
Abstract
Lessons learnt from the COVID-19 pandemic include increased awareness of the potential for zoonoses and emerging infectious diseases that can adversely affect human health. Although emergent viruses are currently in the spotlight, we must not forget the ongoing toll of morbidity and mortality owing to antimicrobial resistance in bacterial pathogens and to vector-borne, foodborne and waterborne diseases. Population growth, planetary change, international travel and medical tourism all contribute to the increasing frequency of infectious disease outbreaks. Surveillance is therefore of crucial importance, but the diversity of microbial pathogens, coupled with resource-intensive methods, compromises our ability to scale-up such efforts. Innovative technologies that are both easy to use and able to simultaneously identify diverse microorganisms (viral, bacterial or fungal) with precision are necessary to enable informed public health decisions. Metagenomics-enabled surveillance methods offer the opportunity to improve detection of both known and yet-to-emerge pathogens.
Similar content being viewed by others
Main
Pathogen surveillance strategies have varying aims, ranging from identifying species and strains to detecting antibiotic resistance genes or plasmids. In this Perspective, we focus on the potential for the application of metagenomics to improve pathogen surveillance for public health decision-making. This differs from metagenomics for clinical diagnostics, in which the primary aim is to determine the presence of disease-causing pathogens in a sample (or samples) from an individual for the purpose of clinical management. Pathogen surveillance for public health is routinely carried out in hospitals, water-purification plants and farms, and is specifically applied in locations thought to be a probable source of emergent disease. The overall aims of public health pathogen surveillance are to monitor known and yet-to-emerge pathogens and to drive early risk mitigation programmes to protect human and animal health.
Traditional pathogen surveillance for public health
Existing public health pathogen surveillance methods include syndrome-based infection event monitoring, active surveillance for specific pathogens, for example, methicillin-resistant Staphylococcus aureus, and laboratory-based surveillance1. Most laboratory-based surveillance efforts focus on monitoring for clinically relevant features, such as antimicrobial resistance (AMR) and disease-causing serotypes in cultured isolates. In addition, specific rooms or wards in hospitals, for example, augmented care units, are routinely surveyed for a predetermined list of pathogens using culture-based methods2,3,4,5. When environmental fomites are epidemiologically implicated in investigations of outbreaks, targeted sampling of air, water and surfaces is recommended to establish potential sources5. Cultivated isolates from investigations of outbreaks are usually subjected to various typing schemes, for example, multilocus sequence typing (MLST), to establish disease transmission. As a result, a wide variety of laboratory techniques, equipment, reagents and associated specialized skills are required to carry out surveillance of a subset of known human pathogens in hospitals.
Pathogen surveillance in communities is mainly carried out in hospitals, water-purification plants and farms, and a variety of strategies is used. These conventional approaches are summarized below.
Prevention and management of outbreaks
Public health and sentinel laboratories support infectious disease surveillance and investigations of outbreaks by participating in disease notification, monitoring of known human pathogens and detection of pathogens in patient samples6 (Table 1). In addition to pathogen identification by culturing and molecular testing, surveillance can include strain typing using different assays, for example, serotyping for Neisseria meningitidis, MLST for S. aureus and antibiotic resistance testing. Traditional surveillance protocols often require a wide range of laboratory capabilities and assays (Fig. 1), which makes it challenging to integrate information at a local or national level, let alone globally, despite attempts to standardize processes (for example, WHO/CDS/CSR/ISR/99.2)6. Furthermore, techniques such as serotyping require manipulation of viable cultures of bacteria, some of which pose substantial risks of laboratory-acquired infections (LAIs), for example, N. meningitidis or Salmonella enterica serotype typhi. The use of whole-genome sequencing (WGS) for bacteria, viruses and, to a certain extent, fungi, is gaining traction but has not become the standard for most sentinel laboratories. Although WGS is well suited to this purpose, upfront resource and human capital requirements have been a barrier to adoption, particularly in low- and middle-income countries.

Existing laboratory-based surveillance efforts focus on monitoring characteristics of culture isolates, such as AMR, serotype and virulence factors. Typically, the culturing step is followed by various typing and phenotypic assays that require specialized equipment and techniques and significant time and resources. The time taken to obtaining a pure isolate varies based on the growth rate of each species (two days and up to several weeks), and the resultant isolate is subjected to downstream phenotypic and molecular analysis based on specific requirements of each sector and species. BSL-3, biosafety level 3.
Food and water safety
Traditional microbiological safety monitoring for food relies on risk assessment and subsequent targeted detection of pathogens and hygiene or safety indicator organisms in food7,8. A wide variety of culture methods are required to enrich and select for specific species from complex food matrices (Table 1). The monitoring of foodborne AMR is based on susceptibility testing of selected bacterial foodborne pathogens9,10. For drinking water, monitoring of microbial contamination is similarly risk based11. Although monitoring of indicator bacterial organisms (for example, Escherichia coli) is widely adopted, waterborne viral and protozoan pathogens are not consistently addressed11. Since the early 2010s, WGS has also been effectively deployed in high-income countries to track foodborne pathogens in investigations of outbreaks12. Of note, although the burden of foodborne disease is highest in low-income countries13, these countries often lack the resources to adopt WGS for mitigating outbreaks.
Human–animal–ecosystem interface
Livestock has been implicated in various zoonotic infectious disease outbreaks, including those caused by avian influenza, swine influenza and Nipah virus14,15. With the intensification of livestock farming, surveillance will probably play an increasingly important part in our preparedness against epidemics and pandemics. Surveillance for zoonotic diseases requires human health, animal health and agricultural sectors to effectively communicate about disease trends, as well as taking part in collaborative testing of samples from human, animal (wildlife, livestock and companion animals), environment (both soil and water), vector and food sources16. For example, zoonotic influenza A viruses, such as the highly pathogenic avian influenza strain H5N1, which is panzootic among poultry and wild birds, are priority pathogens. The Global Influenza Surveillance and Response System (GISRS) supports detection and whole-genome characterization of influenza in human clinical samples around the world17, and these data are integrated by the World Health Organization (WHO) to monitor influenza activity and to guide vaccine composition. For influenza viruses in animals, the Global Early Warning System for Major Animal Diseases (GLEWS) combines and coordinates alert mechanisms of the WHO, the Food and Agriculture Organization of the United Nations (FAO) and the World Organization for Animal Health (OIE) for surveillance of wildlife, livestock, poultry and the environment. However, the coverage remains variable and is not adequately resourced in most countries (Table 1). The public health threat posed by zoonotic and emerging diseases is further highlighted by the ongoing coronavirus disease 2019 (COVID-19) pandemic, in which a low barrier to cross-species transmission is evidenced by the increasing number of animal species reported to be infected by reverse zoonosis18.
Community health
Monitoring of wastewater (sewage) treatment systems has frequently been mooted as a promising way to assess community health, and has been adopted in polio eradication programmes in various countries such as Egypt, India, Israel, Pakistan, Afghanistan and Nigeria19 (Table 1). The COVID-19 pandemic has accelerated efforts to improve sample processing and sensitivity of assays20, as well as serving to further highlight the usefulness of wastewater monitoring to uncover and interrupt outbreak clusters21 (Fig. 1). However, virus isolation from wastewater requires specialized laboratory setup and skilled personnel. In the case of high-risk pathogens such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), manipulation of samples beyond routine clinical diagnostics necessitates biosafety level 3 containment, which is only accessible in well-resourced centres with highly skilled personnel.
Healthcare-associated infections
In addition to surveillance for clinically relevant bacterial and fungal isolates, healthcare environments, particularly air and water, are routinely sampled for pathogens and other microbial contamination22,23 (Fig. 1). For example, in augmented care units, periodic culture of potable water for Legionella species is performed24, and water used for haemodialysis is monitored for total viable bacterial cell counts and endotoxin levels22. Some healthcare guidelines advocate monitoring for Pseudomonas aeruginosa if the water used is in direct contact with patients in augmented care25. Routine surveillance of water used in healthcare settings can be technically demanding and requires a variety of culture media and culture conditions as well as trained personnel (Table 1).
AMR surveillance
The global antimicrobial resistance surveillance systems (GLASS) network has fostered AMR surveillance and enabled the collection, integrated analysis and sharing of standardized human bacterial pathogen AMR data by participating countries around the world26. AMR surveillance in its current state relies on the successful culture and isolation of the pathogens of interest (Table 1). The culture isolate is then subjected to standardized antimicrobial susceptibility testing for a predetermined panel of antibiotics. AMR present in the food chain, crops, animals and environment can be mobilized into bacterial pathogens of humans and animals through horizontal gene transfer and thereby affect human and animal health. AMR surveillance in these non-healthcare domains, however, is less established and standardized.
Integrated pathogen surveillance
Current initiatives for integrated surveillance, including guidelines, laboratory methods, data collection and analyses, continue to be segregated by One Health sectors (for example, human health, animal health and plant health). Given the diversity of sample types and microbial species targeted, a range of processing and culture methods are required for optimal recovery and isolation. As known human pathogens are prioritized and selected for during the culture process, any novel, emerging or atypical pathogens and their associated AMR determinants will probably not be detected. The overall expense, lack of consistent workflows and outcomes, targeted nature of many assays (in which you only find what you are looking for) and paucity of cost-effective options that are less biased27 remain substantial hurdles to success. Correspondingly, pathogen surveillance, particularly outside healthcare sectors, remains under-resourced, and the complexity and requirements for multiple assays will probably contribute to the overall suboptimal surveillance of pathogens.
The availability of inexpensive, high-throughput sequencing technologies has paved the way for the development of a new generation of pathogen surveillance methods28. Metagenomics—the interrogation of genetic material from a sample containing multiple species—could enable culture-free methods and workflows for the detection of known and unknown viruses, bacteria and fungi. Metagenomic studies are often either done using a targeted amplicon approach that is based on marker genes (for example, 16S rRNA gene sequencing) or shotgun metagenomic sequencing to probe the entire genetic content of a sample. Metagenomic sequencing of samples can also enable the detection of virulence and resistance determinants outside their genome contexts. With improvements in ease of analysis and cost, the majority of biosurveillance efforts across various sectors would benefit from the adoption of metagenomic approaches. Despite its promise, there are technical and analytical complexities in metagenomic workflows that either need to be addressed or accounted for before integration into routine laboratory-based surveillance.
Next, we review the challenges in sample preparation, sequencing and bioinformatic analyses in metagenomics, examine examples of real-world applications of metagenomics in pathogen surveillance and evaluate the advances needed to enable adoption of these methods. We also discuss the potential for metagenomics-based surveillance to accelerate and transform global efforts to detect risks to human health within a One Health framework.
Metagenomics for pathogen surveillance
Metagenomic workflows for pathogen surveillance29 are unified by the use of sequencing as a read-out. Long-read and short-read sequencing technologies have both been used for metagenomics, in which sequence data can be sifted using computational approaches to reconstruct exquisitely detailed outputs that are not constrained by the availability of cultures or references genomes.
Nevertheless, there are several considerations related to sample acquisition and nucleic acid processing that are invariably application-specific and affect downstream steps. Metagenomic workflows need to take into account surveillance objectives, resource availability, operational feasibilities and technical compatibilities, as highlighted in Table 2. The following sections outline the main components of a metagenomic workflow and we illustrate their application using recent case studies. Although some of the challenges for sample manipulation and processing (for example, dealing with PCR inhibitors) are applicable across molecular assays, different metagenomic sequencing technologies (particularly long-read sequencing) impose additional constraints (for example, DNA purity, molecular weight, among others).
Sample collection
This step has the greatest variability in metagenomic workflows and needs to account for a variety of sample matrices30 and their physical and chemical properties. These include swabs and tapes (for example, skin, nasal and environmental), filters, for example, for water, sludge and semi-solids (for example, stool, sewage, soil or sediment and food) and fluids (for example, saliva). Some samples, such as sludge, may need to be collected in large volumes to obtain sufficient biomass, and associated leakage concerns can pose logistical challenges to safe sampling. Other considerations include the need to prevent cross-contamination, a need for culture stocks and whether cold-chain storage or transport in stabilization medium is needed to minimize overgrowth or nucleic acid degradation (Table 2).
Nucleic acid extraction
The choice of nucleic acid extraction protocols is primarily determined by the trade-off between the ability to effectively lyse cells31 using harsh physical disruption, such as bead beating, versus the importance of preserving DNA integrity through treatment by enzyme cocktails such as MetaPolyzme, which is a mixture of achromopeptidase, chitinase, lyticase, lysostaphin, lysozyme and mutanolysin. Certain sample types are inherently more challenging to process, for example, stool, food and soil samples, owing to PCR inhibitors and the need to separate nucleic acids from complex matrices with debris of varying sizes and consistency. Correspondingly, some sequencing protocols can be less forgiving, requiring suitable nucleic acid quantity and quality to enable optimal throughput32.
An additional aspect that affects this step is the relative proportion of microbial nucleic acids in the sample, a factor that influences the cost, sensitivity and feasibility of analysis33. Microbial enrichment can either be done before nucleic acid extraction (for example, through centrifugation, filtration, culture enrichment or flow cytometry) or as part of the protocol, for example, using selective lysis steps to deplete eukaryotic cells34 or using targeted probes to enrich and amplify microbial DNA35 (Table 2).
Library preparation and sequencing
Current sequencing technologies are either second-generation systems that provide short reads at low cost (~100 base pairs, for example, Illumina) or third-generation instruments with longer reads but with cost and accuracy trade-offs36 (for example, Pacific Biosciences and Oxford Nanopore Technologies). Applications in which accurate quantification or higher sensitivity is needed through deeper sequencing generally use short-read platforms, whereas de novo assembly is greatly improved by the inclusion of long reads. Other features that can affect the choice of a platform for surveillance include portability, cost of acquisition and maintenance, and time-to-answer (Table 2). Recent developments in real-time nanopore sequencing using a portable thumb-drive-sized device37 have massively expanded the feasibility of in-field sequencing-based surveillance programmes. Although library preparation workflows are largely determined by sequencing instruments, advancements in linked read38 and Hi-C39 protocols have broadly expanded our ability to generate high-quality genomes from metagenomic data.
Bioinformatic analyses
The analysis of metagenomic data can be done at various resolutions, from identifying and quantifying specific taxa using taxonomic classifiers to analyses of genes and pathways40,41,42,43, and finally to whole-genome reconstruction, annotation and phylogenetic analyses. This is facilitated by a range of specialized tools, such as taxonomic classifiers, read mappers, pathway reconstruction tools and genome assembly programs40,41,42,43. In Table 3, we highlight some of the more popular pipelines and webtools that integrate these capabilities into user-friendly workflows accessible to non-specialists. In addition, there are cloud-deployable clinical metagenomic computational workflows such as SURPI44,45 and IDseq46 for pathogen detection and identification.
For metagenomic surveillance, improved capabilities in generating metagenome-assembled genomes (MAGs) have been a transformative factor as they enable finer-grained transmission analysis and more comprehensive antibiotic resistance predictions. These include improved binning strategies that retrieve bins of short-read contigs that belong to the same genome47 and long-read48 and hybrid assembly49 programs that circumvent higher error rates to produce more contiguous assemblies. Near-complete genomes can now be reconstructed from metagenomic datasets, although sensitivity and accuracy improvements still remain to be made, particularly in cases in which multiple similar strains of a species are present in a community50.
After the generation of MAGs, phylogenetic analysis can be done using two main approaches to aid epidemiological tracing and transmission analysis51,52: (1) multilocus (multi-gene) alignment and (2) whole-genome comparisons. Although whole-genome analysis provides higher resolution in principle53, horizontal gene transfer, recombination events and higher error rates in repeat regions can affect results. Frequently, average nucleotide identity (ANI) thresholds are used to infer transmission, but these need to be adjusted to evolutionary rates of specific pathogens54. The analysis of AMR genes is usually done through mapping and alignment to reference databases such as CARD55, ARG-ANNOT56 and MEGARes2.057. As these approaches can have false negatives and arbitrary coverage and identity cut-off values, deep-learning approaches have been proposed as an alternative58. Additionally, the analysis of mobile and extrachromosomal AMR genes has been boosted by the use of Hi-C techniques to link them to their host59.
Learning by example from case studies
In the following paragraphs, we discuss case studies that reveal how metagenomic surveillance has been used, including the specific choices made and lessons learnt and how this can guide future efforts to operationalize metagenomic surveillance.
Urban environments and transportation systems
As the world becomes increasingly urbanized, surveillance of public spaces in cities, particularly mass-transport systems, represents an efficient way to map our microbial exposomes. One of the first applications of metagenomic surveillance at scale was conducted in 2013–2014 in the subways of New York City, United States60. The results highlighted the feasibility and scalability of this strategy to detect pathogens and AMR genes, as well as potential pitfalls of read-level analysis61. In subsequent work, the international MetaSUB consortium expanded this study to more than 3,700 samples from 58 cities, building a global reference map for microbial diversity and AMR genes62. As environmental swabs often have low biomass, standardized sample collection and inclusion of positive and negative controls were used to minimize kitome artefacts (that is, reagent- and laboratory-associated contaminants). In addition, de novo assembly of short-read sequencing data was used to identify novel species and AMR genes that were missing from existing reference databases, thereby reducing the effect of this confounding factor in reference-based analyses.
AMR tracking from sewage
Metagenomic surveillance for AMR has been performed based on sewage and livestock fecal slurry in a few large-scale studies63,64. In a global sewage surveillance project64, untreated sewage from 74 cities in 60 countries was shipped frozen and centrally processed to obtain short-read metagenomic data and assemblies. In total, 1,625 different AMR genes belonging to 408 gene classes were identified from these assemblies, including blaNDM, mcr and optrA genes, which are AMR determinants for last-resort antibiotics64. The study found that total AMR abundance varies by region, and values were highly correlated with socioeconomic factors such as sanitation. Large-scale sewage surveillance may therefore provide a convenient, low-cost method of AMR monitoring at the population level. These studies highlight that sampling and transportation logistics can be addressed, and that further automation in collection and processing would make this approach attractive for surveillance.
Healthcare environments
Large-scale metagenomic mapping of healthcare environments was pioneered by Lax et al.65 to determine microbial colonization and transmission patterns in a newly built hospital. Owing to their importance as a high-risk setting, Brooks et al.66 focused on neonatal intensive care units, using shotgun metagenomic sequencing and MAGs to demonstrate the direct interchange of microorganisms between patients and the environment. In addition, their work demonstrated the persistence of some strains in the intensive care unit environment throughout the one-year study period. In recent work, we showed the enhanced persistence and widespread distribution of multidrug-resistant organisms in hospital environments67 based on hybrid metagenomic assemblies. In addition, comparisons with patient isolates up to eight years apart showed high similarity, which highlighted the persistence of AMR reservoirs. This study used quasi-metagenomics (that is, metagenomics of intentionally modified microbiomes, for example, via culture enrichment) to enrich for antibiotic resistant organisms and to acquire a large number of high-quality MAGs (>2,000) from a few hundred samples. Collection curve analysis indicated that as few as 50 samples may be sufficient for routine strain and AMR surveillance in hospitals. Further improvements in analysis of low-biomass samples and real-time sequencing are likely to make these approaches even more attractive in healthcare settings in which time-to-answer is critical. Sandbox trials would also be needed to provide evidence for its ability to rapidly track outbreaks and prevent infections to make the case for routine adoption.
Zoonotic viral reservoirs
Metagenomic approaches have been used to detect and characterize zoonotic pathogens in various potential reservoirs, such as arthropod vectors68,69,70, bats71 and wild ducks72. Known, emerging and novel viral pathogens have been detected from ticks69,70, mosquitos68, bat gastrointestinal excreta71 and wild duck fecal droppings72, thereby highlighting the utility of metagenomic surveillance of zoonotic viral reservoirs. Correspondingly, the Global Virome Project73 was initiated as a large-scale effort to develop a global atlas of potential zoonotic viral pathogens and to improve our capacity to discover, detect and diagnose viruses that may threaten human health. Viral metagenomics poses significant technical challenges74, including safety concerns, low viral loads in most samples, RNA degradation and quasi-species diversity. Addressing these concerns through the development of new automation, nucleic acid extraction and bioinformatics tools would therefore be key to improving our ability to monitor and understand the vast virosphere (especially coronaviruses, influenza viruses and ebolavirus) through metagenomics.
Taken together, these case studies highlight the unparalleled breadth and depth of survey that is now feasible in various domains of interest using metagenomics. The ability to unify analysis of diverse pathogens, to integrate information across kingdoms in a single high-throughput assay and to carry out surveillance for AMR genes provides analytical opportunities that were previously not feasible. In addition, the generation of high-quality metagenome-assembled genomes in some studies further enabled whole-genome investigations62,67 and high-resolution phylogenetics that were previously only feasible through a more low-throughput process of culturing and isolating genome sequencing. There is therefore strong interest in performing routine metagenomic surveillance because the data generated can be used to answer biological research questions.
Challenges to widespread adoption of metagenomics surveillance
Although there is a robust rationale for implementing metagenomic workflows in surveillance, there are challenges to the broad adoption of this technology. In this section, we highlight some of these issues and discuss ongoing developments that will smooth the path to routine application.
Sensitivity, specificity and standardization
An important consideration for adoption of metagenomics methods is the sensitivity, specificity and reproducibility of an assay, an aspect that was not investigated in detail in the metagenomic case studies discussed here. Other studies have sought to determine the sensitivity and specificity of shotgun metagenomic assays relative to conventional testing approaches44,75. Similar to other molecular methods such as PCR, the sensitivity and specificity of metagenomic analytics depends on the optimization of protocol workflows that include several factors, such as different target pathogens and different (sometimes parallel) sample processing workflows for maximizing assay sensitivity and specificity. Depending on the resources and sample biomass available, this may or may not be feasible to implement, which poses a bottleneck for adoption. Progress in the development of molecular biology techniques and adoption of robotics and laboratory automation (see the section ‘Breakthrough technologies’) would greatly address this pertinent challenge and facilitate widespread application.
Furthermore, the wide range of sample matrices and multi-step custom laboratory and analytical workflows are sources of variability that complicate metagenomic assay implementation. Despite these challenges, there are studies suggesting that high intra-laboratory reproducibility can be achieved with rigorous optimization, standardization and validation44,75. Overall, efforts towards standardization of workflows and quality assurance are already gaining momentum75,76,77, and should be aided by automation (ease of use), reduction in analysis cost (sequencing cost and multiplexing) and efforts to benchmark bioinformatics tools78. Crucially, sequencing costs are already close to the threshold (less than US$100) where they are comparable with overall costs for conventional surveillance protocols (incorporating labour costs). End-to-end standardization (including bioinformatics) and validation of metagenomic workflows may enable better uptake.
Despite the inherent complexity of metagenomic data collection and interpretation, the increasing adoption of shotgun metagenomic approaches in clinical diagnostics indicates that such assays can be standardized and validated44,76,79. Nevertheless, as surveillance programmes affect public health decision-making, acceptance of metagenomics is influenced by considerations beyond sensitivity, specificity and economic feasibility. For example, concerns can range from the impact of microbial contamination on the results80,81,82 and the fidelity of bioinformatic analysis for related species62 to the viability and infectivity of detected pathogens and the ability to estimate the public health risk that is posed83. Thus, in addition to stringent protocol standardization, end-to-end guidance for common sample types, inclusion of various controls, quality assurance and pipeline verification with proficiency samples and the use of reference standards84,85, further studies are needed to develop public health risk models based on metagenomic surveillance data. As the main objective of pathogen surveillance is to provide early intelligence on potential public health threats, this intelligence will require follow-up confirmation. Furthermore, the regulatory implications for surveillance will be complex and determined by each jurisdiction based on local resources available, local surveillance needs, epidemiology and local policies.
Technological hurdles
One advance that could lower the barrier to adoption of metagenomic surveillance is the availability of kits and protocols for linked reads, synthetic long reads and Hi-C reads86,87. In parallel, rapid developments in sequencing technology, particularly in generating highly accurate long reads88 and ultralong reads89, as well as the ability to do real-time, field-deployable whole-genome analysis, are making metagenomics surveillance a realistic option90. For routine work, inexpensive and robust sequencing kits with long shelf-life are important areas of ongoing development91.
In tandem, bioinformatic algorithms that enable sensitive classification of long reads, such as MetaMaps92, fast and accurate metagenomic assembly49,93, and composition and coverage-based assembly binning94, are bringing robust metagenomic-surveillance workflows within reach. Significant research investment is needed to improve strain-resolved assemblies of metagenomes. Specifically, the utility and reliability of metagenome assemblies remain limited by a failure to assemble difficult-to-assemble genomic content such as plasmids, 16S rRNA and repetitive DNA elements, and the generation of incomplete and mixed or composite genome bins. The ability to obtain near-complete metagenome-assembled genomes, in particular, is a key development for metagenomic surveillance as it enables more complete mapping of AMR genes and the use of these genomes for transmission analysis. A growing set of research studies have exploited this capability to expand our understanding of microbial diversity and have resulted in new MAG databases such as UHGG, GTDB and IGGdb, which can be useful resources for future microbial surveillance efforts95,96,97. Community-driven initiatives such as the Genomic Standards Consortium98 are driving data and metadata standardization efforts that would be valuable for the use of MAGs in surveillance99,100. Curated, quality-controlled reference genome databases, such as FDA-ARGOS, will also be instrumental for improving the accuracy of metagenomic analyses.
Breakthrough technologies
Several recent advances will make a difference to accelerate surveillance efforts.
Robotics and laboratory automation
The streamlined workflow of metagenomic assays makes them amenable to various forms of automation, thereby improving scalability, safety and reproducibility over traditional surveillance strategies. In particular, open-source automation platforms (for example, Opentron and potentially the VoITRAX) enable seamless wet-bench standardization through sharing of digital protocols that can be directly deployed to automated systems of participating laboratories.
Genome analysis on mobile applications
The time required for data synthesis and reporting can be a major bottleneck in transdisciplinary biosurveillance efforts. There is therefore a need to couple portable sequencing technologies, such as Oxford Nanopore’s MinION, with applications that enable genomic analysis in the field, for example, the MinKNOW app and the iGenomics app101.
Adaptive sequencing
Nanopore sequencing has the feature that DNA templates can be ejected, which provides the ability to select sequenced DNA in real time. Recent applications of this method have demonstrated its utility to do enrichment sequencing of target regions in a metagenome without the need for specialized sample preparation, thereby reducing turnaround time and resource requirements102,103. Further developments of this technology could involve selecting for specific mutations or adaptively choosing a virulence marker gene to search for once a particular pathogen signature has been observed.
Citizen science
With increasingly accessible scientific tools such as the MinION sequencer and the iGenomics app, broader participation of citizen scientists in environmental surveillance is now becoming feasible. Consortiums such as MetaSUB and the American Gut have taken a first step in this direction by crowd-sourcing samples and funds104. In addition to aiding in wider and more representative sampling, these efforts helped engage a broader community in their scientific efforts, thus promoting a better understanding of the underlying science in matters of public concern such as AMR.
Outlook
The COVID-19 pandemic has shown that infectious diseases do not respect borders and that nobody is safe until everyone is safe. Although the pandemic has spurred a greater appreciation for the need to strengthen global sequencing capacity, to share genomic data and to coordinate surveillance efforts, these have not been high-priority areas until recently. Importantly, we run the risk that future efforts may be limited to surveillance for viruses similar to SARS-CoV-2 or solely for respiratory viruses. By broadening the aperture of surveys, metagenomics goes beyond a technological upgrade. It aspires to unify microbial surveillance and transform public health efforts to proactively screen for threats, involve the very citizens that it seeks to protect and be harmonized on a global scale in step with the globalized world that we live in. We anticipate that a future model of microbial surveillance utilizing metagenomics will have several key elements, including the following:
-
(1)
Standardized processes and analysis that a range of facilities and individuals can apply, including well-resourced reference and sentinel laboratories to interested citizen scientists. This would include pre-set requirements for sampling, significant investments to improve molecular biology workflows and low-cost liquid handling automation to enable programmable automated sample processing and sequencing, and provenance-tracked validated pipelines that submit data to global repositories in a standardized format (Fig. 2).
Fig. 2: Harmonized pathogen surveillance using metagenomics. 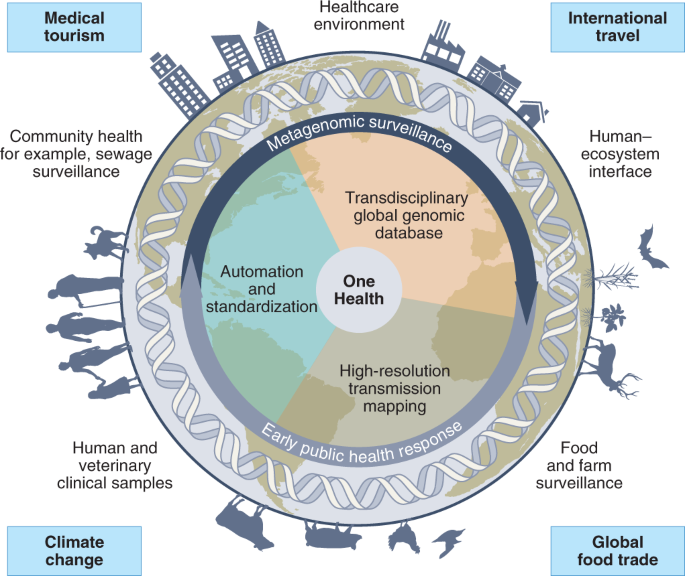
Schematic illustrating the opportunities for transdisciplinary One Health surveillance, utilizing metagenomic sequencing as a way to unify workflows and harmonize data. Key drivers for this integration are highlighted, including climate change and its impact on zoonotic reservoirs, globalization and international travel, medical tourism and global food trade. Created with BioRender.com.
-
(2)
Unification in a global One Health network, whereby genomic data across domains (for example, clinical, food and water safety, and vector control) and countries can be jointly analysed with appropriate privacy, security and data-ownership safeguards (for example, GISAID105; Fig. 2). This would involve automated analyses integrating epidemiological (for example, the TraceTogether app, https://www.tracetogether.gov.sg/) and environmental information to flag events of concern (for example, AMR or pathogen transmission). We envisage that such efforts would need to be integrated and coordinated at the national and global level through multinational agencies such as the WHO to effectively meet its goals.
-
(3)
High-resolution transmission mapping, whereby availability of whole-genomes (MAGs or isolates) would facilitate detailed source tracking, identification of reservoirs of concern and targeted mitigation efforts (Fig. 2).
-
(4)
Boost infectious disease research, whereby similar to precision medicine efforts worldwide, the availability of massive genetic datasets with rich metadata will facilitate biological and genetic research into virulence genes, microbial evolution, plasmid and phage diversity, in turn feeding back to support rational therapy for infections.
-
(5)
Data-informed public policy and practices, whereby the integration of clinical, epidemiological and metagenomic data will be routinely used to determine policy choices for desired public health outcomes. The implementation of such evidence-backed policies will need to become more commonplace to improve our chances to avoid future pandemics106.

The importance of better international surveillance and information sharing is clear in the WHO’s review of the global COVID-19 response, in which the panel recommends key transformational changes “to establish a new global system for surveillance, based on full transparency by all parties, using state-of-the-art digital tools to connect information centres around the world and including animal and environmental health surveillance”107. This would go beyond the current national efforts to update therapeutic guidelines and antimicrobial stewardship programmes, and could foster a new era of international cooperation supported by a multilateral agreement similar to the World Trade Organization. The harmonization of pathogen surveillance workflows in a metagenomics-enabled unified One Health strategy108,109 would have the added benefit that other public health priority pathogens would not be relegated in times of crisis.
References
2021 NHSN Patient Safety Component Manual (Centers for Disease Control and Prevention, 2021).
(HTM 03-01) Specialised Ventilation for Healthcare Buildings (NHS England, 2021).
(HTM 04-01) Safe Water in Healthcare Premises. Part A: Design, Installation and Commissioning (NHS England, 2016).
(HTM 04-01) Safe Water in Healthcare Premises. Part B: Operational Management (NHS England, 2016).
Environmental Infection Control Guidelines (Centers for Disease Control and Prevention, 2019).
WHO Recommended Surveillance Standards 2nd edn (WHO, 1999).
Carolissen, V. Principles and Guidelines for the Establishment and Application of Microbiological Criteria Related to Foods CAC/GL 21-1997, Codex Alimentarius 6 (FAO and WHO, 1997).
Principle and Guidelines for the Conduct of Microbiological Risk Assessment CAC/GL 30-1999, Amendments 2012, 2014, Codex Alimentarius 7 (FAO and WHO, 1999).
Guidelines for Risk Analysis of Foodborne Antimicrobial Resistance. CAC/GL 77-2011, Codex Alimentarius (FAO and WHO, 2011)
OIE Standards, Guidelines and Resolutions on Antimicrobial Resistance and the use of Antimicrobial Agents (OIE, 2020).
Assessing Microbial Safety of Drinking Water. Improving Approaches and Methods (WHO and OECD, 2003).
Jackson, K. A. et al. Use of whole genome sequencing and patient interviews to link a case of sporadic listeriosis to consumption of prepackaged lettuce. J. Food Prot. 79, 806–809 (2016).
Havelaar, A. H. et al. World Health Organization global estimates and regional comparisons of the burden of foodborne disease in 2010. PLoS Med. 12, e1001923 (2015).
Lam, S. K. & Chua, K. B. Nipah virus encephalitis outbreak in Malaysia. Clin. Infect. Dis. 34, S48–S51 (2002).
Widdowson, M.-A., Bresee, J. S. & Jernigan, D. B. The global threat of animal influenza viruses of zoonotic concern: then and now. J. Infect. Dis. 216, S493–S498 (2017).
Taking a Multisectoral, One Health Approach: A Tripartite Guide to Addressing Zoonotic Diseases in Countries (WHO, OIE and FAO, 2019).
Deng, Y.-M. et al. A simplified Sanger sequencing method for full genome sequencing of multiple subtypes of human influenza A viruses. J. Clin. Virol. 68, 43–48 (2015).
Jia, P., Dai, S., Wu, T. & Yang, S. New approaches to anticipate the risk of reverse zoonosis. Trends Ecol. Evol. 36, 580–590 (2021).
Asghar, H. et al. Environmental surveillance for polioviruses in the Global Polio Eradication Initiative. J. Infect. Dis. 210, S294–S303 (2014).
Status of environmental surveillance for SARS-CoV-2 virus. WHO https://www.who.int/news-room/commentaries/detail/status-of-environmental-surveillance-for-sars-cov-2-virus (2020).
Ting, V. Hong Kong fourth wave: sewage tests for coronavirus to be expanded, aim for ‘gold standard’. South China Morning Post https://www.scmp.com/news/hong-kong/health-environment/article/3117041/hong-kong-fourth-wave-sewage-tests-coronavirus-be (2021).
Sehulster, L. & Chinn, R. Y. W. Guidelines for environmental infection control in health-care facilities. Recommendations of CDC and the Healthcare Infection Control Practices Advisory Committee (HICPAC). MMWR Recomm. Rep. 52, 1–42 (2003).
(HTMM 04-01) Safe Water in Healthcare Premises. Part C: Pseudomonas aeruginosa, Advice for Augmented Care Units (NHS England, 2016).
Legionnaires’ Disease. The Control of Legionella Bacteria in Water Systems: Approved Code of Practice and Guidance on Regulations (Health and Safety Executive, 2013).
(HTM 04-01) Safe Water in Healthcare Premises (NHS England, 2017).
Global Antimicrobial Resistance Surveillance System (GLASS) (WHO, 2015); http://www.who.int/glass/en/
Hassibi, A. et al. Multiplexed identification, quantification and genotyping of infectious agents using a semiconductor biochip. Nat. Biotechnol. 36, 738–745 (2018).
Gardy, J. L. & Loman, N. J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 19, 9–20 (2018).
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J. & Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844 (2017).
Gu, W. et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat. Med. 27, 115–124 (2021).
Greathouse, K. L., Sinha, R. & Vogtmann, E. DNA extraction for human microbiome studies: the issue of standardization. Genome Biol. 20, 212 (2019).
Schalamun, M. et al. Harnessing the MinION: an example of how to establish long-read sequencing in a laboratory using challenging plant tissue from Eucalyptus pauciflora. Mol. Ecol. Resour. 19, 77–89 (2019).
Pereira-Marques, J. et al. Impact of host DNA and sequencing depth on the taxonomic resolution of whole metagenome sequencing for microbiome analysis. Front. Microbiol. 10, 1277 (2019).
Street, T. L. et al. Optimizing DNA extraction methods for nanopore sequencing of Neisseria gonorrhoeae directly from urine samples. J. Clin. Microbiol. 58, e01822-19 (2020).
Quick, J. et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276 (2017).
Levy, S. E. & Myers, R. M. Advancements in next-generation sequencing. Annu. Rev. Genomics Hum. Genet. 17, 95–115 (2016).
Charalampous, T. et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 37, 783–792 (2019).
Bishara, A. et al. High-quality genome sequences of uncultured microbes by assembly of read clouds. Nat. Biotechnol. 36, 1067–1075 (2018).
Stewart, R. D. et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 9, 870 (2018).
Knight, R. et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422 (2018).
Roumpeka, D. D., Wallace, R. J., Escalettes, F., Fotheringham, I. & Watson, M. A review of bioinformatics tools for bio-prospecting from metagenomic sequence data. Front. Genet. 8, 23 (2017).
Niu, S.-Y. et al. Bioinformatics tools for quantitative and functional metagenome and metatranscriptome data analysis in microbes. Brief. Bioinform. 19, 1415–1429 (2018).
Breitwieser, F. P., Lu, J. & Salzberg, S. L. A review of methods and databases for metagenomic classification and assembly. Brief. Bioinform. 20, 1125–1136 (2019).
Miller, S. et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 29, 831–842 (2019).
Naccache, S. N. et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 24, 1180–1192 (2014).
Kalantar, K. L. et al. IDseq—an open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. GigaScience 9, giaa111 (2020).
Sedlar, K., Kupkova, K. & Provaznik, I. Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics. Comput. Struct. Biotechnol. J. 15, 48–55 (2017).
Watson, M. & Warr, A. Errors in long-read assemblies can critically affect protein prediction. Nat. Biotechnol. 37, 124–126 (2019).
Bertrand, D. et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol. 37, 937–944 (2019).
Segata, N. On the road to strain-resolved comparative metagenomics. mSystems 3, e00190–17 (2018).
Grubaugh, N. D. et al. Tracking virus outbreaks in the twenty-first century. Nat. Microbiol. 4, 10–19 (2019).
Klinkenberg, D., Backer, J. A., Didelot, X., Colijn, C. & Wallinga, J. Simultaneous inference of phylogenetic and transmission trees in infectious disease outbreaks. PLoS Comput. Biol. 13, e1005495 (2017).
Klemm, E. & Dougan, G. Advances in understanding bacterial pathogenesis gained from whole-genome sequencing and phylogenetics. Cell Host Microbe 19, 599–610 (2016).
Stimson, J. et al. Beyond the SNP threshold: identifying outbreak clusters using inferred transmissions. Mol. Biol. Evol. 36, 587–603 (2019).
McArthur, A. G. et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357 (2013).
Gupta, S. K. et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220 (2014).
Doster, E. et al. MEGARes 2.0: a database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res. 48, D561–D569 (2020).
Arango-Argoty, G. et al. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 6, 23 (2018).
Stalder, T., Press, M. O., Sullivan, S., Liachko, I. & Top, E. M. Linking the resistome and plasmidome to the microbiome. ISME J. 13, 2437–2446 (2019).
Afshinnekoo, E. et al. Geospatial resolution of human and bacterial diversity with city-scale metagenomics. Cell Syst. 1, 72–87 (2015); erratum 1, 97–97.e3 (2015).
Ackelsberg, J. et al. Lack of evidence for plague or anthrax on the New York City Subway. Cell Syst. 1, 4–5 (2015).
Danko, D. et al. A global metagenomic map of urban microbiomes and antimicrobial resistance. Cell 184, 3376–3393.e17 (2021).
Munk, P. et al. Abundance and diversity of the faecal resistome in slaughter pigs and broilers in nine European countries. Nat. Microbiol. 3, 898–908 (2018).
Hendriksen, R. S. et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 10, 1124 (2019).
Lax, S. et al. Bacterial colonization and succession in a newly opened hospital. Sci. Transl. Med. 9, eaah6500 (2017).
Brooks, B. et al. Strain-resolved analysis of hospital rooms and infants reveals overlap between the human and room microbiome. Nat. Commun. 8, 1814 (2017).
Chng, K. R. et al. Cartography of opportunistic pathogens and antibiotic resistance genes in a tertiary hospital environment. Nat. Med. 26, 941–951 (2020).
Atoni, E. et al. Metagenomic virome analysis of Culex mosquitoes from Kenya and China. Viruses 10, 30 (2018).
Bouquet, J. et al. Metagenomic-based surveillance of Pacific Coast tick Dermacentor occidentalis identifies two novel Bunyaviruses and an emerging human ricksettsial pathogen. Sci. Rep. 7, 12234 (2017).
Tokarz, R. et al. Microbiome analysis of Ixodes scapularis ticks from New York and Connecticut. Ticks Tick Borne Dis. 10, 894–900 (2019).
Geldenhuys, M. et al. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLoS ONE 13, e0194527 (2018).
Vibin, J., Chamings, A., Klaassen, M., Bhatta, T. R. & Alexandersen, S. Metagenomic characterisation of avian parvoviruses and picornaviruses from Australian wild ducks. Sci. Rep. 10, 12800 (2020).
Carroll, D. et al. The Global Virome Project. Science 359, 872–874 (2018).
Rose, R., Constantinides, B., Tapinos, A., Robertson, D. L. & Prosperi, M. Challenges in the analysis of viral metagenomes. Virus Evol. 2, vew022 (2016).
Blauwkamp, T. A. et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat. Microbiol 4, 663–674 (2019).
Schlaberg, R. et al. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch. Pathol. Lab. Med. 141, 776–786 (2017).
Bharucha, T. et al. STROBE-metagenomics: a STROBE extension statement to guide the reporting of metagenomics studies. Lancet Infect. Dis. 20, e251–e260 (2020).
Sczyrba, A. et al. Critical assessment of metagenome interpretation—a benchmark of metagenomics software. Nat. Methods 14, 1063–1071 (2017).
Simner, P. J. et al. Development and optimization of metagenomic next-generation sequencing methods for cerebrospinal fluid diagnostics. J. Clin. Microbiol. 56, e00472-18 (2018).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014).
Strong, M. J. et al. Microbial contamination in next generation sequencing: implications for sequence-based analysis of clinical samples. PLoS Pathog. 10, e1004437 (2014).
Park, S.-J. et al. A systematic sequencing-based approach for microbial contaminant detection and functional inference. BMC Biol. 17, 72 (2019).
National Wastewater Surveillance System (Centers for Disease Control and Prevention, 2020).
Hardwick, S. A., Deveson, I. W. & Mercer, T. R. Reference standards for next-generation sequencing. Nat. Rev. Genet. 18, 473–484 (2017).
Cleveland, M. H., Zook, J. M., Salit, M. & Vallone, P. M. Determining performance metrics for targeted next-generation sequencing panels using reference materials. J. Mol. Diagn. 20, 583–590 (2018).
Logsdon, G. A., Vollger, M. R. & Eichler, E. E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 21, 597–614 (2020).
Bickhart, D. M. et al. Generating lineage-resolved, complete metagenome-assembled genomes from complex microbial communities. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-01130-z (2022).
Hon, T. et al. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci. Data 7, 399 (2020).
Quick, J. Ultra-long read nanopore sequencing methods for metagenomics. J. Biomol. Tech. 30, S63 (2019).
Razook, Z. et al. Real time, field-deployable whole genome sequencing of malaria parasites using nanopore technology. Preprint at bioRxiv https://doi.org/10.1101/2020.12.17.423341 (2020).
Flongle (Oxford Nanopore Technologies, 2022); http://nanoporetech.com/products/flongle
Dilthey, A. T., Jain, C., Koren, S. & Phillippy, A. M. Strain-level metagenomic assignment and compositional estimation for long reads with MetaMaps. Nat. Commun. 10, 3066 (2019).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110 (2020).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Almeida, A. et al. A new genomic blueprint of the human gut microbiota. Nature 568, 499–504 (2019).
Pasolli, E. et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662.e20 (2019).
Nayfach, S., Shi, Z. J., Seshadri, R., Pollard, K. S. & Kyrpides, N. C. New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505–510 (2019).
Field, D. et al. The Genomic Standards Consortium. PLoS Biol. 9, e1001088 (2011).
ten Hoopen, P. et al. The metagenomic data life-cycle: standards and best practices. GigaScience 6, 1–11 (2017).
Public Health Alliance for Genomic Epidemiology (PHA4GE, 2022); https://pha4ge.org/
Palatnick, A., Zhou, B., Ghedin, E. & Schatz, M. C. iGenomics: Comprehensive DNA sequence analysis on your Smartphone. GigaScience 9, giaa138 (2020).
Kovaka, S., Fan, Y., Ni, B., Timp, W. & Schatz, M. C. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat. Biotechnol. 39, 431–441 (2021).
Payne, A. et al. Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat. Biotechnol. 39, 442–450 (2021).
McDonald, D. et al. American Gut: an open platform for citizen science microbiome research. mSystems 3, e00031-18 (2018).
GISAID Phylodynamics (GISAID, 2022); https://www.gisaid.org/phylodynamics/
Main Report & Accompanying Work (The Independent Panel for Pandemic Preparedness and Response, 2021).
Sirleaf, E. J. & Clark, H. Report of the Independent Panel for Pandemic Preparedness and Response: making COVID-19 the last pandemic. Lancet 398, 101–103 (2021).
Cook, R., Karesh, W. & Osofsky, S. Conference Summary: One World, One Health: Building Interdisciplinary Bridges to Health in a Globalized World (One World, One Health, 2004).
King, L. J. et al. Executive summary of the AVMA One Health Initiative Task Force report. J. Am. Vet. Med. Assoc. 233, 259–261 (2008).
National Enhanced Surveillance of Vaccination Programmes Targeting Invasive Meningococcal Disease in England. Public Health England Immunisation Department and Meningococcal Reference Unit PHE Publication Gateway Number: 2015294 version number 1.2 10/12/2015 (Public Health England, 2015).
Centers for Disease Control and Prevention (CDC) Laboratory-acquired meningococcal disease—United States, 2000. MMWR Morb. Mortal. Wkly Rep. 51, 141–144 (2002).
Unemo, M. et al. World Health Organization global gonococcal antimicrobial surveillance program (WHO GASP): review of new data and evidence to inform international collaborative actions and research efforts. Sex. Health 16, 412–425 (2019).
Grimont, P. A. D., Weill, F. X. WHO Collaborating Centre for Reference and Research on Salmonella. Antigenic Formulae of the Salmonella serovars 9th edn (WHO, 2007).
Smith, A. M. et al. Laboratory-acquired infections of Salmonella enterica serotype typhi in South Africa: phenotypic and genotypic analysis of isolates. BMC Infect. Dis. 17, 656 (2017).
Bacteriological Analytical Manual (BAM) (US FDA, 2021).
Global Influenza Surveillance and Response System (GISRS) (WHO, 2022); https://www.who.int/initiatives/global-influenza-surveillance-and-response-system
Guidelines for Environmental Surveillance of Poliovirus Circulation (WHO, 2003).
Lasken, R. S. & Stockwell, T. B. Mechanism of chimera formation during the multiple displacement amplification reaction. BMC Biotechnol. 7, 19 (2007).
Binga, E. K., Lasken, R. S. & Neufeld, J. D. Something from (almost) nothing: the impact of multiple displacement amplification on microbial ecology. ISME J. 2, 233–241 (2008).
Keegan, K. P., Glass, E. M. & Meyer, F. MG-RAST, a metagenomics service for analysis of microbial community structure and function. Methods Mol. Biol. 1399, 207–233 (2016).
Chen, I.-M. A. et al. The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res. 49, D751–D763 (2021).
Mitchell, A. L. et al. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 48, D570–D578 (2020).
Batut, B. et al. ASaiM: a Galaxy-based framework to analyze microbiota data. GigaScience 7, giy057 (2018).
Li, P.-E. et al. Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform. Nucleic Acids Res. 45, 67–80 (2017).
Acknowledgements
K.K.K.K. is supported by the Singapore National Medical Research Council Research Training Fellowship. We thank C. Suphavilai, T. H. Koh and A. L. Tan for their valuable comments on the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Joshua Quick, Charles Chiu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ko, K.K.K., Chng, K.R. & Nagarajan, N. Metagenomics-enabled microbial surveillance. Nat Microbiol 7, 486–496 (2022). https://doi.org/10.1038/s41564-022-01089-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-022-01089-w
This article is cited by
-
Application of metagenomics for diagnosis of broilers displaying neurological symptoms
BMC Veterinary Research (2023)
-
Prevalence of fever of unidentified aetiology in East African adolescents and adults: a systematic review and meta-analysis
Infectious Diseases of Poverty (2023)
-
Development of a portable on-site applicable metagenomic data generation workflow for enhanced pathogen and antimicrobial resistance surveillance
Scientific Reports (2023)
-
Virome-wide detection of natural infection events and the associated antibody dynamics using longitudinal highly-multiplexed serology
Nature Communications (2023)
-
Modelling the effectiveness of surveillance based on metagenomics in detecting, monitoring, and forecasting antimicrobial resistance in livestock production under economic constraints
Scientific Reports (2023)
