndrome has not resulted in a protocol for risk stratification and management other than according to established guidelines21,106. Coronary microembolization remains the conceptual ‘missing link’ between the culprit plaque lesion in the epicardial coronary artery (diagnosed by intravascular imaging) and the clinical presentation of NSTEMI with an increased level of troponin but without ST-segment elevation, with the consequent lack of an urgent need for PCI in many instances and an indication for proactive antiplatelet and anti-inflammatory treatment. In a sense, iatrogenically induced coronary microembolization during a coronary intervention can be viewed as a human model of a minor acute coronary syndrome and might facilitate its mechanistic understanding.
Spontaneous coronary microembolization
Spontaneous coronary microembolization in the absence of PCI can be seen only at autopsy. In a systematic search for microthrombi in patients with heart disease, mostly of ischaemic origin, microthrombi rich in platelets and fibrin were seen in the coronary microcirculation, and platelet-rich microthrombi were more common in younger patients (aged <45 years) who had suffered sudden cardiac death than in older patients4. The combination of epicardial plaque rupture or fissure together with microvascular atherothrombotic embolization in patients with ischaemic heart disease who had suffered sudden cardiac death was subsequently established through autopsy studies. The microemboli consisted of atherosclerotic plaque material, including cholesterol1, platelet aggregates, platelet–leukocyte aggregates3,5,6, hyalin3 and fibrin5,6, and caused microinfarcts with an inflammatory response3,5,97. These microinfarcts with inflammation were reminiscent of those seen in animal experiments with embolization induced by inert microspheres (discussed above). Subsequently, Schwartz et al. demonstrated that plaque erosion caused coronary microembolization more frequently than plaque rupture, microemboli were unrelated to stenosis severity of the culprit coronary artery and emboli were mostly seen in microvessels of <120 µm in diameter19.
Periprocedural coronary microembolization
Biomarkers
Transient increases in serum levels of creatine kinase, creatine kinase-MB (CK-MB), and troponin I or troponin T after PCI are characteristic of periprocedural myocardial injury107,108. Preferably, high-sensitivity cardiac troponin assays are used98,99. According to the Fourth Universal Definition of Myocardial Infarction109, an increase in cardiac troponin levels (measured by high-sensitivity assay) to above the 99th percentile upper reference limit in patients with normal baseline values or an increase by >20% in patients with elevated baseline values is defined as procedural myocardial injury (PMI). Major PMI and type 4a MI are defined by increases in cardiac troponin levels to above the fivefold 99th percentile upper reference limit and only such an increase carries an adverse prognosis for clinical outcome110. However, the assessment of PMI from an elevation in troponin levels cannot distinguish between injury from side-branch occlusion and injury from distal coronary microembolization during PCI. The magnitude of biomarker release depends on the clinical situation of the patient110 (greater in patients with diabetes mellitus111 or chronic kidney disease112), the nature of the vessel undergoing PCI (greater in saphenous vein grafts (SVGs) than in native coronary arteries)113,114 and the type of procedure (greater with rotablation than simple stenting)115. The difference between elective and primary PCI in terms of resultant coronary microembolization is simply the iatrogenic versus spontaneous nature of its origin in the culprit lesion.
Imaging
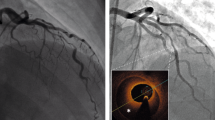
Distal embolization can initially be visualized by angiography as an abrupt filling defect in a peripheral coronary branch116,117. Imaging during PCI can identify the origin of the embolizing material in the epicardial coronary vascular wall by intravascular ultrasonography (IVUS)118 or optical coherence tomography (OCT)119. IVUS has greater penetration into the vascular wall than OCT and allows the different plaque components (that is, calcified, fatty, fibrotic and necrotic) to be distinguished by radiofrequency-based virtual histology. OCT has greater spatial resolution than IVUS and permits more detailed visualization of the endothelial layer and fibrous plaque cap14,16. Therefore, OCT is particularly well suited to distinguishing between plaque rupture (a thin fibrous cap with rupture) and plaque erosion (a thick fibrous cap without rupture), both of which can result in coronary microembolization17. Plaque volume on IVUS correlates with TNF release into the aspirate from an SVG during PCI120, and the volume of the plaque necrotic core in native coronary arteries correlates with the release of creatine kinase and troponin into the systemic circulation following PCI118. Showers of microemboli during elective PCI of native coronary arteries in patients with stable angina were visualized by intracoronary Doppler and correlated with the postprocedural elevation in serum troponin levels121 (Fig. 4). In patients with diabetes undergoing elective PCI, the Doppler-derived amount of microembolization correlated with the incidence of major adverse cardiovascular events (MACE) at 2-year follow-up122. A typical finding indicating coronary microembolization, also reported in experimental studies using intracoronary injection of microspheres48,123, is an increase in baseline coronary blood flow and a reduction in coronary reserve immediately after PCI124,125. The reduction in coronary reserve correlated with postprocedural serum creatine kinase and troponin levels125.

a | Intravascular ultrasonography of a ruptured epicardial coronary atherosclerotic plaque after percutaneous coronary intervention (PCI). Arrows point to the ruptured cap and to the plaque, the debris from which is emptied into the coronary microcirculation. b | In the same vessel 2 mm distally, the thin cap is still intact but the plaque is empty. c | Optical coherence tomography of a thin and ruptured fibrous cap overlying an atherosclerotic plaque following PCI. d | Section of the same image at higher magnification. e | Intracoronary Doppler imaging of coronary blood flow velocity with high-intensity signals at baseline (arrow). f | After PCI, blood flow velocity is increased and more high-intensity signals (arrows) reflect showers of microemboli. Parts a and b adapted with permission from ref.233, Deutscher Aerzte-Verlag GmbH. Parts c and d adapted with permission from ref.234, EuroIntervention. Parts e and f adapted with permission from ref.122, Springer Nature Limited.
As in animal experiments with intracoronary injection of microspheres60,65,66,67, local myocardial lesions resulting from microembolization in patients undergoing PCI can be visualized on postprocedural MRI126,127,128,129. Quantification of patchy areas with delayed gadolinium contrast hyperenhancement correlated with postprocedural serum troponin levels126 and a reduction in plaque volume on IVUS127. Areas with delayed hyperenhancement also had decreased local perfusion reserve, reflecting coronary microvascular obstruction128. The amount of postprocedural hyperenhancement on MRI between 24 h and 6 days after PCI or coronary artery bypass graft surgery correlated with clinical outcome during a 3-year follow-up period129. A disadvantage of MRI is that it cannot be used to distinguish between myocardial injury from coronary microembolization and other types of local injury such as side-branch occlusion.
Interventional prevention and treatment
In patients undergoing elective PCI, remote ischaemic preconditioning by one or more cycles of blood pressure cuff inflation and deflation on the arm reduced biomarker-defined PMI130,131,132 and the incidence of adverse events in several133 but not all134 studies. These studies differ from the experimental models discussed earlier in which local ischaemic preconditioning did not attenuate coronary microembolization. Whether remote ischaemic preconditioning stabilized the plaque or attenuated the myocardial injury from distal embolization is unclear. Possibly, remote ischaemic preconditioning induces the release of systemically circulating anti-inflammatory and cardioprotective substances, whereas local preconditioning does not135,136. Reduced microvascular obstruction by remote ischaemic preconditioning in patients undergoing PCI for acute MI has also been reported137,138,139,140 but whether this benefit resulted from attenuated coronary microembolization or is a secondary effect of reduced infarct size is unknown83.
Direct stenting reduced coronary microvascular resistance and tended to decrease postprocedural troponin release in patients with stable angina141. In patients with acute MI, this procedure improved thrombolysis in myocardial infarction (TIMI) flow142 and reduced microvascular obstruction and infarct size on MRI in a subanalysis of a larger trial143. The most likely mechanism for this reduction in microvascular obstruction is through the prevention of coronary microembolization from the culprit lesion.
The recognition of periprocedural coronary microembolization as a complication of PCI has stimulated the development of protection devices — filters or occlusion/aspiration systems to capture atherothrombotic debris, soluble substances or both that are released from the culprit lesion during PCI. The use of distal protection devices during elective PCI in native coronary arteries or SVGs was established as safe and feasible114,144,145,146 (Fig. 5). In the SAFER trial147 of patients undergoing PCI of SVGs under distal protection by an occlusion/aspiration device, TIMI flow was improved, the incidence of no-reflow was reduced, and the primary composite clinical end point of death, MI, emergency bypass and target-vessel revascularization after 30 days was reduced. In the FIRE trial148, no significant difference in procedural outcome or the primary composite end point of death, MI or target-vessel revascularization was seen in patients undergoing PCI of SVGs under protection with a filter device as compared with a balloon occlusion/aspiration device. Similarly, no significant difference in procedural and clinical end points was evident in the AMEthyst trial149 in patients undergoing PCI of SVGs under protection with a novel filter device (AVE Interceptor; Medtronic) as compared with a balloon occlusion/aspiration device. In the PROXIMAL trial150, no significant difference was seen in postprocedural creatine kinase level or in the primary clinical end point of death, MI and target-vessel revascularization between a proximal and a distal balloon occlusion/aspiration device in patients undergoing PCI of SVGs. Finally, in the CANARY trial151, patients undergoing PCI of native coronary arteries under protection from a filter device had no benefit in terms of postprocedural troponin or creatine kinase levels. Therefore, evidence for the use of a protection device exists only in PCI of SVGs and essentially comes only from the results of the SAFER trial.

a | Angiography of a saphenous vein graft (sternal wires from earlier coronary artery bypass graft surgery), with a stent balloon and a distal filter device. b | A photograph of the device with the captured debris. c | Haematoxylin and eosin-stained histology of particulate debris from the aspirate with amorphic lipid substance (ALS), a foam cell (FC), cholesterol crystals (arrows) and a thrombus (T). Parts a and b courtesy of Michael Haude (Rheinlandklinikum Neuss, Germany). Part c adapted with permission from ref.235, Springer Nature Limited.
Several small, randomized, controlled clinical trials in patients with STEMI undergoing primary PCI have demonstrated the benefits of manual or device-based thrombus aspiration in terms of attenuated coronary microvascular and myocardial injury and clinical outcome152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167 (Table 1). In the large TOTAL trial168 of patients with STEMI, investigators reported better ST-segment resolution and less distal embolization on angiography from PCI with manual thrombus aspiration versus primary PCI alone. However, clinical outcome (cardiovascular death, re-infarction, cardiogenic shock or NYHA class IV heart failure) was not significantly improved, and a safety signal for stroke was reported with the use of thrombus aspiration168. Patients with STEMI who presented late (≥12 and ≤48 h) after symptom onset also did not derive a benefit from thrombus aspiration169. Therefore, in the 2018 European Society of Cardiology guidelines for the management of acute MI, routine use of thrombus aspiration is not recommended but can be considered in cases of large thrombus burden106. By contrast, in patients with STEMI and culprit lesions that are particularly vulnerable to rupture (defined on IVUS), the combination of thrombus aspiration with a distal filter could be protective for the coronary microcirculation and improve clinical outcome170. In the TATORT-NSTEMI trial171, patients with NSTEMI and thrombus-containing culprit lesions had no better TIMI flow or reduced enzymatic infarct size with thrombus aspiration than with standard PCI, but a reduction in the primary composite end point of all-cause death, re-infarction and new heart failure with thrombus aspiration was reported. No guideline recommendation exists for the use of thrombus aspiration in patients undergoing PCI for NSTEMI but the positive clinical outcome data from the TATORT-NSTEMI trial171 suggest a greater potential for thrombus aspiration in NSTEMI than STEMI, indicating that coronary microembolization could have a greater role in NSTEMI than in STEMI.
Analysis of coronary aspirates
Filter and aspiration devices are used not only to protect the coronary microcirculation during PCI but also to retrieve particulate debris and soluble factors for analysis172. Debris consists of plaque material, including foam cells114 and cholesterol crystals173, and thrombus material rich in platelets and fibrin146,174,175. The aspirate from stented native coronary arteries contains less particulate debris113 and the retrieved embolic material in native coronary arteries is smaller in volume and particle size174,175 than that from SVGs (plaque volume 135 ± 16 versus 176 ± 19 mm3; particulate debris 66 ± 10 versus 146 ± 23 mg)113. In other studies, patients with diabetes111 or chronic kidney disease112 had more particulate debris (and more calcium for the latter group of patients112) in the aspirate from their stented SVGs than patients without these conditions. The coronary aspirate from native coronary arteries and SVGs can also contain microparticles derived from platelets and the endothelium, with no significant difference in their concentration between native coronary arteries and SVGs176. In patients undergoing PCI for STEMI, increased numbers (as compared with peripheral arterial blood) of neutrophil–platelet aggregates were retrieved by thrombus aspiration177. Notably, thrombi also contained high levels of neutrophil extracellular traps (NETs), which correlated with infarct size as determined by CK-MB levels and MRI177. Again, in patients undergoing PCI for STEMI, increased levels of double-stranded DNA (as a surrogate for NETs) and IL-6 were retrieved by thromboaspiration from the culprit site and correlated with coronary microvascular obstruction on MRI 4 ± 2 days later178.
Soluble substances in the aspirate from stented native coronary arteries or SVGs have been determined biochemically or by functional bioassays. An increased release of tissue factor113,173,179, endothelin113,179, serotonin179,180, thromboxane113,180 and TNF113,120,180 has been reported from the aspirate of stented coronary arteries and SVGs. The level of endothelin was greater in aspirate from native coronary arteries than from SVGs, with no significant differences in the other soluble substances between the two types of vessel113. Aspiration from ruptured atherosclerotic plaque retrieved from patients undergoing primary PCI also had increased concentrations of IL-6 and serum amyloid181. In functional bioassays using rat isolated mesenteric arteries, the aspirate from stented SVGs induced profound vasoconstriction180,182, which was mediated by serotonin and thromboxane and facilitated by the endothelial dysfunction caused by TNF180,182. Accordingly, blockade of 5-HT (serotonin) and thromboxane receptors abrogated the intense vasoconstriction, as did more unspecific vasodilatation by verapamil and less so by nitroprusside180. In patients with severely stenotic SVGs, the release of TNF into SVG aspirate correlated with a reduction in plaque volume (measured by IVUS) and with the development of restenosis (measured by angiography) after 6 months120 and a correlation between TNF release and restenosis was also seen in patients with diabetes111.
In the OPTICO-ACS trial183, 170 consecutive patients undergoing PCI for an acute coronary syndrome had an increased concentration of CD8+ lymphocytes and cytotoxic effector molecules (granzyme A, granulysin and perforin) and a lower concentration of TNF in the aspirate distal to the culprit lesion when characterized by OCT as plaque erosion than when characterized as plaque rupture.
Collectively, these data from analyses of coronary aspirate support the notion that active prothrombotic, pro-inflammatory and vasoconstrictor substances are a major cause of microvascular and myocardial injury from microembolization. The exact cellular (vascular and inflammatory cells within the atherosclerotic plaque, intravascular cells in the thrombotic material) and biochemical sources of these substances are still unclear. Given that the myocardial perfusion territory of a vessel with physical obstruction by atherosclerotic debris can probably not be rescued from infarction, greater attention should be directed to the pathogenetic soluble factors and their pharmacological antagonism.
Prevention and treatment
Herrmann et al. were the first to report that patients who were statin naive undergoing elective PCI had greater increases in postprocedural serum creatine kinase levels than patients treated with statins184, and this finding was subsequently confirmed185. Whether long-term statin treatment improved plaque stability186 and prevented coronary microembolization or instead attenuated myocardial injury from coronary microembolization was unclear. Indeed, acute pretreatment with statins of patients who were statin naive before elective PCI also decreased postprocedural serum creatine kinase and troponin elevations187,188. Attenuation of intercellular adhesion molecule 1 (ICAM1) and E-selectin concentrations in the plasma reflected reduced endothelial inflammation but, again, whether this finding reflected proximal coronary plaque stabilization or reduced distal microvascular and myocardial injury was uncertain189. In any event, acute pretreatment with statins just before PCI also reduced the rate of postprocedural MACE190. The observation of reduced release of serum creatine kinase and troponin and a reduced incidence of MACE with statin reload a few hours before elective PCI191,192 supports the notion that statins also attenuate distal microvascular and myocardial injury. Less coronary microvascular and myocardial injury was also seen in mouse isolated hearts with ischaemia–reperfusion193,194 and in anaesthetized pigs with coronary occlusion and reperfusion195.
Antiplatelet and antithrombotic agents are routinely used before any cardiac catheterization and PCI procedure and are standard and guideline-recommended treatments for any acute coronary syndrome196,197. Therefore, evaluating the effect of antiplatelet agents on periprocedural coronary microembolization is impossible. However, in the ARMYDA-2 trial198, an increased loading dose of clopidogrel (600 mg) attenuated the increases in serum CK-MB, troponin I and myoglobin levels more than the standard dose (300 mg). Antiplatelet agents not only reduce intravascular formation and embolization of platelet aggregates but also have direct cardioprotective effects to attenuate myocardial injury199,200,201. However, not all antiplatelet agents are equal. In the ISAR-REACT 5 trial202, the regimen with prasugrel was superior to that with ticagrelor in reducing the composite end point of death, MI or stroke in patients with acute coronary syndromes after 1 year. The reasons for the difference between the two platelet inhibitors in their effects on coronary microembolization are not understood202. The INFUSE-AMI trial166 demonstrated that intracoronary abciximab (a glycoprotein IIb/IIIa inhibitor) reduced microvascular obstruction and infarct size in patients undergoing primary PCI for MI. Routine administration of glycoprotein IIb/IIIa inhibitors resulted in better TIMI flow, reduced ischaemic events and reduced mortality in patients with reperfused STEMI in a large meta-analysis of 8,585 patients203. However, most of the trials included in this analysis were conducted before the era of prasugrel and ticagrelor. By contrast, fibrinolysis with intracoronary infusion of alteplase did not attenuate microvascular obstruction in patients with STEMI undergoing PCI within 6 h of symptom onset204. Therefore, whether intracoronary fibrinolysis confers additional benefit to PCI and antiplatelet agents in the attenuation of coronary microvascular obstruction is still unclear and under investigation105.
The use of coronary vasodilators, including adenosine, nitroprusside and verapamil, in the attenuation of coronary microvascular obstruction has not been convincing. The effects of adenosine on microvascular obstruction per se205,206 and on clinical benefit31,105,207 are ambiguous. The ex vivo vasoconstrictor effects of coronary aspirate from SVGs, when analysed in isolated rat mesenteric arteries, were attenuated to a greater extent by verapamil than by nitroprusside, whereas adenosine had a negligible effect180. Verapamil also improved TIMI flow to a greater extent than nitroglycerin in patients undergoing PCI of degenerated SVGs208.
Anti-inflammatory interventions to prevent and treat coronary microembolization warrant further study. The CANTOS trial209 addressed the pathogenetic role of IL-1β in patients with previous MI and increased plasma C-reactive protein (CRP) concentrations. A reduced rate of cardiovascular events, including unstable angina requiring urgent revascularization, was reported with subcutaneous administration of the human monoclonal IL-1β-neutralizing antibody canakinumab209. Mechanistically, canakinumab might blunt the IL-1β-associated increase in NETs and levels of tissue factor found in patients with MI and increased plasma CRP concentration210. Patients who received canakinumab in the CANTOS trial had a reduced cardiovascular event rate associated with reductions in plasma IL-6 concentrations211. Long-term antagonism of IL-1β and IL-6 could stabilize epicardial atherosclerotic plaques and prevent their eventual rupture or erosion. In the more acute situation of an interventional procedure for a stable or acute coronary syndrome, in which coronary microembolization can occur and anti-inflammatory action in the coronary microcirculation might be important, the benefit of interleukin receptor antagonists and other anti-inflammatory drugs is less clear212. Indeed, the monoclonal IL-1β receptor antagonist anakinra given subcutaneously to patients within 48 h of PCI for NSTEMI reduced plasma CRP levels but did not provide clinical benefit213. By contrast, the IL-6 receptor antagonist tocilizumab given intravenously in patients undergoing PCI for NSTEMI reduced plasma concentrations of both CRP and troponin T (measured by high-sensitivity assay)214. In addition, reduced myocardial injury was seen in patients who were resuscitated after cardiac arrest215. Notably, in patients with STEMI, intravenous tocilizumab during PCI reduced microvascular obstruction and improved the salvage index on MRI 3–7 days after the procedure, supporting the notion that inflammatory microembolization is a contributing factor in microvascular obstruction216. However, the protection by tocilizumab was significant but small in magnitude, and no outcome data for tocilizumab in patients with NSTEMI or STEMI are yet available. In the SELECT-ACS trial217, preprocedural infusion of inclacumab (a monoclonal antibody to the endothelial adhesion molecule P-selectin) reduced circulating P-selectin and troponin I concentrations in patients with NSTEMI undergoing angiography and ad hoc PCI. Colchicine given orally to patients with acute coronary syndrome within hours to days of PCI218,219,220,221,222,223 reduced plasma CRP and interleukin concentrations in some218,219 but not all220,221,223 studies. Similarly, colchicine reduced biomarkers of myocardial injury in some218,223 but not all222 studies. Therefore, the clinical benefit of periprocedural anti-inflammatory treatment with PCI is still uncertain but these agents warrant further investigation. The prevention and treatment of coronary microembolization are summarized in Box 2.
Coronary microembolization in COVID-19
Coronavirus disease 2019 (COVID-19) is characterized by endothelial infection by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), with consequent endothelial injury224 and systematic thromboinflammation224,225 involving cytokine release, increased platelet activation and platelet–leukocyte aggregation226. The theory that IL-6 has a pathogenetic role in COVID-19 is supported by the finding that the IL-6 receptor antagonist tocilizumab (with aspirin or a P2Y12 inhibitor) blunted platelet activation after ex vivo exposure of platelets and leukocytes from healthy volunteers to COVID-19 plasma226. These data support the notion that inhibition of IL-6 not only stabilizes atherosclerotic plaques but also attenuates the downstream effects of platelet aggregation and inflammation. Thromboembolization, particularly in patients with pre-existing endothelial dysfunction, is a typical complication of COVID-19 and could also affect the coronary circulation, even in the absence of pre-existing coronary artery disease. Indeed, human coronary endothelial cells are particularly sensitive to SARS-CoV-2 infection227. Thrombi rich in fibrin, platelets and leukocytes in the coronary microcirculation were seen at autopsy in 10–20% of patients who had died from COVID-19 (refs228,229). In a pathology analysis, intracoronary microthrombi, which were particularly rich in fibrin and complement, were seen in 35% of patients who had died from COVID-19 and had evidence of myocardial necrosis, irrespective of whether or not they had pre-existing coronary artery disease230. In patients with STEMI with or without COVID-19, thromboaspiration retrieved thrombi containing fibrin, leukocytes and NETs231. In patients with COVID-19, no plaque fragments were observed and a greater density of NETs was present in the aspirate than in patients without COVID-19 (ref.231).
Conclusions
The secular trends in a shift from plaque rupture to plaque erosion as the trigger event17 and from STEMI to NSTEMI in clinical presentation18 suggest the increasing pathophysiological importance of distal embolization in the coronary circulation. Indeed, coronary microemboli are more frequently seen at autopsy after plaque erosion than after plaque rupture19. There is certainly no stoichiometric one-to-one relationship between plaque erosion, coronary microembolization and NSTEMI. Nevertheless, the observed association advocates for greater awareness of coronary microembolization in the assessment and treatment of these patients. Prevention of spontaneous coronary microembolization entails the use of all anti-atherosclerotic medications. Statins and inhibitors of platelet aggregation are effective for the prevention of coronary microembolization and to treat its consequences. Studies of protection devices to prevent procedural coronary microembolization have been largely disappointing, and their use is limited to situations of large atherothrombotic plaque burden in SVGs.
Future therapies should target the interaction between inflammation and platelet aggregation232 at the epicardial culprit lesion and in the coronary microcirculation. Further analyses of coronary aspirate or transcoronary gradients from patients with acute coronary syndromes183 could uncover more detail on the pathophysiological mechanisms of coronary microembolization and help to develop targeted therapies.
References
Falk, E. Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br. Heart J. 50, 127–134 (1983).
Davies, M. J. & Thomas, A. C. Plaque fissuring- the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina. Br. Heart J. 53, 363–373 (1985).
Falk, E. Unstable angina with fatal outcome: dynamic coronary thrombosis leading to infarction and/or sudden death. Autopsy evidence of recurrent mural thrombosis with peripheral embolization culminating in total vascular occlusion. Circulation 71, 699–708 (1985).
El-Maraghi, N. & Genton, E. The relevance of platelet and fibrin thrombembolism of the coronary microcirculation, with special reference to sudden cardiac death. Circulation 62, 936–944 (1980).
Davies, M. J., Thomas, A. C., Knapman, P. A. & Hangartner, J. R. Intramyocardial platelet aggregation in patients with unstable angina suffering sudden ischemic cardiac death. Circulation 73, 418–427 (1986).
Frink, R. J., Rooney, P. A., Trowbridge, J. O. & Rose, J. P. Coronary thrombosis and platelet/fibrin microemboli in death associated with acute myocardial infarction. Br. Heart J. 59, 196–200 (1988).
Folts, J. D., Gallagher, K. & Rowe, G. G. Blood flow reductions in stenosed canine coronary arteries: vasospasm or platelet aggregation? Circulation 65, 248–255 (1985).
Eidt, J. F. et al. Thrombin is an important mediator of platelet aggregation in stenosed canine coronary arteries with endothelial injury. J. Clin. Invest. 84, 18–27 (1989).
Golino, P., Buja, M., Sheng-Kun, Y., McNatt, J. & Willerson, J. T. Failure of nitroglycerin and diltiazem to reduce platelet-mediated vasoconstriction in dogs with coronary artery stenosis and endothelial injury: further evidence for thromboxane A2 and serotonin as mediators of coronary artery vasoconstriction in vivo. J. Am. Coll. Cardiol. 15, 718–726 (1990).
Folts, J. D. Deleterious hemodynamic effects of thrombotic/embolic materials on the distal myocardial vasculature. Cardiovasc. Res. 42, 6–7 (1999).
Erbel, R. & Heusch, G. Brief review: coronary microembolization. J. Am. Coll. Cardiol. 36, 22–24 (2000).
Topol, E. J. & Yadav, J. S. Recognition of the importance of embolization in atherosclerotic vascular disease. Circulation 101, 570–580 (2000).
Gregorini, L., Marco, J. & Heusch, G. Peri-interventional coronary vasomotion. J. Mol. Cell. Cardiol. 52, 883–889 (2012).
Libby, P., Pasterkamp, G., Crea, F. & Jang, I. K. Reassessing the mechanisms of acute coronary syndromes. Circ. Res. 124, 150–160 (2019).
Fahed, A. C. & Jang, I. K. Plaque erosion and acute coronary syndromes: phenotype, molecular characteristics and future directions. Nat. Rev. Cardiol. 18, 724–734 (2021).
Kolte, D., Libby, P. & Jang, I. K. New insights into plaque erosion as a mechanism of acute coronary syndromes. JAMA 325, 1043–1044 (2021).
Libby, P. The changing landscape of atherosclerosis. Nature 592, 524–533 (2021).
Puymirat, E. et al. Acute myocardial infarction: changes in patient characteristics, management, and 6-month outcomes over a period of 20 years in the FAST-MI Program (French Registry of Acute ST-Elevation or Non-ST-elevation Myocardial Infarction) 1995 to 2015. Circulation 14, 1908–1919 (2017).
Schwartz, R. S. et al. Microemboli and microvascular obstruction in acute coronary thrombosis and sudden coronary death relation to epicardial plaque histopathology. J. Am. Coll. Cardiol. 54, 2167–2173 (2009).
Partida, R. A., Libby, P., Crea, F. & Jang, I. K. Plaque erosion: a new in vivo diagnosis and a potential major shift in the management of patients with acute coronary syndromes. Eur. Heart J. 39, 2070–2076 (2018).
Collet, J. P. et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 42, 1289–1367 (2021).
Hamburger, W. W., Priest, W. S., Bettman, R. B. & Howard, H. C. Experimental coronary embolism. Am. J. Med. Sci. 171, 168–185 (1926).
Agress, C. M. et al. Protracted shock in the closed-chest dog following coronary embolization with graded microspheres. Am. J. Physiol. 170, 536–549 (1952).
Bing, R. J., Castellanos, A., Gradel, E., Lupton, C. & Siegel, A. Experimental myocardial infarction: circulatory biochemical and pathologic changes. Am. J. Med. Sci. 232, 533–554 (1956).
Marcus, E. et al. The production of myocardial infarction. Chronic coronary insufficiency and chronic coronary heart disease in the dog. Acta Cardiol. 13, 190–198 (1958).
Guzman, S. V., Swenson, E. & Jones, M. Intercoronary reflex. Demonstration by coronary angiography. Circ. Res. 10, 739–745 (1962).
Jacoby, J. A., Taylor, W. J., Smith, G. T., Gorlin, R. & Harken, D. E. A new therapeutic approach to acute coronary occlusion. Production of standardized coronary occlusion with microspheres. Am. J. Cardiol. 9, 60–73 (1962).
Herzberg, R. M., Rubio, R. & Berne, R. M. Coronary occlusion and embolization: effect on blood flow in adjacent arteries. Am. J. Physiol. 210, 169–175 (1966).
Monroe, R. G., LaFarge, C. G., Gamble, W. J., Kumar, A. E. & Manasek, F. J. Left ventricular performance and coronary flow after coronary embolization with plastic microspheres. J. Clin. Invest. 50, 1656–1665 (1971).
Heusch, G., Schulz, R., Baumgart, D., Haude, M. & Erbel, R. Coronary microembolization. Prog. Cardiovasc. Dis. 44, 217–230 (2001).
Heusch, G. et al. Coronary microembolization: from bedside to bench and back to bedside. Circulation 120, 1822–1836 (2009).
Franciosa, J. A., Heckel, R., Limas, C. & Cohn, J. N. Progressive myocardial dysfunction associated with increased vascular resistance. Am. J. Physiol. 239, H477–H482 (1980).
Sabbah, H. N. et al. A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. Am. J. Physiol. 29, H1379–H1384 (1991).
Eng, C., Cho, S., Factor, S. M., Sonnenblick, E. H. & Kirk, E. S. Myocardial micronecrosis produced by microsphere embolization. Role of an alpha-adrenergic tonic influence on the coronary microcirculation. Circ. Res. 54, 74–82 (1984).
Hori, M. et al. Role of adenosine in hyperemic response of coronary blood flow in microcirculation. Am. J. Physiol. 250, H509–H518 (1986).
Dörge, H. et al. Perfusion-contraction mismatch with coronary microvascular obstruction: role of inflammation. Am. J. Physiol. Heart Circ. Physiol. 279, H2587–H2592 (2000).
Grund, F., Sommerschild, H. T., Lyberg, T., Kirkeboen, K. & Ilebekk, A. Microembolization in pigs: effects on coronary blood flow and myocardial ischemic tolerance. Am. J. Physiol. 277, H533–H542 (1999).
Thielmann, M. et al. Myocardial dysfunction with coronary microembolization: signal transduction through a sequence of nitric oxide, tumor necrosis factor-a and sphingosine. Circ. Res. 90, 807–813 (2002).
Malyar, N. M., Lerman, L. O., Gössl, M., Beighley, P. E. & Ritman, E. L. Relation of nonperfused myocardial volume and surface area to left ventricular performance in coronary microembolization. Circulation 110, 1946–1952 (2004).
Carlsson, M., Wilson, M., Martin, A. J. & Saeed, M. Myocardial microinfarction after coronary microembolization in swine: MR imaging characterization. Radiology 250, 703–713 (2009).
Dispersyn, G. D. et al. Dissociation of cardiomyocyte apoptosis and dedifferentiation in infarct border zones. Eur. Heart J. 23, 849–857 (2002).
Cao, Y. Y. et al. Establishment of a novel mouse model of coronary microembolization. Chin. Med. J. 129, 2951–2957 (2016).
Li, L., Zhao, X., Lu, Y., Huang, W. & Wen, W. Altered expression of pro- and anti-inflammatory cytokines is associated with reduced cardiac function in rats following coronary microembolization. Mol. Cell. Biochem. 342, 183–190 (2010).
Li, S. et al. Blockade of NF-kappaB by pyrrolidine dithiocarbamate attenuates myocardial inflammatory response and ventricular dysfunction following coronary microembolization induced by homologous microthrombi in rats. Basic Res. Cardiol. 105, 139–150 (2009).
Bikou, O. et al. A novel large animal model of thrombogenic coronary microembolization. Front. Cardiovasc. Med. 6, 157 (2019).
Gallagher, K. P., Osakada, G., Kemper, W. S. & Ross, J. Jr Cyclical coronary flow reductions in conscious dogs equipped with ameroid constrictors to produce severe coronary narrowing. Basic Res. Cardiol. 80, 100–106 (1985).
Barrabés, J. A. et al. Dynamic intracoronary thrombosis does not cause significant downstream platelet embolization. Cardiovasc. Res. 47, 265–273 (2000).
Skyschally, A., Schulz, R., Erbel, R. & Heusch, G. Reduced coronary and inotropic reserves with coronary microembolization. Am. J. Physiol. Heart Circ. Physiol. 282, H611–H614 (2002).
Agress, C. M., Rosenburg, M. J., Binder, M. J., Schneiderman, A. & Clark, W. G. Blood volume changes in protracted shock resulting from experimental myocardial infarction. Am. J. Physiol. 166, 603–609 (1951).
Heusch, G. The regional myocardial flow-function relationship: a framework for an understanding of acute ischemia, hibernation, stunning and coronary microembolization. Circ. Res. 112, 1535–1537 (2013).
Arras, M. et al. Tumor necrosis factor-α is expressed by monocytes/macrophages following cardiac microembolization and is antagonized by cyclosporine. Basic Res. Cardiol. 93, 97–107 (1998).
Su, Q. et al. Effect of metoprolol on myocardial apoptosis and caspase-9 activation after coronary microembolization in rats. Exp. Clin. Cardiol. 18, 161–165 (2013).
Liu, Y. C., Li, L., Su, Q., Liu, T. & Tang, Z. L. Trimetazidine pretreatment inhibits myocardial apoptosis and improves cardiac function in a swine model of coronary microembolization. Cardiology 130, 130–136 (2015).
Mao, Q., Liang, X., Wu, Y. & Lu, Y. Nobiletin protects against myocardial injury and myocardial apoptosis following coronary microembolization via activating PI3K/Akt pathway in rats. Naunyn Schmiedebergs Arch. Pharmacol. 392, 1121–1130 (2019).
Mao, Q., Liang, X., Wu, Y. & Lu, Y. Resveratrol attenuates cardiomyocyte apoptosis in rats induced by coronary microembolization through SIRT1-mediated deacetylation of p53. J. Cardiovasc. Pharmacol. Ther. 24, 551–558 (2019).
Yuan, Y., Li, B., Peng, W. & Xu, Z. Protective effect of glycyrrhizin on coronary microembolization-induced myocardial dysfunction in rats. Pharmacol. Res. Perspect. 9, e00714 (2021).
Chen, Z.-Q. et al. miR-200a-3p attenuates coronary microembolization-induced myocardial injury in rats by inhibiting TXNIP/NLRP3-mediated cardiomyocyte pyroptosis. Front. Cardiovasc. Med. 8, 693257 (2021).
Zhou, Y. et al. Overexpression of lncRNA TUG1 alleviates NLRP3 inflammasome-mediated cardiomyocyte pyroptosis through targeting the miR-186-5p/XIAP axis in coronary microembolization-induced myocardial damage. Front. Immunol. 12, 637598 (2021).
Hori, M. et al. Role of oxygen-derived free radicals in myocardial edema and ischemia in coronary microvascular embolization. Circulation 84, 828–840 (1991).
Saeed, M. et al. Evaluation of the acute effects of distal coronary microembolization using multidetector computed tomography and magnetic resonance imaging. Magn. Reson. Med. 67, 1747–1757 (2012).
Dörge, H. et al. Coronary microembolization: the role of TNFa in contractile dysfunction. J. Mol. Cell. Cardiol. 34, 51–62 (2002).
Li, L., Li, D. H., Qu, N., Wen, W. M. & Huang, W. Q. Signaling pathway in coronary microembolization-induced rat myocardial inflammation and injury. Cardiology 117, 207–215 (2010).
Kluge, A. et al. Insulin-like growth factor I is involved in inflammation linked angiogenic processes after microembolisation in porcine heart. Cardiovasc. Res. 29, 404–415 (1995).
Chilian, W. M. et al. Microvascular occlusions promote coronary collateral growth. Am. J. Physiol. 258, H1103–H1111 (1990).
Nassenstein, K. et al. How much myocardial damage is necessary to enable detection of focal late gadolinium enhancement at cardiac MR imaging? Radiology 249, 829–835 (2008).
Breuckmann, F. et al. Systematic analysis of functional and structural changes after coronary microembolization: a cardiac magnetic resonance imaging study. JACC Imaging 2, 121–130 (2009).
Carlsson, M., Jablonowski, R., Martin, A. J., Ursell, P. C. & Saeed, M. Coronary microembolization causes long-term detrimental effects on regional left ventricular function. Scand. Cardiovasc. J. 45, 205–214 (2011).
Skyschally, A. et al. Glucocorticoid treatment prevents progressive myocardial dysfunction resulting from experimental coronary microembolization. Circulation 109, 2337–2342 (2004).
Canton, M. et al. Oxidative modification of tropomyosin and myocardial dysfunction following coronary microembolization. Eur. Heart J. 27, 875–881 (2006).
Skyschally, A. et al. Reduced calcium responsiveness characterizes contractile dysfunction following coronary microembolization. Basic Res. Cardiol. 103, 552–559 (2008).
Chen, Z. Q. et al. Breviscapine pretreatment inhibits myocardial inflammation and apoptosis in rats after coronary microembolization by activating the PI3K/Akt/GSK-3β signaling pathway. Drug Des. Devel. Ther. 15, 843–855 (2021).
Chen, Z. Q. et al. Puerarin pretreatment attenuates cardiomyocyte apoptosis induced by coronary microembolization in rats by activating the PI3K/Akt/GSK-3β signaling pathway. Korean J. Physiol. Pharmacol. 25, 147–157 (2021).
Zheng, J. et al. Nicorandil inhibits cardiomyocyte apoptosis and improves cardiac function by suppressing the HtrA2/XIAP/PARP signaling after coronary microembolization in rats. Pharmacol. Res. Perspect. 9, e00699 (2021).
Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 17, 773–789 (2020).
Schmermund, A. et al. Effects of acute and chronic angiotensin receptor blockade on myocardial vascular blood volume and perfusion in a pig model of coronary microembolization. Am. J. Hypertens. 13, 827–837 (2000).
Skyschally, A. et al. Bidirectional role of tumor necrosis factor-α in coronary microembolization: progressive contractile dysfunction versus delayed protection against infarction. Circ. Res. 100, 140–146 (2007).
Skyschally, A. et al. Coronary microembolization does not induce acute preconditioning against infarction in pigs – the role of adenosine. Cardiovasc. Res. 63, 313–322 (2004).
Skyschally, A. et al. Preinfarction angina: no interference of coronary microembolization with acute ischemic preconditioning. J. Mol. Cell. Cardiol. 39, 355–361 (2005).
Skyschally, A., Walter, B. & Heusch, G. Coronary microembolization during early reperfusion–infarct extension, but protection by ischemic postconditioning. Eur. Heart J. 34, 3314–3321 (2013).
Krug, A., du Mesnil de Rochemont, W. & Korb, G. Blood supply of the myocardium after temporary coronary occlusion. Circ. Res. 19, 57–62 (1966).
Kloner, R. A., Ganote, C. E. & Jennings, R. B. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J. Clin. Invest. 54, 1496–1508 (1974).
Heusch, G. The coronary circulation as a target of cardioprotection. Circ. Res. 118, 1643–1658 (2016).
Heusch, G. Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Res. Cardiol. 114, 45 (2019).
Kogaki, S. et al. Selectin on activated platelets enhances neutrophil endothelial adherence in myocardial reperfusion injury. Cardiovasc. Res. 43, 968–973 (1999).
Barrabes, J. A. et al. Antagonism of selectin function attenuates microvascular platelet deposition and platelet-mediated myocardial injury after transient ischemia. J. Am. Coll. Cardiol. 45, 293–299 (2005).
Barrabes, J. A. et al. Platelet deposition in remote cardiac regions after coronary occlusion. Eur. J. Clin. Invest. 37, 939–946 (2007).
Barrabes, J. A. et al. Microvascular thrombosis: an exciting but elusive therapeutic target in reperfused acute myocardial infarction. Cardiovasc. Hematol. Disord. Drug Targets 10, 273–283 (2010).
Driesen, R. B. et al. Histological correlate of a cardiac magnetic resonance imaged microvascular obstruction in a porcine model of ischemia-reperfusion. Cardiovasc. Pathol. 21, 129–131 (2012).
Zhu, H. et al. Prostaglandin E1 protects coronary microvascular function via the glycogen synthase kinase 3beta-mitochondrial permeability transition pore pathway in rat hearts subjected to sodium laurate-induced coronary microembolization. Am. J. Transl. Res. 9, 2520–2534 (2017).
Sharov, V. G. et al. Evidence of cardiocyte apoptosis in myocardium of dogs with chronic heart failure. Am. J. Pathol. 148, 141–149 (1997).
Sabbah, H. N. et al. Effects of long-term monotherapy with enalapril, metoprolol, and digoxin on the progression of left ventricular dysfunction and dilation in dogs with reduced ejection fraction. Circulation 89, 2852–2859 (1994).
He, K.-L. et al. Mechanisms of heart failure with well preserved ejection fraction in dogs following limited coronary microembolization. Cardiovasc. Res. 64, 72–83 (2004).
Ikram, H. et al. An ovine model of acute myocardial infarction and chronic left ventricular dysfunction. Angiology 48, 679–688 (1997).
George, I., Sabbah, H. N., Xu, K., Wang, N. & Wang, J. β-Adrenergic receptor blockade reduces endoplasmic reticulum stress and normalizes calcium handling in a coronary embolization model of heart failure in canines. Cardiovasc. Res. 91, 447–455 (2011).
Sabbah, H. N. et al. Chronic therapy with metoprolol attenuates cardiomyocyte apoptosis in dogs with heart failure. J. Am. Coll. Cardiol. 36, 1698–1705 (2000).
Suzuki, G., Young, R. F., Leiker, M. M. & Suzuki, T. Heart-derived stem cells in miniature swine with coronary microembolization: Novel ischemic cardiomyopathy model to assess the efficacy of cell-based therapy. Stem Cell Int. 2016, 6940195 (2016).
Leach, I. H., Blundell, J. W., Rowley, J. M. & Turner, D. R. Acute ischaemic lesions in death due to ischaemic heart disease. An autopsy study of 333 cases of out-of-hospital death. Eur. Heart J. 16, 1181–1185 (1995).
Haller, P. M., Beer, B. N., Tonkin, A. M., Blankenberg, S. & Neumann, J. T. Role of cardiac biomarkers in epidemiology and risk outcomes. Clin. Chem. 67, 96–106 (2021).
Park, K. C., Gaze, D. C., Collinson, P. O. & Marber, M. S. Cardiac troponins: from myocardial infarction to chronic disease. Cardiovasc. Res. 113, 1708–1718 (2017).
Raphael, C. E. et al. Coronary embolus: An underappreciated cause of acute coronary syndromes. JACC Cardiovasc. Interv. 11, 172–180 (2018).
Saber, R. S. et al. Coronary embolization after balloon angioplasty or thrombolytic therapy: an autopsy study of 32 cases. J. Am. Coll. Cardiol. 22, 1283–1288 (1993).
de Waha, S. et al. Relationship between microvascular obstruction and adverse events following primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: an individual patient data pooled analysis from seven randomized trials. Eur. Heart J. 38, 3502–3510 (2017).
Durante, A. et al. Identification of high-risk patients after ST-segment-elevation myocardial infarction: Comparison between angiographic and magnetic resonance parameters. Circ. Cardiovasc. Imaging 10, e005841 (2017).
Pezel, T. et al. Predictive value of early cardiac magnetic resonance imaging functional and geometric indexes for adverse left ventricular remodelling in patients with anterior ST-segment elevation myocardial infarction: a report from the CIRCUS study. Arch. Cardiovasc. Dis. 113, 710–720 (2020).
Niccoli, G. et al. Optimized treatment of ST-elevation myocardial infarction: the unmet need to target coronary microvascular obstruction as primary treatment goal to further improve prognosis. Circ. Res. 125, 245–258 (2019).
Ibanez, B. et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 39, 119–177 (2018).
Herrmann, J. Peri-procedural myocardial injury: 2005 update. Eur. Heart J. 26, 2493–2519 (2005).
Ganesha Babu, G., Malcolm, W. J., Yellon, D. M. & Hausenloy, D. J. Peri-procedural myocardial injury during percutaneous coronary intervention: an important target for cardioprotection. Eur. Heart J. 32, 23–31 (2011).
Thygesen, K. et al. Fourth universal definition of myocardial infarction (2018). Circulation 138, e618–e651 (2018).
Bulluck, H. et al. Prognostically relevant periprocedural myocardial injury and infarction associated with percutaneous coronary interventions: a consensus document of the ESC Working Group on Cellular Biology of the Heart and European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur. Heart J. 42, 2630–2642 (2021).
Baars, T. et al. Coronary aspirate TNFalpha reflects saphenous vein bypass graft restenosis risk in diabetic patients. Cardiovasc. Diabetol. 12, 12 (2013).
Baars, T. et al. Saphenous vein aorto-coronary graft atherosclerosis in patients with chronic kidney disease: more plaque calcification and necrosis, but less vasoconstrictor potential. Basic Res. Cardiol. 107, 303 (2012).
Kleinbongard, P. et al. Aspirate from human stented native coronary arteries vs. saphenous vein grafts: more endothelin but less particulate debris. Am. J. Physiol. Heart Circ. Physiol. 305, H1222–H1229 (2013).
Grube, E. et al. Prevention of distal embolization during coronary angioplasty in saphenous vein grafts and native vessels using porous filter protection. Circulation 104, 2436–2441 (2001).
Kini, A. et al. Incidence and mechanism of creatine kinase-MB enzyme elevation after coronary intervention with different devices. Catheter. Cardiovasc. Interv. 48, 123–129 (1999).
Hong, M. K. et al. Clinical significance of distal embolization after transluminal extraction atherectomy in diffusely diseased saphenous vein grafts. Am. Heart J. 127, 1496–1503 (1994).
Henriques, J. P. S. et al. Incidence and clinical significance of distal embolization during primary angioplasty for acute myocardial infarction. Eur. Heart J. 23, 1112–1117 (2002).
Boese, D. et al. Impact of atherosclerotic plaque composition on coronary microembolization during percutaneous coronary interventions. Basic Res. Cardiol. 103, 587–597 (2008).
Hildebrandt, H. A. et al. Fingerprints of periprocedural coronary microembolization on multimodality intravascular imaging. Herz 39, 115–118 (2013).
Boese, D. et al. Release of TNF-a during stent implantation into saphenous vein aortocoronary bypass grafts and its relation to plaque extrusion and restenosis. Am. J. Physiol. Heart Circ. Physiol. 292, H2295–H2299 (2007).
Bahrmann, P. et al. Detection of coronary microembolization by Doppler ultrasound in patients with stable angina pectoris undergoing elective percutaneous coronary interventions. Circulation 115, 600–608 (2007).
Otto, S. et al. Microembolization and myonecrosis during elective percutaneous coronary interventions in diabetic patients: an intracoronary Doppler ultrasound study with 2-year clinical follow-up. Basic Res. Cardiol. 107, 289 (2012).
Hori, M. et al. Adenosine-induced hyperemia attenuates myocardial ischemia in coronary microembolization in dogs. Am. J. Physiol. 257, H244–H251 (1989).
Nanto, S. et al. Temporal increase in resting coronary blood flow causes an impairment of coronary flow reserve after coronary angioplasty. Am. Heart J. 123, 28–36 (1992).
Herrmann, J. et al. Abnormal coronary flow velocity reserve following coronary intervention is associated with cardiac marker elevation. Circulation 103, 2339–2345 (2001).
Selvanayagam, J. B. et al. Troponin elavation after percutaneous coronary intervention directly represents the extent of irreversible myocardial injury. Insights of cardiovascular magnetic resonance imaging. Circulation 111, 127–1032 (2005).
Porto, I. et al. Plaque volume and occurence and location of periprocedural myocardial necrosis after percutaneous coronary intervention. Insights from delayed-enhancement magnetic resonance imaging, thrombolysis in myocardial infarction myocardial perfusion grade analysis, and intravascular ultrasound. Circulation 114, 662–669 (2006).
Selvanayagam, J. B. et al. Effect of distal embolization on myocardial perfusion reserve after percutaneous coronary intervention: a quantitative magnetic resonance perfusion study. Circulation 116, 1458–1464 (2007).
Rahimi, K. et al. Prognostic value of coronary revascularisation-related myocardial injury: a cardiac magnetic resonance imaging study. Heart 95, 1937–1943 (2009).
Davies, W. R. et al. Remote ischemic preconditioning improves outcome at 6 years after elective percutaneous coronary intervention: The CRISP stent trial long-term follow-up. Circ. Cardiovasc. Interv. 6, 246–251 (2013).
Luo, S. J. et al. Remote ischemic preconditioning reduces myocardial injury in patients undergoing coronary stent implantation. Can. J. Cardiol. 29, 1084–1089 (2013).
Zografos, T. A., Katritsis, G. D. & Katritsis, D. G. Remote ischemic preconditioning reduces peri-procedural myocardial injury in elective percutaneous coronary intervention: a meta-analysis. Int. J. Cardiol. 173, 530–532 (2014).
Pei, H. et al. Remote ischemic preconditioning reduces perioperative cardiac and renal events in patients undergoing elective coronary intervention: a meta-analysis of 11 randomized trials. PLoS One 9, e115500 (2014).
Prasad, A. et al. Remote ischemic preconditioning immediately before percutaneous coronary intervention does not impact myocardial necrosis, inflammatory response, and circulating endothelial progenitor cell counts: a single center randomized sham controlled trial. Catheter. Cardiovasc. Interv. 81, 930–936 (2013).
Skyschally, A. et al. Across-species transfer of protection by remote ischemic preconditioning with species-specific myocardial signal transduction by reperfusion injury salvage kinase and survival activating factor enhancement pathways. Circ. Res. 117, 279–288 (2015).
Lieder, H. R. et al. Vago-splenic axis in signal transduction of remote ischemic preconditioning in pigs and rats. Circ. Res. 123, 1152–1163 (2018).
Crimi, G. et al. Remote ischemic post-conditioning of the lower limb during primary percutaneous coronary intervention safely reduces enzymatic infarct size in anterior myocardial infarction: a randomized controlled trial. JACC Cardiovasc. Interv. 6, 1055–1063 (2013).
Eitel, I. et al. Cardioprotection by combined intrahospital remote ischaemic perconditioning and postconditioning in ST-elevation myocardial infarction: the randomized LIPSIA CONDITIONING trial. Eur. Heart J. 36, 3049–3057 (2015).
White, S. K. et al. Remote ischemic conditioning reduces myocardial infarct size and edema in patients with ST-segment elevation myocardial infarction. JACC Cardiovasc. Interv. 8, 178–188 (2015).
Liu, Z., Zhao, L., Hong, D. & Gao, J. Remote ischaemic preconditioning reduces myocardial ischaemic reperfusion injury in patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention. Acta Cardiol. 71, 596–603 (2016).
Cuisset, T. et al. Direct stenting for stable angina pectoris is associated with reduced periprocedural microcirculatory injury compared with stenting after pre-dilation. J. Am. Coll. Cardiol. 51, 1060–1065 (2008).
Loubeyre, C. et al. A randomized comparison of direct stenting with conventional stent implantation in selected patients with acute myocardial infarction. J. Am. Coll. Cardiol. 39, 15–21 (2002).
Saad, M. et al. Impact of direct stenting on myocardial injury assessed by cardiac magnetic resonance imaging and prognosis in ST-elevation myocardial infarction. Int. J. Cardiol. 283, 88–92 (2019).
Popma, J. J. et al. Initial clinical experience with distal protection using the FilterWire in patients undergoing coronary artery and saphenous vein graft percutaneous intervention. Catheter. Cardiovasc. Interv. 57, 125–134 (2002).
Angelini, A. et al. Distal protection with a filter device during coronary stenting in patients with stable and unstable angina. Circulation 110, 515–521 (2004).
Limbruno, U. et al. Distal embolization during primary angioplasty: histopathologic features and predictability. Am. Heart J. 150, 102–108 (2005).
Baim, D. S. et al. Randomized trial of a distal embolic protection device during percutaneous intervention of saphenous vein aorto-coronary bypass grafts. Circulation 105, 1285–1290 (2002).
Stone, G. W. et al. Randomized comparison of distal protection with a filter-based catheter and a balloon occlusion and aspiration system during percutaneous intervention of diseased saphenous vein aorto-coronary bypass grafts. Circulation 108, 548–553 (2003).
Kereiakes, D. J. et al. A novel filter-based distal embolic protection device for percutaneous intervention of saphenous vein graft lesions. JACC Cardiovasc. Interv. 1, 248–257 (2008).
Mauri, L. et al. The PROXIMAL trial: proximal protection during saphenous vein graft intervention using the Proxis Embolic Protection System: a randomized, prospective, multicenter clinical trial. J. Am. Coll. Cardiol. 50, 1442–1449 (2007).
Stone, G. W. et al. Plaque characterization to inform the prediction and prevention of periprocedural myocardial infarction during percutaneous coronary intervention: the CANARY Trial (Coronary Assessment by Near-infrared of Atherosclerotic Rupture-prone Yellow). JACC Cardiovasc. Interv. 8, 927–936 (2015).
Burzotta, F. et al. Manual thrombus-aspiration improves myocardial reperfusion. The randomized evaluation of the effect of mechanical reduction of distal embolization by thrombus-aspiration in primary and rescue angioplasty (REMEDIA) trial. J. Am. Coll. Cardiol. 46, 371–376 (2005).
Stone, G. W. et al. Distal microcirculatory protection during percutaneous coronary intervention in acute ST-segment elevation myocardial infarction: a randomized controlled trial. JAMA 293, 1063–1072 (2005).
Silva-Orrego, P. et al. Thrombus aspiration before primary angioplasty improves myocardial reperfusion in acute myocardial infarction: the DEAR-MI (Dethrombosis to Enhance Acute Reperfusion in Myocardial Infarction) study. J. Am. Coll. Cardiol. 48, 1552–1559 (2006).
Cura, F. A. et al. Protection of distal embolization in high-risk patients with acute ST-segment elevation myocardial infarction (PREMIAR). Am. J. Cardiol. 99, 357–363 (2007).
Muramatsu, T. et al. Comparison of myocardial perfusion by distal protection before and after primary stenting for acute myocardial infarction: angiographic and clinical results of a randomized controlled trial. Catheter. Cardiovasc. Interv. 70, 677–682 (2007).
Kelbaek, H. et al. Randomized comparison of distal protection versus conventional treatment in primary percutaneous coronary intervention: the drug elution and distal protection in ST-elevation myocardial infarction (DEDICATION) trial. J. Am. Coll. Cardiol. 51, 899–905 (2008).
Svilaas, T. et al. Thrombus aspiration during primary percutaneous coronary intervention. N. Engl. J. Med. 358, 557–567 (2008).
Tahk, S. J. et al. Distal protection device protects microvascular integrity during primary percutaneous intervention in acute myocardial infarction: a prospective, randomized, multicenter trial. Int. J. Cardiol. 123, 162–168 (2008).
Haeck, J. D. E. et al. Randomized comparison of primary percutaneous coronary intervention with combined proximal embolic protection and thrombus aspiration versus primary percutaneous coronary intervention alone in ST-segment elevation myocardial infarction. The PREPARE (PRoximal Embolic Protection in Acute myocardial infarction and Resolution of ST-Elevation) Study. JACC Cardiovasc. Interv. 2, 934–943 (2009).
Sardella, G. et al. Thrombus aspiration during primary percutaneous coronary intervention improves myocardial reperfusion and reduces infarct size: the EXPIRA (thrombectomy with export catheter in infarct-related artery during primary percutaneous coronary intervention) prospective, randomized trial. J. Am. Coll. Cardiol. 53, 309–315 (2009).
Dudek, D. et al. Thrombus aspiration followed by direct stenting: a novel strategy of primary percutaneous coronary intervention in ST-segment elevation myocardial infarction. Results of the Polish-Italian-Hungarian randomized ThrombEctomy Trial (PIHRATE Trial). Am. Heart J. 160, 966–972 (2010).
Migliorini, A. et al. Comparison of angioJet rheolytic thrombectomy before direct infarct artery stenting with direct stenting alone in patients with acute myocardial infarction. The JETSTENT trial. J. Am. Coll. Cardiol. 56, 1298–1306 (2010).
Ciszewski, M. et al. Aspiration coronary thrombectomy for acute myocardial infarction increases myocardial salvage: single center randomized study. Catheter. Cardiovasc. Interv. 78, 523–531 (2011).
De Carlo, M. et al. A prospective randomized trial of thrombectomy versus no thrombectomy in patients with ST-segment elevation myocardial infarction and thrombus-rich lesions: MUSTELA (multidevice thrombectomy in acute ST-segment elevation acute myocardial infarction) trial. JACC Cardiovasc. Interv. 5, 1223–1230 (2012).
Stone, G. W. et al. Intracoronary abciximab and aspiration thrombectomy in patients with large anterior myocardial infarction: the INFUSE-AMI randomized trial. JAMA 307, 1817–1826 (2012).
Fröbert, O. et al. Thrombus aspiration during ST-segment elevation myocardial infarction. N. Engl. J. Med. 369, 1587–1597 (2013).
Jolly, S. S. et al. Randomized trial of primary PCI with or without routine manual thrombectomy. N. Engl. J. Med. 372, 1389–1398 (2015).
Desch, S. et al. Thrombus aspiration in patients with ST-segment elevation myocardial infarction presenting late after symptom onset. JACC Cardiovasc. Interv. 9, 113–122 (2016).
Hibi, K. et al. A randomized study of distal filter protection versus conventional treatment during percutaneous coronary intervention in patients with attenuated plaque identified by intravascular ultrasound. JACC Cardiovasc. Interv. 11, 1545–1555 (2018).
Feistritzer, H. J. et al. Long-term outcome after thrombus aspiration in non-ST-elevation myocardial infarction: results from the TATORT-NSTEMI trial: thrombus aspiration in acute myocardial infarction. Clin. Res. Cardiol. 109, 1223–1231 (2020).
Kleinbongard, P. et al. Lessons from human coronary aspirate. J. Mol. Cell. Cardiol. 52, 890–896 (2012).
Bonderman, D. et al. Coronary no-reflow is caused by shedding of active tissue factor from dissected atherosclerotic plaque. Blood 99, 2794–2800 (2002).
Rogers, C. et al. Embolic protection with filtering or occlusion balloons during saphenous vein graft stenting retrieves identical volumes and sizes of particulate debris. Circulation 109, 1735–1740 (2004).
Quan, V. H. et al. Morphometric analysis of particulate debris extracted by four different embolic protection devices from coronary arteries, aortocoronary saphenous vein conduits, and carotid arteries. Am. J. Cardiol. 95, 1415–1419 (2005).
Horn, P. et al. Release of intracoronary microparticles during stent implantation into stable atherosclerotic lesions under protection with an aspiration device. PLoS One 10, e0124904 (2015).
Mangold, A. et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 116, 1182–1192 (2015).
Mangold, A. et al. Culprit site extracellular DNA and microvascular obstruction in ST-elevation myocardial infarction. Cardiovasc. Res. https://doi.org/10.1093/cvr/cvab217 (2021).
Salloum, J., Tharpe, C., Vaughan, D. & Zhao, D. X. Release and elimination of soluble vasoactive factors during percutaneous coronary intervention of saphenous vein grafts: analysis using the PercuSurge GuardWire distal protection device. J. Invasive Cardiol. 17, 575–579 (2005).
Kleinbongard, P. et al. Vasoconstrictor potential of coronary aspirate from patients undergoing stenting of saphenous vein aortocoronary bypass grafts and its pharmacological attenuation. Circ. Res. 108, 344–352 (2011).
Maier, W. et al. Inflammatory markers at the site of ruptured plaque in acute myocardial infarction. Locally increased interleukin-6 and serum amyloid A but decreased c-reactive protein. Circulation 111, 1355–1361 (2005).
Leineweber, K. et al. Intense vasoconstriction in response to aspirate from stented saphenous vein aortocoronary bypass grafts. J. Am. Coll. Cardiol. 47, 981–986 (2006).
Leistner, D. M. et al. Differential immunological signature at the culprit site distinguishes acute coronary syndrome with intact from acute coronary syndrome with ruptured fibrous cap: results from the prospective translational OPTICO-ACS study. Eur. Heart J. 41, 3549–3560 (2020).
Herrmann, J. et al. Preprocedural statin medication reduces the extent of periprocedural non-Q-wave myocardial infarction. Circulation 106, 2180–2183 (2002).
Mulukutla, S. R. et al. Effect of statin therapy prior to elective percutaneous coronary intervention on frequency of periprocedural myocardial injury. Am. J. Cardiol. 94, 1363–1366 (2004).
Sukhova, G. K., Williams, K. & Libby, P. Statins reduce inflammation in atheroma of nonhuman primates independent of effects on serum cholesterol. Arterioscler. Thromb. Vasc. Biol. 22, 1452–1458 (2002).
Briguori, C. et al. Statin administration before percutaneous coronary intervention: impact on periprocedural myocardial infarction. Eur. Heart J. 25, 1822–1828 (2004).
Pasceri, V. et al. Randomized trial of atorvastatin for reduction of myocardial damage during coronary intervention. Results from the ARMYDA (atorvastatin for reduction of myocardial damage during angioplasty) study. Circulation 110, 674–678 (2004).
Patti, G. et al. Protection from procedural myocardial injury by atorvastatin is associated with lower levels of adhesion molecules after percutaneous coronary intervention: results from the ARMYDA-CAMs (atorvastatin for reduction of myocardial damage during angioplasty-cell adhesion molecules) substudy. J. Am. Coll. Cardiol. 48, 1560–1566 (2006).
Patti, G. et al. Atorvastatin pretreatment improves outcomes in patients with acute coronary syndromes undergoing early percutaneous coronary intervention: results of the ARMYDA-ACS randomized trial. J. Am. Coll. Cardiol. 49, 1272–1278 (2007).
Di Sciascio, G. et al. Efficacy of atorvastatin reload in patients on chronic statin therapy undergoing percutaneous coronary intervention. Results of the ARMYDA-RECAPTURE (atorvastatin for reduction of myocardial damage during angioplasty) randomized trial. J. Am. Coll. Cardiol. 54, 558–565 (2009).
Patti, G. et al. Clinical benefit of statin pretreatment in patients undergoing percutaneous coronary intervention: a collaborative patient-level meta-analysis of 13 randomized studies. Circulation 123, 1622–1632 (2011).
Bell, R. M. & Yellon, D. M. Atorvastatin, administered at the onset of reperfusion, and independent of lipid lowering, protects the myocardium by up-regulating a pro-survival pathway. J. Am. Coll. Cardiol. 41, 508–515 (2003).
Efthymiou, C. A., Mocanu, M. M. & Yellon, D. M. Atorvastatin and myocardial reperfusion injury. New pleiotropic effect implicating multiple prosurvival signaling. J. Cardiovasc. Pharmacol. 45, 247–252 (2005).
Mendieta, G. et al. Intravenous statin administration during ischemia exerts cardioprotective effects. J. Am. Coll. Cardiol. 74, 475–477 (2019).
Valgimigli, M. et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease developed in collaboration with EACTS: the Task Force for dual antiplatelet therapy in coronary artery disease of the European Society of Cardiology (ESC) and of the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 39, 213–260 (2018).
Kamran, H. et al. Oral antiplatelet therapy after acute coronary syndrome: a review. JAMA 325, 1545–1555 (2021).
Patti, G. et al. Randomized trial of high loading dose of clopidogrel for reduction of periprocedural myocardial infarction in patients undergoing coronary intervention. Results from the ARMYDA-2 (antiplatelet therapy for reduction of myocardial damage during angioplasty) study. Circulation 111, 2099–2106 (2005).
Yang, X. M. et al. Platelet P2Y(1)(2) blockers confer direct postconditioning-like protection in reperfused rabbit hearts. J. Cardiovasc. Pharmacol. Ther. 18, 251–262 (2013).
Cohen, M. V. & Downey, J. M. What are optimal P2Y12 inhibitor and schedule of administration in patients with acute coronary syndrome? J. Cardiovasc. Pharmacol. Ther. 25, 121–130 (2020).
He, Z., Davidson, S. M. & Yellon, D. M. The importance of clinically relevant background therapy in cardioprotective studies. Basic Res. Cardiol. 115, 69 (2020).
Schuepke, S. et al. Ticagrelor or prasugrel in patients with acute coronary syndromes. N. Engl. J. Med. 381, 1524–1534 (2019).
Karathanos, A. et al. Routine glycoprotein IIb/IIIa Iinhibitor therapy in ST-segment elevation myocardial infarction: a meta-analysis. Can. J. Cardiol. 35, 1576–1588 (2019).
McCartney, P. J. et al. Effect of low-dose intracoronary alteplase during primary percutaneous coronary intervention on microvascular obstruction in patients with acute myocardial infarction: a randomized clinical trial. JAMA 321, 56–68 (2019).
Niccoli, G. et al. Open-label, randomized, placebo-controlled evaluation of intracoronary adenosine or nitroprusside after thrombus aspiration during primary percutaneous coronary intervention for the prevention of microvascular obstruction in acute myocardial infarction: the REOPEN-AMI study. JACC Cardiovasc. Interv. 6, 580–589 (2013).
Nazir, S. A. et al. Strategies to attenuate micro-vascular obstruction during P-PCI: the randomized reperfusion facilitated by local adjunctive therapy in ST-elevation myocardial infarction trial. Eur. Heart J. 37, 1910–1919 (2016).
Bulluck, H., Sirker, A., Loke, Y. K., Garcia-Dorado, D. & Hausenloy, D. J. Clinical benefit of adenosine as an adjunct to reperfusion in ST-elevation myocardial infarction patients: an updated meta-analysis of randomized controlled trials. Int. J. Cardiol. 202, 228–237 (2016).
Kaplan, B. M. et al. Treatment of no-reflow in degenerated saphenous vein graft interventions: comparison of intracoronary verapamil and nitroglycerin. Cathet. Cardiovasc. Diagn. 39, 113–118 (1996).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Liberale, L. et al. Interleukin-1beta mediates arterial thrombus formation via NET-associated tissue factor. J. Clin. Med. 8, 2072 (2019).
Ridker, P. M. et al. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Heart J. 39, 3499–3507 (2018).
Cao, D., Chiarito, M. & Mehran, R. Treating inflammation prior to percutaneous coronary intervention: does the heart care? Circ. Cardiovasc. Interv. 13, e009127 (2020).
Morton, A. C. et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA heart study. Eur. Heart J. 36, 377–384 (2015).
Kleveland, O. et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 37, 2406–2413 (2016).
Meyer, M. A. S. et al. Treatment effects of interleukin-6 receptor antibodies for modulating the systemic inflammatory response after out-of-hospital cardiac arrest (The IMICA Trial): a double-blinded, placebo-controlled, single-center, randomized, clinical trial. Circulation 143, 1841–1851 (2021).
Broch, K. et al. Randomized trial of interleukin-6 receptor inhibition in patients with acute ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 77, 1845–1855 (2021).
Tardif, J. C. et al. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-ST-segment elevation myocardial infarction: results of the SELECT-ACS trial. J. Am. Coll. Cardiol. 61, 2048–2055 (2013).
Deftereos, S. et al. Anti-inflammatory treatment with colchicine in acute myocardial infarction: a pilot study. Circulation 132, 1395–1403 (2015).
Martinez, G. J. et al. Colchicine acutely suppresses local cardiac production of inflammatory cytokines in patients with an acute coronary syndrome. J. Am. Heart Assoc. 4, e002128 (2015).
Akodad, M. et al. COLIN trial: value of colchicine in the treatment of patients with acute myocardial infarction and inflammatory response. Arch. Cardiovasc. Dis. 110, 395–402 (2017).
Hennessy, T. et al. The low dose colchicine after myocardial infarction (LoDoCo-MI) study: a pilot randomized placebo controlled trial of colchicine following acute myocardial infarction. Am. Heart J. 215, 62–69 (2019).
Shah, B. et al. Effects of ccute colchicine administration prior to percutaneous coronary intervention: COLCHICINE-PCI randomized trial. Circ. Cardiovasc. Interv. 13, e008717 (2020).
Cole, J. et al. Colchicine to prevent periprocedural myocardial injury in percutaneous coronary intervention: the COPE-PCI pilot trial. Circ. Cardiovasc. Interv. 14, e009992 (2021).
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418 (2020).
Gu, S. X. et al. Thrombocytopathy and endotheliopathy: crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 18, 194–209 (2021).
Canzano, P. et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic Transl. Sci. 6, 202–218 (2021).
Wagner, J. U. G. et al. Increased susceptibility of human endothelial cells to infections by SARS-CoV-2 variants. Basic Res. Cardiol. 116, 42 (2021).
Basso, C. et al. Pathological features of COVID-19-associated myocardial injury: a multicentre cardiovascular pathology study. Eur. Heart J. 41, 3827–3835 (2020).
Halushka, M. K. & Vander Heide, R. S. Myocarditis is rare in COVID-19 autopsies: cardiovascular findings across 277 postmortem examinations. Cardiovasc. Pathol. 50, 107300 (2021).
Pellegrini, D. et al. Microthrombi as a major cause of cardiac injury in COVID-19: a pathologic study. Circulation 143, 1031–1042 (2021).
Blasco, A. et al. Assessment of neutrophil extracellular traps in coronary thrombus of a case series of patients with COVID-19 and myocardial infarction. JAMA Cardiol. 6, 469–474 (2021).
Stark, K. & Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 18, 666–682 (2021).
Heusch, G. & Erbel, R. Koronare mikroembolisation. Dtsch. Ärztebl. 99, A2200–A2202 (2002).
Hildebrandt, H. A. et al. Quantification and characterization of released plaque material during bioresorbable vascular scaffold implantation into right coronary artery lesions by multimodality intracoronary imaging. EuroIntervention 12, 1481–1489 (2016).
Erbel, R. & Heusch, G. Coronary microembolization - its role in acute coronary syndromes and interventions. Herz 24, 558–575 (1999).
Acknowledgements
P.K. and G.H. receive support from the German Research Foundation (SFB 1116 B8) and the European Union COST ACTION (CA 16225). The authors are grateful to their former colleagues Raimund Erbel (University of Essen Medical School, Germany) and Michael Haude (Rheinlandklinikum Neuss, Germany) for excellent collaborations in this field.
Author information
Authors and Affiliations
Contributions
P.K. and G.H. researched data for the article and discussed its content. G.H. wrote the manuscript, and both authors reviewed and edited it before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Cardiology thanks Niels van Royen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kleinbongard, P., Heusch, G. A fresh look at coronary microembolization. Nat Rev Cardiol 19, 265–280 (2022). https://doi.org/10.1038/s41569-021-00632-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-021-00632-2
This article is cited by
-
Colchicine ameliorates myocardial injury induced by coronary microembolization through suppressing pyroptosis via the AMPK/SIRT1/NLRP3 signaling pathway
BMC Cardiovascular Disorders (2024)
-
Coronary microvascular obstruction and dysfunction in patients with acute myocardial infarction
Nature Reviews Cardiology (2024)
-
Thrombolysis in Stroke-Heart Syndrome: a useful tool for neurocardiac wellness?
Journal of Neurology (2024)
-
Pinacidil ameliorates cardiac microvascular ischemia–reperfusion injury by inhibiting chaperone-mediated autophagy of calreticulin
Basic Research in Cardiology (2024)
-
Macrophage profiling in atherosclerosis: understanding the unstable plaque
Basic Research in Cardiology (2024)
