
Abstract
To explore favourable niches while avoiding threats, many bacteria use a chemotaxis navigation system. Despite decades of studies on chemotaxis, most signals and sensory proteins are still unknown. Many bacterial species release d-amino acids to the environment; however, their function remains largely unrecognized. Here we reveal that d-arginine and d-lysine are chemotactic repellent signals for the cholera pathogen Vibrio cholerae. These d-amino acids are sensed by a single chemoreceptor MCPDRK co-transcribed with the racemase enzyme that synthesizes them under the control of the stress-response sigma factor RpoS. Structural characterization of this chemoreceptor bound to either d-arginine or d-lysine allowed us to pinpoint the residues defining its specificity. Interestingly, the specificity for these d-amino acids appears to be restricted to those MCPDRK orthologues transcriptionally linked to the racemase. Our results suggest that d-amino acids can shape the biodiversity and structure of complex microbial communities under adverse conditions.
Similar content being viewed by others

Main
Most bacteria can sense and respond to a wide variety of signals to thrive in changing environments. One such adaptive response is chemotaxis, by which motile bacteria monitor changes in local concentrations of certain substances and initiate a signalling cascade that, over time, biases bacterial movement towards favourable conditions (for example, nutrients) or away from toxic compounds1.
Albeit species-specific components exist, the core of the chemotaxis signalling pathway consists of chemoreceptors, known as methyl-accepting chemotaxis proteins (MCPs), the adaptor protein CheW, the histidine kinase CheA and the response regulator CheY. In some species such as Escherichia coli, the MCPs form a stable ternary sensory complex with CheW and CheA that typically clusters at cell poles. Upon ligand binding or other environmental perturbations, the MCPs change conformation and modulate CheA kinase activity with the help of CheW. CheA in turn phosphorylates CheY, thereby allowing it to bind to the flagellar motor to induce clockwise rotation and, thus, cell tumbling2,3. To ensure a rapid response to chemotactic stimuli, the phosphorylation of CheY can be controlled by a specific phosphatase4. Although the chemotaxis signalling core components are highly conserved among bacteria and archaea, the number and type of chemoreceptors are highly variable between species and their specificities remain largely uncharacterized5,6,7.
Compared to E. coli, which has a single chemosensory system and 5 MCPs, the facultative pathogen Vibrio cholerae has a very sophisticated chemotaxis system organized into three sets of chemosensory pathways (Che systems I/F9, II/F6 and III/F7) and at least 45 chemoreceptors8. Thus far, only system II/F6 has been demonstrated to control motility9, while the function of the others remains unclear. The high number of MCPs has been suggested to reflect the complexity of V. cholerae’s life cycle10; however, except for a handful of input signals (for example, l-amino acids11, taurine12, oxygen13) there is almost no information about the ligand specificity of most MCPs.
V. cholerae releases millimolar concentrations of diverse d-amino acid (DAAs) into the environment14,15 (Fig. 1a). DAAs are produced from their l-amino acid (LAA) counterparts by the broad-spectrum racemase BsrV, which is conserved in many bacterial species14. These molecules regulate diverse cellular processes (for example, cell wall biogenesis16,17, biofilm integrity18,19,20,21, spore germination22 or bacteria–bacteria interactions15) but overall, the physiological role of extracellular DAAs is mainly dependent on each specific d-amino acid type and bacterial species. In V. cholerae, it is known that d-Met and d-Leu modulate cell wall biogenesis, but the function of other DAAs is unclear.

a, Schematic representation of DAA racemization. The most abundant amino acids produced are shown (open circles, l-amino acids; filled circles, d-amino acids). OM, outer membrane; IM, inner membrane; CYT, cytoplasm. b, Representative images of V. cholerae wild type (wt) and ΔbsrV mutant expanding in 0.3% soft agar. Images are representative of experiments repeated at least three times. c, Motility and d-amino acid production analysis of BsrV derivatives; grey bars indicate relative motility compared to wt or ΔbsrV knockout mutant, catalytic site mutant (BsrV K95A) and the complemented strain harbouring bsrV under an aTc inducible promoter (pbsrV); green dots represent the amount of secreted d-amino acids. pØ, empty plasmid. Error bars represent mean ± s.d. of 3 biologically independent replicates. Significant differences (paired t-test) are indicated by **P < 0.01 or ***P < 0.001. d, Motility of ΔbsrV mutant in soft-agar plates that are chemically complemented with 5 mM d-amino acids are shown relative to the wild-type strain. Error bars represent mean ± s.d. of 6 biologically independent replicates examined over 2 independent experiments. Significant differences (one-way analysis of variance (ANOVA)) are indicated by ****P < 0.0001. e, Chemotactic response to 0.1 mM d-amino acids in capillary assays. Chemotaxis ratio (CR) was calculated relative to the control capillary containing no stimulus. CR > 1, attractants; CR < 1, repellents; CR = 1, no response. Black diamonds represent the mean of 3 independent biological replicates. Significant differences (unpaired t-test) are indicated by *P < 0.05 or ***P < 0.001. f, Thermal proteome profiling analysis of V. cholerae cell extracts exposed to d-Arg. Stabilized and destabilized proteins in the presence of d-Arg are highlighted in red and green, respectively. From the 45 MCPs found in V. cholerae’s genome (black), only 3 interacted with d-Arg: VC2161, VC1313 and VC1406. g, Chemotactic response of selected MCP candidates to d-Arg. Double mutants (ΔbsrV Δmcp) were constructed and the ability to respond to d-Arg was tested by capillary assays. ΔbsrV strain was used as background. LAA, l-amino acids; DAA, d-amino acids. Black diamonds represent the mean of 3 independent biological replicates. Significant differences (unpaired t-test) are indicated by ***P < 0.001.
Results
d-arginine and d-lysine are chemorepellent signals
While screening for DAA-specific phenotypes, we found that a V. cholerae strain defective in d-amino acid production (ΔbsrV) exhibited a 40% motility reduction compared to wild-type cells in soft-agar plates. This reduction was fully restored upon ectopic genetic complementation with the bsrV racemase (Fig. 1b,c). ΔbsrV’s motility defect was recapitulated by a catalytically inactive mutant (BsrV K95A, ref. 23), thus suggesting that this phenotype was directly associated with BsrV’s activity to produce d-amino acids. To ascertain that the absence of secreted d-amino acids accounted for the motility defect that we found, we chemically complemented the ΔbsrV mutant by supplementing the soft-agar plates with innocuous concentrations of individual DAAs. Their corresponding l-enantiomers were used as controls (Extended Data Fig. 1). We found that only d-arginine (d-Arg) and d-lysine (d-Lys) restored the motility of the ΔbsrV mutant to wild-type levels (Fig. 1d), suggesting that these specific DAAs play a specific role in V. cholerae’s ability to spread in soft agar.
To narrow down the role of d-Arg and d-Lys in V. cholerae’s motility, we first assessed the integrity of the flagellum. However, ΔbsrV cells showed no significant differences in flagellum composition or structure compared to the wild type (Supplementary Fig. 1). As motility is functionally related to chemotaxis, we reasoned that d-Arg and d-Lys modulate V. cholerae’s chemotactic response. In this line, we tested the activity of diverse DAAs in capillary assays. We used the ΔbsrV genetic background (control hereafter) to prevent BsrV-mediated racemization of the amino acids during the assays. Importantly, this strain has wild-type chemotactic and swimming ability towards previously characterized stimuli (for example, l-Arginine, α-aminoisobutyric acid; γ-aminobutyric acid, succinate) (Extended Data Fig. 2). Interestingly, we found that both d-Arg and d-Lys promote repulsive chemotactic responses in V. cholerae ΔbsrV, while the other DAAs did not affect the bacteria (Fig. 1e). To confirm that the motility defect of V. cholerae ΔbsrV is exclusively related to chemotaxis signalling and not due to a change in V. cholerae’s swimming capacity, we used a permanently active phosphomimetic cheY3D16K,Y109K mutant (hereafter referred to as CheY3**). As previously described24,25, CheY3** cells are non-chemotactic because they are locked in tumbling mode but maintain the ability to swim. Our results show that the spread of a ΔcheY3 strain complemented with CheY3** on soft-agar plates was unaffected by d-Arg (Extended Data Fig. 2c), thus indicating that this DAA indeed does not regulate V. cholerae swimming. Collectively, these results show that d-Arg and d-Lys sensing induces a repellent chemotactic response in V. cholerae.
VC1313 is the chemoreceptor of d-Arg and d-Lys
The V. cholerae chemotaxis machinery comprises at least 45 putative MCPs8, some of which are known to be functionally redundant (for example, Mlp24 and Mlp37, which sense several LAAs)11,12. Therefore, to screen for MCP candidates sensing d-Arg and d-Lys, we opted for two-dimensional thermal proteome profiling (2D-TPP) (refs. 26,27) over mutagenesis. TPP is based on the principle that protein thermal stability can be affected by interactions with ligands28. In our TPP experiment, we treated V. cholerae cells with different concentrations of d-amino acids and quantified the resulting changes in protein thermal stability using mass spectrometry (Extended Data Fig. 3a). Considering the structural similarity and comparable chemotactic responses of d-Arg and d-Lys, we focused on d-Arg for our study. Our results revealed ~223 proteins affected in their thermal stability (false discovery rate, FDR <0.01) by d-Arg (Fig. 1f, Extended Data Fig. 3b, and Supplementary Tables 1 and 2). Some 185 proteins presented a decreased thermal stability, while 38 displayed increased thermal stability, including three putative MCPs: VC2161, VC1406 and VC1313. To further investigate whether any of these chemoreceptors reacts to d-Arg, we generated individual MCP knockouts in the ΔbsrV background and examined their chemotactic response to d-Arg. Remarkably, we found that the d-Arg response of the Δvc2161 and Δvc1406 strains was comparable to that of the control, while the Δvc1313 mutant did not respond to d-Arg. Notably, the d-Arg response in this mutant was fully restored upon genetic complementation with vc1313 (Fig. 1g). We found that ΔbsrV Δvc1313 cells were also unresponsive to d-Lys (Extended Data Fig. 4a), indicating that this MCP senses both DAAs. Additional capillary and microfluidic-based assays29 with a broader range of amino acid concentrations narrowed VC1313’s repellent response to d-Arg concentrations in the 0.01 mM to 1 mM range (Extended Data Fig. 4). Interestingly, 1 mM d-Arg showed a slightly positive chemotactic response in ΔbsrV Δvc1313 cells. This suggests that alternative lower-affinity chemoreceptors sense d-Arg as an attractant. However, our results show that vc1313 encodes the MCP that senses d-Arg and d-Lys to initiate a repulsive chemotactic response in V. cholerae; therefore, we renamed this protein as MCPDRK for MCP sensing d-Arg (R) and d-Lys (K).
RpoS sigma factor regulates MCPDRK
The mcpDRK locus is localized in the vicinity of vc1312, which encodes BsrV—the broad-spectrum racemase that produces d-Arg and d-Lys (Extended Data Fig. 5a). Analysis of the expression profiles across different publicly available RNAseq datasets for V. cholerae strongly suggests that these two genes are co-regulated and could form an operon (Extended Data Fig. 5b). Using promoter-probe plasmids, we found a promoter region upstream of vc1313 (Pvc1313) but not of vc1312 (Extended Data Fig. 5c). Moreover, Pvc1313 activity increases in stationary phase, consistent with a previous study showing that BsrV is induced under the same conditions, in an RpoS-dependent fashion30 (Extended Data Fig. 5d).
To confirm this result, we decided to monitor the expression and localization of MCPDRK labelled with super-folding green fluorescence protein (sfGFP-MCPDRK) throughout V. cholerae growth. As predicted, cultures inoculated with stationary phase cells displayed polar MCPDRK localization in nearly 90% of the cells. This percentage rapidly dropped as the cells started to grow until no foci were detected at the onset of exponential growth (Fig. 2a). As cultures plateaued, polar foci reappeared, reaching the starting levels. We found a direct correlation between sfGFP-MCPDRK foci and protein levels at different optical densities by western blot (Fig. 2b). This demonstrates that expression of MCPDRK in V. cholerae is indeed linked to the stationary phase. As RpoS is highly induced during glucose starvation, we cultured V. cholerae on a mixture with glucose and succinate and monitored expression and localization of MCPDRK. Similar to other bacteria, V. cholerae cells follow a diauxic growth caused by catabolite repression; cells consume d-glucose first, followed by a short pause, then resuming growth by metabolizing succinate until it is spent30 (Fig. 2c). We again observed that the stationary phase cells from the inoculum exhibited polar sfGFP-MCPDRK foci in basically all cells, but these vanished when growth started. Interestingly, foci rapidly increased to maximum levels when cells entered the first growth arrest caused by glucose depletion. When cells resumed growth on succinate, foci decreased to ~60% and rose up again when succinate was consumed. These results indicate that the induction and polar localization of MCPDRK respond to growth arrest rather than high population densities.

a, Left: localization of sfGFP-MCPDRK expressed from its native locus and promoter at exponential and stationary phase. Representative micrographs of 3 independent replicates are shown; at least 3 images were acquired per timepoint. Scale bar, 2 μm. Right: demographic analysis of the fluorescence intensity of sfGFP-MCPDRK relative to cell length (n > 500 cells). The colour bar represents fluorescence intensity. b, Bottom: growth-phase dependent subcellular localization of sfGFP-MCPDRK. Filled grey circles, OD600 of V. cholerae grown in TB; open tradewind-blue circles, percentage of cells with sfGFP-MCPDRK polar foci. Top: western blot against sfGFP-MCPDRK using anti-GFP-specific antibody at the indicated OD600. Samples were normalized and loaded with equal protein amount. c, Growth curve of V. cholerae grown in MSR6 minimal medium supplemented with 0.04% w/v d-glucose and 0.4% w/v succinate (grey line) at the indicated timepoints. The corresponding percentage of cells with sfGFP-MCPDRK foci (open tradewind-blue circles) is also indicated. Red (glucose depletion) and purple (succinate depletion) arrows indicate carbon starvation. Quantification of % of cells with polar foci in b and c was based on a single experiment where >500 cells were counted from 3 different images per timepoint. d, Left: micrographs showing the expression of sfGFP-MCPDRK in a ΔrpoS mutant background. Representative micrographs of 3 independent replicates are shown; at least 3 images were acquired per timepoint: scale bar, 2 μm. Right: demographic analysis of the fluorescence intensity of sfGFP-MCPDRK relative to cell length. The colour bar represents fluorescence intensity. e, Chemotaxis response of ΔrpoS mutant to d-Arg. Black diamonds represent the mean of 3 independent biological replicates. Error bars represent mean ± s.d. of 3 biologically independent replicates. Significant differences (unpaired t-test) are indicated by ***P < 0.001. f, DAA production by the different strains. Significant differences (unpaired t-test) are indicated by **P < 0.01 or ***P < 0.001.
To confirm that MCPDRK’s expression depended on RpoS, we cloned the sfGFP-MCPDRK reporter into a ΔrpoS mutant background. In contrast to wild-type stationary phase cultures where most cells displayed foci, the ΔrpoS cells showed no polar localization of the MCP (Fig. 2d). Consistently, ΔrpoS phenocopied the lack of DAA production and taxis response to d-Arg of the ΔbsrV and ΔmcpDRK mutants (Fig. 2e,f), respectively. These results demonstrate that V. cholerae displays a synchronic RpoS-dependent DAA production and MCPDRK-chemotactic response during nutrient deprivation stresses that lead to growth arrest.
MCPDRK forms part of the chemotaxis system II/F6
Similar to V. cholerae chemosensory cluster III/F7 proteins30, MCPDRK also forms polar foci during the stationary phase and its expression is RpoS-dependent. However, based on domain length and sequence conservation, MCPDRK belongs to the 40H (heptad) class of chemoreceptors31. These are membrane-bound, polarly localized MCPs predicted to signal through the chemotaxis system II/F6—the only one so far related to motility in this bacterium9,32. To experimentally determine the chemotaxis system that transduces the MCPDRK signalling, we constructed mutants with complete deletions of the I/F9, II/F6 and III/F7 chemotaxis systems and analysed their response to d-Arg. Neither deletion of I/F9 nor of III/F7 reduced V. cholerae’s negative chemotactic response to d-Arg, strongly suggesting MCPDRK association to the II/F6 chemosensory system (Extended Data Fig. 6a). Further evidence was obtained by investigating the localization of the MCPDRK chemoreceptor in these mutant backgrounds. MCPDRK foci overlap with those of CheY3 of the chemotaxis cluster II (Extended Data Fig. 6b) and are lost in the II/F6 but not in the III/F7 or I/F9 mutants, confirming that MCPDRK belongs to the chemotaxis system II/F6 (Extended Data Fig. 6c).
Structure of the sensory module of MCPDRK
MCPDRK is a 59.1 kDa protein (Uniprot: Q9KSE4) that is predicted to have the typical transmembrane chemoreceptor domain architecture. MCPDRK contains a ligand binding domain (LBD) predicted to belong to the 4HB_MCP protein family (Pfam PF12729) as a sensory module, a transmembrane HAMP domain (Pfam PF00672) and a cytoplasmic MCP-signalling domain (Pfam PF00015) as output module (Fig. 3a). To elucidate the molecular mechanisms of ligand recognition by MCPDRK, we purified the LBD domain of MCPDRK (20 kDa) with a Glutathione S-transferase (GST) tag that was cleaved off before the final size-exclusion chromatography (SEC) step. This purified LBD was monomeric in solution, both in the absence and presence of its d-Arg ligand, as determined by multi-angle laser light scattering (SEC-MALS, see Supplementary Fig. 2). We solved two X-ray crystal structures of MCPDRK’s LBD in complex with either d-Arg or d-Lys at 1.8 Å resolution (PDB 8BSA and 8BSB, Supplementary Table 3). The crystals belonged to space group P1 and contained two LBD molecules, organized head-to-tail, in the asymmetric unit. This organization in the crystal is highly unlikely to be a physiologically relevant assembly since the membrane-proximal subdomain of one subunit packs against the membrane-distal subdomain of the other. As the monomers in the asymmetric unit were virtually identical and both were ligand bound, we here only show and discuss the A-chain of each structure. The final structure models correspond to residues 32–187 or 32–189 for the d-Arg and d-Lys-bound structures, respectively.

a, Domain architecture of MCPDRK protein generated by alphafold40,41,42, showing one subunit in rainbow colours, with the N terminus in blue and C terminus in red. The full-length MCPDRK transmembrane chemoreceptor contains a ligand binding domain (LBD), a transmembrane (TM) and a signalling domain. b, MCPDRK-LBD monomers bound to d-Arg (purple spheres) or d-Lys (orange spheres) are coloured in rainbow colours from the N terminus (blue) to the C terminus (red). c, Superimposition of the LBD domain of MCPDRK crystallized with d-Arg (in purple) and d-Lys (in orange) with the aspartate-bound Tar receptor from E. coli (PDB ID: 4z9i in green)81, the citrate bound MCP2201 chemoreceptor from C. testosterone (PDB ID: 5XUB, in blue)34 and the more promiscuous quinate-bound PcaY_PP chemoreceptor from P. putida (PDB ID: 6S38, in yellow)35. The Tar and the PcaY_PP receptor ligand binding sites are located at the dimer interface and are therefore shown as dimers. d, The binding site of MCPDRK-LBD bound to d-Arg (in purple). e, The binding site of MCPDRK-LBD bound to d-Lys (in orange). f, Functional analysis of the d-Arg binding pocket was performed by substituting residues Asn43, Asp47, Thr48, Thr105, W107, Glu111 and Asn176 with an alanine. Chemotactic response to d-Arg was then analysed for ΔmcpDKR cells expressing either wild-type or mutant MCPDRK under its native promoter. ΔbsrV strain was used as background. −, not complemented ΔmcpDRK. Black diamonds represent the mean of 3 independent biological replicates. Significant differences (unpaired t-test) are indicated by *P < 0.05 or **P < 0.01.
Overall, MCPDRK-LBD forms an antiparallel four-α-helix bundle structure (4HB) (Fig. 3b). This fold is distinct from the dCACHE LBD-type fold that is found in other chemoreceptors from V. cholerae that respond to l-amino acids (Mlp24, Mlp37) (refs. 11,12), but has been found in various LBDs from other Gram-negative bacteria. A structure-based homology search via the DALI server33 showed that MCPDRK-LBD was structurally most similar to the citrate-binding chemoreceptor MCP2201 from Comamonas testosterone (PDB ID: 5XUB, RMSD of 1.7 Å) (ref. 34) and the more promiscuous PcaY_PP chemoreceptor from Pseudomonas putida (PDB ID: 6S38, RMSD of 2.1 Å) (ref. 35), but also showed similarity to the well-characterized aspartate-binding Tar receptor from E. coli (PDB ID: 4Z9I, RMSD of 2.5 Å) (ref. 36). While these LBDs share a similar overall fold, their ligand binding sites are at entirely different locations (Fig. 3c). For instance, the model receptors Tar and Tsr bind ligands at the dimer interface with negative cooperativity37,38,39, while MCPDRK binds d-Arg and d-Lys centrally and remains as monomer instead (Fig. 3c and Supplementary Fig. 3). As for MCPDRK, the chemoreceptor MCP2201 from C. testosterone also binds the ligand to a single protein chain, but at a different location (Fig. 3c)34.
In the MCPDRK-LBD structures, both d-Arg and d-Lys bind at the same site, at the bottom of the second and the third helix (Fig. 3b). To bind the ligands, Asp47 forms an ionic bond with the positively charged α-amine group of both DAAs. Further, Glu111 and Asp47 interact with the delocalized positive charge of the guanidino group from d-Arg; in a similar manner, the positively charged side-chain amine of d-Lys forms a salt bridge with Glu111. The ligands are further stabilized by hydrogen bonds with residues from all helices, namely Asn43, Thr48, Thr105, Glu111 and Asn176 (Fig. 3d,e). Trp107, from the short loop between helix 2 and 3, forms a lid that closes off the binding site and has a stacking interaction with the ligands.
To further characterize the precise mechanism of ligand specificity, we generated seven individual mutants in which each amino acid residue that interacts with the ligands according to the crystal structures was replaced with alanine (N43A, D47A, T48A, T105A, W107A, E111A and N176A). Complementation assays were performed by introducing these mutant alleles under the control of mcpDRK native promoter interrupting the lacZ locus in the ΔmcpDRK strain. Chemotaxis assays demonstrated the essentiality of 5 of the 7 MCPDRK-LBD residues tested (N43, T48, W107, E111 and N176) to promote a repulsive response to d-Arg in V. cholerae (Fig. 3f). d-Arg-mediated taxis was not affected by the N43A and T105A mutations, suggesting a less relevant role for these residues in stabilizing the d-Arg and d-Lys. These in vivo assays thus confirmed the location of the ligand binding site that was found in the crystal structure.
Conservation of MCPDRK
To analyse the conservation of MCPDRK, we built a phylogenetic tree on the basis of sequence similarity (Fig. 4a). We found that this chemoreceptor is widely conserved in the Gamma-proteobacteria orders Aeromonadales, Alteromonadales and Vibrionales, which are known to be polar flagellated bacteria that are commonly found in aquatic environments. MCPDRK-like proteins cluster into three groups on the basis of their percentage of homology and genomic association with the BsrV racemase (Extended Data Fig. 7). MCPDRK orthologues with the highest protein identity (>50%) cluster together with a BsrV-like racemase (for example, many Vibrio species and Moritella sp.), suggesting coevolution of d-amino acid production and chemotactic recognition. In these orthologues, the d-Arg/d-Lys binding site residues are either fully conserved or have only subtle changes (for example, V. metschnikovii, V. cincinnatiensis and V. anguillarum; see Extended Data Fig. 7). Species with MCPDRK orthologues of slightly lower identity (48–51%) (for example, Aeromonas and Aliivibrio) form a single clade and typically show a substantial change in two important binding site residues: D > S in position 47, resulting in a loss of the salt bridge with the ligand, and W > G in position 107, which removes the stacking interaction with the ligand. Remarkably, in these species, the BsrV-like racemase does not form an operon with the chemoreceptor but is encoded elsewhere in the genome. Finally, the species encoding MCPDRK homologues of lower identity (<45%) (for example, V. owensii) contain neither the d-Arg/d-Lys binding residues nor a BsrV-like locus.

a, Tree of MCPDRK orthologues. The cladogram was constructed on the basis of MCPDRK sequence homology. Orthologues selected for an extended comparison are highlighted in bold. b, Left: selection of MCPDRK orthologue representatives and the conservation of d-Arg binding residues. Only the residues that differ from the reference protein (VC1313, that is, MCPDRK from V. cholerae El Tor strain N16961, boxed) are labelled. Coloured circles indicate the amino acid type (N, asparagine; D, aspartic acid; T, threonine; W, tryptophan; E, glutamic acid; S, serine; A, alanine; G, glycine). Right: genetic context of the chemoreceptor in several species. Both the MCP and the racemase are highlighted. The percentage of total protein identity (%) is shown. c, Chemotactic response of MCPDRK orthologue proteins to d-Arg. The chemotactic response to d-Arg was tested for ΔmcpDKR cells complemented with several orthologue MCPs expressed under the mcpDKR native promoter. ΔbsrV strain was used as background. −, not complemented ΔmcpDRK; VC, V. cholerae; VV, V. vulnificus; VPA, V. parahaemolyticus; VFU, V. furnissii; VA, V. anguillarum; AF, Aliivibrio fischeri; AH, Aeromonas hydrophyla; VO, V. owensii. Black diamonds represent the mean of 4 independent biological replicates. Significant differences (unpaired t-test) are indicated by **P < 0.01.
To assess the potential functional divergency of the MCPDRK chemoreceptor across species, we selected three orthologues from the pool of representative genomes that we used to build the phylogenetic tree. These were V. anguillarum VAA_1440 and V. parahaemolyticus VPA_1000, which cluster with a BsrV-like racemase and show a conserved binding site. We also selected A. fischeri VF_A1069, which does not form an operon with the racemase and has a partially conserved binding site (Fig. 4b). LBD model analysis on models generated by alphafold40,41,42 predicted defective d-Arg stabilization caused by changes in the VF_A1069 binding site as compared to the canonical one (that is, V. cholerae) (Supplementary Fig. 3). To study this, we monitored by capillary assays the response to d-Arg of the ΔmcpDRK mutant strain complemented with the selected orthologues expressed under the mcpDRK native promoter. As expected, we found that expression of VAA_1440 (68.6% identity, 100% conserved binding site) and VPA_1000 (65.7% identity, 100% conserved binding site) restored the ability of the ΔmcpDRK mutant to sense d-Arg. The expression of VF_A1069 (48.1% identity, 71% conserved binding site) did not complement the chemotactic response to d-Arg (Fig. 4c), probably because this orthologue does not conserve the D47 and W107 residues (Supplementary Fig. 3) that were found to be essential for d-Arg binding (Fig. 3). Together, these results suggest that MCPDRK-like proteins specific for d-Arg and d-Lys are those encoded nearby a bsr locus.
Discussion
Bacterial chemotaxis has been the subject of decades of research. Although the signal transduction components have been extensively investigated, less is known about the identity and specificity of the chemoreceptors that bacteria use to sense their surroundings43. Given the intimate association between MCPs and a bacterium’s lifestyle, these chemoreceptors are generally numerous and specific, suggesting that many MCP–ligand pairs remain uncharacterized6.
We previously reported that many bacteria release high concentrations of a variety of d-amino acids to the environment. d-amino acids display different biological functions depending on their chemical properties. For instance, stationary-phase V. cholerae cells produce d-Met to adapt their cell wall biogenesis to growth arrest mode17 while they use d-Arg to inhibit the growth of nearby competitors15. While d-Arg is lethal to many species, V. cholerae’s physiology appears to be unaffected by this d-amino acid15. Using a mutant defective in d-amino acid production, we report here that d-Arg is sensed by a previously unrecognized MCP to move the V. cholerae population away from stressful environments.
It is interesting that among all the d-amino acids produced by V. cholerae, only the toxic d-Arg (and d-Lys) induce negative chemotaxis. This means that d-Arg plays multifaceted roles in shaping the biodiversity and structure of microbial communities when nutrients are scarce by clearing the environment of potential competitors while also migrating the community towards more favourable niches. For this ‘fight and flight’ strategy, V. cholerae has evolved to arrange the genes encoding the d-Arg-sensing MCP (MCPDRK) and the broad-spectrum racemase (BsrV) together in one operon whose expression is controlled by the stress sigma factor RpoS. By synchronizing the production of d-Arg and its MCP, V. cholerae builds an efficient stress-dependent response while simultaneously preventing its futile activation under favourable conditions (Fig. 5). In fact, when the MCPDRK is not expressed, d-Arg stimulates a mild chemotactic attraction that suggests the existence of other MCPs with lower affinity for this ligand. Indeed, our TPP experiments identified potential MCP candidates whose stability is affected by d-Arg, such as VC2161 (Mlp24) and VC1406 (Fig. 1f). Chemotaxis towards d-Arg may have a nutritional purpose. This idea is supported by results in E. coli showing that certain compounds with low nutritional value can be chemoattractants44 and by the presence of a catabolic d-amino acid dehydrogenase enzyme45.

Under certain environmental stresses (that is, starvation), the RpoS response induces the expression of both the broad-spectrum racemase BsrV and the dedicated d-Arg/d-Lys chemoreceptor MCPDRK. Among the d-amino acids (DAAs) produced by BsrV, d-Arg and d-Lys stand out as warning signals sensed by V. cholerae as well as other MCPDRK-encoding species. Chemotactic run-away response to these d-amino acids enables these bacterial communities to move away and explore more favourable niches.
The crystal structures of the LBD of MCPDRK showed that it has a typical antiparallel 4HB structure, which is conserved in various receptors46. Interestingly, this relatively simple fold has evolved to recognize a variety of substrates by using different binding sites. The binding site of Tar, Tsr and the PcaY_PP receptors is located at the dimer interface, while MCPDRK and MCP2201 bind their ligands centrally within the 4HB (Fig. 3c). MCPDRK’s d-Arg-binding pocket is located at the bottom of the second and the third helix, where only five residues are essential for recognizing this d-amino acid and similar compounds such as d-Lys. Contrary to the 4HB-containing model receptors, that is, Tar and Tsr, our purified MCPDRK-LBD construct does not oligomerize in the presence of the ligand. This is also in contrast with MCP2201, where ligand binding inside the monomer induces a structural rearrangement that prevents dimerization of the LBD, instead stimulating trimerization34.
Interestingly, although MCPDRK is broadly conserved among species from three Gamma-proteobacteria orders, the specificity for d-Arg/Lys seems to be evolutionarily restricted to receptors that are transcriptionally linked to broad-spectrum racemases such as BsrV. As some of these species are both tolerant to d-Arg toxicity15 and inhabit aquatic habitats, d-Arg/d-Lys produced by one of these species could simultaneously coordinate the chemotactic response for many inhabitants of the same niche. Conversely, species that lack a Bsr homologue or encode it somewhere else in the genome display changes in the ligand binding domain that are likely to disable the chemotactic response to d-Arg/d-Lys (Fig. 4 and Extended Data Fig. 7). While some of these changes imply critical structural variations in the binding site (Supplementary Fig. 3), more subtle ones could instead recognize alternative signals that are chemically related to d-Arg. Indeed, multiple arginine analogues are known to be naturally produced, such as l-homoarginine in humans and animals47 and l-canavanine and l-indospicine produced by certain plants48. Interestingly, canavanine can be racemized by microbial Bsr enzymes, suggesting that it should be possible to find both l- and d-enantiomers of arginine analogues in the natural environment49. Future studies should investigate how interspecies and interkingdom competing signals for the same MCP may impact chemotaxis regulation and hence bacterial capacity to respond to stress in complex polymicrobial environments.
The role of d-amino acids as chemotactic repellent signals adds to previously reported roles for these molecules in stationary-phase adaptation. An obvious ecological scenario where the d-Arg run-away response could be beneficial is abandoning spent environments. It is known that V. cholerae forms biofilms during the aquatic and intestinal phases of its life cycle50; when the biofilm ages, RpoS controls biofilm detachment51. In a host environment, RpoS-dependent activation of chemotaxis and motility is linked to mucosal escape. Given that this response coincides with a reduction in the expression of virulence genes, it has been proposed as a preparation of V. cholerae for the next stage in its life cycle52. While several degradative proteins contributing to biofilm disassembly have been reported in recent years (for example, in Vibrio25), the signal(s) promoting biofilm dispersal remain enigmatic. Therefore, an exciting question for the future will be whether d-Arg’s dual function as a broad-spectrum toxin and chemotactic warning signal could play a role in biofilm destablization and dispersal. In this line, a study recently reported that the MCPDRK-BsrV operon (vc1313-1312) is part of the in-biofilm repressome and that constitutive expression of some of these genes interferes with biofilm formation53. However, the loss of MCPDRK was reported to have no effect on V. cholerae’s fitness in infection or dissemination54. While the effect of d-Arg chemotaxis on virulence and transmission remains to be studied, the widespread presence of this chemoreceptor in other marine non-pathogenic bacteria suggests that this response could be an important and overlooked aspect of bacteria’s ability to assess niche habitability.
Methods
Bacterial growth conditions
Strains are summarized in Supplementary Table 4. All V. cholerae strains are derivatives of the sequenced N16961, an El Tor clinical isolate55.
Bacteria were grown using standard laboratory conditions. Cultures were streaked onto LB plates (1% tryptone, 0.5% yeast extract, 1% NaCl, 15% bacto-agar) supplemented with antibiotics or amino acids (see below), and single colonies were selected and grown overnight at 30 °C in tryptone broth (TB) (1% tryptone, 0.5% NaCl) and subcultured in fresh TB until the desired growth phase, unless otherwise specified. Antibiotics were added when necessary at the following concentrations: 200 μg ml−1 streptomycin (Sm, Duchefa Biochem, S0148), 100 μg ml−1 carbenicillin (Cb, Duchefa Biochem, C0109) and 50 μg ml−1 kanamycin (Kn, Duchefa Biochem, K0126). Soft-agar plates were supplemented with 5 mM amino acids (Sigma). Optical density (OD) was measured in cuvettes using an Ultrospec 7000 spectrophotometer (GE Healthcare). For experiments in minimal medium, V. cholerae was grown in either M9 minimal medium supplemented with 1 μg ml−1 thiamine or MSR6 (50 mM KH2PO4, 50 mM Na2HPO4, 7.5 mM (NH4)2SO4, 2 mM MgSO4, 0.1 mM CaCl2, 25 μM FeSO4, 0.04% w/v d-glucose and 0.4% w/v succinate).
For growth curves, stationary-phase cultures were normalized to an OD of 0.04 and 20 μl were used for inoculating 96-well plates containing 180 μl of fresh medium. At least 3 replicates per strain and condition were inoculated in 2 independent experiments. Optical density was monitored using Biotek Gen5 (v.08) in an Eon Biotek plate reader (Biotek) at 5 min intervals at 30 °C.
Construction of mutants and plasmids
All mutants were generated by in vivo allelic exchange as previously described56. In brief, 1 Kb flanking regions were cloned into XbaI-digested pCVD442 to generate recombinant suicide vectors. For conjugations into V. cholerae, stationary-phase recipients and Sm10 λpir donor strains were washed of antibiotics, mixed in equal ratios and spotted onto LB without selection. After 4–6 h incubation at 37 °C, cells were streaked onto Cb/Sm-supplemented LB plates to select for transconjugants. Finally, cells were cured through a counterselection round on salt-free 10% sucrose plates. The chromosomal fluorescent sfGFP-vc1313 reporter was constructed in a similar way, directly inserting sfGFP upstream of vc1313 in-frame.
Plasmids and oligonucleotides used are summarized in Supplementary Tables 5 and 6, respectively. Plasmids were constructed by standard DNA cloning techniques. bsrV was PCR amplified using the appropriate primers including a strong ribosomal binding site (RBS) and cloned into restriction-digested pXB300 by isothermal assembly, yielding plasmid pbsrV. Similarly, for the generation of the phosphomimetic non-chemotactic mutant, cheY3 was PCR amplified with primers carrying the desired substitutions (CheY3D16K, Y109K), and the fragment was isothermally assembled into restriction-digested pXB300 to generate pcheY3**. Plasmids pbsrV and pcheY3** were introduced into ΔbsrV and ΔcheY3, respectively, via electroporation.
The DNA fragment encoding the entire periplasmic domain (residues Ser32–Ser191) of vc1313 (herein referred to as vc1313-LBD) was PCR amplified and subcloned into BamHI/EcoRI-digested expression vector pGEX-6P-2 by isothermal assembly so that the N-terminal GST-encoding sequence was fused in-frame to yield pGEX-vc1313-LBD. The stop codon located downstream of the multicloning region was used, therefore the resulting protein contained an additional C-terminal 12-amino acid arginine-rich tail.
Δvc1313 complementation was achieved by expressing a copy of vc1313 under the control of its native promoter introduced in neutral locus lacZ. vc1313 fragment was PCR amplified (including 500 bp upstream of the promoter region) and isothermal assembled into restriction-digested pJL1, a suicide plasmid that permits allele insertion into V. cholerae lacZ57. Successful integration was first verified by blue–white screening on plates containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal, 40 μg ml−1, Sigma, B4252) and later checked by PCR. Site-directed mutagenesis of the vc1313 gene was carried out following Q5 site-directed mutagenesis protocol (NEB) using pJL1::vc1313 plasmid as template.
The fidelity of all generated mutants and plasmids was confirmed by DNA sequencing.
Soft-agar motility assay
Single colonies of V. cholerae strains were grown overnight in LB broth at 30 °C with shaking, subcultured 1:100 in fresh broth and grown until exponential phase. A 2 μl volume of the normalized cultures were then spotted into 0.3% soft-agar plates and drops were allowed to dry at room temperature. When indicated, plates were supplemented with 5 mM of amino acids. After inoculation, plates were incubated for 8 h at 30 °C and then imaged in a LAS-3000 imaging system (Fuji). Image analysis and processing were performed using Fiji/ImageJ58 and results were plotted using GraphPad Prism (v.8.0).
d-amino acids quantification
Total d-amino acid concentration was determined by a colorimetric d-amino acid oxidase (DAAO) assay adapted to a 96-well plate format as previously described14,15. Briefly, 20 µl of sample were mixed with 60 µl of reaction buffer (sodium phosphate buffer 33 mM, pH 7.5, Trigonopsis variabilis DAAO (ref. 59) 0.04 mg ml−1, horseradish peroxidase 0.04 mg ml−1, o-phenylenediamine 83 ng ml−1 and flavin adenine dinucleotide (FAD) 8.3 ng ml−1) and incubated for 1 h at 37 °C. The reactions were inactivated by adding 160 μl 2 M HCl and measurements were taken at 292 nm in an Eon Biotek plate reader (Biotek).
Capillary assays
Chemotactic ability was examined using previously described capillary assays60, with some modifications. An overnight culture of V. cholerae was diluted 1:200 into fresh M9 minimal medium and incubated until late exponential phase at 30 °C with shaking. Cells were collected by low-speed centrifugation, washed twice with motility buffer (MB) (50 mM HEPES (pH 7.4), 300 mM NaCl, 10 mM glucose, 5 mM MgCl2) and finally resuspended to a final OD of 0.1 in MB. Tween20 was added to a final concentration of 0.01% to avoid cell adhesion. After preincubation of cells at r.t. for 1 h, pre-filled 1 µl microcapillary tubes (Sigma, P1421) containing the amino acids of interest (500 µM, unless specified otherwise) were placed in contact with the cell suspension and the suspension was incubated at r.t. for an additional 2 h. Finally, the number of bacteria (colony-forming units per ml) in each capillary was counted by plating serial dilutions on LB agar, and chemotaxis ratio was calculated relative to a control capillary.
Microfluidic chemotaxis experiments
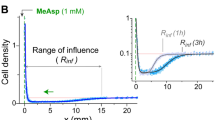
Microfluidics chambers were fabricated as previously described29. Briefly, the SU8-based negative master mold of the microfluidic devices was produced by standard photolithography techniques. Microfluidics devices were produced from the mold using polydimethylsiloxane (1:10 crosslinker-to-base ratio, thermal-cured overnight at 65 °C), which was cut to shape and bound to glass slides via oxygen plasma treatment. After production, devices were stored short-term in sterile deionised water to retain their hydrophilic properties. V. cholerae strains were grown overnight in TB at 30 °C from glycerol stocks, diluted 1:100 in 10 ml M9 medium and grown for 4.5 h at 30 °C with shaking until an OD of ~0.3. The day culture was washed three times in motility buffer and diluted to a final OD of 0.1. The microfluidic device consists of two large chambers linked by a small straight channel (length = 2 mm, width = 1 mm, height = 70 µm). The suspension of cells at OD = 0.1 was introduced in one chamber, while the second was filled with MB supplemented with indicated amounts of the tested compound. The device’s inputs were then sealed with microscopy grease to prevent evaporative flows. A gradient of the compound forms rapidly (~1 h) in the connecting channel. The resulting chemotactic motion of the cells was measured in the centre of the channel 1, 2 and 3 h after loading the device. To this end, the sample was observed under phase contrast microscopy at ×10 magnification (NA 0.3) and a 10,000-frames movie was recorded at a rate of 200 frames per second using a Mikrotron Eosens 4CXP CMOS camera with a 717 × 717 µm² (512 × 512 px²) field of view. A control with the same cell suspension but only MB in the second chamber was always performed simultaneously.
Swimming speed experiments
V. cholerae strains were grown overnight in TB (supplemented with 500 µM d-Arg if indicated) at 30 °C from glycerol stocks, diluted 1:200 in 20 ml TB (+500 µM d-Arg if indicated) and grown for 3 h at 30 °C with shaking until an OD of ~0.6. The cells were then washed into fresh TB. A small volume (3 µl) of the suspension of cells was sandwiched between two coverslips maintained 100–200 µm apart by microscopy grease. Cell motion was recorded at mid-height between the coverslips using the same microscopy protocol as described above.
Swimming speed and chemotactic bias measurement
Movies were analysed offline using the differential dynamic microscopy (DDM) and phase differential microscopy (PhiDM) methods implemented as plugins in ImageJ (https://github.com/croelmiyn/FourierImageAnalysis) to measure swimming speed61 and chemotactic drift62, respectively. The DDM algorithm computes the time autocorrelation of the spatial Fourier components of the images, which are then fitted with a model assuming that a fraction of the cells is non-motile and diffusing, and the other is motile, with swimming speeds being Schulz distributed around a mean. Consistent fits across the different Fourier components yield the fraction of swimming cells \(\Phi\) and their average swimming speed \({v}_{0}\). With PhiDM, the temporal shift in the phase of the same Fourier components is fitted to measure the population-averaged drift velocity of the cells in the field of view, \({v}_{d}\). In the absence of external flows, this drift is solely due to the chemotactic motion of the motile cells since the non-motile cells do not drift on average. The chemotactic drift velocity of the motile cells was therefore computed as \({v}_{{\mathrm{ch}}}={v}_{d}/\Phi\). Since the chemotactic drift velocity is expected to scale with the swimming speed of the cells, we also computed the chemotactic bias \(b={v}_{{\mathrm{ch}}}/{v}_{0}={v}_{d}/\Phi {v}_{0}\), which must lie in the range (−1, 1) and estimates the angular motion bias of the cells up (b > 0) or down (b < 0) the gradient.
Thermal proteome profiling
V. cholerae cells were grown overnight at 37 °C in LB and diluted 100-fold into 100 ml of fresh LB. Cultures were grown aerobically at 37 °C with shaking until an OD of ~2. Cells were pelleted at 4,000 × g for 5 min, washed with 10 ml PBS and resuspended in 1.2 ml of lysis buffer (final concentration: 50 μg ml−1 lysozyme, 250 U ml−1 benzonase and 1 mM MgCl2 in PBS). Cells were lysed by 5 repeated cycles of freeze–thawing (freezing in liquid nitrogen, followed by 5 min at 25 °C with shaking until complete thawing). Then, d-Arg was added at 4 different concentrations (0, 0.05, 0.4, 3.2 mM) and 20 μl was aliquoted to a PCR plate and immediately subjected to a temperature gradient for 3 min in a PCR machine (Agilent SureCycler 8800), followed by 3 min at room temperature. NP40 was then added to all conditions to a final concentration of 0.8%. The plate was then centrifuged at 2,000 × g for 5 min to remove cell debris and the supernatant was filtered at 500 × g for 5 min through a 0.45 μm 96-well filter plate (Millipore, MSHVN4550) to remove protein aggregates. The flow-through was mixed 1:1 with 2× sample buffer (180 mM Tris (pH 6.8), 4% SDS, 20% glycerol, 0.1 g bromophenol blue) and kept at −20 °C until analysis. Mass spectrometry-based proteomics (using tandem mass tags) was then used to quantify the amount of protein in each condition, as previously described63. Data were analysed by a method to detect dose-dependent changes in protein thermal stability27.
Western blotting to determine presence of vc1313
An overnight culture of V. cholerae sfGFP-vc1313 was diluted 1:100 into fresh LB minimal medium and incubated at 37 °C with shaking. OD600 was measured and samples were taken as a function of time. Samples were then normalized to total protein amount and analysed by SDS–PAGE. Western blotting was performed using specific antibodies against GFP-tag (1:500, Thermo Fisher, A-11122). GFP signal was detected using Amersham Imager 600 (GE) and analysed using Fiji/ImageJ (v.1.53f51) (ref. 58).
Time-lapse microscopy
Bacterial cells were immobilized onto agarose pads (1% agarose w/v, 20% v/v PBS and 20% v/v LB) on microscope slides. Phase contrast microscopy was performed using a Zeiss Axio Imager.Z2 microscope (Zeiss) equipped with a Plan-Apochromat ×63 phase contrast objective lens and an ORCA-Flash 4.0 LT digital CMOS camera (Hamamatsu Photonics) using the Zeiss Zen 2 Blue edition (v2.0.0.0) software. Image analysis and processing were performed using Fiji/ImageJ (v.1.53) (ref. 58) and MicrobeJ plugin (v.5.13) (ref. 64).
To follow the detailed localization of sfGFP-MCPDRK during growth, cells were added into 50 ml of TB medium and growth was followed by measuring optical density. For each datapoint, samples were analysed by fluorescence microscopy and the percentage of cells with sfGFP-MCPDRK foci was counted. For the carbon catabolite repression assays, cells were grown in MSR6 medium supplemented with 0.04% w/v d-glucose and 0.4% w/v succinate, and a similar procedure was followed.
Demographic analysis of microscopy data
Demographic analysis was performed in several steps. Fluorescence intensity profiles of sfGFP-VC1313 cells were first measured using Fiji/ImageJ (v.1.53f51) (ref. 58). The generated data were then processed using the cellProfiles package (v.3.0.1; developed by Todd Cameron) in RStudio (v.1.4.1106; www.rstudio.com) to sort cells by length and normalize the generated intensity profiles as an average of each cell’s fluorescence. Finally, the ggplot2 package was used to plot the demographics (v.3.3.5) (ref. 65).
Expression and purification of MCPDRK-LBD
E. coli strain BL21 (DE3) (Novagen) carrying pGEX-vc1313-LBD was cultured in Terrific Broth (24 g l−1 yeast extract, 20 g l−1 tryptone, 4 ml l−1 glycerol, 0.017 M KH2PO4, 0.072 M K2HPO4) containing 50 μg ml−1 carbenicillin at 30 °C until cell density reached an OD600 of ~1. Isopropyl β-d-1-thiogalactopyranoside (Sigma, I6758) was then added to a final concentration of 0.1 mM to induce protein expression, and the culture was incubated for an additional 16 h at 18 °C. Cells were collected by centrifugation, suspended in 1x PBS (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 1.8 mM KH2PO4 at pH 7.3) and disrupted using a pressure cell homogenizer FC600 (Julabo) followed by centrifugation at 70,000 × g at 4 °C for 1 h. After removing cell debris, VC1313-LBD was purified from the soluble fraction by affinity chromatography using Glutathione Sepharose 4B resin (Cytiva) previously equilibrated with PBS. Following 3 washing steps with cleavage buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol (pH 7)), GST-VC1313 was eluted with 50 mM Tris-HCl (pH 8.0) buffer containing 10 mM reduced glutathione. Cleavage buffer was added to a final 1x concentration and the N-terminal GST tag was then cleaved using PreScission Protease (Cytiva) following manufacturer instructions (16 h at 4 °C). The resulting sample was then dialysed in 2 l dialysis buffer (50 mM Tris-HCl (pH 7.0), 150 mM NaCl, 1 mM dithiothreitol and 1 mM EDTA) at 4 °C for 16 h using Slide-A-Lyzer dialysis cassettes, 7K MWCO (ThermoFisher,66370). Afterwards, the protein mixture was incubated with Glutathione Sepharose 4B resin to remove cleaved GST, PreScission protease and the remaining GST-tagged VC1313. The flow-through was loaded on a Superdex 200 Increase 10/300 GL column (Cytiva) in 10 mM Tris-HCl and 150 mM NaCl (pH7.4). The purity of MCPDRK-LBD was analysed by SDS–PAGE.
Crystallization and structure determination
Purified MCPDRK-LBD was loaded on a Superdex 200 Increase 10/300 GL (Cytiva) equilibrated in 10 mM Tris and 150 mM NaCl (pH 7.4). Protein peak fractions were concentrated to 20–30 mg ml−1 on a 10 kDa cut-off Amicon Ultra centrifugal filter (Merck–Millipore).
Before setting up the drops, 13 mg ml−1 MCPDRK-LBD was mixed with either 10 mM d-arginine or 10 mM d-lysine, both from a 200 mM stock. The protein–ligand mixtures were mixed with reservoir solution at a 3:1 ratio. All crystals were grown at 20 °C by sitting-drop vapour diffusion in conditions containing 0.1 M Tris (base) bicine (pH 8.6–8.8), 17–21% v/v poly(ethylene glycol) methyl ether 500 (PEG 500 MME) and 8–10% w/v PEG 20000. Before flash freezing the crystal in liquid nitrogen, the PEG concentrations were raised to 23% v/v PEG 500 MME and 12% w/v PEG 20000.
X-ray diffraction data of the different crystals were collected on the beamlines ID30B and ID23-2 at the European Synchrotron Radiation Facility in Grenoble, France66. The data were processed using XDS67. The d-lysine and d-arginine crystals both belonged to the P1 space group and contained 2 molecules in the asymmetric unit. The crystallographic phase problem of the MCPDRK-LBD bound to d-arginine was solved by Arcimboldo68 using 8 helices of 14 residues as a search model. The structures of MCPDRK-LBD bound to d-lysine were solved by PHASER69 using the MCPDRK-LBD:d-Arg structure as a molecular replacement model. Coot (0.9.5) (ref. 70) was used to build the models and the structures were refined using Refmac5 (v.5.8.0267) (ref. 71) and PHENIX refine (v.1.13) (ref. 72). For complete data collection and refinement statistics, see Supplementary Table 3. The final models of the MCPDRK-LBD structures were validated using MolProbity73. Atomic coordinates and structure factors of the MCPDRK-LBD crystal structures (in complex with d-Arg or d-Lys) have been deposited in the Protein Data Bank (PDB codes 8BSA and 8BSB, respectively).
Phylogenetic analysis
Nucleotide and amino acid sequences were obtained from the National Center for Biotechnology Information (NCBI)74 and the Biocyc database75. MCPDRK homologues were identified using the BlastP algorithm76 against a non-redundant protein database, with an E-value threshold of 10−10 and with V. cholerae VC1313 as reference sequence. The MUSCLE algorithm77 was used to generate the multiple sequence alignment. The resulting alignment was then filtered by removing 90% of redundancy, and unique representative species were selected for further analysis. The Jalview (v.2.11.2.0) toolkit78 was used to visualize all the alignments and analyse the conservancy of residues of interest. The phylogenetic tree was generated using ClustalW2 (ref. 79), and Interactive Tree of Life (iTOL, v.5) (ref. 80) was then used to further customize the phylogenetic tree and add additional information. The genetic context analysis was carried out by comparing upstream/downstream regions of the selected representatives.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD038312.
Atomic coordinates and structure factors of the MCPDRK-LBD crystal structures (in complex with d-Arg (PDB: 8BSA) and d-Lys (PDB:8BSB)) have been deposited in the Protein Data Bank. Source data are provided with this paper.
References
Toews, M. L., Goy, M. F., Springer, M. S. & Adler, J. Attractants and repellents control demethylation of methylated chemotaxis proteins in Escherichia coli. Proc. Natl Acad. Sci. USA 76, 5544–5548 (1979).
Scharf, B. E., Fahrner, K. A., Turner, L. & Berg, H. C. Control of direction of flagellar rotation in bacterial chemotaxis. Proc. Natl Acad. Sci. USA 95, 201–206 (1998).
Sourjik, V. & Wingreen, N. S. Responding to chemical gradients: bacterial chemotaxis. Curr. Opin. Cell Biol. 24, 262–268 (2012).
Sourjik, V. Receptor clustering and signal processing in E. coli chemotaxis. Trends Microbiol. 12, 569–576 (2004).
Martín-Mora, D. et al. Functional annotation of bacterial signal transduction systems: progress and challenges. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19123755 (2018).
Ortega, Á., Zhulin, I. B. & Krell, T. Sensory repertoire of bacterial chemoreceptors. Microbiol. Mol. Biol. Rev. 81, e00033-17 (2017).
Gavira, J. A. et al. How bacterial chemoreceptors evolve novel ligand specificities. mBio 11, e03066-19 (2020).
Boin, M. A., Austin, M. J. & Häse, C. C. Chemotaxis in Vibrio cholerae. FEMS Microbiol. Lett. 239, 1–8 (2004).
Gosink, K. K., Kobayashi, R., Kawagishi, I. & Häse, C. C. Analyses of the roles of the three cheA homologs in chemotaxis of Vibrio cholerae. J. Bacteriol. 184, 1767–1771 (2002).
Ortega, D. R., Kjær, A. & Briegel, A. The chemosensory systems of Vibrio cholerae. Mol. Microbiol. 114, 367–376 (2020).
Nishiyama, S. et al. Mlp24 (McpX) of Vibrio cholerae implicated in pathogenicity functions as a chemoreceptor for multiple amino acids. Infect. Immun. 80, 3170–3178 (2012).
Nishiyama, S. I. et al. Identification of a Vibrio cholerae chemoreceptor that senses taurine and amino acids as attractants. Sci. Rep. 6, 20866 (2016).
Boin, M. A. & Häse, C. C. Characterization of Vibrio cholerae aerotaxis. FEMS Microbiol. Lett. 276, 193–201 (2007).
Espaillat, A. et al. Structural basis for the broad specificity of a new family of amino-acid racemases. Acta Crystallogr. D 70, 79–90 (2014).
Alvarez, L., Aliashkevich, A., de Pedro, M. A. & Cava, F. Bacterial secretion of d-arginine controls environmental microbial biodiversity. ISME J. 12, 438–450 (2017).
Cava, F., de Pedro, M. A., Lam, H., Davis, B. M. & Waldor, M. K. Distinct pathways for modification of the bacterial cell wall by non-canonical d-amino acids. EMBO J. 30, 3442–3453 (2011).
Lam, H. et al. d-amino acids govern stationary phase cell wall remodeling in bacteria. Science 325, 1552–1555 (2009).
Kolodkin-Gal, I. et al. d-amino acids trigger biofilm disassembly. Science 328, 627–629 (2010).
Rumbo, C. et al. Assessment of antivirulence activity of several d-amino acids against Acinetobacter baumannii and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 71, 3473–3481 (2016).
Bucher, T., Oppenheimer-Shaanan, Y., Savidor, A., Bloom-Ackermann, Z. & Kolodkin-Gal, I. Disturbance of the bacterial cell wall specifically interferes with biofilm formation. Environ. Microbiol. Rep. 7, 990–1004 (2015).
Leiman, S. A. et al. d-amino acids indirectly inhibit biofilm formation in Bacillus subtilis by interfering with protein synthesis. J. Bacteriol. 195, 5391–5395 (2013).
Hills, G. Chemical factors in the germination of spore-bearing aerobes; the effects of amino acids on the germination of Bacillus anthracis, with some observations on the relation of optical form to biological activity. Biochem. J. 45, 363–370 (1949).
Wu, D. et al. Residues Asp164 and Glu165 at the substrate entryway function potently in substrate orientation of alanine racemase from E. coli: enzymatic characterization with crystal structure analysis. Protein Sci. 17, 1066–1076 (2008).
Schuster, M., Zhao, R., Bourret, R. B. & Collins, E. J. Correlated switch binding and signaling in bacterial chemotaxis. J. Biol. Chem. 275, 19752–19758 (2000).
Bridges, A. A., Fei, C. & Bassler, B. L. Identification of signaling pathways, matrix-digestion enzymes, and motility components controlling Vibrio cholerae biofilm dispersal. Proc. Natl Acad. Sci. USA 117, 32639–32647 (2020).
Mateus, A. et al. Thermal proteome profiling in bacteria: probing protein state in vivo. Mol. Syst. Biol. 14, e8242 (2018).
Kurzawa, N. et al. A computational method for detection of ligand-binding proteins from dose range thermal proteome profiles. Nat. Commun. 11, 5783 (2020).
Mateus, A. et al. Thermal proteome profiling for interrogating protein interactions. Mol. Syst. Biol. 16, e9232 (2020).
Colin, R., Drescher, K. & Sourjik, V. Chemotactic behaviour of Escherichia coli at high cell density. Nat. Commun. 10, 5329 (2019).
Ringgaard, S., Hubbard, T., Mandlik, A., Davis, B. M. & Waldor, M. K. RpoS and quorum sensing control expression and polar localization of Vibrio cholerae chemotaxis cluster III proteins in vitro and in vivo. Mol. Microbiol. 97, 660–675 (2015).
Alexander, R. P. & Zhulin, I. B. Evolutionary genomics reveals conserved structural determinants of signaling and adaptation in microbial chemoreceptors. Proc. Natl Acad. Sci. USA 104, 2885–2890 (2007).
Briegel, A. et al. Chemotaxis cluster 1 proteins form cytoplasmic arrays in Vibrio cholerae and are stabilized by a double signaling domain receptor DosM. Proc. Natl Acad. Sci. USA 113, 10412–10417 (2016).
Holm, L. Using Dali for protein structure comparison. Methods Mol. Biol. 2112, 29–42 (2020).
Hong, Y. et al. The ligand-binding domain of a chemoreceptor from Comamonas testosteroni has a previously unknown homotrimeric structure. Mol. Microbiol. 112, 906–917 (2019).
Gavira, J. A., Matilla, M. A., Fernández, M. & Krell, T. The structural basis for signal promiscuity in a bacterial chemoreceptor. FEBS J. 288, 2294–2310 (2021).
Mise, T., Matsunami, H., Samatey, F. A. & Maruyama, I. N. Crystallization and preliminary X-ray diffraction analysis of the periplasmic domain of the Escherichia coli aspartate receptor Tar and its complex with aspartate. Acta Crystallogr. F 70, 1219–1223 (2014).
Milligans, D. L. & Koshland, D. E. Purification and characterization of the periplasmic domain of the aspartate chemoreceptor. J. Biol. Chem. 268, 19991–19997 (1993).
Biemann, H.-P. & Koshland, D. E. Aspartate receptors of Escherichia coli and Salmonella typhimurium bind ligand with negative and half-of-the-sites cooperativity. Biochemistry https://doi.org/10.1021/bi00169a002 (1994).
Tajima, H. et al. Ligand specificity determined by differentially arranged common ligand-binding residues in bacterial amino acid chemoreceptors Tsr and Tar. J. Biol. Chem. 286, 42200–42210 (2011).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Bi, S. & Sourjik, V. Stimulus sensing and signal processing in bacterial chemotaxis. Curr. Opin. Microbiol. 45, 22–29 (2018).
Mesibov, R. & Adler, J. Chemotaxis toward amino acids in Escherichia coli. J. Bacteriol. 112, 315–326 (1972).
Tsukada, K. d-amino acid dehydrogenases of Pseudomonas fluorescens. J. Biol. Chem. 241, 4522–4528 (1966).
Ulrich, L. E. & Zhulin, I. B. Four-helix bundle: a ubiquitous sensory module in prokaryotic signal transduction. Bioinformatics 21, 45–48 (2005).
Tsikas, D. & Wu, G. Homoarginine, arginine, and relatives: analysis, metabolism, transport, physiology, and pathology. Amino Acids 47, 1697–1702 (2015).
Staszek, P., Weston, L. A., Ciacka, K., Krasuska, U. & Gniazdowska, A. l-canavanine: how does a simple non-protein amino acid inhibit cellular function in a diverse living system? Phytochem. Rev. 16, 1269–1282 (2017).
Aliashkevich, A., Howell, M., Brown, P. J. B. & Cava, F. d-canavanine affects peptidoglycan structure, morphogenesis and fitness in Rhizobiales. Environ. Microbiol. 23, 5823–5836 (2021).
Teschler, J. K. et al. Living in the matrix: assembly and control of Vibrio cholerae biofilms. Nat. Rev. Microbiol. 13, 255–268 (2015).
Silva, A. J. & Benitez, J. A. Vibrio cholerae biofilms and cholera pathogenesis. PLoS Negl. Trop. Dis. 10, e0004330 (2016).
Nielsen, A. T. et al. RpoS controls the Vibrio cholerae mucosal escape response. PLoS Pathog. 2, 0933–0948 (2006).
Pombo, J. P., Ebenberger, S. P., Müller, A. M., Wolinski, H. & Schild, S. Impact of gene repression on biofilm formation of Vibrio cholerae. Front. Microbiol. 13, 2033 (2022).
Kamp, H. D., Patimalla-Dipali, B., Lazinski, D. W., Wallace-Gadsden, F. & Camilli, A. Gene fitness landscapes of Vibrio cholerae at important stages of its life cycle. PLoS Pathog. 9, e1003800 (2013).
Heidelberg, J. F. et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406, 477–483 (2000).
Donnenberg, M. S. & Kaper, J. B. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59, 4310–4317 (1991).
Murphy, S. G., Johnson, B. A., Ledoux, C. M. & Dörr, T. Vibrio cholerae’s mysterious Seventh Pandemic island (VSP-II) encodes novel Zur-regulated zinc starvation genes involved in chemotaxis and cell congregation. PLoS Genet. 17, e1009624 (2021).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Komarova, N. V., Golubev, I. V., Khoronenkova, S. V., Chubar, T. A. & Tishkov, V. I. Engineering of substrate specificity of d-amino acid oxidase from the yeast Trigonopsis variabilis: directed mutagenesis of phe258 residUe. Biochemistry 77, 1181–1189 (2012).
Adler, J. A method for measuring chemotaxis and use of the method to determine optimum conditions for chemotaxis by Escherichia coli. J. Gen. Microbiol. 74, 77–91 (1973).
Wilson, L. G. et al. Differential dynamic microscopy of bacterial motility. Phys. Rev. Lett. 106, 018101 (2011).
Colin, R., Zhang, R. & Wilson, L. G. Fast, high-throughput measurement of collective behaviour in a bacterial population. J. R. Soc. Interface 11, 20140486 (2014).
Mateus, A. et al. The functional proteome landscape of Escherichia coli. Nature 588, 473–478 (2020).
Ducret, A., Quardokus, E. M. & Brun, Y. V. MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat. Microbiol. 1, 16077 (2016).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
Gabadinho, J. et al. MxCuBE: a synchrotron beamline control environment customized for macromolecular crystallography experiments. J. Synchrotron Radiat. 17, 700–707 (2010).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
Rodríguez, D. D. et al. Crystallographic ab initio protein structure solution below atomic resolution. Nat. Methods 6, 651–653 (2009).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367 (2011).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Agarwala, R. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 44, D7–D19 (2016).
Karp, P. D. et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 20, 1085–1093 (2019).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5, 113 (2004).
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M. & Barton, G. J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Mise, T. Structural analysis of the ligand-binding domain of the aspartate receptor Tar from Escherichia coli. Biochemistry 55, 3708–3713 (2016).
Acknowledgements
We thank E. Bouvet for helping with strains and plasmids construction, A. Espaillat for all the insightful discussions, and all Cava lab members for their support and feedback. We also thank UCEM-NMI for the SEM Facility, and especially N. Lee for technical support and guidance. Research in the Cava lab is funded by the Knut and Alice Wallenberg Foundation (KAW2012.0184, F.C.), the Laboratory of Molecular Infection Medicine Sweden (MIMS), Kempestiftelserna and the Swedish Research Council (2018-02823 and 2018-05882, F.C.). Research in the Berntsson lab is funded by the Swedish Research Council (2016-03599, R.P.-A.B.), the Knut and Alice Wallenberg Foundation and Kempestiftelserna (SMK-1869, R.P.-A.B.). R.C. and V.S. acknowledge support by the Max Planck Society. R.C. acknowledges support from the Deutsche Forschungsgemeinschaft (CO 1813/2-1, R.C.).
Funding
Open access funding provided by Umea University.
Author information
Authors and Affiliations
Contributions
O.I. and F.C. conceived the study. O.I., J.t.B., L.A., A.M. and R.C. designed and performed experiments with supervision from A.T., M.M.S., V.S., R.P.-A.B. and F.C. All authors participated in data analysis and interpretation. O.I. and F.C. wrote the draft manuscript, with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Tino Krell, Joseph Dillard and Jing Yan for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Effect of D-amino acids in motility.
a, Motility analysis of V. cholerae in soft agar in presence of L- and D-amino acids. Grey bars indicate relative motility of BsrV knockout mutant compared to wild-type (wt) both in presence and absence of 5 mM amino acids. b, Effect of D-amino acids (DAAs) in growth. Survival rate was calculated as relative maximum growth in late stationary phase using the not supplemented culture as reference. Cultures were supplemented with 5 mM DAAs. c, Spot assay for the indicated V. cholerae strains. Cells were grown in TB until stationary phase, serially diluted and spotted on LB agar plates supplemented with 5 mM DAAs. Error bars in a and b represent mean values ± SD of 6 and 3 biologically independent replicates. Significant differences (One-way ANOVA, p-values were adjusted for multiple comparison using Bonferroni-Dunn method) are indicated by **** (p < 0.0001).
Extended Data Fig. 2 Comparison of chemotactic ability of V. cholerae wild-type and ΔbsrV strains.
a, Chemotactic ability of ΔbsrV mutant. Both chemotactic bias and swimming speed were analysed in presence of several known chemoeffectors to compare the behaviour of wild-type (wt) and ΔbsrV mutant cells. Chemotaxis was measured in microfluidic devices, monitoring cell motion from a reservoir filled with cells in motility buffer to a reservoir filled only with 100 µM of indicated compound in motility buffer. Blank, no attractant in second reservoir; L-Arg, L-arginine; AiBu, α-aminoisobutiric acid; GABA, γ-aminobutyric acid; Succ, succinate. Each data point represents the average over the 3 time points for a given experiment (n = 4) and the line represents the mean. Significant differences (unpaired t-test) are indicated by *** (p < 0.001) and was evaluated relative to the blank for the given strains. b, Swimming speed analysis of V. cholerae grown in presence of D-Arg. Cells were grown in TB (± 500 μM D-Arg) and transferred to fresh TB for swimming speed measurements. Each point represents the mean and error bars indicate the SD over 3 biological replicates. c, Motility analysis of non-chemotactic V. cholerae cells in presence and absence of D-Arg. Relative motility on soft-agar plates supplemented or not with 5 mM D-Arg of several strains compared to wild-type (wt). ΔbsrV and ΔcheY3 strains are included as controls. Error bars in c represent mean values ± SD of 3 biologically independent replicates. Significant differences (unpaired t-test, adjusted for multiple comparison using Bonferroni-Dunn method) are indicated by *** (p < 0.001). See source data.
Extended Data Fig. 3 Thermal proteome profiling.
a, Schematic representation of Thermal Proteome Profiling (2D-TPP) experimental setting. 4 different conditions were tested: 0, 0.05, 0.4, 3.2 mM D-Arg. Cell extracts were prepared and treated with a range of D-Arg concentrations (1); the samples were heated to a range of temperatures (2); then soluble proteins fraction was extracted and digested to analyze by mass-spectrometry (3); finally the effect of ligand on thermal stability of each protein was evaluated by comparing the remaining soluble fraction at each ligand concentration with the untreated control for each temperature (4). b, Heatmaps for selected candidates targeting D-Arg in V. cholerae. The legend represents the log2-transformed fold-change protein abundance for a given temperature and D-Arg concentration relative to the no D-Arg control.
Extended Data Fig. 4 Chemotactic ability of V. cholerae vc1313 mutant towards Arg.
a, Comparison between D-Arg and D-Lys chemotaxis responses in V. cholerae. b, Chemotaxis response to L- and D-Arg increasing concentrations for V. cholerae ΔbsrV and ΔbsrV Δvc1313 double mutant. Significant differences (unpaired t-test) are indicated by *** (p < 0.001), n = 3. c, Chemotactic bias of V. cholerae ΔbsrV and ΔbsrV Δvc1313, measured in microfluidics devices, towards a reservoir containing either 500 µM D-Arg in motility medium or only motility medium (blank control), 1, 2 and 3 h after the preparation of the device. d, Difference between the chemotactic bias (that is, the ratio vch/v0 of the chemotactic drift velocity vch over the cell swimming speed v0, see Methods) towards D-Arg and the corresponding control of the experiments of (b) as a function of the time since the preparation of the microfluidic device. e, Population averaged swimming speed measured in the experiments of (b, c). Each data point of in c, d and e represents the mean and error bars ± SEM of n = 6 biologically independent replicates. Significant differences (unpaired t-test) are indicated by ns (not-significant), * (p < 0.05) ** (p < 0.01) or *** (p < 0.001). See source data.
Extended Data Fig. 5 MCPDRK genetic context and promoter transcriptional activity.
a, Genetic context of vc1313. b, Relative expression of vc1313-vc1312 and adjacent genes across 307 available V. cholerae RNAseq experiments. c, Transcriptional activity of the putative promoter regions of the genes forming the operon (vc1313, vc1312 and vc1311) in wild-type (wt) and ΔbsrV mutant backgrounds. β-galactosidase assay was performed in cultures carrying pCB192N-derivatives grown for 8 hours at 37 °C in LB. β-galactosidase activity is measured in Miller units. d, Transcriptional activity of the Pvc1313 putative promoter region at different timepoints. β-galactosidase assay was performed in cultures of wild-type (wt) and ΔbsrV mutant backgrounds carrying pCB192N-Pvc1313, grown at 37 °C in LB for the indicated time. β-galactosidase activity is measured in Miller units. Error bars in c and d represent mean values ± SD of 3 biologically independent samples.
Extended Data Fig. 6 MCPDRK forms part of the chemotaxis F6 system.
a, Chemotaxis response to D-Arg of chemotaxis system mutants. ΔI/F9 and ΔIII/F7 showed similar response to D-Arg as control strain. No response was observed for ΔII/F6 system mutant. ΔbsrV strain was used as background. Black diamond represents the mean of 3 independent biological replicates. Significant differences (unpaired t-test) are indicated by *** (p < 0.001), n = 3. b, Co-localization of sfGFP-MCPDRK and CheY3-dTtomato (chemotaxis system II/F6) fluorescence reporters during stationary phase. c, Localization of sfGFP-MCPDRK in the three chemotaxis systems knockout backgrounds. Representative micrographs of all experiments are shown. Representative micrographs of 3 independent replicates are shown in b and c, at least 3 images were acquired per condition: scale bar, 2 μm.
Extended Data Fig. 7 Tree of MCPDRK orthologues.
The cladogram was constructed based on MCPDRK sequence homology. Representative species and strains were downloaded from Biocyc database. We included the non-related E. coli Tar chemoreceptor to help root the tree (light gray). In the first column, the presence of a Bsr ortholog is represented as full (presence) and empty (absence) circles. The presence of Bsr ortholog clustered together with the racemase is also represented in a similar way in the second column. Conservancy of the LBD residues implied in D-Arg binding is also shown (residue alignment). The bar chart represents the complete protein identity percentage (%). Bacteria were clustered in 3 different groups (cluster I, II, III) depending on the presence/absence of Bsr and MCPDRK orthologs.
Supplementary information
Supplementary Information
Supplementary Figs. 1–3, Methods and Source Data.
Supplementary Tables 1–6
Supplementary Table 1. Thermal protein profiling results. The complete thermal protein profiling dataset is available in ProteomeXchange Consortium under identifier PXD038312. Table 2. Thermal protein profiling statistical analysis. Table 3. Crystallization data collection and refinement statistics. Table 4. List of strains. Table 5. List of plasmids. Table 6. List of oligonucleotides.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Unprocessed western blot for Fig. 2b.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Irazoki, O., ter Beek, J., Alvarez, L. et al. d-amino acids signal a stress-dependent run-away response in Vibrio cholerae. Nat Microbiol 8, 1549–1560 (2023). https://doi.org/10.1038/s41564-023-01419-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-023-01419-6
This article is cited by
-
Run Vibrio, run!
Nature Reviews Microbiology (2023)

{kind=link}