
Abstract
Metagenomic next-generation sequencing (mNGS), the shotgun sequencing of RNA and DNA from clinical samples, has proved useful for broad-spectrum pathogen detection and the genomic surveillance of viral outbreaks. An additional target enrichment step is generally needed for high-sensitivity pathogen identification in low-titre infections, yet available methods using PCR or capture probes can be limited by high cost, narrow scope of detection, lengthy protocols and/or cross-contamination. Here, we developed metagenomic sequencing with spiked primer enrichment (MSSPE), a method for enriching targeted RNA viral sequences while simultaneously retaining metagenomic sensitivity for other pathogens. We evaluated MSSPE for 14 different viruses, yielding a median tenfold enrichment and mean 47% (±16%) increase in the breadth of genome coverage over mNGS alone. Virus detection using MSSPE arboviral or haemorrhagic fever viral panels was comparable in sensitivity to specific PCR, demonstrating 95% accuracy for the detection of Zika, Ebola, dengue, chikungunya and yellow fever viruses in plasma samples from infected patients. Notably, sequences from re-emerging and/or co-infecting viruses that have not been specifically targeted a priori, including Powassan and Usutu, were successfully enriched using MSSPE. MSSPE is simple, low cost, fast and deployable on either benchtop or portable nanopore sequencers, making this method directly applicable for diagnostic laboratory and field use.
Similar content being viewed by others


Main
The threat from new or re-emerging viruses has markedly increased in recent decades due to population growth, urbanization and the expansion of global travel, facilitating the rapid spread of infection during an outbreak1. Over the past four decades, we have encountered epidemics from human immunodeficiency virus (HIV; 1981–present), severe acute respiratory syndrome (SARS; 2002–2004) and Middle East respiratory syndrome (MERS; 2012–present) coronaviruses, 2009 pandemic influenza H1N1 and avian influenza viruses (1996–present), Ebola virus (EBOV) in West Africa (2013–2016) and Central Africa (1976–present) and Zika virus (ZIKV) in the Americas (2015–2016)2. The initial identification and containment of these outbreaks were hindered by their occurrence in resource-poor settings3 and the unavailability of diagnostic assays that could detect a novel, unanticipated viral strain. This lack of preparedness underscores the critical need for the deployment of effective tools able to rapidly diagnose emerging viral infections in febrile patients and to sequence genomes that can inform public health interventions to curb transmission.
A high sensitivity of detection is essential for assays that are used in clinical and public health settings. PCR-based assays for individual viruses have been widely deployed for diagnostic and surveillance applications due to their high sensitivity (1–10 copies (cp) per ml) and low cost4. However, these assays are limited by the requirement for a priori knowledge of the viruses to be targeted and the limited number of targets that can be typically multiplexed in a single test. The primers used in virus-specific PCR assays also invariably suffer from sequence-signature erosion over time due to genomic divergence, which occurs rapidly in RNA viruses encoded by an error-prone polymerase5.
Metagenomic next-generation sequencing (mNGS) is a promising candidate approach for broad-spectrum pathogen identification in clinical samples, as nearly all potential pathogens (viruses, bacteria, fungi and parasites) can be detected by uniquely identifying DNA and/or RNA shotgun sequences6,7. This method has been successfully applied for the clinical diagnosis of infectious diseases8,9, outbreak surveillance10,11 and pathogen discovery12. As it does not require a priori targeting of pathogens that may suddenly emerge in a new geographic region, such as EBOV in West Africa13, mNGS is a potentially useful diagnostic tool for addressing unknown viral outbreaks. However, issues related to cost, sequencing depth and background contamination14,15 can limit the accuracy of mNGS-based diagnostics relative to specific PCR testing. In particular, although unenriched mNGS using a nanopore sequencer is useful for sequencing known-positive viruses from clinical samples at moderate–high titres16,17, it is challenging to use it for the metagenomic analysis of clinical samples from patients with low-titre or undiagnosed viral infections.
Viral genome sequencing is essential for outbreak management, as it enables origin determination and monitoring of viral transmission patterns10. In general, target enrichment of NGS libraries is required to obtain sufficient viral genome coverage for phylogenetic and molecular clock analyses6. The enrichment of libraries using multiplex PCR of tiled amplicons (tiling multiplex PCR) and/or capture probe enrichment has been successfully used for the genomic surveillance of EBOV18,19, ZIKV20,21,22 and yellow fever virus (YFV) outbreaks23. Tiling multiplex PCR usually targets only a single circulating viral strain at a time; this requires that infection from that strain be established a priori (generally by previous virus-specific PCR testing) and it is thus less practical for viruses that exhibit high sequence divergence and/or recombination (for example HIV)24, are present as co-infections25 or comprise multiple genotypes (for example, hepatitis C virus (HCV), dengue (DENV)). Large panels containing millions of probes have been developed to capture viral diversity and potentially enrich for hundreds of different viruses26,27,28, but the relatively high cost, complex protocols and prolonged turnaround times (6–24 h for the hybridization step alone) needed for efficient capture probe hybridization hinder the broad application of this approach. For both methods, cross-contamination is a serious concern when pooling enriched samples together during multiplexing, as high-titre samples commonly contaminate low-titre or negative samples on the same sequencing run, with ~0.05% cross-contamination reported28.
Here we propose a target enrichment strategy, termed metagenomic sequencing with spiked primer enrichment (MSSPE), for simple, low-cost (0.10–0.34 USD per sample) enrichment of viral reads in sequencing libraries that adds no extra time to existing protocols yet retains the breadth of detection afforded by mNGS. We previously combined MSSPE with capture probe enrichment to recover whole-genome sequences of ZIKV from infected patients in Central America and Mexico29, revealing the introduction of the virus from Brazil via Honduras and the largely undetected spread throughout the region in 2014. In the current study, we expand the applications of MSSPE for use in the simultaneous detection and genome recovery of a wide range of emerging viruses associated with blood-borne infections, including vector-borne (arbovirus; ArboV)-related febrile illness and haemorrhagic fever. We also validate the method on both benchtop Illumina and portable nanopore sequencing platforms using primary clinical samples from infected patients.
Results
MSSPE for viral pathogen detection
We designed short 13-nucleotide spiked primers for 15 viruses (14 of which are evaluated in the current study; Fig. 1a,b). The number of primers per kilobase (kb) of viral genome reflected the relative diversity of a virus species, ranging from 10.8 for measles virus (MeV) to 46.9, 72.5 and 136.5 for Lassa virus (LASV), HIV and HCV, respectively (Supplementary Table 1).

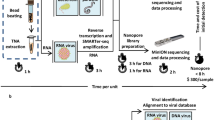
a, An algorithm for the design of viral spiked primers (SP). Sets of viral reference genomes (n = 60–3,571 for each virus) were aligned using MAFFT multiple sequence alignment software49, followed by the partitioning of each genome into 300–500-nt overlapping segments. Forward and reverse 13-nt primers were selected and filtered according to specific criteria (rounded rectangular box). Unique primer sequences are individually coloured in red, blue, orange and green. Using this algorithm, primers were designed for 15 RNA viruses. SP panels for ArboV (n = 4), HFV (n = 6) and CombV (n = 13, excluding HCV and JCV SP) were also constructed. b, The metagenomic sequencing workflow. MSSPE primers (red) were added (spiked) to a reaction mix containing random primers (blue) during the reverse transcription step of cDNA synthesis, without adding to the overall turnaround time for the subsequent transposase-based library amplification with adapter primers (brown) and sequencing analysis protocols. The MSSPE workflow is compatible with subsequent enrichment using tiling multiplex PCR and/or capture probes (dashed lines). Metagenomic sequence data were analysed for pathogen identification using SURPI (ref. 50; also see Methods). MARV, Marburg virus; RVFV, Rift Valley fever virus; HEV, hepatitis E virus; and Tm, melting temperature.
First, we evaluated the enrichment effect of virus-specific spiked primers for ZIKV and West Nile virus (WNV) detection on Illumina MiSeq. For these experiments, we used either viral culture supernatant (ZIKV, DENV, EBOV) or a high-titre clinical sample (WNV) spiked at defined concentrations into a negative plasma donor matrix. At a spiked primer concentration of 1 μM, the maximum concentration recommended for specific PCR30, the degree of ZIKV enrichment in contrived samples containing ZIKV and HCV as an off-target virus was highest (4–6×) at 5:1 and 10:1 molar ratios of spiked to random hexamer (RH) primers (Supplementary Table 2). There was no or minimal loss of detection sensitivity for off-target HCV. Increasing the molar ratio of spiked to RH primers to 100:1 from 10:1 did not result in increased enrichment of WNV reads using an ArboV spiked primer panel at 1 μM concentration (Supplementary Table 3). For spiked primers targeting individual viruses, a comparison of concentrations (1, 4 and 10 μM) at a molar ratio of 10:1 found that the degree of enrichment peaked at 4 μM (Supplementary Table 4).
Next, we tested spiked primer concentrations ranging from 1 μM to 40–80 μM for the enrichment of ZIKV, DENV and EBOV using spiked primer panels for the detection of ArboV, haemorrhagic fever viruses (HFV) and 13 viruses combined (CombV). The peak performance of the ArboV panel was at a primer concentration of 10–20 μM, yielding a 12-fold enrichment in ZIKV and 6-fold enrichment in DENV reads (Fig. 2a). Metagenomic detection of off-target viruses (EBOV) was not impaired. The optimal primer concentration for the HFV panel was found to be 20 μM (Fig. 2b), yielding a 7× enrichment for EBOV. The CombV panel at the optimal 10–20 μM primer concentration yielded 29×, 15× and 6× enrichment for ZIKV, DENV and EBOV, respectively (Fig. 2c). As the degree of enrichment was noted to be higher at lower viral titres (Fig. 2a,c), we next tested virus-specific primers (4 μM) and expanded panels (10–20 μM) for enrichment of ZIKV, DENV and EBOV using optimal concentrations. Notably, the degree of fold enrichment of ZIKV, DENV and EBOV using virus-specific primers versus larger primer panels (ArboV, HFV and CombV) was comparable (Fig. 2d,e).

a–c, Plots of the fold enrichment achieved for contrived samples containing ZIKV, DENV and/or EBOV at defined titres and using RH primers only or SP concentrations ranging from 1 μM to 40–80 μM. The enrichment of ZIKV and DENV using an ArboV SP panel (a). The asterisk denotes EBOV as an off-target virus when using the ArboV SP panel. The enrichment of EBOV using a HFV SP panel (b) and the enrichment of ZIKV, DENV and EBOV using a CombV SP panel (c). Dashed lines denote 1× or no enrichment. d–h, Box-and-whisker plots of the fold enrichment achieved using MSSPE compared to using RH only. The box outlines denote the IQR, the solid line in the box denotes median fold enrichment, the dashed line denotes mean (µ) fold enrichment and the whiskers outside of the box extend to the minimum and maximum fold enrichment points. The fold enrichment for DENV and ZIKV using virus-specific primers, ArboV panels or CombV panels (d). The fold enrichment for EBOV using virus-specific primers, HFV panels or CombV panels (e). The overall fold enrichment, including all 92 pairwise comparisons (with and without MSSPE) of contrived and clinical samples (f). The fold enrichment for 65 pairwise comparisons of contrived samples (g). The fold enrichment for 27 pairwise comparisons of clinical samples (h). The degree of fold enrichment at <100 cp ml−1 is significantly higher than at other titres (paired two-sided Student’s t test; P = 0.008 between groups <100 cp ml−1 and 100–1,000 cp ml−1; P = 0.0002 between groups <100 cp ml−1 and >1,000 cp ml−1).
Next, we evaluated the overall MSSPE enrichment effect across 14 viruses and 92 paired mNGS runs from both contrived and clinical samples at titres ranging from 8 to 112,201 cp ml−1 on the Illumina MiSeq platform. For this, and all subsequent sequencing experiments, final spiked primer concentrations of 4 μM for individual viruses, 10 μM for the ArboV panel and CombV panel and 20 μM for the HFV panel were used, all mixed with RH primers at a ratio of 10:1. The overall median fold enrichment was 10×, with 6–17× enrichment within the interquartile range (IQR) (Fig. 2f). Among contrived samples, a trend of the highest median fold enrichment at the lowest titre (median 16× at 10 cp ml−1) was observed, with less enrichment (median 7–11×) at titres between 10 and 10,000 cp ml−1 (Fig. 2g).
The performance of the spiked primer panels was then evaluated on the portable MinION nanopore sequencing platform (Oxford Nanopore Technologies). Overall levels of ZIKV, EBOV and DENV enrichment at viral titres ranging from 10–1,000 cp ml−1 for the MinION were comparable to those for the Illumina MiSeq, with median enrichment of 13× and 16×, respectively (Supplementary Table 5). The use of MSSPE enabled both nanopore- and Illumina-based metagenomic detection of ZIKV and EBOV down to 10 cp ml−1, or 2 viral cp per complementary DNA reaction, near the limits of detection for virus-specific PCR31,32 (Supplementary Table 5). Probit analysis of contrived samples of ZIKV spiked into donor plasma matrix at serial dilutions of 104 to 1 cp ml−1 using the ArboV primer panel and mean sequencing depth of 1,348,588 (±369,170 s.d.) reads yielded an analytical limit of detection of 48 cp ml−1 (Supplementary Table 6).
We next evaluated the performance of the ArboV and HFV panels on a portable nanopore sequencer using clinical blood samples from 19 patients infected with chikungunya virus (CHIKV; n = 3), DENV (n = 4), ZIKV (n = 3, from Brazil), EBOV (n = 3, from the Democratic Republic of the Congo (DRC)) and YFV (n = 6, from Angola) (Table 1). A median 14× fold enrichment (IQR: 7–17×) was observed across different viruses at titres ranging from 15 to 5,500 cp ml−1 when comparing spiked primers to random primers alone. Consistent with the results using contrived samples, clinical samples with lower viral titres of <100 cp ml−1 produced more robust enrichment than higher titre samples (P < 0.05 by paired t test) (Fig. 2h).
To evaluate the clinical performance of MSSPE testing for the detection of ArboV (ZIKV, DENV and CHIKV) and HFV (YFV and EBOV) using ArboV and HFV primer panels, respectively, we also tested 21 plasma samples from infected patients in parallel with 18 negative control samples on the nanopore sequencer (Supplementary Table 7). The overall sensitivity, specificity, positive predictive value and negative predictive value of the assay were 95.0%, 94.8%, 92.7% and 96.5%, respectively (Supplementary Table 8). Three false-positive cases were attributed to barcode misassignments when demultiplexing samples due to a nanopore sequencing error (Supplementary Table 7). No cross-contamination of viral reads was observed in either water or donor plasma matrix samples that were processed and sequenced along with contrived or clinical samples at estimated titres ranging from 101–107 cp ml−1 (Supplementary Tables 6 and 7).
Next, we tested whether spiked primers could detect and potentially enrich sequences from emerging flaviviruses in clinical samples from infected patients, including Powassan virus (POWV) and Usutu virus (USUV) (Table 2). Notably, these viruses had not been specifically targeted a priori in the initial spiked primer design. In cerebrospinal fluid (CSF) from a patient with tick-borne POWV meningoencephalitis, the use of ArboV spiked primers enriched for POWV reads by 15× and improved viral genome coverage by 43% (Table 2 and Fig. 3d). The alignment of ArboV spiked primers to the POWV genome (accession no. NC_003687), tolerating at most one mismatch, identified 39 mapping to the genome (Supplementary Fig. 1a). As part of an ongoing HIV genomic surveillance study, we incidentally detected reads mapping to USUV in a plasma sample from an HIV-1-infected patient using mNGS. Experimental testing on an Illumina MiSeq at a limited throughput of ~1 million raw reads resulted in a failure to detect USUV reads using RH primers alone, versus detection of six USUV reads using ArboV spiked primers (Supplementary Table 9). Follow-up sequencing on an Illumina HiSeq at a higher sequencing depth of ~123 million reads revealed that the degree of enrichment of USUV reads using the ArboV panel was ~7.5× (Table 2), with a corresponding increase in genome coverage of 17.5%. Of the spiked primers in the ArboV panel, 64 mapped to the USUV genome (Supplementary Fig. 1b).

a, Genome coverage of the ZIKV MRC766 (Uganda) strain (mapped to accession no. LC002520) at 1,000 cp ml−1 with no enrichment (top) or MSSPE enrichment using ZIKV SP (second from top), an ArboV SP panel (third from top) or a CombV SP panel (bottom). With no enrichment, there were 50 reads and 45% coverage; with ZIKV SP, there were 456 reads and 97.6% coverage; with ArboV SP, 528 reads and 100% coverage; with CombV SP, there were 254 reads and 93.9% coverage. b, Genome coverage of an HIV-1 Group M, CRF01 strain (mapped to accession no. KY580709) at 1,000 cp ml−1 with no enrichment (left) or using HIV-1 SP (right). With no enrichment, there were 35 reads and 23.2% coverage; with HIV-1 SP, there were 289 reads and 92.8% coverage. c, Genome coverage of an HCV genotype 4 strain (mapped to accession no. KM587625) at 10,000 cp ml−1 with no enrichment (left) or using HCV SP (right). With no enrichment, there were 63 reads and 31.5% coverage; with HCV SP, there were 686 reads and 80% coverage. d, Genome coverage of a POWV strain identified in CSF from an infected patient with tick-borne meningoencephalitis (mapped to accession no. NC_003687) at <1,000 cp ml−1 with no enrichment (left) or using the ArboV SP panel (right). With no enrichment, there were 48 reads and 37.1% coverage; with ArboV SP, there were 209 reads and 88.0% coverage. e, Genome coverage of a contrived sample of LASV (Josiah strain) spiked into donor plasma matrix at a titre of 10 cp ml−1 (mapped to accession nos. AY628202 and NC_004296) with no enrichment (left) or using the HFV SP panel (right). With no enrichment, there were 4 reads and 3.8% coverage; with HFV SP, there were 154 reads and 67.9% coverage. f, Genome coverage of a contrived sample of CCHFV (mapped to accession nos. AY389508, U39455 and U88410) spiked into donor plasma matrix at a titre of 2,500 cp ml−1 with no enrichment (left) or using the HFV SP panel (right). With no enrichment, there were 69 reads and 23.3% coverage; with HFV SP, there were 2,636 reads and 100% coverage. g, Genome coverage of a strain from a patient from Mexico with acute ZIKV infection during the 2013–2016 outbreak (ZIKV/Homo sapiens/MEX/2016/mex30; mapped to accession no. KX879603) at ~2,000 cp ml−1 with no enrichment (top) or enrichment using MSSPE with ZIKV SP (second from top), tiling multiplex PCR (third from top), capture probes (fourth from top, using random primers alone) or MSSPE with ZIKV SP followed by capture probes (bottom). With no enrichment, there were 33 reads and 26.5% coverage; with ZIKV SP, there were 260 reads and 87.5% coverage; with tiling multiplex PCR, there were 158,243 reads and 88.2% coverage (75.0% ≥10× coverage); with capture probes, there were 49,927 reads and 49.1% coverage (29.6% ≥10× coverage); and with ZIKV SP plus capture probes, there were 275,105 reads and 99.8% coverage (95.6% ≥10× coverage). The red bars below the coverage plots show nucleotide regions with coverage of ≥10×, at a threshold to minimize the inclusion of cross-contaminating reads36. For each graph in a–g, the number of reads is normalized to the total number of viral reads obtained with no enrichment. bp, base pairs; L, large segment; M, medium segment, S, small segment.
To assess whether metagenomic detection sensitivity for off-target DNA viral and non-viral pathogens was retained using MSSPE, we evaluated the method using a representative mixture of seven organisms developed as a standardized positive control for a clinical metagenomic assay from CSF33. The normalized reads per million (RPM) corresponding to each of the seven pathogen types were comparable when using the ArboV or HFV spiked primer panel versus RH primers alone (Supplementary Table 10). Furthermore, the MSSPE method did not enrich reads corresponding to off-target viruses associated with laboratory and/or reagent contamination, such as murine leukaemia virus34. To determine whether the human host background affects the viral enrichment effect by MSSPE, we compared the proportion of human reads among clinical and contrived samples at varying levels of background (25–99.4%). The efficiency of MSSPE enrichment did not decrease with higher levels of background, since robust enrichment (5–39×) was still observed in CSF or brain tissue samples containing >99% human reads (Supplementary Fig. 2).
MSSPE for viral genome sequencing
We hypothesized that the increased proportion of viral reads obtained using the MSSPE method would improve genome coverage. MSSPE primers were used to enrich contrived samples of ZIKV, DENV, EBOV, MeV (Edmonston strain), LASV (Josiah strain from Sierra Leone) and Crimean–Congo haemorrhagic fever virus (CCHFV, strain IbAr10200 from Nigeria) spiked into donor plasma matrix (Fig. 3a; Supplementary Tables 11 and 12). On average, 45% (±16% s.d.) absolute percentage increases in the breadth of genome coverage from deduplicated reads were achieved in contrived samples for the non-segmented viruses using MSSPE relative to RH primers (Supplementary Table 11). For the segmented CCHFV and LASV viruses, MSSPE using virus-specific spiked primers improved genome coverage by 69% (±12% s.d.) and 30% (±10% s.d.) for the L and S segments of LASV, respectively, and by 58% (±19% s.d.), 62% (±33% s.d.) and 66% (±21% s.d.) for the L, S and M segments of CCHFV, respectively (Fig. 3e,f; Supplementary Table 12). MSSPE primers were then used to enrich clinical samples of HIV-1 (divergent and recombinant strains from Cameroon and the DRC; Fig. 3b), HCV (genotypes 2, 4 and 6 from California, United States; Fig. 3c), ZIKV, CHIKV, YFV, DENV and EBOV (haemorrhagic fever patients during the 2014 EBOV Boende outbreak) at low–moderate viral titres ranging from 43 to 16,512 cp ml−1 (Table 3). For these clinical samples, there was a mean 50% (±16% s.d.) increase in genome coverage using MSSPE. We also used MSSPE to enrich for Jamestown Canyon virus (JCV) in post-mortem brain tissue samples from a patient who developed a fatal viral encephalitis following a mosquito bite. Virus-specific enrichment for JCV increased the number of viral reads by ~40× versus no enrichment using ZIKV primers (Supplementary Table 13). Overall, for the non-segmented viruses, the increase in genome coverage using MSSPE across all contrived and clinical samples was 47% (±16% s.d.) (calculated from all samples in Table 3 and Supplementary Table 11).
Despite robust fold enrichment of 16–55×, no substantial gains in genome coverage were observed with clinical samples of ZIKV and EBOV at 10 cp ml−1 (Supplementary Table 14), a finding attributed to insufficient sequencing depth at low viral titres. To evaluate the ability of MSSPE to discriminate between different viral subtypes for the purpose of outbreak tracking, we tested ten clinical samples of divergent HIV and HCV viruses with confirmed genotypes by Sanger sequencing-based assays (Supplementary Table 15). All ten samples (100%) were identified as the correct viral subtype using MSSPE and genome assembly of mapped reads (average depth >30×, ranging from 36× to 6,000×), suggesting that MSSPE does not bias the consensus genome assembly.
Comparison of MSSPE with other target enrichment methods
We performed head-to-head comparisons of MSSPE with both capture probe35 and tiling multiplex PCR36 methods for the enrichment of viral reads from ZIKV-positive clinical samples at low titres (310–28,200 cp ml−1). The degree of improvement in genome coverage using MSSPE was comparable to or better than capture probe and tiling multiplex PCR methods. However, cross-contamination was observed using tiling multiplex PCR and capture probe enrichment, versus no cross-contamination using MSSPE (Supplementary Table 16). Furthermore, these two methods generated 80–95% duplicate reads versus ~20% for MSSPE (Supplementary Table 17). Tiling multiplex PCR for ZIKV was negative when testing a contrived ZIKV sample containing the 1947 prototype Uganda strain MR766, probably due to sequence divergence from the American ZIKV strains from the 2015–2018 outbreak used in the initial multiplex PCR primer design36.
Next, we evaluated the combined performance of MSSPE and subsequent tiling multiplex PCR or capture probe enrichment on low-titre contrived and clinical ZIKV samples (666–3,340 cp ml−1). The use of spiked primers further increased the number of ZIKV reads by 6× and corresponding genome coverage by 25–80% (average 58.5 ± 21.5%), as compared to RH primers alone when used in combination with tiling multiplex PCR or capture probe enrichment (Supplementary Table 18 and Fig. 3g). Notably, MSSPE was critical for ZIKV genome recovery in the two low-titre samples tested by tiling multiplex PCR, as multiplex PCR with standard RH priming failed to yield a distinct band on gel electrophoresis.
Discussion
In this study, we developed MSSPE as a universal robust target enrichment method that is simple, low cost, fast (incurring no extra turnaround time), compatible with different library preparation protocols (transposon or adapter-ligation based) and deployable on benchtop or portable sequencers. We found that the MSSPE method produced a median 10× enrichment across 14 different viruses in low-titre clinical samples (10–10,000 cp ml−1), improving detection sensitivity to 10 cp ml−1 and increasing mean genome coverage by 47% (±16%), while preserving broad metagenomic sensitivity for pathogen detection. Enrichment was possible across a wide range of potential targets, from single viruses with varying sequence diversity to expanded panels (ArboV and haemorrhagic fever) to the inclusion of a combined panel of 4,792 spiked primers from 13 viral species. Notably, MSSPE demonstrated an analytical limit of detection of 48 genomic cp ml−1 for ZIKV by probit analysis and compared favourably to gold-standard quantitative PCR with reverse transcription (qRT–PCR) testing for the detection of ZIKV, CHIKV, DENV, EBOV and YFV in clinical samples from febrile patients with viral infection, with analytic performance metrics (sensitivity, specificity, positive predictive value and negative predictive value) all >92%. Taken together, these results demonstrate the utility of the MSSPE method for simultaneous viral diagnosis, genome recovery and metagenomic surveillance in laboratory or field settings.
In comparison to other NGS enrichment strategies, such as tiling multiplex PCR and capture probe enrichment, we found that MSSPE had less biased amplification (~20% duplicate reads versus 80–95% for the other methods) and less sample cross-contamination. In particular, minimal or no cross-contamination was observed with MSSPE when testing contrived and clinical samples at virus titres ranging from 10 to 107 cp ml−1. Nevertheless, MSSPE was complementary to these other strategies, further increasing the yield of viral reads for detection and genome recovery when used in combination.
An alternative approach for pathogen enrichment in mNGS libraries is the use of a host depletion method. Such methods include differential lysis37, the removal of abundant human ribosomal and mitochondrial RNA sequences using antibody hybridization or depletion of abundant sequences by hybridization (DASH)38 and nuclease treatment before extraction39,40. Aside from a post-extraction DNase step, we did not incorporate host depletion as part of MSSPE, given that these methods can be cumbersome, with additional steps, and difficult to standardize. Furthermore, host depletion methods can bias detection towards specific pathogen types, such as the enrichment of bacteria and fungi with cell walls with differential lysis37, or encapsidated viruses with pre-extraction nuclease treatment39,40. Host depletion can also be performed at the RNA level by the use of non-human primers to bias against abundant human host ribosomal and/or mitochondrial RNA41,42. These methods have been primarily applied to samples such as cellular lysates and throat swabs, which contain significant amounts of human ribosomal RNA. For cell-free fluids such as the clinical plasma, CSF and serum samples analysed here, the lower proportions of human ribosomal RNA can make these methods less effective. The impact of non-human primers on the proportion of contaminant sequences or metagenomic detection sensitivity for non-viral pathogens is also unknown.
To enrich viruses from unknown clinical samples, we developed spiked primer panels to detect viruses associated with ArboV infection or haemorrhagic fever. The use of customized spiked primer panels allows RNA viral pathogens associated with a geographic region to be targeted (for example, HFV for testing in the DRC and ArboV for testing in Brazil). In the future, regional public health surveillance data can be leveraged in the composition of real-time updated spiked primer panels targeting actively circulating pathogens. This is especially important to facilitate the rapid development of diagnostic tests for a viral pathogen that is newly introduced to a geographic region (for example, EBOV in West Africa, ZIKV in Brazil). Of note, we have shown here that detection and enrichment of unexpected re-emerging and/or co-infecting viruses, such as USUV and POWV, is possible with MSSPE, even if these viruses had not been specifically targeted a priori. Enrichment was due to conserved flaviviral primers in the ArboV panel exhibiting incidental sequence homology to these off-target viruses.
On average, genome recovery for eight different viruses using virus-specific spiked primers and expanded panels improved by 47% (±16%). The minimum threshold that has been proposed for completion of a draft viral genome is ≥50% (ref. 43). The percentage of samples with ≥50% coverage increased from 15.6% (7 of 45) using mNGS alone to 86.7% (39 of 45) using MSSPE primers (P = 0.0001 by McNemar’s paired two-sided test). Our previously published simulations and analyses undertaken in the context of ZIKV genomic epidemiology in Central America showed that 40–60% genome coverage is sufficient for phylogenetic analysis and accurate determination of viral lineage (Supplementary Fig. 1 in ref. 29). Notably, the MSSPE method was effective in the genome sequencing of recombinant HIV viruses, in both circulating and unknown recombinant forms44, which exhibit sequence divergence of up to 35% in the env gene45, as well as multiple HCV genotypes and divergent LASV and CCHFV viruses. Robust improvements in genome coverage were also observed when testing clinical samples of YFV, CHIKV and DENV on the portable nanopore sequencer, underscoring the potential utility of MSSPE for the rapid detection and monitoring of virus outbreaks.
Our study had some limitations. First, we did not test respiratory viruses of outbreak importance, such as coronaviruses, enterovirus D68 (ref. 16) and influenza viruses. Given the success with all RNA viruses tested to date, it is likely that the MSSPE method will work in a similar way for additional RNA viruses. Second, we did not formally test MSSPE for double-stranded RNA viruses, DNA viruses or non-viral pathogens. Third, the degree of enrichment by MSSPE varied among the different viruses (range: 4–55×), in a similar range to that observed in previous large-panel capture probe enrichment studies26,27,28. The varying enrichment across different sample concentrations is probably due to several factors, including the number of spiked primers, viral titres in the sample and the diversity of viral targets. Fourth, the variable level of enrichment and coverage at a given nucleotide position achieved using MSSPE might preclude it from high-fidelity viral quasispecies analysis or whole-genome assembly at high coverage depth, especially on the nanopore sequencer given current sequencing error rates of ~5–10% (ref. 46). For these applications, MSSPE may be synergistic with complementary tiled multiplex PCR or capture probe enrichment approaches, as demonstrated here and previously29.
Methods
Ethics statement
Clinical ZIKV serum samples from Mexico were collected as part of the national epidemiological surveillance programme of the Instituto Mexicano del Seguro Social (IMSS), a branch of the Ministry of Health, as previously described29. Samples and ancillary clinical and epidemiological data were de-identified before analysis and are thus considered exempt from human subject regulations, with a waiver of informed consent according to 45 CFR 46.101(b) of the US Department of Health and Human Services. Analysis of whole blood samples from patients with EBOV disease was approved by the Ministry of Health in the DRC. Patients in the 2014 Boende EBOV outbreak from 13 August 2014 to 8 September 2014 provided oral consent for study enrolment and the collection and analysis of their blood. Consent was obtained at the homes of patients or in hospital isolation wards by a team that included staff members of the Ministry of Health. Plasma samples from patients with HIV-1 and/or USUV infection were provided by the Abbott Global HIV-1 Surveillance Program. Briefly, informed consent was obtained for the collection of HIV-1 infected blood donations from blood banks in Cameroon and analysis for viral load determination and sequencing under protocols approved by local ethics committees47. Clinical samples were analysed at the University of California San Francisco (UCSF) under protocols approved by the UCSF Institutional Review Board (protocol no. 11-05519).
Sample collection
Viral cultures of ZIKV (Uganda strain), DENV (type 1) and MS2 bacteriophage were purchased from the American Type Culture Collection (ATCC). Ebola cultures (Kikwit strain) in TRIzol LS (Thermo Fisher Scientific) were provided by J. Patterson at Texas Biomedical Research Institute. Donor plasma matrix consisting of pooled plasma from multiple de-identified blood donors that tested negative for infection by blood-borne pathogens, including HIV, HBV, HCV and Treponema pallidum, was obtained from Golden West Biologicals, Inc.
Clinical ZIKV serum samples were provided by the Central Laboratory of Epidemiology (CLE), IMSS in Mexico City, Mexico and Blood Systems Research Institute. Forward and reverse primers (ZIKV 1086 and ZIKV 1162c, respectively) and carboxyfluorescein (FAM)-labelled probes (ZIKV 1107-FAM) were used as previously described48. Clinical CHIKV and DENV samples from febrile returning travellers were provided by the California Department of Public Health. Clinical Ebola samples collected from patients in the 2014 Boende outbreak were provided by INRB in Kinshasa, DRC. Real-time qRT–PCR testing was used for the determination of viral titres for ZIKV (refs. 25,48), DENV and EBOV by standard curve analysis (Supplementary Table 19). Clinical HIV and hepatitis C plasma samples were obtained from the UCSF Clinical Microbiology Laboratory (San Francisco, USA). Clinical samples from HIV-infected patients in Cameroon were provided by the Universities of Yaounde, Bamenda and Montanges in Cameroon and Abbott Laboratories, Inc. Clinical yellow fever samples collected from patients in the 2015–2016 yellow fever outbreak in Angola were provided by the Angolan National Institute of Public Health. The CSF sample from a patient with POWV meningoencephalitis was provided by Boston Children’s Hospital. Post-mortem brain biopsy tissue from a patient who died of JCV encephalitis was provided by Massachussetts General Hospital. The positive control of seven organisms was obtained from the UCSF Clinical Microbiology Laboratory. A representative mixture of seven organisms included an RNA virus (HIV), a DNA virus (cytomegalovirus), a Gram-positive bacterium (Streptococcus agalactiae), a Gram-negative bacterium (Klebsiella pneumoniae), a yeast (Cryptococcus neoformans), a mould (Aspergillus niger) and a parasite (Toxoplasma gondii).
MSSPE viral spiked primer design
We sought to develop a general method for combining metagenomic viral detection with enrichment and genome recovery from clinical samples. We required that the method should: be applicable for targeted viruses with varying numbers of reference genomes/genome segments in the database (for example, from 60 to 3,571) (Fig. 1a); preserve broad metagenomic sensitivity for comprehensive detection of off-target viruses and/or viral co-infections; not impact turnaround times for sample processing; enrich mNGS libraries sufficiently to allow robust viral genome recovery from low-titre clinical samples. Specifically, we designed an automated computational algorithm that took an arbitrary set of reference genomes as an input and constructed a minimal panel of short, 13-nucleotide spiked primers to cover these genomes (Fig. 1a), which were to be added during the cDNA synthesis (reverse transcription) step of mNGS library preparation (Fig. 1b).
To design specific spiked primers for a virus, multiple sequence alignment of complete viral genomes (downloaded from the National Center for Biotechnology Information (NCBI) GenBank nucleotide database as of September 2017) was performed using multiple alignment using fast Fourier transform (MAFFT) (v.7.388) at default parameters (algorithm = “Auto”, scoring matrix = "200PAM/k = 2”, gap open penalty = 1.53, off-set value = 0.123)49. An in-house bioinformatics pipeline named MSSPE-design (v.1.0) was developed on an Ubuntu Linux computational server for the automated design of spiked primers. Briefly, the multiple sequence alignment-aligned genomes were partitioned into overlapping 500-nucleotide segments with 250-nucleotide overlap using PYFASTA (http://pypi.python.org/pypi/pyfasta/). Forward or reverse 13-nucleotide primers were selected from 50-nucleotide regions at the ends of each segment by iteratively ranking candidate 13mer (kmer) sequences in reverse order by frequency, selecting the top kmer shared by the most segments and not containing any ambiguous nucleotides and then removing segments sharing that 13mer before repeating the process on the remaining segments. To decrease overall spiked primer costs, the iterations were repeated until the number of remaining segments containing a shared kmer was below a predesignated threshold (ranging from n = 1 for viruses with only a limited number of genomes/genome segments, such as CCHFV, to n = 10 for viruses comprising thousands of genomes and multiple genotypes, such as DENV). Spiked primers were filtered by the removal of primers with melting temperatures greater than 2 s.d. from the mean or that were predicted to self-dimerize or cross-dimerize with a ΔG value (standard free energy of DNA duplex formation) equal to or more negative than −9 kcal mol−1.
Spiked primers were synthesized by Integrated DNA Technologies Inc. Forward or reverse spiked primer oligonucleotides targeting individual viruses were synthesized on a 10 nmol scale in 96-well plates with standard desalting and 6 nmol of each individual oligonucleotide was mixed and then resuspended to a final volume of 500 μl in IDTE pH 8.0. Spiked primer panels (ArboV, HFV and CombV) were designed by mixing the spiked primers for each individual virus in equimolar ratios and then diluting with Tris–EDTA buffer to the desired concentration. The estimated cost per sample for 1 million reads using the Illumina MiSeq is approximately US$100 for random metagenomic sequencing. For tenfold more viral reads, approximately 10–20 million raw sequencing reads are needed, with an estimated cost of US$250 per sample using an Illumina HiSeq platform. By comparison, the MSSPE approach can obtain 10× average viral enrichment at low incremental costs of US$0.06–US$0.08 per sample using individual virus-specific primers or US$0.17–US$0.34 using spiked primer panels (Supplementary Table 20).
Construction of metagenomic sequencing libraries
To minimize sample and exogenous laboratory cross-contamination during library preparation, strict measures were implemented, including unidirectional workflow, separation of pre-PCR and post-PCR workspaces using different rooms and rigorous decontamination of biosafety cabinets and work benches using 10% bleach and/or 70% ethanol. Potential contamination was monitored by the processing of negative water and donor plasma matrix controls in parallel with samples for a subset of runs.
For the preparation of metagenomic sequencing libraries, viral RNA was first extracted from 400 µl of contrived or clinical patient samples using the EZ1 Advanced XL BioRobot and EZ1 Virus Mini Kit (Qiagen), with the exception of EBOV RNA, which was extracted manually in the viral haemorrhagic fever reference laboratory in INRB, Kinshasa using the Direct-zol RNA MiniPrep Kit (Zymo Research). Final RNA was eluted in 60 μl AVE buffer. 25 μl of nucleic acid extract was treated with DNase (3 μl Turbo DNase, 1 μl Baseline, 5 μl Turbo buffer and 16 μl nuclease-free water) and incubated on an Eppendorf ThermoMixer at 37 °C, 600 r.p.m. for 30 min. The Zymo RNA Clean and Concentrator kit (Zymo Research) was used to clean up DNase-treated RNA and the final RNA was eluted in 32 μl water. The RNA was then mixed with RH alone (1 μM) or spiked primer plus RH in a 10:1 ratio of spiked primer to RH and heated to 65 °C for 5 min. The reverse transcription master mix (10 μl SuperScript III buffer, 5 μl dNTP (12.5 mM), 2.5 μl DTT (0.1 M), 1 μl SuperScript III enzyme) was added to each sample and incubated at 25 °C for 5 min, followed by 42 °C for 30 min and 94 °C for 2 min. After cooling to 10 °C, a second-strand synthesis master mix (3.7 μl Sequenase buffer, 0.225 μl Sequenase enzyme and 1.1 μl water) was added to each reaction, followed by a slow 2 min ramp to 37 °C and an 8-min incubation. The resulting cDNA was cleaned up using the Zymo DNA Clean and Concentrator kit (Zymo Research), with the addition of 10 μl linear acrylamide to each sample, and eluted in 7.5 μl water. Using the Illumina Nextera XT kit, 2.5 μl sample cDNA was incubated at 55 °C for 5 min in tagmentation mix (10 μl TD buffer and 5 μl ATM enzyme) and immediately neutralized with 2.5 μl NT buffer. 12.5 μl tagmented DNA was then transferred to the reaction tube containing indexing mix (7.5 μl Nextera XT PCR master mix, 2.5 μl N-7XX primer and 2.5 μl S-5XX primer from Illumina) for the barcoding of individual samples. PCR amplification was then performed using the following conditions: an initial denaturation step of 95 °C for 30 s, followed by 16 cycles of denaturation (95 °C for 10 s), annealing (55 °C for 30 s) and extension (72 °C for 30 s), with a final extension at 72 °C for 5 min. After PCR, 3 μl of PCR product was analysed by 2% gel electrophoresis to check for library size and band intensity. If no band or only a very faint band was observed on the gel, another round of recovery PCR was performed. For recovery PCR, the library was washed using 0.9X AMPure XT beads (Beckman Coulter) and 5 μl clean library was mixed with 45 μl master mix (10 μl buffer, 2.5 μl 10 μM Nextera general primers (forward: 5′-AATGATACGGCGACCACCGA-3′; reverse: 5′-CAAGCAGAAGACGGCATACG-3′), 1 μl dNTP, 0.5 μl Phusion DNA polymerase enzyme and 31 μl water), followed by a 95 °C incubation for 30 s and 10 cycles of PCR (95 °C for 30 s denaturation, 60 °C for 30 s annealing and 72 °C for 30 s extension), with a final extension at 72 °C for 5 min. The final cDNA library was eluted in 20 μl EB buffer after a wash step using 0.9X AMPure beads.
Metagenomic sequencing
The cDNA libraries were quantified using a Qubit fluorometer (Thermo Fisher Scientific) and the sizes of the libraries were measured using an Agilent Bioanalyzer (Agilent Technologies). Up to 16 samples were mixed and pooled together and the final multiplexed library was quantified again using a Qubit fluorometer. Illumina sequencing was performed on a MiSeq instrument using 150-nucleotide single-end runs according to the manufacturer’s protocol. For nanopore sequencing, three to five individually barcoded cDNA Nextera libraries were mixed in equimolar amounts, with the final mix containing 200–800 ng of DNA. The DNA was then end-repaired and ligated with adapter and motor proteins using the 1D Ligation Sequencing Kit (Oxford Nanopore Technologies). For nanopore sequencing, metagenomic libraries were run on R9.4 or R9.5 flow cells, using either a MinION MK1B or GridION X5 instrument (Oxford Nanopore Technologies). Up to five barcoded sample libraries were prepared using the 1D Ligation Sequencing Kit (Oxford Nanopore Technologies), quantified and pooled together in an identical fashion to the Illumina Nextera libraries and loaded on a single flow cell for sequencing.
Capture probe enrichment for ZIKV samples
The xGen Lockdown Kit (IDT Technologies) was used for capture probe enrichment of ZIKV. Briefly, barcoded amplified cDNA libraries corresponding to each sample were mixed in equimolar proportions to generate a 500-ng pooled library. The pooled library was then added to a hybridization mix containing ZIKV xGen Lockdown probes and the hybridization reaction was performed by incubation at 65 °C for 16 h, followed by streptavidin bead capture for 45 min. Beads containing captured cDNA were resuspended in an amplification reaction mix (25 μl KAPA HiFi HotStart ReadyMix, 1.25 μl xGen primer and 3.75 μl water) and post-capture PCR was performed (98 °C for 45 s, followed by 10 cycles of denaturing (98 °C for 15 s), annealing (60 °C for 30 s) and extension (72 °C for 30 s), with a final extension at 72 °C for 1 min). PCR amplicons were purified using a 1.5× volume of AMPure XP beads and finally eluted in 20 μl EB buffer. Purified PCR products were analysed by 2% gel electrophoresis to check the library size and DNA concentration was estimated using a Qubit fluorometer. The capture probe enriched library was run on an Illumina MiSeq instrument using 150-nucleotide single-end runs according to the manufacturer’s protocol.
Tiling multiplex PCR enrichment for ZIKV
Tiling multiplex PCR for ZIKV enrichment was performed according to the Primal protocol described in ref. 36, except for libraries prepared using both MSSPE and tiling multiplex PCR, for which an AMPure bead wash of 1.2× was performed immediately after cDNA synthesis (before adding multiplexed primers) to remove residual ZIKV MSSPE primers (4 μM) that had been added during the reverse transcription step. After visualization of a PCR band of the expected size (400 nucleotides) by 2% gel electrophoresis, barcoded sequencing libraries were prepared using the NEBNext Ultra II DNA Library Preparation Kit (New England BioLabs, Inc.) and sequenced on an Illumina MiSeq instrument using 250-nucleotide paired-end runs according to the manufacturer’s protocol
Bioinformatics pipelines for viral detection and reference genome alignment
Sequencing data from Illumina MiSeq or HiSeq instruments were analysed for viruses using the sequence-based ultra-rapid pathogen identification (SURPI+ v.1.0) computational pipeline (UCSF), a modified version of a previously published bioinformatics analysis pipeline for pathogen identification from mNGS sequence data33,50. Specifically, the SURPI+ pipeline modifications include: updated reference databases based on the NCBI nucleotide database (March 2015 build); a filtering algorithm for the exclusion of false-positive hits from database misannotations; and taxonomic classification for species-level identification. After SURPI+ identification of viral reads, viral reads were trimmed by 13 nucleotides at the 5′ and 3′ end to remove the spiked primers before mapping to the most closely matched reference genomes and visualization using SURPI+33,50 or Geneious 11.1.3 (ref. 51). Duplicate viral reads were removed using Prinseq (v.0.20.4)52 with the “-12345” parameter before genome mapping and assembly. To estimate the percentage of human reads (human background) in clinical samples or contrived samples spiked into donor plasma matrix, Illumina preprocessed reads were aligned to human reference genome hg38 (accession no. GRCh38.p12) and the proportion of human reads was determined using the SURPI+ pipeline.
Nanopore sequencing was run on either a Mk1b MinION or GridION instrument. Nanopore raw fast5 files were basecalled using Albacore (MinION) or Guppy (GridION) in real-time mode without polishing. For virus detection from nanopore reads, we used a pipeline developed in-house called SURPI real time (SURPIrt v.1.0). Briefly, for multiplexed samples, FASTQ files were first computationally separated by barcode, followed by preprocessing for trimming of adapters and low-complexity sequences. After partitioning the first 450 nucleotides of the preprocessed nanopore read into three 150-nucleotide segments, viral reads were identified using Bowtie 2 (ref. 53) alignment with a minimum alignment score cut-off of 100. Filtering and taxonomic classification algorithms were then applied as described above. Viral reads were trimmed by 13 nucleotides at the 5′ and 3′ end to remove any spiked primers and PCR duplicate reads were removed before mapping to the most closely matched reference genome using GraphMap (v.0.5.2)54 and visualization using Geneious (v.11.1.13)51.
Quantification and statistical analysis
For Illumina sequencing, the RPM metric was calculated as the number of viral species-specific reads divided by the number of preprocessed reads (after trimming, low-quality filtering and low-complexity filtering of raw reads) × 1 million. For nanopore sequencing, the RPM was calculated as the number of viral species-specific reads divided by the number of basecalled reads × 1 million. Enrichment was defined as the RPM obtained for a target virus using MSSPE divided by the RPM obtained using RH priming only. The median fold change and IQR are given for non-normally distributed data. The percentage increase in genome coverage is the genome coverage obtained using RH alone subtracted from that obtained using MSSPE. McNemar’s paired test was used to compare two proportions for paired conditions (before and after using MSSPE), with a P value of less than 0.05 considered statistically significant. A paired t test was used to compare the mean fold enrichment between groups, with a P value of less than 0.05 considered statistically significant. Probit analysis for determination of limits of detection was performed using StatPlus software (AnalystSoft, Inc., v.6.2.30).
For the comparison between the use of spiked primers and random primers only, normalization across barcoded runs was performed by: randomly selecting a subset of preprocessed reads, with the size fixed to the run with the fewest number of reads; determining the number of viral reads within each equally sized subset of preprocessed reads; and tabulating the viral reads for each subset and using them for downstream genome assembly.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Sequence data were deposited in the NCBI Sequence Read Archive after removal of human genomic reads (NCBI BioProject accession no. PRJNA578816, umbrella BioProject accession no. PRJNA171119). The data that support the findings of the study are available from the corresponding author on reasonable request. MSSPE primer sequences tested in this study are provided in Supplementary Table 21. Source data for Fig. 2 are presented with this paper.
Code availability
SURPI+, SURPIrt and MSSPE-design have been deposited on Github and are available for download for research use only at https://github.com/chiulab/SURPI-plus-dist, https://github.com/chiulab/SURPIrt-dist and https://github.com/chiulab/MSSPE-design, respectively.
Change history
21 January 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41564-020-0671-7
References
Bloom, D. E., Black, S. & Rappuoli, R. Emerging infectious diseases: a proactive approach. Proc. Natl Acad. Sci. USA 114, 4055–4059 (2017).
Reperant, L. A. & Osterhaus, A. AIDS, Avian flu, SARS, MERS, Ebola, Zika… what next? Vaccine 35, 4470–4474 (2017).
Shorten, R. J. et al. Diagnostics in Ebola virus disease in resource-rich and resource-limited settings. PLoS Negl. Trop. Dis. 10, e0004948 (2016).
Rasmussen, A. L. & Katze, M. G. Genomic signatures of emerging viruses: a new era of systems epidemiology. Cell Host Microbe 19, 611–618 (2016).
Sozhamannan, S. et al. Evaluation of signature erosion in Ebola virus due to genomic drift and its impact on the performance of diagnostic assays. Viruses 7, 3130–3154 (2015).
Chiu, C. Y. & Miller, S. A. Clinical metagenomics. Nat. Rev. Genet. 20, 341–355 (2019).
Wilson, M. R. et al. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N. Engl. J. Med. 380, 2327–2340 (2019).
Simner, P. J., Miller, S. & Carroll, K. C. Understanding the promises and hurdles of metagenomic next-generation sequencing as a diagnostic tool for infectious diseases. Clin. Infect. Dis. 66, 778–788 (2018).
Wilson, M. R. et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N. Engl. J. Med. 370, 2408–2417 (2014).
Gardy, J. L. & Loman, N. J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 19, 9–20 (2018).
Gire, S. K. et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345, 1369–1372 (2014).
Chiu, C. Y. Viral pathogen discovery. Curr. Opin. Microbiol. 16, 468–478 (2013).
Pollock, N. R. & Wonderly, B. Evaluating novel diagnostics in an outbreak setting: lessons learned from Ebola. J. Clin. Microbiol. 55, 1255–1261 (2017).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014).
Strong, M. J. et al. Microbial contamination in next generation sequencing: implications for sequence-based analysis of clinical samples. PLoS Pathog. 10, e1004437 (2014).
Greninger, A. L. et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 7, 99 (2015).
Kafetzopoulou, L. E. et al. Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak. Science 363, 74–77 (2019).
Koehler, J. W. et al. Development and evaluation of a panel of filovirus sequence capture probes for pathogen detection by next-generation sequencing. PLoS ONE 9, e107007 (2014).
Quick, J. et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 530, 228–232 (2016).
Faria, N. R. et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 546, 406–410 (2017).
Grubaugh, N. D. et al. Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature 546, 401–405 (2017).
Metsky, H. C. et al. Zika virus evolution and spread in the Americas. Nature 546, 411–415 (2017).
Faria, N. R. et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science 361, 894–899 (2018).
Song, H. et al. Tracking HIV-1 recombination to resolve its contribution to HIV-1 evolution in natural infection. Nat. Commun. 9, 1928 (2018).
Sardi, S. I. et al. Coinfections of Zika and chikungunya viruses in Bahia, Brazil, identified by metagenomic next-generation sequencing. J. Clin. Microbiol. 54, 2348–2353 (2016).
Briese, T. et al. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 6, e01491-15 (2015).
Metsky, H. C. et al. Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat. Biotechnol. 37, 160–168 (2019).
Wylie, T. N., Wylie, K. M., Herter, B. N. & Storch, G. A. Enhanced virome sequencing using targeted sequence capture. Genome Res. 25, 1910–1920 (2015).
Thézé, J. et al. Genomic epidemiology reconstructs the introduction and spread of Zika virus in Central America and Mexico. Cell Host Microbe 23, 855–864 (2018).
Lorenz, T. C. Polymerase chain reaction: basic protocol plus troubleshooting and optimization strategies. J. Vis. Exp. 63, e3998 (2012).
Cherpillod, P. et al. Ebola virus disease diagnosis by real-time RT-PCR: a comparative study of 11 different procedures. J. Clin. Virol. 77, 9–14 (2016).
Corman, V. M. et al. Assay optimization for molecular detection of Zika virus. Bull. World Health Organ. 94, 880–892 (2016).
Miller, S. et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 29, 831–842 (2019).
Erlwein, O. et al. DNA extraction columns contaminated with murine sequences. PLoS ONE 6, e23484 (2011).
Naccache, S. N. et al. Distinct Zika virus lineage in Salvador, Bahia, Brazil. Emerg. Infect. Dis. 22, 1788–1792 (2016).
Quick, J. et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276 (2017).
Hasan, M. R. et al. Depletion of human DNA in spiked clinical specimens for improvement of sensitivity of pathogen detection by next-generation sequencing. J. Clin. Microbiol. 54, 919–927 (2016).
Gu, W. et al. Depletion of abundant sequences by hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 17, 41 (2016).
Stang, A., Korn, K., Wildner, O. & Uberla, K. Characterization of virus isolates by particle-associated nucleic acid PCR. J. Clin. Microbiol. 43, 716–720 (2005).
Temmam, S. et al. Host-associated metagenomics: a guide to generating infectious RNA viromes. PLoS ONE 10, e0139810 (2015).
Endoh, D. et al. Species-independent detection of RNA virus by representational difference analysis using non-ribosomal hexanucleotides for reverse transcription. Nucleic Acids Res. 33, e65 (2005).
Nguyen, A. T. et al. Development and evaluation of a non-ribosomal random PCR and next-generation sequencing based assay for detection and sequencing of hand, foot and mouth disease pathogens. Virol. J. 13, 125 (2016).
Ladner, J. T. et al. Standards for sequencing viral genomes in the era of high-throughput sequencing. mBio 5, e01360-14 (2014).
Robertson, D. L., Hahn, B. H. & Sharp, P. M. Recombination in AIDS viruses. J. Mol. Evol. 40, 249–259 (1995).
Lynch, R. M., Shen, T., Gnanakaran, S. & Derdeyn, C. A. Appreciating HIV type 1 diversity: subtype differences in Env. AIDS Res. Hum. Retroviruses 25, 237–248 (2009).
Tyler, A. D. et al. Evaluation of Oxford Nanopore’s MinION sequencing device for microbial whole genome sequencing applications. Sci. Rep. 8, 10931 (2018).
Berg, M. G. et al. A pan-HIV strategy for complete genome sequencing. J. Clin. Microbiol. 54, 868–882 (2016).
Lanciotti, R. S. et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 14, 1232–1239 (2008).
Katoh, K. & Standley, D. M. MAFFT: iterative refinement and additional methods. Methods Mol. Biol. 1079, 131–146 (2014).
Naccache, S. N. et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 24, 1180–1192 (2014).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Sovic, I. et al. Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nat. Commun. 7, 11307 (2016).
Acknowledgements
We thank N. Loman and J. Quick at the University of Birmingham for providing ZIKV tiling multiplex PCR primers. The following viral RNA extracts were obtained through Biodefense and Emerging Infections Resources, the National Institute of Allergy and Infectious Diseases (NIAID) and the National Institutes of Health (NIH): CCHFV, IbAr10200, NR-37382; LASV, Josiah, NR-31821; Rift Valley Fever Virus, ZH501, NR-37379; MeV, Edmonston strain, NR-44104. This work was also funded in part by Abbott Laboratories (C.Y.C.), NIH grant no. R33-AI129455 (C.Y.C.) from the NIAID, NIH grant no. R01-HL105704 (C.Y.C.) from the National Heart, Lung, and Blood Institute, the California Initiative to Advance Precision Medicine (C.Y.C.), the Charles and Helen Schwab Foundation (C.Y.C.), the Steven and Alexandra Cohen Foundation (C.Y.C.), United States Department of Defense award W81XWH-17-1-0681 (C.Y.C.), the Wellcome Trust and Royal Society/Sir Henry Dale Fellowship grant no. 204311/Z/16/Z (N.R.F.), the Global Challenges Research Fund grant no. 005073 (N.R.F.), the Oxford John Fell Research Fund grant no. 005166 (N.R.F.) and Africa Oxford grant no. AfiOx-48 (N.R.F.).
Author information
Authors and Affiliations
Contributions
C.Y.C. conceived, designed, and supervised the study, developed MSSPE-design software and SURPIrt pathogen identification software for nanopore sequencing and analysed data. X.D. coordinated the study, performed experiments and analysed data. A.A., G.Y. and S.S. performed experiments. S.F. and J.T. performed the bioinformatics analysis of sequence data. I.B., N.R.F., O.G.P., Z.N., J.M. and N.T. collected YFV samples from patients and extracted the viral RNA. S.Y., K.H., S. Me. and D.A.W. collected CHIKV and DENV samples from febrile travellers returning to the United States and extracted the viral RNA. P.M.-K., J.K., S.A.-M. and J.-J.M.-T. collected Ebola samples from patients and extracted the viral RNA. A.A.A. collected a clinical CSF sample from a patient with POWV meningoencephalitis. V.G. collected a clinical CSF sample from a patient with JCV meningoencephalitis. M.T. and J.L.P. cultured the Ebola Kikwit strain for use in MSSPE experiments. N.N., D.M., L.K., C.M., M.R., G.C. and J.R.H.Jr. collected clinical HIV samples from patients in Cameroon, genotyped the strains and performed qRT–PCR for viral titre estimates. J.E.M.-M., C.R.G.-B., S.L. and C.F.A. collected clinical ZIKV samples from patients in Mexico. S.A. and S. Mi. provided clinical HCV samples from patients in California, USA. M.S. and M.B. collected ZIKV and DENV samples from infected blood donors. C.Y.C. and X.D. wrote the manuscript. C.Y.C., X.D., M.R. and G.C. edited the manuscript. All authors read the manuscript and agreed to its contents.
Corresponding author
Ethics declarations
Competing interests
C.Y.C. is the director of the UCSF–Abbott Viral Diagnostics and Discovery Center and receives research support funding from Abbott Laboratories, Inc. X.D. and C.Y.C. are inventors on a patent application titled ‘Spiked Primer Design for Targeted Enrichment of Metagenomic Libraries’ (US application no. 62/667,344, filed 4 May 2018 by the University of California San Francisco) that includes a description of the methods and primer sets presented in this paper. A.A.A. is an employee of Karius, Inc.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2 and Supplementary Tables 1–18.
Supplementary Tables
Supplementary Tables 19–21.
Source data
Source Data Fig. 2
Statistical source data.
Rights and permissions
About this article

Cite this article
Deng, X., Achari, A., Federman, S. et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat Microbiol 5, 443–454 (2020). https://doi.org/10.1038/s41564-019-0637-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-019-0637-9
This article is cited by
-
Antimicrobial resistance prediction by clinical metagenomics in pediatric severe pneumonia patients
Annals of Clinical Microbiology and Antimicrobials (2024)
-
Nanopore DNA sequencing technologies and their applications towards single-molecule proteomics
Nature Chemistry (2024)
-
Increase in the incidence of invasive fungal infections due to Volvariella Volvacea
European Journal of Clinical Microbiology & Infectious Diseases (2024)
-
Despite low viral titer in saliva samples, Sanger-based SARS-CoV-2 spike gene sequencing is highly applicable for the variant identification
BMC Medical Genomics (2023)
-
Amazon Biobank: a collaborative genetic database for bioeconomy development
Functional & Integrative Genomics (2023)
