
Abstract
The number of old people is rising worldwide, and advancing age is a major risk factor for atherosclerotic cardiovascular disease. However, the mechanisms underlying this phenomenon remain unclear. In this Review, we discuss vascular intrinsic and extrinsic mechanisms of how ageing influences the pathology of atherosclerosis. First, we focus on factors that are extrinsic to the vasculature. We discuss how ageing affects the development of myeloid cells leading to the expansion of certain myeloid cell clones and induces changes in myeloid cell functions that promote atherosclerosis via inflammation, including a potential role for IL-6. Next, we describe vascular intrinsic factors by which ageing promotes atherogenesis — in particular, the effects on mitochondrial function. Studies in mice and humans have shown that ageing leads to a decline in vascular mitochondrial function and impaired mitophagy. In mice, ageing is associated with an elevation in the levels of the inflammatory cytokine IL-6 in the aorta, which participates in a positive feedback loop with the impaired vascular mitochondrial function to accelerate atherogenesis. We speculate that vascular and myeloid cell ageing synergize, via IL-6 signalling, to accelerate atherosclerosis. Finally, we propose future avenues of clinical investigation and potential therapeutic approaches to reduce the burden of atherosclerosis in old people.
Key points
-
Ageing-related alterations in the bone marrow increase the phenomenon of clonal haematopoiesis of indeterminate potential (CHIP) and promote a skewing towards myeloid cell differentiation, both of which can accelerate atherosclerosis.
-
The increased risk of atherosclerotic cardiovascular diseases associated with the presence of CHIP might be mediated by IL-6 signalling and/or inflammasome activation.
-
Ageing is associated with a decline in mitochondrial function and an increase in IL-6 levels in the vasculature, and both effects probably accelerate atherosclerosis independently of chronic hyperlipidaemia.
-
The role of the vasculature and myeloid cells of the immune system in promoting age-related atherosclerosis might be mediated by shared inflammatory pathways, in particular IL-6 signalling.
Similar content being viewed by others

Introduction
The number of old people (aged >65 years) is rising worldwide, and cardiovascular diseases are the largest contributor to morbidity and mortality in this population1,2. Changes in diet and lifestyle contribute to the high cardiovascular morbidity and mortality in old individuals, but many biological processes that are altered with ageing also contribute to this increased cardiovascular risk. As a result, therapies for cardiovascular disease that are effective in young and middle-aged people might be less effective in older people. Additionally, novel therapies might be required to improve disease management specifically in old people. Deciphering the mechanisms by which ageing promotes atherosclerotic cardiovascular disease will be fundamental for the development of novel therapies to reduce the burden of atherosclerosis with ageing. The development of new therapies is especially relevant with the coronavirus disease 2019 (COVID-19) pandemic, because old people and particularly those with cardiovascular diseases are at a substantially higher risk of morbidity and death3,4.
In this Review, we describe two major areas by which ageing promotes atherosclerosis. First, we discuss age-related factors that are extrinsic to the vasculature, focusing on the effects of ageing on myeloid cells of the immune system. Age-related effects in the bone marrow skew the differentiation of haematopoietic cells towards the myeloid cell lineage. Ageing also promotes the generation of clones of haematopoietic cells without clear development of haematopoietic malignancy or other known clonal disorder, a phenomenon known as clonal haematopoiesis of indeterminate potential (CHIP)5,6,7,8. Clinical studies from the past decade have revealed that the presence of CHIP increases the risk of cardiovascular diseases6,7. Intriguingly, the increased risk of cardiovascular disease associated with the presence of CHIP is abrogated in patients with a loss-of-function mutation in IL6 (ref.9). However, the precise mechanisms by which CHIP promotes the development of cardiovascular diseases are yet to be fully clarified.
We next address vascular intrinsic factors, discussing how ageing impairs vascular bioenergetics by compromising mitochondrial function and how this alteration connects with inflammatory pathways within the vasculature to promote atherosclerosis10,11. We describe studies in mice and humans showing that, in the aorta, ageing impairs both mitochondrial function and the removal of damaged mitochondria (mitophagy)10,11. We describe experimental evidence reported in 2020 demonstrating that the age-mediated increase in the levels of the pleiotropic cytokine IL-6 in the aorta occurs in a positive feedback loop with vascular mitochondrial dysfunction and that these alterations promote atherosclerosis11.
The CANTOS study12 demonstrated that IL-1β blockade reduces the risk of recurrent cardiovascular events in patients aged >60 years. Importantly, the greatest benefit of IL-1β blockade was seen in patients who had low plasma IL-6 levels13. This pivotal clinical study indicates that chronic inflammation, potentially via IL-6 signalling, is a major contributor to age-related atherosclerosis. Given this observation, we speculate that increased atherosclerosis with ageing could result from a synergy between myeloid cells of the immune system and the vasculature via IL-6 signalling (Fig. 1). This mechanism is especially important because clinically-approved agents targeting this pathway (such as anti-IL-6 therapies) are already available and could reduce the risk of cardiovascular disease in old people. Finally, we propose that future experimental and clinical investigation will be required to determine the contribution of this inflammatory pathway in age-related atherosclerosis. We acknowledge that other inflammatory pathways and cytokines could contribute to age-related atherosclerosis, and the source of these cytokines (including IL-6) could be senescent adipocytes. A detailed discussion of the contribution of ageing to senescence and atherosclerosis has been published previously14.

Ageing promotes the development and progression of atherosclerosis through different mechanisms, which might be related to age-induced elevations in circulating and intracellular IL-6 levels. During ageing, IL-6 signalling in bone marrow adipocytes increases. This increased IL-6 signalling might skew haematopoietic stem cells towards myeloid cell differentiation and increase the risk of mutations in genes encoding transcriptional regulators, such as TET2, that can result in the positive selection and expansion of clones of haematopoietic cells without clear development of malignancy or other known clonal disorder, a phenomenon known as clonal haematopoiesis of indeterminate potential (CHIP). Ageing can also have direct effects on haematopoietic stem cells (HSCs) that lead to CHIP. Clones of myeloid cells with a TET2 mutation show an increased production of IL-6 and IL-1β, which can contribute to accelerated atherosclerosis. Ageing can also have pro-atherogenic effects directly on the vasculature. Ageing is associated with an increase in the levels of IL-6, possibly mediated by increased production by vascular smooth muscle cells (VSMCs), and mitochondrial genomic instability and with a decline in mitochondrial function in the vasculature. The reduced mitochondrial function alters mitophagy and increases IL-6 levels, creating a positive feedback loop that accelerates atherogenesis. Vascular ageing also leads to the production of chemoattractants that increase myeloid cell recruitment into the arterial wall, further promoting atherosclerosis. Impaired mitochondrial function combined with reduced mitophagy might lead to increased levels of reactive oxygen species (ROS).
Ageing affects the immune system in complex ways (as reviewed previously15,16,17), and various components of the immune system contribute to atherosclerosis18,19. This Review focuses on clones of myeloid cells that increase with ageing and how these clones contribute to atherosclerosis. We do not describe how ageing affects other cells of the immune system, which has been reviewed previously (for example, B cells20, T cells21, eosinophils or dendritic cells22). In addition, we focus on vascular mitochondrial function and how mitochondrial dysfunction could influence inflammatory pathways within the vasculature. However, given that most of the available evidence indicates that oxidative stress is not a major driver of biological ageing23,24,25, and given the complex roles that oxidative stress has in atherosclerosis26, we do not describe in detail the contributions of oxidative stress in age-related atherosclerosis. Neither do we describe in detail how ageing affects other processes within the arterial wall, such as extracellular matrix remodelling or production of pro-fibrotic and pro-calcific factors, which can promote atherosclerosis indirectly via increasing arterial stiffness and hypertension27,28, and which have been previously reviewed29,30.
Vascular extrinsic mechanisms
Effects of ageing on myeloid cell production
Numerous subpopulations of immune cells of various lineages have been implicated in atherosclerosis, including macrophages31, dendritic cells32, T helper 1 (TH1) cells33 and B cells34 (reviewed previously18,35), all of which are affected by ageing. Immune cells are generated in the bone marrow via haematopoiesis from regenerative haematopoietic stem cells (HSCs)36,37. Monocytes, macrophages and neutrophils are derived from myeloid-biased HSCs. With ageing, although the absolute number of HSCs increases38, HSCs lose their regenerative capacity39,40. This loss of regenerative potential is accompanied by an expansion of the number of HSCs that are committed to the platelet (megakaryocytes) and myeloid lineages38,41. Competitive bone marrow transplantation studies in mice have demonstrated that aged HSCs have a reduced repopulation capacity, with an imbalance towards myeloid cell differentiation, compared with young HSCs38,42. Several major biological pathways contribute to ageing, including DNA damage, mitochondrial dysfunction, cell senescence, impaired autophagy, epigenetic alterations and gene transcription dysregulation25. Transcriptomic studies in mice have shown that with ageing, HSCs upregulate stress responses and inflammatory pathways and downregulate the expression of genes related to genetic stability43. With ageing, HSCs exhibit an increase in epigenetic dysregulation, specifically downregulation in chromatin remodelling and transcriptional silencing43, and increases in DNA methylation (as reviewed previously44). These epigenetic alterations are accompanied by functional defects in HSCs, including a reduction in HSC homing to the bone marrow and HSC proliferation43,45. Importantly, mutations in genes such as IDH2, which alter epigenetic regulation, lead to impairments in haematopoietic progenitors in mice46 and are associated with T cell lymphomas in humans47, a malignancy that increases with ageing. Although whether HSCs, or stem cells in general, undergo senescence is questionable48, the clearance of senescent cells improves HSC engraftment in bone marrow transplantation mouse models and reduces myeloid skewing49. Autophagy-deficient young mice have increased mitochondrial content and metabolism that lead to mitochondrial stress in HSCs compared with wild-type young mice50. These features are also observed in aged wild-type mice and are associated with a skewing towards the myeloid lineage and a reduced proliferative capacity of HSCs50,51. Loss of microRNA-146a (miR-146a) in HSCs with ageing also promotes a myeloid bias52. Furthermore, myeloid cells derived from miR-146a-deficient HSCs have elevated levels of both IL-6 and tumour necrosis factor (TNF)52, which connects altered regulation of transcription in HSCs to inflammation. Overall, these studies indicate that ageing has effects on HSCs via several complex pathways that lead to reduced HSC function.
Ageing also alters haematopoiesis by influencing the bone marrow niche independently of the direct effects on HSCs. The bone marrow niche provides a supporting environment for HSC function and includes mesenchymal cells and endothelial cells53. How ageing affects the bone marrow niche is not clear, but the presence of chronic systemic inflammation might contribute. Ageing leads to a chronic systemic low-grade inflammatory state54,55, which might be mediated by cellular senescence that leads to the production of inflammatory mediators (termed the senescence-associated secretory phenotype (SASP))56,57. One source of senescent inflammatory cells is the adipose tissue, which typically increases in size with ageing58,59. The number of adipocytes also increases in the bone marrow with ageing, accompanied by an elevation in the levels of pro-inflammatory cytokines, including IL-6 (refs60,61). These cytokines promote a skewing towards myeloid cell differentiation and an increase in platelet production, the latter of which could contribute to thrombosis60,61. Importantly, adipocytes arising from leptin-receptor-positive progenitors in the bone marrow, but not within other fat depots, synthesize stem cell factor, which promotes HSC regeneration62,63. Senescent stromal cells in the bone marrow are another potential source of inflammation64. These cells can differentiate into adipocytes in the ageing bone marrow65 and further promote an inflammatory environment. The function of bone marrow endothelial cells also declines with ageing66. Furthermore, the number of vascular niches in the bone marrow that support HSC regeneration decreases with ageing, but can be restored in aged mice by activating Notch signalling in endothelial cells53.
Activation of the innate immune receptor Toll-like receptor 4 (TLR4) induces myeloid differentiation in HSCs in mice67. Ageing is associated with alterations in gut microbiota68, which could act as a microbial source for TLR4 stimulation (for example, lipopolysaccharide (LPS) from Gram-negative bacteria activates TLR4). TLR4 activation could then increase the imbalance of HSC differentiation towards the myeloid lineage. In a 2019 study in mice, β2-adrenergic receptor signalling in the bone marrow niche was found to increase with ageing in association with increased generation of myeloid cells and platelets through an IL-6-dependent mechanism60. This study also demonstrated that the bone marrow niche switches from an endosteal to a non-endosteal niche with ageing, indicating that ageing shifts myeloid cell production away from the bone tissue to further within the bone marrow60. This study also found that a mouse model of Hutchinson–Gilford progeria syndrome, which is associated with accelerated ageing69, had an imbalance favouring myeloid cells over lymphoid cells in the peripheral blood60. This effect was mitigated by administration of a β3-adrenergic receptor agonist60. Overall, clear evidence indicates that ageing alters the bone marrow niche via multiple mechanisms to impair HSC function and promote myeloid cell differentiation.
Clonal haematopoiesis and cardiovascular disease: clinical correlation
The positive selection and expansion of clones of HSCs carrying certain somatic mutations, known as clonal haematopoiesis, occurs commonly with ageing. Approximately 10% of individuals aged >70 years carry mutations associated with clonal haematopoiesis, whereas these mutations are rare in individuals aged <40 years7,70,71. These clones of haematopoietic cells harbour single somatic mutations most commonly in genes associated with haematological malignancies, such as DNMT3A, TET2 and ASXL1. Individuals with mutations in these genes have an increased risk of developing haematological malignancies (HR 11–12, depending on the study)7,70,71. All-cause mortality is increased in individuals with any somatic mutation associated with clonal haematopoiesis (HR 1–2) compared with those with no mutations7. Interestingly, the cause of the increased mortality in these individuals is not only the higher rate of haematological malignancies but also a higher rate of adverse cardiovascular events7,70,71,72. The association between clonal haematopoiesis and the risk of adverse cardiovascular events remained even after adjustment for traditional cardiovascular risk factors, such as diabetes mellitus, hypertension, smoking and BMI, in multivariate analyses6. As a result of these studies, the term CHIP was coined to distinguish the phenomenon of clonal haematopoiesis without clear development of haematopoietic malignancy or other known clonal disorder from the pre-malignant clonal haematopoiesis of clinical importance73.
A follow-up clinical study provided further evidence of the association between cardiovascular disease and CHIP6. In particular, old individuals (aged 60–70 years) with CHIP had an approximately twofold higher risk of incident coronary artery disease, a fourfold higher risk of early-onset myocardial infarction and a threefold higher coronary artery calcium score than similarly aged individuals without CHIP6. Importantly, the size of the CHIP clone, defined as the variant allele frequency (VAF), correlates with the risk of cardiovascular disease. Specifically, individuals with a CHIP clone with a VAF of >10% have a 12-fold increased risk of cardiovascular disease compared with individuals with no mutations, whereas the risk of cardiovascular disease is not significantly increased in CHIP carriers with a VAF of <10%6. This study has established that CHIP is associated with the risk of cardiovascular diseases and has developed a potential new paradigm that certain clones of haematopoietic cells accelerate atherogenesis6. However, to date, the presence of CHIP can be used only as a biomarker of atherosclerosis and is not therapeutically actionable.
CHIP and ageing increase atherogenesis via myeloid cells
Mutations in TET2 are the second most prevalent somatic mutations associated with CHIP after those in DNMT3A. Mouse models have been used to elucidate the mechanistic contributions of TET2 mutations to atherogenesis. In irradiated, atheroprone Ldlr−/− mice, those reconstituted with either Tet2−/− or Tet2+/− bone marrow had increased atherosclerotic lesion size compared with mice receiving wild-type bone marrow6,74. These data imply that deletion of one copy of the Tet2 gene is sufficient to increase atherosclerosis in mice. Further studies in mice showed that myeloid-cell-specific TET2 deficiency increases atherosclerotic plaque size74. Interestingly, TET2 deficiency in bone marrow-derived macrophages leads to an elevated secretion of IL-6 and IL-1β (a signature cytokine produced by inflammasome activation)75 in response to various stimuli (such as LDL, LPS and IFNγ) in vitro74. Furthermore, the increased atherogenic potential of Tet2−/− bone marrow cells is reduced when bone marrow transplantation recipients are treated with a small-molecule inhibitor of the NLRP3 inflammasome74. The effect of TET2 deficiency might not be limited to vascular diseases, because experimental studies have demonstrated that transfer of Tet2−/− bone marrow cells into non-irradiated mice accelerates the development of age-related cardiac hypertrophy and fibrosis76, and TET2 deficiency in myeloid cells worsens the development of heart failure in mice after acute injury77.
The contribution of IL-6 to the cardiovascular risk in individuals with large CHIP clones (VAF >10%) was evaluated in 35,416 individuals without prevalent cardiovascular disease enrolled in the UK Biobank registry9. The investigators examined whether an IL6R coding mutation that leads to reduced IL-6 signalling alters the association between CHIP and the risk of adverse cardiovascular events (myocardial infarction, coronary artery disease revascularization, stroke or death). The study revealed that the presence of the IL6R mutation mitigated the increased risk of adverse cardiovascular events in individuals with large CHIP clones but not in individuals without CHIP9. These data indicate that IL-6 signalling is causally linked to the increased risk of cardiovascular disease associated with CHIP.
IL-6 is released following inflammasome activation78; therefore, the observed link between IL-6 and CHIP suggests that inflammasome activation is a mechanism by which CHIP promotes the development of cardiovascular diseases. This concept is compatible with the study in mice discussed above, showing that the NLRP3 inflammasome contributes to the increased atherosclerotic burden induced by Tet2−/− bone marrow transplantation74. The contribution of IL-1β to cardiovascular disease in humans was demonstrated in the CANTOS study12,13, which showed that a monoclonal antibody against IL-1β reduces the risk of recurrent cardiovascular events in patients with previous myocardial infarction. The effects of IL-1β blockade in the CANTOS trial were greater in patients who had lower circulating IL-6 levels after IL-1β blockade than in those with higher circulating IL-6 levels13. However, the role of IL-1β and IL-6 in experimental models of atherosclerosis is not completely clear because data indicating that each of these cytokines has atheroprotective effects have been reported79,80. However, these studies were performed in young mice, so these cytokines might have increasingly pathogenic roles with ageing.
Monocytes and macrophages contribute to both the initiation of the chronic inflammatory process of atherosclerosis and the resolution of the chronic vascular inflammation81. Ageing directly influences the function of monocytes and macrophages16. Human monocytes have lower levels of TLRs and a reduction in TLR-dependent pro-inflammatory cytokine production with ageing16. A study comparing bone marrow-derived monocytes from young and aged atheroprone Ldlr−/− mice showed that ageing leads to a downregulation in the expression of Tnf and Il1b but monocyte chemotaxis is preserved82. Aged (6-month-old) atherosclerotic Apoe−/− mice have a reduction in the number of vascular progenitor cells in the bone marrow compared with 1-month-old atherosclerotic Apoe−/− mice83. Furthermore, administration of bone marrow-derived HSCs from young non-atherosclerotic mice to non-irradiated 6-month-old Apoe−/− mice reduced atherogenesis after feeding a high-fat diet83. This finding suggests that ageing is accompanied by a reduction in the number of atheroprotective progenitor cells in the bone marrow. Aged (18–21-month-old) mice with chronic or induced acute hyperlipidaemia have more macrophage infiltration into atherosclerotic lesions than young mice11,82. Furthermore, the aortas of aged atherosclerotic mice (12-months-old) and rats (30-months-old) have higher levels of macrophage-attracting chemokines and IL-6 than the aortas of young atherosclerotic mice (2-months-old) and rats(10-months-old)82,84. Although macrophages and monocytes can have an increased basal secretion of inflammatory cytokines, such as IL-1β, IL-6 and IL-8, with ageing85 (possibly owing to senescence)57, whether these cells are the major contributors to the increased vascular production of IL-6 with ageing during atherogenesis is unclear. Vascular cells such as vascular smooth muscle cells (VSMCs) have been shown in animal models to have an elevated IL-6 production with ageing before any signs of atherosclerosis development86,87.
Efferocytosis is a crucial mechanism for resolving plaque inflammation and reducing atherosclerosis progression88. In vivo and in vitro assays have indicated that the phagocytic function of tissue alveolar macrophages to take up apoptotic neutrophils declines with ageing89 and is associated with reduced expression of scavenger receptor CD204 (ref.90). In a mouse model of peritonitis, ageing led to reduced resolution of acute inflammation and was associated with reduced levels of pro-resolution lipid mediators, specifically resolvins91. Resolution of inflammation was also delayed with ageing in a human model of skin blistering92. This phenotype is related to reduced expression of the efferocytotic receptor TIM4 in macrophages. Reduced TIM4 expression with ageing was caused by elevations in p38 mitogen-activated protein kinase activity in macrophages, and treatment with an oral p38 inhibitor increased the resolution of blister inflammation in old individuals92. Overall, macrophages show impaired inflammation resolution properties with ageing; however, whether this impaired macrophage function contributes to increased atherosclerosis is not yet clear.
Vascular intrinsic mechanisms
Vascular mitochondrial dysfunction with ageing before atherogenesis initiation
Ageing affects the vasculature before the development of atherosclerosis. Generally, ageing is associated with remodelling of the arterial wall, with evidence of reduced endothelial cell function, increased collagen deposition, fibrosis and functionally stiffer vessels28,93,94. In addition, VSMCs acquire a more proliferative and synthetic function with ageing86. VSMCs also show an increased generation of reactive oxygen species (ROS) and high oxidative damage95. Endothelial cells also have a dysregulated antioxidant capacity with ageing (mediated by the disruption of nuclear factor erythroid 2-related factor 2 signalling), thereby contributing to vascular ageing96,97. All these effects of ageing can contribute to the development of hypertension, a major risk factor for cardiovascular disease.
Most studies on vascular ageing in rodent models have been performed in normolipidaemic animals. These studies provide evidence that mitochondrial dysfunction, a known hallmark of ageing25, contributes to vascular ageing before the initiation of atherogenesis. Disease-free, normolipidaemic mice develop mitochondrial dysfunction in the aorta as they age, first detected at 11 months of age (measured as a decline in oxygen consumption rate (OCR)) and becoming more evident as the mice reach 18 months of age98. The reduction in OCR is accompanied by an increase in mitochondrial DNA (mtDNA) damage98, a sign of mitochondrial genomic instability, which is another hallmark of ageing25. Furthermore, reduced vascular mitochondrial function with ageing is accompanied by a decrease in the expression of the mtDNA helicase Twinkle98, an enzyme involved in preserving mtDNA integrity. Aged transgenic mice expressing high levels of Twinkle show delayed vascular ageing; in particular, the decrease in aortic compliance and the increase in aortic stiffness are delayed in these mice compared with aged wild-type mice98. Overall, experimental evidence indicates that mitochondrial dysfunction and mitochondrial genomic instability contribute to vascular ageing.
Vascular mitochondrial dysfunction during atherogenesis
In humans, atherosclerotic plaques show evidence of damage to mtDNA, which is associated with reduced mitochondrial function, specifically lower OCR in the fibrous cap and core regions of the atherosclerotic plaque than in the shoulder region of the plaque or in non-overtly diseased regions of the aorta10. These findings are compatible with those of previous experimental work indicating that Apoe−/− mice fed a low-fat, standard chow diet have increased vascular mtDNA damage but not nuclear DNA damage as the mice age99,100. Furthermore, human atherosclerotic plaques have lower levels of mitochondrial complex I and complex II than non-diseased aortic regions10. Similar findings are noted in atherosclerotic Apoe−/− mice fed a high-fat diet10. Apoe−/− mice overexpressing Twinkle have a reduced necrotic core area in atherosclerotic plaques compared with control Apoe−/− mice10. Mitochondrial dysfunction probably has a central role in ROS generation but the interaction between these two factors is complex. For example, low levels of ROS might improve cell fitness and promote survival, a concept known as mitohormesis25,101. However, higher levels of ROS might contribute to age-related chronic vascular diseases. The complex interaction between mitochondrial dysfunction and ROS might explain why disruption of some mitochondrial enzymatic pathways (such as NADPH oxidase 1 (NOX1) and NOX2 signalling) in atherosclerotic mice has no effect on age-related atherosclerosis102, whereas partial deficiency of ROS-scavenging enzymes (such as superoxide dismutase)103 in atherosclerotic mice contributes to atherosclerosis99. However, one study found that mtDNA damage occurs in both VSMCs and monocytes and correlates with atherosclerotic burden in humans but without evidence of alterations in ROS levels100. Furthermore, clinical trials on antioxidants have yet to reveal a beneficial effect in patients with atherosclerotic cardiovascular disease104,105. Overall, mitochondrial dysfunction occurs during chronic hyperlipidaemia and atherogenesis, and this mitochondrial dysfunction promotes atherosclerosis. However, the precise role of ROS in this context is complex and requires further investigation.
Dissecting the role of vascular ageing and chronic hyperlipidaemia
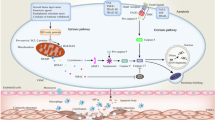
Part of the challenge of using standard mouse models of atherosclerosis (such as Ldlr−/− or Apoe−/− mice) to understand the role of ageing on atherogenesis is that even when fed a standard low-fat diet, these mice age with chronic hyperlipidaemia. Therefore, the effects of ageing cannot be dissected from the effects of chronic hyperlipidaemia. A study in mice published in 2020 circumvented this issue by first examining mitochondrial function in the aortas of young and aged wild-type mice without hyperlipidaemia or vascular diseases11. Consistent with previous studies, aged mice had evidence of reduced OCR in the aortas compared with young mice11. This OCR reduction in the vasculature from aged mice was accompanied by increased expression of the mitophagy protein Parkin and increased basal mitophagy (Box 1), a macroautophagy process to remove damaged mitochondria. Altered mitochondrial quality control in the ageing vasculature without hyperlipidaemia is linked to arterial stiffening in mice106. The mitochondrial dysfunction and elevated Parkin levels with ageing in the mouse aorta are accompanied by an increase in TLR9, MYD88 and IL-6 levels11. Importantly, blocking IL-6 in aged mouse aortas in vitro increased the OCR and reduced Parkin levels. This study identified a positive feedback loop in which mitochondrial dysfunction and elevated IL-6 levels coexist and positively influence each other11. However, the exact identity of the IL-6-producing and IL-6-responsive cell(s) has yet to be identified, although evidence suggests that VSMCs secrete more IL-6 with ageing87.
To study the link between the changes occurring with normolipidaemia in the aged aorta and atherogenesis, young and aged wild-type mice were made acutely hyperlipidaemic by inducing a decrease in LDL receptor levels with adeno-associated virus vector-mediated delivery of Pcsk9 and by feeding the mice a high-fat diet for 10 weeks11, which is an established technique107. With this protocol, young and aged mice had similar and durable levels of hyperlipidaemia; however, aged hyperlipidaemic mice had larger atherosclerotic lesions with larger necrotic cores than young hyperlipidaemic mice11. Importantly, administering spermidine, an agent that increases macroautophagy and mitophagy, to aged hyperlipidaemic mice reduced the levels of both IL-6 and Parkin in the aorta and reduced the size of atherosclerotic plaques11. This finding is consistent with previous studies showing that treatment with spermidine or trehalose, an agent that increases mitophagy, reduces stiffness in the aged vasculature in normolipidaemic, non-atherosclerotic aged mice106,108.

Dysfunctional mitochondria activate inflammation
The CANTOS study12 showed that in old patients (aged >60 years) with cardiovascular disease, IL-1β blockade reduces the risk of recurrent cardiovascular events, indicating that chronic inflammation is a major contributor to age-related atherosclerosis. As described above, mitochondrial dysfunction might coexist in a positive feedback loop with IL-6 signalling11 to increase chronic inflammation in vascular ageing. Furthermore, mitochondrial components that are released to the cytosol after mitochondrial damage can stimulate innate immune responses109. Mitochondrial injury in turn can be induced by TLR stimulation leading to the activation of caspase 4 and caspase 5 in humans or caspase 11 in mice110. These inflammatory caspases cleave gasdermin D, which enables gasdermin D to form pores in the outer mitochondrial membrane, leading to impaired mitochondrial membrane potential and further increasing mitochondrial injury110. Whether this pathway is activated during ageing and in particular vascular ageing is unclear. Nevertheless, the involvement of such a pathway could explain why chronic TLR activation, via either microbial products or sterile inflammatory mediators, could lead to a chronic basal inflammatory state and mitochondrial dysfunction in the vasculature with ageing.
Mitochondrial injury leads to the release of mitochondrial components, known as mitochondrial damage-associated molecular patterns (mtDAMPs), including mtDNA, that when in the cytosol, can activate intracellular innate immune signalling pathways, such as the DNA-sensing receptor cyclic GMP–AMP synthase and the inflammasome75,110,111,112. Transfer of mitochondrial components into endosomes also activates the TLR9 inflammatory pathway111, but the detailed mechanisms are not fully elucidated. Mitochondria also contain N-formylated peptides that induce inflammation via engaging the N-formyl peptide receptor 1 to increase neutrophil chemoattraction113, arterial injury and ROS release114. Cardiolipin, a component of mitochondrial membranes, can directly bind to NLRP3 and activate the NLRP3 inflammasome115. If chronically activated, all these pathways could promote vascular ageing and also diminish mitochondrial function, although ascertaining the definitive contributions of each pathway requires future investigation.
Synergistic mechanisms
Shared inflammatory pathways between myeloid cells and the vasculature
Age-related atherosclerosis might be mediated by alterations in the vasculature and myeloid cells via a shared inflammatory pathway. A potential candidate pathway is IL-6 signalling because available evidence indicates that the level of IL-6 is elevated with ageing in both the immune system and the vasculature. In the bone marrow niche in mice, IL-6 levels increase with ageing, which is probably mediated by increased β2-adrenergic receptor signalling and increased numbers of adipocytes60 (Fig. 1). IL-6 directly acts on HSCs to promote a bias towards myeloid cell differentiation60. In mouse macrophages, TET2 deficiency, which is one of the most common genetic alterations found in the age-related condition CHIP, increases IL-6 secretion in vitro74. Importantly, the atherosclerosis-promoting effects of CHIP seem to be abrogated in individuals with a loss-of-function IL6 genetic polymorphism9. In the vasculature, the level of IL-6 increases with ageing, which is at least in part mediated by IL-6 production by VSMCs87.
IL-6 is associated with ageing in general and is part of the ‘inflammageing’ phenotype15,16,116. Why ageing leads to elevated basal secretion of inflammatory cytokines (not solely IL-6 but also other inflammatory mediators such as TNF) is not clear but might be caused by alterations in the microbiota117, increased adiposity118, and changes in the immune system17 and the vasculature29,30. Elevated cytokine levels with ageing could also be a manifestation of chronic, latent infections such as with herpesviruses119, of cellular senescence57,86,87 or, potentially, of mitochondrial dysfunction.
The role of IL-6 in young animal models of atherosclerosis remains unclear and might relate to the complexities of IL-6 signalling (Box 2). Specifically, signalling via the classic IL-6 pathway occurs in a restricted number of cells (such as hepatocytes and some immune cells) and involves IL-6 binding to the membrane-bound IL-6 receptor (IL-6R), with subsequent association with the signal-transducing IL-6R subunit β (also known as gp130). Evidence indicates that classic IL-6 signalling is important for tissue homeostasis, regeneration and host defence (as reviewed previously120). Soluble IL-6R can also engage IL-6 in the circulation and activate a broader range of cells than the classic pathway, via membrane activation of gp130. This pathway is termed IL-6 trans-signalling (Box 2) and can result in chronic inflammation120. These different IL-6 signalling pathways might explain the pleiotropic effects of IL-6 in different tissues and cellular compartments and also the divergent role of IL-6 in experimental atherosclerotic models. For instance, one study in Apoe−/− mice showed that administration of exogenous IL-6 worsens atherosclerosis121. By contrast, another study in Apoe+/− mice showed that IL-6 deficiency worsens atherosclerosis80, indicating that IL-6 might have atheroprotective effects. Neither of these studies distinguished between the classic and trans-signalling pathways of IL-6. However, a third study specifically inhibited the IL-6 trans-signalling with a fusion protein that blocks the soluble form of gp130 in atherosclerotic Ldlr−/− mice122. The study found that inhibiting IL-6 trans-signalling reduced atherosclerosis122, indicating that IL-6 trans-signalling might have a pathogenic role in atherosclerosis. Therefore, clinical therapeutics to reduce atherosclerosis should focus on this IL-6 pathway.
Whether IL-6 has a causal role in age-related atherosclerosis is not known yet. The contribution of IL-6 to age-related atherosclerosis should be investigated in the future and should determine the main IL-6-producing and IL-6-responding cells. Furthermore, the identification of the major IL-6-producing cells (Fig. 2) and whether IL-6 activation occurs via the classic or trans-signalling pathway with ageing could lead to more targeted therapeutics for atherosclerosis, especially given the availability of clinically-approved agents to target IL-6 (refs123,124). Importantly, the risk–benefit balance of targeting IL-6 in atherosclerosis will need to be determined, given that anti-IL-6 therapies in human studies increased the risk of infections120, similar to other biological agents (such as anti-IL-1β antibodies) that have been used to reduce atherosclerosis12,13. However, other biological agents such as TNF inhibitors125 might be beneficial for the treatment of atherosclerotic cardiovascular disease and should be investigated in age-related atherosclerosis. Finally, other inflammatory cytokines (such as TNF, C-C motif chemokine 2 and IL-18, which are all part of the SASP)56 might have a pathogenic role in age-related atherosclerosis and should be assessed in future studies.

IL-6 is upregulated in multiple tissues that have important roles in the increase in atherogenesis with ageing. Therefore, blockade of IL-6 might be an effective therapeutic strategy to reduce atherosclerosis development and progression during ageing. Blocking IL-6 might interfere with the increased IL-6 signalling in bone marrow adipocytes that occurs with ageing (which promotes a skewing towards myeloid cell differentiation), thereby reducing the risk of clonal haematopoiesis of indeterminate potential (CHIP). IL-6 blockade might also reduce the inflammatory potential of clones of myeloid cells associated with CHIP. IL-6 blockade might reduce atherosclerosis burden, although direct comparisons of efficacy and safety with IL-1β inhibition requires future investigation. IL-6 blockade might increase mitochondrial function and reduce the expression of Parkin, a mitochondrial stress protein, which might also contribute to reducing atherogenesis during ageing. HSC, haematopoietic stem cell; VSMC, vascular smooth muscle cell.
Therapies that can mitigate some of the detrimental biological effects of ageing, such as removing senescent cells (including senescent adipocytes)126,127, improving mitochondrial function (for example, with metformin therapy)128, or augmenting macroautophagy (for example, with rapamycin therapy)129 or mitophagy, might reduce the burden of atherosclerosis in old people and should be investigated in future clinical studies. Agents that increase mitophagy, such as spermidine, have been shown in experimental studies to reduce atherosclerosis in both young130 and aged11 mice. Some or all these agents might have pleiotropic effects, which could reduce inflammation. Furthermore, these agents might synergize with specific anti-inflammatory therapies to reduce atherosclerosis with ageing, which will require future clinical investigation.

Conclusions
Ageing influences atherogenesis via multiple complex pathways, and one sole factor is unlikely to be a dominant pathophysiological mechanism. In this Review, we provide an overview of how ageing affects two systems, myeloid cell haematopoiesis and the vasculature, to promote atherosclerosis. We lay a framework of a potential shared inflammatory pathway, mediated by IL-6 signalling, that connects the role of the two systems in age-related atherosclerosis and propose future avenues of investigation to determine whether IL-6 and/or other inflammatory pathways are feasible and effective therapeutic targets to reduce the burden of atherosclerosis in old people. Anti-inflammatory strategies should be considered in the context of other therapies that aim to reduce many of the detrimental biological effects of ageing. Overall, we hope that with the pursuit of further clinical investigation and trials, therapeutic options will be available in the future to reduce the burden of atherosclerosis in the increasing number of old people in our society.
References
Sturlaugsdottir, R. et al. Prevalence and determinants of carotid plaque in the cross-sectional REFINE-Reykjavik study. BMJ Open 6, e012457 (2016).
Benjamin, E. J. et al. Heart disease and stroke statistics–2019 update: a report from the American Heart Association. Circulation 139, e56–e528 (2019).
Wang, D. et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323, 1061–1069 (2020).
Shi, S. et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 5, 802–810 (2020).
Fuster, J. J. & Walsh, K. Somatic mutations and clonal hematopoiesis: unexpected potential new drivers of age-related cardiovascular disease. Circ. Res. 122, 523–532 (2018).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017).
Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014).
Zink, F. et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752 (2017).
Bick, A. G. et al. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation 141, 124–131 (2020).
Yu, E. P. K. et al. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arterioscler. Thromb. Vasc. Biol. 37, 2322–2332 (2017).
Tyrrell, D. J. et al. Age-associated mitochondrial dysfunction accelerates atherogenesis. Circ. Res. 126, 298–314 (2020).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Ridker, P. M. et al. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Heart J. 39, 3499–3507 (2018).
Bennett, M. R. & Clarke, M. C. H. Killing the old: cell senescence in atherosclerosis. Nat. Rev. Cardiol. 14, 8–9 (2017).
McElhaney, J. E., Kuchel, G. A., Zhou, X., Swain, S. L. & Haynes, L. T-cell immunity to influenza in older adults: a pathophysiological framework for development of more effective vaccines. Front. Immunol. 7, 41 (2016).
Shaw, A. C., Goldstein, D. R. & Montgomery, R. R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 13, 875–887 (2013).
Nikolich-Žugich, J. The twilight of immunity: emerging concepts in aging of the immune system. Nat. Immunol. 19, 10–19 (2018).
Hansson, G. R. K. & Libby, P. The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol. 6, 508–519 (2006).
Wolf, D. & Ley, K. Immunity and inflammation in atherosclerosis. Circ. Res. 124, 315–327 (2019).
Ma, S., Wang, C., Mao, X. & Hao, Y. B cell dysfunction associated with aging and autoimmune diseases. Front. Immunol. 10, 318 (2019).
Weyand, C. M. & Goronzy, J. J. Aging of the immune system. Mechanisms and therapeutic targets. Ann. Am. Thorac. Soc. 13, S422–S428 (2016).
Ventura, M. T., Casciaro, M., Gangemi, S. & Buquicchio, R. Immunosenescence in aging: between immune cells depletion and cytokines up-regulation. Clin. Mol. Allergy 15, 21 (2017).
Pérez, V. I. et al. Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 1790, 1005–1014 (2009).
Jang, Y. C. et al. Overexpression of Mn superoxide dismutase does not increase life span in mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 64A, 1114–1125 (2009).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Kattoor, A. J., Pothineni, N. V. K., Palagiri, D. & Mehta, J. L. Oxidative stress in atherosclerosis. Curr. Atheroscler. Rep. 19, 42 (2017).
Franklin, S. S., Khan, S. A., Wong, N. D., Larson, M. G. & Levy, D. Is pulse pressure useful in predicting risk for coronary heart disease? The Framingham Heart Study. Circulation 100, 354–360 (1999).
van Bussel, B. C. et al. Endothelial dysfunction and low-grade inflammation are associated with greater arterial stiffness over a 6-year period. Hypertension 58, 588–595 (2011).
Donato, A. J., Machin, D. R. & Lesniewski, L. A. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ. Res. 123, 825–848 (2018).
Ungvari, Z., Tarantini, S., Donato, A. J., Galvan, V. & Csiszar, A. Mechanisms of vascular aging. Circ. Res. 123, 849–867 (2018).
Swirski, F. K. et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 117, 195–205 (2007).
Bobryshev, Y. V. & Lord, R. S. S-100 positive cells in human arterial intima and in atherosclerotic lesions. Cardiovasc. Res. 29, 689–696 (1995).
Laurat, E. et al. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation 104, 197–202 (2001).
Zhou, X. & Hansson, G. K. Detection of B cells and proinflammatory cytokines in atherosclerotic plaques of hypercholesterolaemic apolipoprotein E knockout mice. Scand. J. Immunol. 50, 25–30 (1999).
Libby, P., Ridker, P. M. & Maseri, A. Inflammation and atherosclerosis. Circulation 105, 1135–1143 (2002).
Lee, J., Yoon, S. R., Choi, I. & Jung, H. Causes and mechanisms of hematopoietic stem cell aging. Int. J. Mol. Sci. 20, 1272 (2019).
Kovtonyuk, L. V., Fritsch, K., Feng, X., Manz, M. G. & Takizawa, H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front. Immunol. 7, 502 (2016).
Beerman, I. et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl Acad. Sci. USA 107, 5465–5470 (2010).
Rossi, D. J. et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl Acad. Sci. USA 102, 9194–9199 (2005).
Sudo, K., Ema, H., Morita, Y. & Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 192, 1273–1280 (2000).
Rundberg Nilsson, A., Soneji, S., Adolfsson, S., Bryder, D. & Pronk, C. J. Human and murine hematopoietic stem cell aging is associated with functional impairments and intrinsic megakaryocytic/erythroid bias. PLoS ONE 11, e0158369 (2016).
Chen, J., Astle, C. M. & Harrison, D. E. Genetic regulation of primitive hematopoietic stem cell senescence. Exp. Hematol. 28, 442–450 (2000).
Chambers, S. M. et al. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 5, e201 (2007).
Kramer, A. & Challen, G. A. The epigenetic basis of hematopoietic stem cell aging. Semin. Hematol. 54, 19–24 (2017).
Dykstra, B., Olthof, S., Schreuder, J., Ritsema, M. & de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 208, 2691–2703 (2011).
Sasaki, M. et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 488, 656–659 (2012).
Wang, C. et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 126, 1741–1752 (2015).
Attema, J. L., Pronk, C. J., Norddahl, G. L., Nygren, J. M. & Bryder, D. Hematopoietic stem cell ageing is uncoupled from p16 INK4A-mediated senescence. Oncogene 28, 2238–2243 (2009).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Ho, T. T. et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210 (2017).
Mohrin, M. et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377 (2015).
Grants, J. M. et al. Altered microRNA expression links IL6 and TNF-induced inflammaging with myeloid malignancy. Blood 135, 2235–2251 (2020).
Kusumbe, A. P. et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 532, 380–384 (2016).
Franceschi, C. et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. NY Acad. Sci. 908, 244–254 (2000).
Harris, T. B. et al. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am. J. Med. 106, 506–512 (1999).
Kirkland, J. L. & Tchkonia, T. Cellular senescence: a translational perspective. EBioMedicine 21, 21–28 (2017).
Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J. & Kirkland, J. L. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972 (2013).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Cesari, M. et al. Sarcopenia, obesity, and inflammation–results from the trial of angiotensin converting enzyme inhibition and novel cardiovascular risk factors study. Am. J. Clin. Nutr. 82, 428–434 (2005).
Ho, Y.-H. et al. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem Cell 25, 407–418.e6 (2019).
Tuljapurkar, S. R. et al. Changes in human bone marrow fat content associated with changes in hematopoietic stem cell numbers and cytokine levels with aging. J. Anat. 219, 574–581 (2011).
Zhou, B. O. et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat. Cell Biol. 19, 891–903 (2017).
Yue, R., Zhou, B. O., Shimada, I. S., Zhao, Z. & Morrison, S. J. Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell 18, 782–796 (2016).
Gnani, D. et al. An early-senescence state in aged mesenchymal stromal cells contributes to hematopoietic stem and progenitor cell clonogenic impairment through the activation of a pro-inflammatory program. Aging Cell 18, e12933 (2019).
Takeshita, S., Fumoto, T., Naoe, Y. & Ikeda, K. Age-related marrow adipogenesis is linked to increased expression of RANKL. J. Biol. Chem. 289, 16699–16710 (2014).
Poulos, M. G. et al. Endothelial transplantation rejuvenates aged hematopoietic stem cell function. J. Clin. Invest. 127, 4163–4178 (2017).
Esplin, B. L. et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J. Immunol. 186, 5367–5375 (2011).
Claesson, M. J. et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184 (2012).
Villa-Bellosta, R. et al. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation 127, 2442–2451 (2013).
Genovese, G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014).
Xie, M. et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014).
Shlush, L. I. Age-related clonal hematopoiesis. Blood 131, 496–504 (2018).
Jaiswal, S. & Ebert, B. L. Clonal hematopoiesis in human aging and disease. Science 366, eaan4673 (2019).
Fuster, J. J. et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017).
Guo, H., Callaway, J. B. & Ting, J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687 (2015).
Wang, Y. et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 5, e135204 (2020).
Sano, S. et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J. Am. Coll. Cardiol. 71, 875–886 (2018).
Calabrese, L. H. & Rose-John, S. IL-6 biology: implications for clinical targeting in rheumatic disease. Nat. Rev. Rheumatol. 10, 720–727 (2014).
Gomez, D. et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 24, 1418–1429 (2018).
Madan, M., Bishayi, B., Hoge, M. & Amar, S. Atheroprotective role of interleukin-6 in diet- and/or pathogen-associated atherosclerosis using an ApoE heterozygote murine model. Atherosclerosis 197, 504–514 (2008).
Moore, K. J., Sheedy, F. J. & Fisher, E. A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13, 709–721 (2013).
Du, W. et al. Age-associated vascular inflammation promotes monocytosis during atherogenesis. Aging Cell 15, 766–777 (2016).
Rauscher, F. M. et al. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation 108, 457–463 (2003).
Belmin, J. et al. Increased production of tumor necrosis factor and interleukin-6 by arterial wall of aged rats. Am. J. Physiol. 268, H2288–H2293 (1995).
Qian, F. et al. Age-associated elevation in TLR5 leads to increased inflammatory responses in the elderly. Aging Cell 11, 104–110 (2011).
Csiszar, A. et al. Age-associated proinflammatory secretory phenotype in vascular smooth muscle cells from the non-human primate Macaca mulatta: reversal by resveratrol treatment. J. Gerontol. A Biol. Sci. Med. Sci. 67, 811–820 (2012).
Song, Y. et al. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 32, 103–109 (2012).
Thorp, E., Subramanian, M. & Tabas, I. The role of macrophages and dendritic cells in the clearance of apoptotic cells in advanced atherosclerosis. Eur. J. Immunol. 41, 2515–2518 (2011).
Boyd, A. R., Shivshankar, P., Jiang, S., Berton, M. T. & Orihuela, C. J. Age-related defects in TLR2 signaling diminish the cytokine response by alveolar macrophages during murine pneumococcal pneumonia. Exp. Gerontol. 47, 507–518 (2012).
Wong, C. K. et al. Aging impairs alveolar macrophage phagocytosis and increases influenza-induced mortality in mice. J. Immunol. 199, 1060–1068 (2017).
Arnardottir, H. H., Dalli, J., Colas, R. A., Shinohara, M. & Serhan, C. N. Aging delays resolution of acute inflammation in mice: reprogramming the host response with novel nano-proresolving medicines. J. Immunol. 193, 4235–4244 (2014).
De Maeyer, R. P. H. et al. Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat. Immunol. 21, 615–625 (2020).
Wu, J. et al. Origin of matrix-producing cells that contribute to aortic fibrosis in hypertension. Hypertension 67, 461–468 (2016).
Shao, J.-S. et al. Vascular calcification and aortic fibrosis: a bifunctional role for osteopontin in diabetic arteriosclerosis. Arterioscler. Thromb. Vasc. Biol. 31, 1821–1833 (2011).
Moon, S.-K. et al. Aging, oxidative responses, and proliferative capacity in cultured mouse aortic smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 280, H2779–H2788 (2001).
Valcarcel-Ares, M. N. et al. Disruption of Nrf2 signaling impairs angiogenic capacity of endothelial cells: implications for microvascular aging. J. Gerontol. A Biol. Sci. Med. Sci. 67, 821–829 (2012).
Ungvari, Z. et al. Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 301, H363–H372 (2011).
Foote, K. et al. Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice. Aging Cell 17, e12773 (2018).
Ballinger, S. W. et al. Mitochondrial integrity and function in atherogenesis. Circulation 106, 544–549 (2002).
Yu, E. et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 128, 702–712 (2013).
Bárcena, C., Mayoral, P. & Quirós, P. M. in International Review of Cell and Molecular Biology Vol. 340 Ch. 2 (eds. López-Otín, C. & Galluzzi, L.) 35–77 (Academic, 2018).
Vendrov, A. E. et al. NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid. Redox Signal. 23, 1389–1409 (2015).
Vendrov, A. E. et al. Attenuated superoxide dismutase 2 activity induces atherosclerotic plaque instability during aging in hyperlipidemic mice. J. Am. Heart Assoc. 6, e006775 (2017).
Goszcz, K. et al. Antioxidants in cardiovascular therapy: panacea or false hope? Front. Cardiovasc. Med. 2, 29 (2015).
Cherubini, A. et al. Role of antioxidants in atherosclerosis: epidemiological and clinical update. Curr. Pharm. Des. 11, 2017–2032 (2005).
LaRocca, T. J., Hearon, C. M. Jr., Henson, G. D. & Seals, D. R. Mitochondrial quality control and age-associated arterial stiffening. Exp. Gerontol. 58, 78–82 (2014).
Bjorklund, M. M. et al. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ. Res. 114, 1684–1689 (2014).
LaRocca, T. J., Gioscia-Ryan, R. A., Hearon, C. M. Jr & Seals, D. R. The autophagy enhancer spermidine reverses arterial aging. Mech. Ageing Dev. 134, 314–320 (2013).
West, A. P., Shadel, G. S. & Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402 (2011).
Huang, L. S. et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52, 475–486.e5 (2020).
Song, Y., Shen, H., Du, W. & Goldstein, D. R. Inhibition of x-box binding protein 1 reduces tunicamycin-induced apoptosis in aged murine macrophages. Aging Cell 12, 794–801 (2013).
Schoggins, J. W. et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695 (2014).
Gao, J. L. et al. F2L, a peptide derived from heme-binding protein, chemoattracts mouse neutrophils by specifically activating Fpr2, the low-affinity N-formylpeptide receptor. J. Immunol. 178, 1450–1456 (2007).
Wenceslau, C. F., McCarthy, C. G., Szasz, T., Goulopoulou, S. & Webb, R. C. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am. J. Physiol. Heart Circ. Physiol. 308, H768–H777 (2015).
Iyer, S. S. et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 (2013).
Baylis, D., Bartlett, D. B., Patel, H. P. & Roberts, H. C. Understanding how we age: insights into inflammaging. Longev. Healthspan 2, 8 (2013).
Thevaranjan, N. et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21, 455–466.e4 (2017).
Addison, O., LaStayo, P. C., Dibble, L. E. & Marcus, R. L. Inflammation, aging, and adiposity: implications for physical therapists. J. Geriatric Phys. Ther. 35, 86–94 (2012).
Wang, G. C. & Casolaro, V. Immunologic changes in frail older adults. Transl. Med. UniSa 9, 1–6 (2014).
Rose-John, S., Winthrop, K. & Calabrese, L. The role of IL-6 in host defence against infections: immunobiology and clinical implications. Nat. Rev. Rheumatol. 13, 399–409 (2017).
Huber, S. A., Sakkinen, P., Conze, D., Hardin, N. & Tracy, R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 19, 2364–2367 (1999).
Schuett, H. et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 32, 281–290 (2012).
Scott, L. J. Tocilizumab: a review in rheumatoid arthritis. Drugs 77, 1865–1879 (2017).
Kang, S., Tanaka, T., Narazaki, M. & Kishimoto, T. Targeting interleukin-6 signaling in clinic. Immunity 50, 1007–1023 (2019).
Pawar, A. et al. Risk of serious infections in tocilizumab versus other biologic drugs in patients with rheumatoid arthritis: a multidatabase cohort study. Ann. Rheum. Dis. 78, 456–464 (2019).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Roos, C. M. et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15, 973–977 (2016).
Soukas, A. A., Hao, H. & Wu, L. Metformin as anti-aging therapy: is it for everyone? Trends Endocrinol. Metab. 30, 745–755 (2019).
Miller, R. A. et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 66A, 191–201 (2011).
Michiels, C. F., Kurdi, A., Timmermans, J.-P., De Meyer, G. R. Y. & Martinet, W. Spermidine reduces lipid accumulation and necrotic core formation in atherosclerotic plaques via induction of autophagy. Atherosclerosis 251, 319–327 (2016).
Acknowledgements
D.J.T. is supported by NIH award F32-HL1400728, and D.R.G. is supported by NIH awards R01-HL127687, R01-AI138347 and K07-AG050096.
Author information
Authors and Affiliations
Contributions
Both authors researched data for the article, discussed its content, wrote the manuscript, and reviewed and edited it before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Cardiology thanks C. Leeuwenburgh, H. Oliveira, A. Tedgui and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Clonal haematopoiesis of indeterminate potential
-
(CHIP). Clonal expansion of haematopoietic stem cells that carry certain somatic mutations that confer a cell proliferation advantage.
- Mitophagy
-
Type of macroautophagy for the removal of damaged or dysfunctional mitochondria.
- Senescence
-
A state of permanent replicative arrest in normally proliferative cells.
- Senescence-associated secretory phenotype
-
(SASP). Secretion of cytokines, chemokines, growth factors and proteases by senescent cells.
- Variant allele frequency
-
(VAF). The proportion of sequences that match a gene mutation divided by the overall coverage at that gene locus.
- Efferocytosis
-
Phagocytosis of apoptotic cells by phagocytic cells.
- Mitochondrial damage-associated molecular patterns
-
(mtDAMPs). Pro-inflammatory components of mitochondria that are released as a result of mitochondrial dysfunction or damage.
Rights and permissions
About this article

Cite this article
Tyrrell, D.J., Goldstein, D.R. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol 18, 58–68 (2021). https://doi.org/10.1038/s41569-020-0431-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-020-0431-7
This article is cited by
-
Clonal expansion of CD8+ T cells in aged mice linked to pro-atherogenic phenotype
Nature Reviews Cardiology (2024)
-
Cytosolic DNA initiates a vicious circle of aging-related endothelial inflammation and mitochondrial dysfunction via STING: the inhibitory effect of Cilostazol
Acta Pharmacologica Sinica (2024)
-
Role of macrophage polarization in periodontitis promoting atherosclerosis
Odontology (2024)
-
Unveiling the role of ABI3 and hub senescence-related genes in macrophage senescence for atherosclerotic plaque progression
Inflammation Research (2024)
-
A novel dammarane triterpenoid alleviates atherosclerosis by activating the LXRα pathway
Chinese Medicine (2023)
