
Abstract
Armillaria species are both devastating forest pathogens and some of the largest terrestrial organisms on Earth. They forage for hosts and achieve immense colony sizes via rhizomorphs, root-like multicellular structures of clonal dispersal. Here, we sequenced and analysed the genomes of four Armillaria species and performed RNA sequencing and quantitative proteomic analysis on the invasive and reproductive developmental stages of A. ostoyae. Comparison with 22 related fungi revealed a significant genome expansion in Armillaria, affecting several pathogenicity-related genes, lignocellulose-degrading enzymes and lineage-specific genes expressed during rhizomorph development. Rhizomorphs express an evolutionarily young transcriptome that shares features with the transcriptomes of both fruiting bodies and vegetative mycelia. Several genes show concomitant upregulation in rhizomorphs and fruiting bodies and share cis-regulatory signatures in their promoters, providing genetic and regulatory insights into complex multicellularity in fungi. Our results suggest that the evolution of the unique dispersal and pathogenicity mechanisms of Armillaria might have drawn upon ancestral genetic toolkits for wood-decay, morphogenesis and complex multicellularity.
Similar content being viewed by others

Main
The genus Armillaria causes root rot disease in both gymno- and angiosperms, in forests, parks, and even vineyards in more than 500 host plant species1 across the world. Most Armillaria species are facultative necrotrophs, which, after colonizing and killing the root cambium, transition to a saprobic phase, decomposing dead woody tissues of the host. As saprotrophs, Armillaria spp. are white rot (WR) fungi, which can efficiently decompose all components of plant cell walls, including lignin, (hemi-)cellulose and pectin2. They produce fleshy fruiting bodies (honey mushrooms) that appear in large clumps around infected plants and produce sexual spores. The vegetative phase of Armillaria is predominantly diploid rather than dikaryotic like most basidiomycetes.
Individuals of Armillaria can reach immense sizes and include the ‘humongous fungus’, one of the largest terrestrial organisms on Earth3, measuring up to 965 hectares and 600 tons4, and can display a mutation rate ≅3 orders of magnitude lower than most filamentous fungi5. Individuals reach this immense size via growing rhizomorphs, dark mycelial strings 1–4 mm wide that allow the fungus to bridge gaps between food sources or host plants1,6 (hence the name shoestring root rot). Rhizomorphs develop through the aggregation and coordinated parallel growth of hyphae, similar to some fruiting body tissues7,8. As migratory and exploratory organs, rhizomorphs can grow approximately 1 m yr−1 and cross several metres underground in search for new hosts, although roles in uptake and long-range translocation of nutrients have also been proposed1,9,10. Root contact by rhizomorphs is the main mode of infection by the fungus, which makes the prevention of recurrent infection in Armillaria-contaminated areas particularly difficult1. Despite their huge impact on forestry, horticulture and agriculture, the genetics of the pathogenicity of Armillaria species is poorly understood. The only -omics data published so far have highlighted a substantial repertoire of plant cell wall degrading enzymes (PCWDE) and secreted proteins, among others, in A. mellea and A. solidipes11,12, while analyses of the genomes of other pathogenic basidiomycetes (such as Moniliophthora13,14, Heterobasidion15 and Rhizoctonia16) identified genes coding for PCWDEs, secreted and effector proteins or secondary metabolism (SM) as putative pathogenicity factors. However, the lifecycle and unique dispersal strategy of Armillaria prefigure other evolutionary routes to pathogenicity, which, along with other potential genomic factors (such as transposable elements17) are not yet known.
Here, we investigate genome evolution and the origin of pathogenicity in Armillaria using comparative genomics, transcriptomics and proteomics. We sequenced the genomes of four Armillaria species to combine with those of related saprotrophic, hemibiotrophic and mycorrhizal fungi. Transcript and proteome profiling of invasive and reproductive developmental stages shed light on the role of rhizomorphs, several putative pathogenicity factors, and the morphogenetic mechanisms of rhizomorph and fruiting body development.
Results
We report the genomes of A. ostoyae, A. cepistipes, A. gallica and A. solidipes sequenced using a combination of PacBio and Illumina technologies (Table 1). Genomes of Armillaria species were assembled to 103–319 scaffolds comprising 58–85 Mb and were predicted to contain 20,811–25,704 genes. In comparison, other sequenced species of the Physalacriaceae, Flammulina velutipes and Cylindrobasidium torrendii have 12,218 (35.6 Mb) and 13,940 (31.5 Mb) genes, respectively, while the sister genus of Armillaria, Guyanagaster necrorhiza, has 14,276 (53.6 Mb) (Fig. 1). Armillaria species share significant synteny, comprising macro- to microsynteny (Supplementary Fig. 1), whereas mesosynteny, which is characteristic of certain fungal groups18, was not observed. The transposable element (TE) content of Armillaria genomes shows a modest expansion relative to other Agaricales and an even distribution along the scaffolds, suggesting that their genome expansion is not driven by transposon proliferation, as observed in other plant pathogens17 (Supplementary Figs. 2–3, Supplementary Table 1).

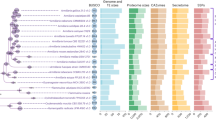
a, Reconstructed gene duplication/loss histories along a time-calibrated phylogeny of 27 fungal species, showing the expansion of protein coding gene repertoires in Armillaria spp. The heat map (right) shows gene copy numbers for PCWDE and pathogenicity-related gene families in the 27 species examined. Numbers in bubbles correspond to the ancestral number of genes inferred in that node. b, Assembly sizes for 27 species broken down by TEs and non-TEs. Data for A. mellea are incomplete due to the highly fragmented available assembly (29,300 scaffolds). Tricholoma matsutake has been truncated for clarity. See Supplementary Fig. 2 for details. Species have been grouped by nutritional mode. BR, brown rot; ECM, ectomycorrhizal; LD, litter decomposer; UN, unknown. c, Numbers of predicted genes by phylogenetic conservation (taxa correspond to those shown in a). d, Numbers of duplicated (+) and lost (−) genes inferred for the Physalacriaceae, showing the net genome expansion in Armillaria spp. and the structure of their mating loci, indicating high levels of synteny. Arrows indicate the location and orientation of genes within the locus. The locus comprises of four homeodomain (HD) transcription factors arranged into MAT-Aα and MAT-Aβ respectively. β-fg and MIP genes anchor the locus. Genes containing carbohydrate esterase (CE), proline racemase (PR), deoxyribose-phosphate aldolase (DPA), metacaspases (MC) and dehydrogenase/reductase (DH) domains (pfam) are located within the locus. For clarity, A. mellea is not displayed as its assembly is highly fragmented. Numbers indicate the genomic coordinates of the mating loci.
Phylogenomic analysis based on 835 conserved single copy genes (188,895 amino acid sites) confirmed the position of Armillaria in the Physalacriaceae, with Guyanagaster and Cylindrobasidium as their closest relatives (Fig. 1a). We estimated the age of pathogenic Armillaria spp. at 21 million years (Myr) and their divergence from Guyanagaster at 42 Myr (Supplementary Fig. 4), coincident with decreasing temperatures and the spread of deciduous forests in the Eocene. Reconstruction of genome-wide gene duplication and loss histories in 27 Agaricales species revealed an early origin for most genes, followed by lineage-specific gene losses in most family and genus level groups, except Armillaria, which showed a net genome expansion: 15,787 protein-coding genes were inferred for the most recent common ancestor (MRCA) of Armillaria (2,012 duplications, 945 losses), as opposed to 14,720 and 14,687 for the MRCA of Armillaria and Guyanagaster and that of Armillaria, Guyanagaster and Cylindrobasidium, respectively (Fig. 1a,d, Supplementary Fig. 5). Further expansion to 19,272 genes was inferred for the MRCA of A. solidipes, A. ostoyae, A. epistipes and A. gallica (3,192 duplications, 607 losses), although the highly fragmented A. mellea assembly might cause some duplications to map to this instead of the preceding node.
Duplicated genes were enriched in functions related to chitin and cellulose binding, polysaccharide metabolism (peroxidase, lyase, hydrolase and oxidoreductase activity), peptidase activity, transmembrane transport, extracellular region and gene expression regulation (p < 0.05, according to a hypergeometric test, HT, Supplementary Table 2). Two hundred and fourteen domains were significantly overrepresented in Armillaria genomes (p < 0.05, HT) relative to other agarics, including several peptidase, glycoside hydrolase (GH) and pectinase domains, LysM (CBM50-s) domains, expansins, multicopper oxidases as well as several pfams related to secondary metabolism (Supplementary Table 3). We found 570 protein clusters specific to Armillaria and Guyanagaster or subclades therein (Supplementary Table 4). Although most of these (70%) had no functional annotations, they included CE4 chitooligosaccharide deacetylases, CBM50-s, iron permeases (FTR1), and 19 transcription factor (TF) families, among others. Taken together, these results suggest that gene family expansion was the predominant mode of genome evolution in Armillaria and that the genome expansion is largely concerned with diverse extracellular functions, including several lineage-specific innovations, some of which had previously been associated with pathogenicity.
Putative pathogenicity-related genes in Armillaria
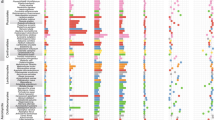
Plant pathogenic fungi possess diverse gene repertoires for invading host plants and modulating their immune systems18,19,20. We catalogued 20 families of putative pathogenesis-related genes to assess whether Armillaria shares expansions of these families with other plant pathogens (Supplementary Table 5). Armillaria species are enriched in expansins (p = 4 × 10−5, Fisher exact test, FET) and possess many cerato-platanin genes, which contribute to unlocking cell-wall polysaccharide complexes and cause cell death in host plants, respectively, and might act as first-line cell-lysis weaponry during invasion (Fig. 1a). Carboxylesterases and salicylate hydroxylases show moderate enrichment in both Armillaria and Moniliophthora species, along with other weakly pathogenic taxa (such as Marasmius fiardii). Salicylate hydroxylases have been implicated in developing a tolerance to salicylic acid, which is released to block the jasmonic acid defense pathway of plants on infection. In contrast, cutinases (CE5) are missing from Armillaria species but are present in Moniliophthora spp.14, which primarily infect through leaves. CBM50 domains are overrepresented in Armillaria compared to other Agaricales (p = 2 × 10−8, FET), while GH75 chitosanases are exclusively found in Armillaria and Moniliophthora species (Fig. 1a, Supplementary Table 5). In other plant pathogens, these are involved in modifying fungal cell wall composition and/or capturing chitin residues to mask chitin-triggered immune signals and evade detection by the host plants21,22,23,24,25, suggesting similar roles in Armillaria.
Homologs of SM genes reported from Heterobasidion15 are overrepresented in Armillaria species (p = 0.03 × 10−13, FET, Fig. 1a, Supplementary Tables 3 and 5). These include terpene cyclases, non-ribosomal peptide synthase-like prenyl transferases, and halogenases, as well as trichodiene and polyprenyl synthases, which have been linked to fungal pathogenesis, virulence and competition with other microbes26. Small secreted proteins (SSPs, <300 amino acids with a secretory signal) can act as effectors in mutualistic and pathogenic interactions in various fungal groups19,27. On average, we found more SSPs (n = 669) in Armillaria species (Fig. 1a) than in saprotrophs (n = 552) or ECM fungi (n = 563), although this is consistent with the larger genomes of Armillaria species (p> 0.05, FET). In contrast, Major Facilitator Superfamily 1 (MFS1) and cytochrome p450 families are expanded in Armillaria species (p = 4 × 10−10 and p = 3 × 10−30). Both superfamilies have been associated with fungal pathogenicity28,29, but also several other cellular functions. Their overrepresentation, nevertheless, suggests unique roles in the biology of these fungi.
Armillaria species as wood-rotting fungi
It has been shown that plant–fungal interactions in both pathogens14,15,18 and mutualists27,28,29 can draw upon the PCWDE repertoire of saprotrophic ancestors. Therefore, we compared the PCWDE complement of Armillaria species to that of other Agaricales with diverse lifestyles. In the saprotrophic phase of their lifecycle, Armillaria species cause WR on wood, which is reflected in their PCWDE repertoire. Their genomes encode lignin-, cellulose-, hemicellulose- and pectin-degrading enzymes, indicating the potential to degrade all plant cell wall (PCW) components (Fig. 1a, Supplementary Table 5). Lignolytic families are underrepresented in Armillaria (p = 0.02 × 10−10, FET), whereas cellulose- and xylan-degrading families generally show similar gene counts to other WR species, with notably higher copy numbers of GH1 (β-glucosidase, p = 3 × 10−6, FET). On the other hand, several pectinolytic families are overrepresented in Armillaria. Pectin-degrading families include GH28, GH78 and GH88, polysaccharide lyase (PL) 1, 3, 4 and 9 as well as carbohydrate esterase 8 (CE8), of which PL3, CE8 and CBM67 rhamnose-binding modules are significantly enriched in Armillaria spp. (p = 0.02 × 10−11, FET) compared to WR Agaricales. The pectinolytic repertoire of Armillaria is unusual for WR fungi30 and might indicate links to dicot pathogenicity20. The PCWDE repertoire of Armillaria species underpins their ability to act as powerful WR decayers and provides a pool to draw upon as necrotrophic pathogens. It might enable them to gain access to wood and avoid competition with other microbes by damaging live trees, a strategy that is unavailable to most WR fungi.
Expression profiles of rhizomorphs
Armillaria species spread either clonally by rhizomorphs or with sexual spores produced on fruiting bodies. Rhizomorphs are unique multicellular structures of Armillaria species, but their functions and morphogenetic origins are debated10. Differential expression analysis of actively growing rhizomorph tips (0–5 cm from the apex) identified 1,303 and 1,610 genes over- and underexpressed relative to vegetative mycelium grown on the same medium (FCVM > 4, p ≤ 0.05, Supplementary Table 6), respectively, marking one of the largest expression changes in our experiments (Fig. 2). Similarly, the highest number of unique proteins (n = 729) was detected in rhizomorphs compared to vegetative mycelia (Fig. 2c,d). Upregulated genes were enriched for Gene Ontology (GO) terms related to carbohydrate, lipid and secondary metabolism, hydrophobins and pectinolysis (Supplementary Table 6). Global expression profiles suggest that rhizomorphs are transitional between vegetative mycelium and fruiting bodies (Supplementary Figs. 6,7). Expression profiles of several PCWDE families and putative pathogenicity-related genes (cerato-platanins, expansins) in rhizomorphs resembles vegetative mycelia (Supplementary Figs 8–12), whereas that of many putative morphogenesis-related genes was shared with fruiting bodies.

a, Gene expression and proteomic map of ten developmental stages and tissue types. Green and red arrows denote the numbers of significantly up- and downregulated genes (p < 0.05, FC > 4), respectively. Bar diagrams show the number of detected proteins common (grey area) to successive developmental stages, and the ones unique to the first (white) or the next (black) stage. The development of rhizomorphs (RH, examples shown for growth in the dark and light, left to right) and that of stage I primordia (P1) from vegetative mycelium (VM) denote the largest expression change, whereas stage II primordia (P2), young fruiting bodies (YFB) and fruiting bodies (FB) show smaller numbers of DEGs (moderated T-test, FDR-corrected p < 0.05, FC > 4). Each sample was sequenced in three biological replicates. Scalebars denote 1 cm, except in P1 (5 mm). b, Phylostratigraphic analysis of 1,303 genes upregulated in rhizomorphs. Inset, Age distribution of all genes in the genome of A. ostoyae. c, Number of detected proteins overlapping in the compared developmental stages. Note the similarity between VM and RH and between all complex multicellular stages (267 proteins). d, Protein abundance profiles in ten developmental stages and tissue types.
Rhizomorphs express an evolutionarily young transcriptome, with most upregulated genes specific to A. ostoyae or the Armillaria clade (including Guyanagaster Fig. 2b). Most of these young genes lack functional annotations but were conserved in Armillaria species, suggesting important functions in the genus. Genes belonging to older phylostrata had comparatively more functional annotation terms. We found 414 genes that had expression maxima in rhizomorphs and were significantly upregulated relative to vegetative mycelium (fold change: FCVM ≥ 4, Supplementary Table 7). The most highly upregulated genes included expansins (log2FCVM = 10.44), bZip and C2H2 transcription factors, three caspase genes, hydrophobins, cytochrome p450s, GHs as well as several unannotated genes. We found an overexpression of 5-oxoprolinases downstream factors of neutralizing intracellular H2O2 and signs of intensified biogenesis and cargo of extracellular proteins (Supplementary Figs 13,14). A moderate set of PCWDE genes was expressed in rhizomorphs, suggestive of assimilative or invasive properties, although the highest PCWDE suite was observed in vegetative mycelium (Supplementary Figs. 8–10).
We observed significant overexpression of three cerato-platanins, three expansins, two carboxylesterases, as well as SM-related trichodiene (five genes), polyketide (six genes) synthases and a polyprenyl synthase, among others (Supplementary Table 8). Notably, all expansins showed upregulation in fruiting bodies too, which could indicate a role in fungal cell wall remodelling instead of cellulose degradation, to which fungal expansin-related genes have mostly been linked31. A gene (ARMOST_01259) coding for a family 6 bacterial extracellular solute-binding protein (SBP) showed an expression peak in rhizomorphs, but low or negligible expression in other stages. Bacterial SBPs associate with ABC transporters to function in Fe3+ transport and are required for the pathogen’s survival in the host. This gene is a member of an Armillaria-specific cluster with homologs in all Armillaria species and bacterial proteins as closest BLAST hits (although maximum likelihood (ML) gene trees don’t support horizontal gene transfer). Its Fe3+ transporting ability and nearly exclusive expression in rhizomorphs suggests a role in substrate and/or host exploitation.
Morphogenesis
The morphogenetic machinery underlying rhizomorph development is among the least known aspects of the biology of Armillaria. As multicellular structures, rhizomorphs express a variety of genes encoding cell-wall proteins, including hydrophobins, pore-forming toxins, two CBM67 and four ricin-B-lectins, an annexin and a cell-wall integrity sensor, among others, indicating several fruiting body-like functions, such as hyphal adhesion, communication or defense (Supplementary Table 9). We found cell-wall biosynthesis genes to be generally but moderately upregulated in rhizomorphs (and stipes) (Supplementary Fig. 15). Further, several GMC oxidoreductases, two mating-type pheromone receptors, CBM50-s, a CBM5_12 and chitooligosaccharide deacetylase genes were significantly overexpressed (the latter two also in stipes). Four hydrophobins reached their highest expression in rhizomorphs whereas two showed high expression in rhizomorphs and stipes, but not in caps or vegetative mycelium (Supplementary Table 9, Supplementary Fig. 16). A homolog of the Cryptococcus red and far-red sensing Tco3 photoreceptor was expressed at high levels (log2FC = 4.36). Although its function in Cryptococcus is unknown32, its overexpression in rhizomorphs and in brown-film forming mycelia of Lentinula33 could suggest morphogenesis-related functions.
We detected 19 significantly upregulated TFs, ten of which had peak expression in rhizomorphs across our experimental conditions. Although based on global TF expression, rhizomorphs are most similar to vegetative mycelium (Supplementary Fig. 17), and several TFs showed shared overexpression in rhizomorphs and fruiting bodies. For example, the expression of ARMOST_01275 peaked in rhizomorphs and stipes, whereas a zf-Mynd TF was highly upregulated in rhizomorphs and all fruiting body stages, and is thus a candidate for governing complex multicellular development in A. ostoyae.
Fruiting bodies
The production of fruiting bodies is probably the largest morphogenetic transition in the fungal lifecycle. It involves the reorganization of hyphal growth patterns and the execution of a complex developmental program. Indeed, we detected the largest number of differentially expressed genes (DEGs) and the second largest change in proteome in stage I primordia (P1, Fig. 2, Supplementary Table 6). Upregulated genes were enriched for gene expression regulation, lipid metabolism, amino acid transport and ribonuclease activity (Fig. 2a, Supplementary Table 10) and included ten TFs and two Dicer-like genes. Following stage I primordia, we tracked expression levels in two cell lineages. While the stipe lineage showed minor changes throughout development (<150 DEGs, < 300 unique proteins), cap differentiation included up to 1,037 DEGs and 646 unique proteins related to signal transduction, carbohydrate metabolism or the regulation of biological processes (Supplementary Table 6). Genes upregulated in gills (n = 502, 56 unique proteins) were enriched for functions related to protein phosphorylation, carbohydrate metabolism, protein kinase activity and ribonucleotide binding.
Hydrophobin genes showed stage and tissue-specific expression patterns, but were generally not expressed in caps (Supplementary Fig. 16), suggesting alternative sources of hydrophobin-related functionalities there. An analysis of cysteine-rich, hydrophobic proteins (excluding hydrophobins) revealed 33 and 13 developmentally regulated genes with expression maxima in fruiting bodies and caps, respectively (Supplementary Table 11). In addition, three cell wall galactomannoproteins show high expression in fruiting bodies (up to logFCVM = 13.5, Supplementary Table 12), two of which were expressed only in caps, whereas ARMOST_19505 was highly expressed throughout development. These genes are homologous to the Aspergillus hydrophobic cell-surface protein HsbA, which has been hypothesized to recruit hydrolytic enzymes during lignocellulose degradation34 and appressorium attachment to host surfaces35. As lignocellulose-related processes are inactive in fruiting bodies, their expression suggests specific roles during cap development, possibly in cell-wall remodelling. Several chitin metabolism-related genes were upregulated in fruiting bodies, including GH88 chitinases, a CBM50-containing gene, a GH75 and six chitooligosaccharide deacetylases (Supplementary Table 12), which might be related to generating development-specific cell-wall architectures. Interestingly, certain HTPs, GMC oxidoreductases (AA3) and pectinolytic genes, generally linked to lignocellulose degradation, were also upregulated in fruiting bodies (Supplementary Fig. 18). Several defense-related genes (pore-forming toxins, lectins and so on)36,37 also showed significant upregulation through development (Supplementary Figure 19, Supplementary Table 13), indicating a phylogenetically and functionally diverse defense arsenal expressed in both fruiting bodies and rhizomorphs.
Shared morphogenetic machineries between rhizomorphs and fruiting bodies
Rhizomorphs share a complex multicellular organization with fruiting bodies. Consistent with this, we observed several genes with similar expression patterns in rhizomorphs and various fruiting body tissues (Fig. 3). These included two mating-type pheromone receptors and the white collar 1–2 genes, which mediate the initiation of fruiting body development and could point to shared developmental origins of rhizomorphs and fruiting bodies. There were 442 genes that showed greater than fourfold elevated expression over vegetative mycelium and relatively constant expression across rhizomorph and fruiting body samples (Fig. 3a, Supplementary Table 14), suggesting they are linked to complex multicellularity in A. ostoyae. A systematic analysis identified 2,225 genes that showed higher expression in rhizomorphs and stipes than in corresponding cap tissues and vegetative mycelium, of which 63 were at least fourfold more abundant in all four pairs of successive developmental stages (Fig. 3b). These included most of the yeast cell-wall biosynthesis pathway homologs (Supplementary Table 15, Supplementary Fig. 20), pore-forming toxins, hydrophobins, TFs and SM genes, among others. For caps and rhizomorphs 1,728 and 28 such genes were found, respectively, including several GMC oxidoreductases, a GH88, a ricin-B-lectin and other SM genes (Fig. 3c, Supplementary Table 15). This indicates that rhizomorph development draws extensively on fruiting body genes, and makes us speculate that fruiting body development could have been the cradle for the evolution of rhizomorphs in Armillaria.

Putative TFBS, UPGMA trees and expression profiles are shown for three co-expressed gene sets. a, 442 genes that are at least four-fold upregulated relative to VM and show relatively constant expression in all complex multicellular structures (P1 to FB). b,c, Expression patterns of 63 and 28 genes with concomitant upregulation in RH and stipe tissues and RH and caps, respectively, showing over four-fold expression difference in RH relative to VM and all four pairs of successive developmental stages. Putative TF genes detected in the co-expressed gene sets are highlighted.
To assess whether co-regulated genes possess a common r-regulatory signature, we searched for putative TF binding sites (TFBS) by de novo motif discovery38 in the promoters of 63 stipe/rhizomorph and 28 cap/rhizomorph co-expressed genes as well as of those that showed constant high expression in all complex multicellular structures. The predicted motifs and their UPGMA trees is shown on Fig. 3. All predicted motifs matched experimentally determined yeast TFBS sequences in the JASPAR database39, which suggests strong biological relevance to our predictions, despite the large phylogenetic distance between S. cerevisiae and A. ostoyae. Taken together, the detected co-expressed genes and the presence of shared motifs in their promoters could suggest common regulatory mechanisms for rhizomorphs and fruiting bodies and probably represent elements of the regulatory networks that govern multicellular development as well as cap and stipe differentiation in Armillaria.
Discussion
Forest pathogens in the genus Armillaria evolved from saprotrophic ancestors in the Agaricales. They have unusually large genomes for WR saprotrophs, which evolved mostly by gene family diversification, in contrast with genome expansions in other plant pathogenic fungi, which are primarily driven by TE proliferation17. We found many lineage-specific genes (including putative pathogenicity factors and pectinolytic families) and coincident overrepresentation of several pathogenicity-related genes with other Basidiomycota (particularly in Moniliophthora species), suggesting convergent origins of pathogenicity in the Agaricales. Armillaria species encode a full complement of PCWDE genes, which comes as no surprise given their ability to cause WR, but provides a resource to draw on in the evolution of pathogenicity. This could give Armillaria spp. early access to dead wood and a strategy to bypass competition with other microbes.
Rhizomorphs are some of the most unique structures of Armillaria spp. that enable them to become the largest terrestrial organism on Earth3,5. They express a wide array of genes involved in secondary metabolism, defense, PCW degradation and to a lesser extent pathogenesis, which indicate active nutrient uptake and adaptations to a soil-borne lifestyle where competition with other microbes and defense against predators are crucial. This underpins their role as exploratory and assimilating organs adapted to bridge the gaps between food sources or potential host plants. In terms of morphogenesis, rhizomorphs resemble fruiting bodies, with many stipe- and to a smaller extent cap-upregulated genes showing concomitant upregulation and shared cis-regulatory signatures. Both rhizomorphs and fruiting bodies show key traits of complex multicellularity, such as three-dimensional organization, cell adhesion or a highly integrated developmental program. We hypothesize that the observed similarity in gene expression indicates common developmental origins and suggest that the evolution of rhizomorphs may have extensively drawn upon the genetic toolkit of fruiting body development in the Agaricomycotina. We identified genes putatively involved in cap and stipe morphogenesis, as well as some co-expressed in all complex multicellular stages and candidate TF-binding motifs within their promoters. These represent putative building blocks of the gene regulatory circuits that govern mushroom development and enabled us to zero in on the genetic bases of complex multicellularity in fungi. This study has provided comparative genomics insights into the evolution, pathogenicity and multicellular development of a group of devastating forest pathogens. It should facilitate further understanding of the biology of Armillaria, which, combined with new genomic resources and in planta interrogation of its pathogenic behaviour, could soon bring the development of efficient strategies for containing the spread and damage of Armillaria root-rot disease in various forest stands within reach.
Methods
Strains and fungal material used for genome sequencing
The diploid A. ostoyae C18 is a field isolate from Switzerland40 and the haploid C18/9 is derived from C18 as a single spore isolate. The A. cepistipes B5 haploid41 was originally isolated from a fruiting body on Fagus sylvatica in Italy. The A. ostoyae C18/9 and A. cepistipes B5 haploid isolates were cultured on Roth and Shaw plates (for 1 l RS: 40 g malt extract, 20 g dextrose, 5 g bacto peptone, 19 g agar) covered with cellophane sheets, and incubated at 25 °C for three weeks. Before genomic DNA extraction, fungal mycelia were detached and harvested from the cellophane sheets, and frozen in liquid nitrogen. The A. gallica 21-2 and A. solidipes 28-4 strains were maintained on 2% malt extract, 2% agar medium. To produce mycelium for DNA extraction, strains were grown in liquid CYM medium (d-glucose 20 g, yeast extract 2 g, peptone 2 g, MgSO4.7H2O 0.5 g, KH2PO4 0.46 g, K2HPO4 1 g, 1 l water) in Petri dishes with 8–10 small pieces of inoculum floated on the air–water interface. Genomic DNA was extracted using the PowerMax MOBIO DNA isolation kit according to the manufacturer’s instructions. Note that a previous study42 proposed that the A. solidipes and A. ostoyae are one species and that the earlier name A. solidipes from a North American collection should always be used in place of A. ostoyae. For consistency with recent historical usage, however, we choose to consider A. ostoyae in Europe as a separate, but closely related species to A. solidipes in North America
We developed an in vitro fruiting system for A. ostoyae C18 based on the protocol published for A. mellea43. A modified RST medium, termed RSTO, was prepared in 720 ml jars and inoculated with A. ostoyae C18, incubated for 28 days at 24 °C in the dark, then placed in a growth chamber at 15 °C with a 10/14 hour light/dark cycle (light intensity: 11 µE m−2 s−1). Vegetative mycelium, rhizomorphs, stage 1 and 2 primordia, young and mature fruiting bodies were harvested as shown on Fig. 1. Fruiting body stages were defined to conform to the general notation44 of mushroom developmental stages as closely as possible. Stage 1 primordia were defined as fruiting body initials up to 1–2 mm tall without a clear differentiation of a pileal section. Stage 2 primordia had a developed button-shaped cap, up to 7–10 mm tall. Young fruiting bodies were 30–50 mm tall with a developed hymenial cavity but prior to cap expansion. From stage 2 primordia, caps and stipes were separated and RNA was extracted separately. Mature fruiting bodies were separated into stipe, gills and cap. RNA was extracted by using the RNEasy Midi kit (QIAGEN) following the manufacturer’s instructions. RNA quality was checked by gel electrophoresis and the Agilent 2200 TapeStation before library preparation. Biological triplicates were analysed for all sample types.
For RNA sequencing (RNA-seq), A. cepistipes was grown on MEA (for 1 l MEA: 20 g malt extract, 0.5 g yeast extract, 15 g agar), RST (15 g Picea abies sawdust, 30 g rice, approximately 100 ml water mixed together in 720 ml jars, sterilized and overlaid with a layer of homogenized tomato approximately 2 cm thick followed by another round of sterilization), RSTO (RST medium with additional 100 g minced orange), Orange media (3 roughly chopped oranges in 720 ml jars) and RNA was extracted using the RNEasy Midi kit (QIAGEN).
Genome sequencing and assembly
The haploid genomes of A. ostoyae and A. cepistipes were sequenced employing the PacBio (Pacific Biosystem) RS II platform (Functional Genomics Center; http://www.fgcz.ch). For preparing the sequencing libraries, 10 μg of gDNA aliquots were mechanically sheared to an average size of 10 kb using the Covaris gTube (KBiosciences p/n 520079) in an Eppendorf microcentrifuge, and the fragment size distributions were assessed applying the Bioanalyzer 2100 12K DNA-Chip assay (Agilent p/n 5067-1508). Five micrograms of the sheared gDNA aliquots were DNA damage repaired, end-repaired and the final SMRTbell templates were created by blunt end ligation and exonuclease treatments. The libraries were quality inspected on an Agilent Bioanalyzer 12K DNA Chip and quantified on a Qubit.1 Fluorimeter (Life Technologies). The SMRTbell was set up by using the DNA Template Prep Kit 2.0 (3 kb to < 10 kb) (Pacific Biosciences p/n 001-540-835). A ready to sequence SMRTbell-Polymerase Complex was arranged by applying the P4 DNA/Polymerase binding kit 2.0 (Pacific Biosciences p/n 100-236-500) according to the manufacturer instructions. Libraries were sequenced on 15 SMRT cell v3.0 (Pacific Biosciences p/n100-171-800), taking 1 movie of 120 minutes each per SMRT cell. The MagBead loading (PacBio p/n 100-133-600) technique served to improve the enrichment for the longer fragments. Final sequencing reports were generated for every cell, via the SMRT portal, to assess the adapter dimer contamination, the sample loading efficiency, the obtained average read length and the number of filtered sub-reads.
The HGAP345 workflow of the SMRT Analysis suite v2.3 was used to create an initial assembly. After removal of redundant contigs, a scaffolding using PBJelly246 and FinisherSC47 followed by polishing via applying the RS Resequencing protocol (SMRT Analysis suite) was performed in four iterations. The final scaffold set was checked for miss-assemblies using the RS BridgeMapper protocol (SMRT Analysis suite) and corrected if necessary. The mitochondrial scaffolds were first identified using a BLASTn search and then circularized by merging and truncating the overlapping ends. To correct PacBio reads, Illumina sequencing was carried out by shotgun sequencing of a 350–450 bp library with paired-end 100 bp reads to 180-fold coverage using HiSeq 2000 (Functional Genomic Center). Finally, Pilon48 was applied to further polish the scaffolds with the genomic Illumina reads.
Draft gene models for A. ostoyae and A. cepistipes were generated by three de novo prediction programs: (1) Fgenesh49 with different matrices (trained on Aspergillus nidulans, Neurospora crassa and a mixed matrix based on different species); (2) GeneMark-ES50; and (3) Augustus51 with RNA-seq-based transcripts as training sets. Annotation was aided by exonerate52 hits of protein sequences from Armillaria species to uncover gene annotation gaps and to validate de novo predictions. Transcripts were assembled on the RNA-seq data sets using Trinity53. The different gene structures and evidences (exonerate mapping, RNA-seq reads and transcripts) were visualized in GBrowse54 allowing manual validation of coding sequences with a focus on chitin, cellulose, pectin, lignin, SM key genes and other genes of interest. The best fitting model per locus was selected manually and gene structures were adjusted by splitting or fusion of gene models and redefining exon–intron boundaries if necessary; tRNAs were predicted using tRNAscan-SE55. The predicted protein sets were searched for highly conserved single- (low) copy genes to assess the completeness of the genomic sequences and gene predictions. Orthologous genes to all 246 single-copy genes were searched for all proteomes by blastp comparisons (eVal: 10-3) against the single-copy families from all 21 species available from the FunyBASE56. Additionally, the proteomes were searched for the 248 core genes commonly present in higher eukaryotes (CEGs) by Blastp comparisons (eVal: 10-3)57. All genomes were analysed using the PEDANT system58.
The genomes and transcriptomes of A. gallica and A. solidipes were sequenced using the Illumina platform at the Joint Genome Institute (JGI). Genomic DNA was sequenced as pairs of Illumina standard and Nextera long mate-pair (LMP) libraries. For the standard libraries, 500 ng of DNA was sheared to 270 bp using the Covaris E220 (Covaris) and size selected using SPRI beads (Beckman Coulter). The fragments were treated with end-repair, A-tailing, and ligation of Illumina compatible adapters (IDT, Inc) using the KAPA-Illumina library creation kit (KAPA biosystems). For LMP, 1 µg of DNA was used to generate the library using the Nextera LMP kit (Illumina). DNA was fragmented and ligated with biotinylated linkers using the Tagmentation enzyme. The fragments were circularized via ligation followed by random shearing using the Covaris LE220 (Covaris). The mate pair fragments were purified using Strepavidin beads (Invitrogen) and treated with end repair, A-tailing, and ligation of Illumina adaptors. The final product was enriched with ten cycles of PCR.
For transcriptomics, stranded cDNA libraries were generated using the Illumina TruSeq Stranded RNA LT kit. Messenger RNA (mRNA) was purified from 1 µg of total RNA using magnetic beads containing poly-T oligos, fragmented and reverse transcribed using random hexamers and SSII (Invitrogen), followed by second strand synthesis. The fragmented cDNA was treated with end-pair, A-tailing, adapter ligation, and ten cycles of PCR.
All libraries were quantified using KAPA Biosystem’s next-generation sequencing library qPCR kit and run on a Roche LightCycler 480 real-time PCR instrument. Except for A. gallica LMP, the quantified libraries were prepared for sequencing on the Illumina HiSeq sequencing platform utilizing a TruSeq paired-end cluster kit, v3 (A. gallica) or v4 (A. ostoyae), and Illumina’s cBot instrument to generate clustered flowcells for sequencing. Sequencing of the flowcells was performed on the Illumina HiSeq2000 or HiSeq2500 sequencer using a TruSeq SBS sequencing kit, v3 or v4, respectively, following a 2 × 150 indexed run recipe. Sequencing of the A. gallica LMP library was performed on the Illumina MiSeq sequencer using a MiSeq Reagent kit, v2, following a 2 × 150 indexed run recipe.
Genomic reads from each pair of libraries were QC filtered for artifact/process contamination and assembled together with AllPathsLG59. Illumina reads of stranded RNA-seq data were used as input for de novo assembly of RNA contigs, assembled into consensus sequences using Rnnotator (v. 3.4)60. Both genomes were annotated using the JGI Annotation Pipeline and made available via the JGI fungal portal MycoCosm61.
CEGMA and BUSCO were used to assess the completeness of the assemblies. We used BUSCO Version 3.0.1 with the lineage specific profile library basidiomycota_odb9 (species:selected 41:25 with 1.335 BUSCO groups) downloaded from http://busco.ezlab.org. We performed the whole analysis in Gene set (proteins) assessment mode. The results were combined in a folder and plotted with the script generate-plot.py.
Phylogenomic analysis
We used whole genomes of five Armillaria spp. and 22 Agaricales encompassing white and brown rot wood decayers, litter decomposers and ECM fungi; species from the Russulales and Boletales were included as outgroups (Supplementary Table 5). An all-versus-all protein BLAST was performed using mpiBLAST-1.6.062 with default parameters to find homologs based on similarity. Homologs were clustered into protein families using the Markov Cluster Algorithm63,64 with an inflation parameter of 2.0. For each cluster, a multiple sequence alignment was inferred using PRANK v.15080365, run with default settings. Subsequently, spuriously aligned regions were excluded with trimAl v1.4.r1566, with a gap threshold of 0.2. Next, we inferred ML gene trees for each cluster of a minimum of four proteins using FastTree v2.1967. FastTree was run with CAT/GAMMA20 model for rate heterogeneity and an improved LG model68 for substitution probabilities. Gene trees were mid-point re-rooted using Phyutility v 2.2.669.
Next, we screened the gene trees with a custom Perl script70 for the presence of deep paralogs and inparalogs. Gene trees with deep paralogs were eliminated, while in trees with inparalogs, the paralog closest to the root was retained in the corresponding alignments. Finally, 835 clusters passed these criteria and had >50 amino acid sites and higher than 60% taxon occupancy were concatenated into a supermatrix with 188,895 sites. We used this supermatrix to infer species tree with RAxML PTHREADS version 7.2.871 using a partitioned WAG+G model, where each data partition represented a single input gene family. To assess branch support of the tree we performed a bootstrap analysis in RAxML with 100 replicates under the same model.
We used penalized likelihood (PL) implemented in the program r8s version 1.7072 for molecular clock dating based on the optimal ML tree, two fossils and one secondary calibration. Archaeomarasmius legettii (94–90 Myr)73 was used to define minimum age constraint (92 Myr) for the origin of marasmioid clade (as used in a previous study74). Palaeoagaricites antiquus (100–110 Myr) is the oldest fossil can be placed within Agaricales75, hence we constrained the MRCAs of this order to a minimum age of 108 Myr. To define the minimum age constraint of the origin of Boletales to 84 Myr, we referred to published analyses27. Maximum age constraints were defined with a wide safety window to allow for the calibrated clades to be inferred at least twice as old as their minimum ages. The MRCA of the Boletales, that of the Agaricales and that of the marasmioid clade were constrained between 84–250 Myr, 108–250 Myr and 92–180 Myr, respectively, as minimum and maximum ages. A cross-validation (CV) analysis was used to identify an optimal smoothing parameter between 10−20 and 109. All analyses were run in four replicates (random number seeds). We applied the additive penalty function and run optimization 25 times initialized from independent starting points. Furthermore after reaching an initial solution of an optimization step, the solution was perturbed and the truncated Newton (TN) optimization was rerun 20 times. We found that the optimal smoothing parameter (λ) is between 10−7 and 10−3, thus we estimated divergence time with five λ ranges between 10−7 and 10−3. By checking the gradients and the estimated ages we found a high similarity between the results of the analyses obtained with different values of λ (Supplementary Fig. 2). By inspecting the CV analyses and the gradient values, λ = 10−6 was chosen as the most adequate and was used to estimate divergence times.
COMPARE analysis
We analysed the evolutionary history of gene families of Armillaria species and closely related Agaricales using the COMPARE pipeline76. To reconstruct gene duplications and losses, a genome-wide collection of 13,821 gene trees (see previous section) was first reconciled with the species tree using Treefix v1.1.1077. Treefix was run with RAxML site-wise likelihood model, Maximum Parsimony Reconciliation model (MPR) and an –alpha threshold of 0.001 to find any alternate gene tree topologies that minimize duplication/loss costs but are statistically equivalent to the ML gene tree. Orthologs were identified and recoded into a presence/absence matrix, as described in a previous study76. We then inferred duplications and losses for each orthogroup along the species tree using Dollo parsimony. Gene trees with less than four proteins were excluded. We also reconstructed the genome size for a given node by summing over gains and losses to the genome size of the MRCA. GO enrichment analysis based on the Fisher exact test with Benjamin–Hochberg correction was performed using Pfam domains mapped to protein clusters and creating GO annotations with pfam2go version 2016/10/0178 followed by enrichment analysis at p < 0.05.
Copy number analyses
We analysed the copy number distribution of selected gene families in Armillaria, Moniliophthora, and Heterobasidion annosum and, non-pathogenic members of Agaricales. For this, predicted proteomes of all the taxa under study were annotated with Pfam domains and IPR signatures. We scanned the protein sequences for pfams using pfamscan.pl v1.5 against the Pfam 30.0 database and InterPro signatures using InterProscan v5. Next, we built search terms for 42 PCWDE families and 25 putative pathogenicity related genes based on evidence from the literature (‘Search_Terms’ in Supplementary Table 5). In few cases, Pfam signatures were either absent or did not yield consistent results. CBM67 copy numbers were obtained based on BLAST hits by counting homologs obtained through blasting proteins annotated by JGI as CBM67. Genomes were searched iteratively for significant hits to avoid the impact of phylogenetic distance on the detection of homologs. We annotated cellobiose dehydrogenases (CDH, AA3_1), alcohol oxidases (Aox, AA3_3) and GMC oxidoreductases (GMT, AA3_2) using a tree-based approach. To this end, proteins of A. gallica (Armga1) were combined with homologs in other genomes, followed by multiple sequence alignment construction using MAFFT v7.27379 with -auto option and the estimation of a gene tree with FastTree v2.19 (as above). Classifications were based on the occurrence as monophyletic groups in the phylogenetic tree. SSPs were defined as proteins with less than 300 amino acids and sequence-based evidence for secretion as inferred by signalP v4.180. Both signalP-noTM and signalP-TM models were used for the detection of signal peptides.
Analysis of TEs
De novo element discovery and annotation
We used the REPET package version 2.581,82 to identify, classify and annotate TEs within the genomes studied. Since the A. mellea genome contained large amounts of ‘chaff’ contigs, we thresholded this assembly to only include contigs larger than 1,000 bp before further analysis.
The REPET de novo pipeline was run using genome self-alignment as well as a search for structural features using LTRHarvest83. Consensus sequences were clustered using Piler v1.084, Recon 1.0885 and Grouper86 and classified using PASTEC82. The resulting consensus sequences were filtered for low-frequency elements, short sequence repeats and sequences that were classified as putative host genes and then clustered into families using Markov clustering64. For annotation, the consensus sequences collected from each species were combined into a pan-species TE library. TE identification was then carried out using the REPET anno pipeline, implementing BLASTER, RepeatMasker 4.06 and CENSOR 4.2.2987. Configuration files for the pipelines containing detailed parameter settings as well as a sample command line script can be found at https://github.com/JackyHess/Armillaria_TE_annotations.
TE organization along the genome in core Armillaria species
To gain an understanding of how TEs are organized among the core Armillaria genomes we investigated TE and gene content density in 50 kb genome windows. Each genome was segmented into 50 kb partitions using bedtools (http://bedtools.readthedocs.io) and for each partition, TE coverage and genic coverage were estimated. Before estimating genic coverage, we filtered annotated CDS for which 20% of the sequence overlapped with TE annotations to remove putative TE-derived genes. This reduced the number of protein-coding genes considered by up to 7,000 (Supplementary Table 1). All analysis scripts can be found at https://github.com/JackyHess/Armillaria_TE_annotations.
In-depth transcriptome analysis of A. ostoyae
Whole transcriptome sequencing was performed using TrueSeq RNA Library Preparation Kit v2 (Illumina) according to the manufacturer’s instructions. Briefly, RNA quality and quantity measurements were performed using RNA ScreenTape and Reagents on TapeStation (all from Agilent) and Qubit (ThermoFisher); only high quality (RIN >8.0) total RNA samples were processed. Next, RNA was DNaseI (ThermoFisher) treated and the mRNA was purified and fragmented. First strand cDNA synthesis was performed using SuperScript II (ThermoFisher) followed by second strand cDNA synthesis, end repair, 3’-end adenylation, adapter ligation and PCR amplification. All purification steps were performed using AmPureXP Beads (Backman Coulter). Final libraries were quality checked using D1000 ScreenTape and Reagents on TapeStation (all from Agilent). Concentration of each library was determined using the QPCR Quantification Kit for Illumina (Agilent). Sequencing was performed on Illumina NextSeq instrument using the NextSeq Series 2 × 150 bp high-output kit (Illumina) generating more tha 20 million clusters for each sample.
Bioinformatic Analysis Draft genome sequence together with genome annotation file was used as a reference for A. ostoyae RNA-seq analysis. Paired-end Illumina NextSeq reads were quality trimmed using CLC Genomics Workbench tool version 9.5.2 (CLC Bio/Qiagen) removing ambiguous nucleotides as well as any low-quality read end using an error probability cutoff value of 0.05 (corresponding to a Phred score of 13). Trimmed reads were mapped using the RNA-Seq Analysis 2.1 package in CLC allowing intergenic read mapping and requiring at least 80% sequence identity over at least 80% of the read lengths; strand specificity was omitted. Reads with less than 30 equally scoring mapping positions were randomly mapped; reads with more than 30 potential mapping positions were considered as uninformative repeat reads and were excluded from the analysis (Supplementary Table 6).
‘Total gene read’ RNA-seq count data was imported from CLC into R version 3.0.2. Genes were filtered on the basis of their expression levels, keeping only those features that were detected by at least five mapped reads in at least 25% of the samples included in the study. Subsequently, ‘calcNormFactors’ from package edgeR version 3.4.288 was used to perform data normalization based on the trimmed mean of M-values (TMM) method89. Log transformation was carried out by the ‘voom’ function of the limma package version 3.18.1390. Linear modelling, empirical Bayes moderation and the calculation of differentially expressed genes were carried out using limma. Genes showing at least four-fold gene expression change with an FDR-corrected p-value below 0.05 were considered as significantly differentially expressed. Multidimensional scaling (‘plotMDS’ function in edgeR) was also applied to visually summarize gene expression profiles to reveal similarities between samples. In addition, unsupervised cluster analysis with Euclidean distance calculation and complete-linkage clustering was carried out on the normalized data using the ‘heatmap.2’ function from R package ‘gplots’.
Proteomics
Whole-cell lysate preparation and trypsin digestion
Two hundred milligrams (±5%) from every developmental stage of A. ostoyae was ground in 600 µL of an ice-cold lysis buffer (100 mM Tris-HCl, 50 mM NaCl, 20 mM EDTA, 10%(v/v) glycerol, 1 mM PMSF and 1 µg ml–1 Pepstatin A pH 7.5) using an IKA T10 basic homogenizer (IKA-Werk GmbH & Co.) and PT 1200 E Ergonomic Homogenizer (POLYTRON PT 1200 E; Kinematica AG), respectively. Samples were sonicated (Bandelin Sonoplus HD2200, Bandelin Elec.) three times on ice (MS73 probe, Cycle 6, 10s, power 20%), lysates were incubated overnight on ice at 4 ˚C and then clarified by centrifugation at 9,700 g for 10 min. The amount of protein was quantified using the Bradford assay and samples were normalized for protein concentration, where possible, per replicate. Samples were brought to 15% (w/v) trichloroacetic acid (TCA) for precipitation, with acetone washes. Samples were resuspended in 6 M urea, 2 M thiourea and 100 mM Tris-HCl pH 8.0 (60–120 µl); protein concentrations were then analysed, as above, and adjusted to 1 M urea final using ammonium bicarbonate before trypsin digestion. Protein samples (10 µg each) were sequentially reduced and alkylated with 5 µM DTT and 15 µM IAA, respectively11, brought to 0.01% (v/v) ProteaseMAX and trypsin added (1.6 µL; 1 µg µl–1) to a final protein amount (10 µg) (37 ˚C, overnight). Samples were acidified by the addition of 1 µL trifluoroacetic acid (TFA). Peptide solutions were then dried in a centrifugal evaporator, resuspended in 0.5% (w/v) TFA and desalted using C18 Ziptips (Millipore Ziptips C18)91,92,93,94.
LC-MS, data processing and interpretation
Peptide mixtures were analysed using a Thermo Fisher Q-Exactive mass spectrometer coupled to a Dionex RSLCnano for LC-MS/MS analysis. LC gradients operated from 3–40%B over 40 min, with data collection using a Top15 method for MS/MS scans93. LC-MS spectra chromatograms were analysed manually using rawMeat software. Raw files corresponding to the aforementioned spectra were then processed against an A. ostoyae predicted protein database using MaxQuant (version 1.3.0.5) and further filtered and visualized in Perseus (Version1.4.1.3)95. MaxQuant parameters were as described in another study94. Peptide intensity values were normalized to log2 values in Perseus. Only samples represented in all replicates of a sample group were taken (n = 1–4). Proteins common to all stages were z–score normalised, averaged and hierarchical clustering was performed to generate heat maps and profile plots. Venn diagrams were prepared for all proteins (unique and abundant proteins inclusive) in the entire data set (http://bioinformatics.psb.ugent.be).
Proteomic analysis revealed the presence of 2,549 proteins, of which 39.7% (1,012 of 2,549) proteins were common to all A. ostoyae developmental stages and 25.2% (643 of 2,549) were unique to individual developmental stages. Proteins were unique to rhizomorphs (n = 286), vegetative mycelium (n = 163), young fruiting bodies (n = 97), stage 1 and 2 primordia (n = 73) and mature fruiting bodies (n = 24). Quantitative changes in protein abundance were also evident between different development stages, albeit to different extents. For instance, comparing rhizomorph to vegetative mycelium stages, individual proteins underwent increased (n = 184) and decreased (n = 125) abundance (total detected n = 2,190), whereas comparative analysis of young fruiting bodies against mature fruiting bodies revealed only eight proteins with increased abundance (total detected n = 1,203). Comparative analysis of all other developmental stages yielded intermediate changes in the abundance of specific proteins.
Motif discovery
We used 1 kb sequences of co-expressed genes (which are most likely to contain their promoter), located upstream of the start codon, including 5’ UTR, to predict putative TFBS. We performed de novo motif discovery in our co-expressed gene sets using Weeder 2.038,96. We created frequency files from promoter regions of all genes of A. ostoyae using the Weeder2.0 frequencymaker. Frequencies were counted on both strands for motif lengths 6, 8 and 10, allowing 1, 2 and 3 mismatches, respectively. Using the inferred frequency counts as reference, both strands of the sequences were scanned using Weeder, allowing the detection of a maximum of 100 motifs. Next, we grouped the motifs based on Pearson correlation co-efficient, aligned using the ungapped Smith–Waterman algorithm and clustered using UPGMA, all executed via the STAMP webtool97. Finally, we constructed familial binding profiles for each motif group and searched for matching DNA motifs in the JASPAR Core (Fungi) 2016 database using the TOMTOM webserver98.
Data availability
Genome assemblies and annotation were deposited at DDBJ/EMBL/GenBank under the following accessions. A. ostoyae: FUEG01000001-FUEG01000106; A. cepistipes: FTRY01000001-FTRY01000182; A. gallica: NKEW00000000; and A. solidipes: NKHM00000000. A Gene Expression Omnibus (GEO) archive of the sequenced A. ostoyae libraries was deposited in the NCBI’s GEO Archive at http://www.ncbi.nlm.nih.gov/geo under accession GSE100213.
Code availability
All analysis scripts can be found at https://github.com/JackyHess/Armillaria_TE_annotations.
Change history
09 February 2018
In the version of this Article originally published, it was incorrectly stated that “16,687 protein-coding genes were inferred for the most recent common ancestor (MRCA) of Armillaria”; the value was incorrect and it should have read “15,787”. This has now been corrected.
References
Baumgartner, K., Coetzee, M. P. & Hoffmeister, D. Secrets of the subterranean pathosystem of Armillaria. Mol. Plant. Pathol. 12, 515–534 (2011).
Floudas, D. et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336, 1715–1719 (2012).
Smith, M. L., Bruhn, J. N. & Anderson, J. B. The fungus Armillaria bulbosa is among the largest and oldest living organisms. Nature 356, 428–431 (1992).
Stanosz, G. R., Patton, R. F. & Spear, R. N. Structure of Armillaria rhizomorphs from Wisconsin aspen stands. Can. J. Bot. 65, 2124–2127 (1987).
Anderson, J. B. & Catona, S. Genomewide mutation dynamic within a long-lived individual of Armillaria gallica. Mycologia 106, 642–648 (2014).
Pareek, M., Cole, L. & Ashford, A. E. Variations in structure of aerial and submerged rhizomorphs of Armillaria luteobubalina indicate that they may be organs of absorption. Mycol. Res. 105, 1377–1387 (2001).
Moore, D. Fungal Morphogenesis (Cambridge Univ. Press, Cambridge, 1998).
Yafetto, L., Davis, D. J. & Money, N. P. Biomechanics of invasive growth by Armillaria rhizomorphs. Fung. Genet. Biol. 46, 688–694 (2009).
Morrison, D. J. Ion uptake by rhizomorphs of Armillaria mellea. Can. J. Bot. 53, 48–51 (1975).
Pareek, M. Structure and Role of Rhizomorphs of Armillaria luteobubalina. PhD thesis, Univ. New South Wales (2006).
Collins, C. et al. Genomic and proteomic dissection of the ubiquitous plant pathogen, Armillaria mellea: toward a new infection model system. J. Proteome Res. 12, 2552–2570 (2013).
Ross-Davis, A. L. et al. Transcriptome of an Armillaria root disease pathogen reveals candidate genes involved in host substrate utilization at the host–pathogen interface. For. Pathol. 43, 468–477 (2013).
Meinhardt, L. W. et al. Genome and secretome analysis of the hemibiotrophic fungal pathogen, Moniliophthora roreri, which causes frosty pod rot disease of cacao: mechanisms of the biotrophic and necrotrophic phases. BMC Genom. 15, 164 (2014).
Mondego, J. M. et al. A genome survey of Moniliophthora perniciosa gives new insights into Witchesȁ; Broom Disease of cacao. BMC Genom. 9, 548 (2008).
Olson, A. et al. Insight into trade-off between wood decay and parasitism from the genome of a fungal forest pathogen. New Phytol. 194, 1001–1013 (2012).
Hane, J. K., Anderson, J. P., Williams, A. H., Sperschneider, J. & Singh, K. B. Genome sequencing and comparative genomics of the broad host-range pathogen Rhizoctonia solani AG8. PLoS Genet. 10, e1004281 (2014).
Raffaele, S. & Kamoun, S. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat. Rev. Microbiol. 10, 417–430 (2012).
Ohm, R. A. et al. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 8, e1003037 (2012).
Giraldo, M. C. & Valent, B. Filamentous plant pathogen effectors in action. Nat. Rev. Microbiol. 11, 800–814 (2013).
Kubicek, C. P., Starr, T. L. & Glass, N. L. Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 52, 427–451 (2014).
de Jonge, R. et al. Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science 329, 953–955 (2010).
Liu, W. et al. Recent progress in understanding PAMP- and effector-triggered immunity against the rice blast fungus. Magnaporthe oryzae. Mol. Plant 6, 605–620 (2013).
Akcapinar, G. B., Kappel, L., Sezerman, O. U. & Seidl-Seiboth, V. Molecular diversity of LysM carbohydrate-binding motifs in fungi. Curr. Genet. 61, 103–113 (2015).
Lee, W. S., Rudd, J. J., Hammond-Kosack, K. E. & Kanyuka, K. Mycosphaerella graminicola LysM effector-mediated stealth pathogenesis subverts recognition through both CERK1 and CEBiP homologues in wheat. Mol. Plant. Microbe Interact. 27, 236–243 (2014).
Liu, H., Zhang, B., Li, C. & Bao, X. Knock down of chitosanase expression in phytopathogenic fungus Fusarium solani and its effect on pathogenicity. Curr. Genet. 56, 275–281 (2010).
Fox, E. M. & Howlett, B. J. Secondary metabolism: regulation and role in fungal biology. Curr. Opin. Microbiol. 11, 481–487 (2008).
Kohler, A. et al. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat. Genet. 47, 410–415 (2015).
Martin, F., Kohler, A., Murat, C., Veneault-Fourrey, C. & Hibbett, D. S. Unearthing the roots of ectomycorrhizal symbioses. Nat. Rev. Microbiol. 14, 760–773 (2016).
Peter, M. et al. Ectomycorrhizal ecology is imprinted in the genome of the dominant symbiotic fungus Cenococcum geophilum. Nat. Commun. 7, 12662 (2016).
Rytioja, J. et al. Plant-polysaccharide-degrading enzymes from Basidiomycetes. Microbiol. Mol. Biol. Rev. 78, 614–649 (2014).
Saloheimo, M. et al. Swollenin, a Trichoderma reesei protein with sequence similarity to the plant expansins, exhibits disruption activity on cellulosic materials. Eur. J. Biochem. 269, 4202–4211 (2002).
Lee, K. T. et al. Systematic functional analysis of kinases in the fungal pathogen Cryptococcus neoformans. Nat. Commun. 7, 12766 (2016).
Tang, L.-H. et al. Transcriptome analysis of candidate genes and signaling path ways associated with light-induced brown film formation in Lentinula edodes. Appl. Microbiol. Biotechnol. 97, 4977–4989 (2013).
Ohtaki, S. et al. Novel hydrophobic surface binding protein, HsbA, produced by Aspergillus oryzae. Appl. Environ. Microbiol. 72, 2407–2413 (2006).
Soanes, D. M., Chakrabarti, A., Paszkiewicz, K. H., Dawe, A. L. & Talbot, N. J. Genome-wide transcriptional profiling of appressorium development by the rice blast fungus Magnaporthe oryzae. PLoS Pathog. 8, e1002514 (2012).
Plaza, D. F., Lin, C.-W., van der Velden, N. S. J., Aebi, M. & Künzler, M. Comparative transcriptomics of the model mushroom Coprinopsis cinerea reveals tissue-specific armories and a conserved circuitry for sexual development. BMC Genom. 15, 492 (2014).
Kunzler, M. Hitting the sweet spot-glycans as targets of fungal defense effector proteins. Molecules 20, 8144–8167 (2015).
Pavesi, G., Mereghetti, P., Mauri, G. & Pesole, G. Weeder Web: discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res. 32, W199–W203 (2004).
Mathelier, A. et al. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 44, D110–D115 (2016).
Prospero, S., Holdenrieder, O. & Rigling, D. Comparison of the virulence of Armillaria cepistipes and Armillaria ostoyae on four Norway spruce provenances. For. Pathol. 34, 1–14 (2004).
Tsykun, T., Rigling, D. & Prospero, S. A new multilocus approach for a reliable DNA-based identification of Armillaria species. Mycologia 105, 1059–1076 (2013).
Burdsall, H. H. & Volk, T. J. Armillaria solidipes, an older name for the fungus called Armillaria ostoyae. N. Am. Fungi 3, 261–267 (2008).
Ford, K. L., Baumgartner, K., Henricot, B., Bailey, A. M. & Foster, G. D. A reliable in vitro fruiting system for Armillaria mellea for evaluation of Agrobacterium tumefaciens transformation vectors. Fungal Biol. 119, 859–869 (2015).
Kues, U. & Navarro-Gonzalez, M. How do Agaricomycetes shape their fruiting bodies? 1. Morphological aspects of development. Fungal Biol. Rev. 29, 63–97 (2015).
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013).
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7, e47768 (2012).
Lam, K. K., LaButti, K., Khalak, A. & Tse, D. FinisherSC: a repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics 31, 3207–3209 (2015).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522 (2000).
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O. & Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990 (2008).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 15, 31 (2005).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29, 644–652 (2011).
Donlin, M.J. Using the Generic Genome Browser. Curr. Protoc. Bioinform. 17, 9.9.1–9.9.24 (2009).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Marthey, S. et al. FUNYBASE: a FUNgal phYlogenomic dataBASE. BMC Bioinform. 9, 456 (2008).
Parra, G., Bradnam, K., Ning, Z., Keane, T. & Korf, I. Assessing the gene space in draft genomes. Nucleic Acids Res. 37, 289–297 (2009).
Walter, M. C. et al. PEDANT covers all complete RefSeq genomes. Nucleic Acids Res. 37, D408–D411 (2009).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA 108, 1513–1518 (2011).
Martin, J. et al. Rnnotator: an automated de novo transcriptome assembly pipeline from stranded RNA-Seq reads. BMC Genom. 11, 663 (2010).
Grigoriev, I. V. et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 42, D699–D704 (2014).
Darling, A. E., Carey, L. & Feng, W.-C. The design, implementation, and evaluation of mpiBLAST. In 4th Int. Conf. Linux Clusters: The HPC Revolution 2003 (Linux Cluster Institute, 2003); http://www.mpiblast.org/downloads/pubs/cwce03.pdf
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 (2002).
van Stijn, D. Graph Clustering by Flow Simulation. PhD thesis, Univ. Utrecht (2000).
Löytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol Biol. 1079, 155–170 (2014).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2009).
Le, S. Q. & Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320 (2008).
Smith, S. A. & Dunn, C. W. Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24, 715–716 (2008).
Nagy, L. G. et al. Genetic bases of fungal wood decay predicted by phylogenomic analyses of gene-phenotype coevolution. Mol. Biol. Evol. 34, 35–44 (2017).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Sanderson, M. J. Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Mol. Biol. Evol. 19, 101–109 (2002).
Hibbett, D., Grimaldi, D. & Donoghue, M. Fossil mushrooms from Miocene and Cretaceous ambers and the evolution of Homobasidiomycetes. Am. J. Bot. 84, 981 (1997).
Matheny, P. B. et al. Major clades of Agaricales: a multilocus phylogenetic overview. Mycologia 98, 982–995 (2006).
Poinar, G. O. Jr & Buckley, R. Evidence of mycoparasitism and hypermycoparasitism in Early Cretaceous amber. Mycol. Res. 111, 503–506 (2007).
Nagy, L. G. et al. Latent homology and convergent regulatory evolution underlies the repeated emergence of yeasts. Nat. Commun. 5, 4471 (2014).
Wu, Y. C., Rasmussen, M. D., Bansal, M. S. & Kellis, M. TreeFix: statistically informed gene tree error correction using species trees. Syst. Biol. 62, 110–120 (2013).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221 (2015).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Petersen, T. N., Brunak, S., von Heijne, G. & Nielsen, H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 (2011).
Flutre, T., Duprat, E., Feuillet, C. & Quesneville, H. Considering transposable element diversification in de novo annotation approaches. PLoS ONE 6, e16526 (2011).
Hoede, C. et al. PASTEC: an automatic transposable element classification tool. PLoS ONE 9, e91929 (2014).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 18 (2008).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21(Suppl. 1), i152–i158 (2005).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002).
Quesneville, H. et al. Combined evidence annotation of transposable elements in genome sequences. PLoS Comput. Biol. 1, 166–175 (2005).
Kohany, O., Gentles, A. J., Hankus, L. & Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 7, 474 (2006).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Dolan, S. K. et al. Regulation of nonribosomal peptide synthesis: bis-thiomethylation attenuates gliotoxin biosynthesis in Aspergillus fumigatus. Chem. Biol. 21, 999–1012 (2014).
Moloney, N. M. et al. The iron-responsive microsomal proteome of Aspergillus fumigatus. J. Proteomics 136, 99–111 (2016).
O’Keeffe, G. et al. RNA-seq reveals the pan-transcriptomic impact of attenuating the gliotoxin self-protection mechanism in Aspergillus fumigatus. BMC Genom. 15, 894 (2014).
Owens, R. A. et al. Interplay between gliotoxin resistance, secretion, and the methyl/methionine cycle in Aspergillus fumigatus. Eukaryot. Cell 14, 941–957 (2015).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Pavesi, G., Mauri, G. & Pesole, G. An algorithm for finding signals of unknown length in DNA sequences. Bioinformatics 17(Suppl. 1), S207–S214 (2001).
Mahony, S. & Benos, P. V. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 35, W253–W258 (2007).
Gupta, S., Stamatoyannopolous, J. A., Bailey, T. & Noble, W. S. Quantifying similarity between motifs. Genome Biol. 8, R24 (2007).
Acknowledgements
This work was supported by the ‘Momentum Program’ of the Hungarian Academy of Sciences under LP2014/12, and an OTKA-ERC_HU #118722 from the NRDI Office to L.G.N. Fungal research at Maynooth University is part-funded by a Science Foundation Ireland Investigator award to SD (12/IP/1695). LC-MS facilities were funded by a competitive award from Science Foundation Ireland (12/RI/2346 (3)). G.S. acknowledges support from a WSL grant for a guest researcher in Switzerland and funding from GINOP-2.3.2-15-2016-00052. The work conducted by the U.S. Department of Energy Joint Genome Institute (JGI), a DOE Office of Science User Facility, was supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. IN was supported by a Janos Bolyai Research Fellowship of the Hungarian Academy of Sciences. We also thank Renate Heinzelmann for verifying the haploid status of the A. ostoyae and A. cepistipes isolates used for genome sequencing and Catherine Aime and Francis Martin for providing access to unpublished genome data produced by JGI.
Author information
Authors and Affiliations
Contributions
G.S. and L.G.N. conceived the study. G.S., K.B., C.S., J.L., U.G., S.M., K.L., A.L., R.W., M.C.W., A.P., I.V.G. and M.M. performed genome sequencing, assembly and annotation, N.M.M., E.O. and S.D. performed the proteomic study, D.F. analysed mating genes, J.H. performed analyses of TE content, A.P., B.K., I.N., B.Bóka, L.K., C.V. and B.Bálint obtained and analysed RNA-Seq data for A. ostoyae, A.P., K.K., J.S., R.R. and L.G.N. performed comparative genomic and phylogenomic analyses, T.V. performed the molecular clock dating. L.G.N, A.P., J.A.B., G.S. wrote the manuscript. D.R. contributed tested strains for sequencing. All authors read and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims inpublished maps and institutional affiliations.
A correction to this article is available online at https://doi.org/10.1038/s41559-018-0469-7.
Supplementary information
Supplementary Figures
Supplementary figures 1–20.
Supplementary Table 1
Transposable element content of Physalacriaceae genomes.
Supplementary Table 2
Enrichment analysis of gene duplicated in the Armillaria clade.
Supplementary Table 3
PFAM enrichment analysis.
Supplementary Table 4
Cluster of protein unique to Armillaria and subclades therein.
Supplementary Table 5
Species used in comparative genomic analyses, the lists of CAZy and pathogenicity-related genes analysed, and the search terms used to identify them.
Supplementary Table 6
RNA-Seq mapping statistics broken down by sample for Armillaria ostoyae enrichment analysis for rhizomorph upregulated genes and DEG analyses across all pairs of developmental stages and tissue types.
Supplementary Table 7
Genes showing an expression peak in Armillaria ostoyae rhizomorphs.
Supplementary Table 8
Notable genes upregulated in rhizomorphs.
Supplementary Table 9
Putative morphogenesis-associated and cell-wall biogenesis genes in rhizomorphs and their expression levels across development.
Supplementary Table 10
Enrichment analysis of genes upregulated in Stage1 primordia relative to VM.
Supplementary Table 11
List and expression patterns of cysteine-rich short secreted proteins identified in Armillaria ostoyae.
Supplementary Table 12
Notable fruiting body upregulated genes and their expression patterns across development.
Supplementary Table 13
Putative defence-related genes and their expression patterns across development.
Supplementary Table 14
Genes showing significant upregulation relative to vegetative mycelium and a close to constant expression in all complex multicellular developmental stages.
Supplementary Table 15
Genes showing rhizomorph-cap and rhizomorph-stipe upregulation patterns.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sipos, G., Prasanna, A.N., Walter, M.C. et al. Genome expansion and lineage-specific genetic innovations in the forest pathogenic fungi Armillaria. Nat Ecol Evol 1, 1931–1941 (2017). https://doi.org/10.1038/s41559-017-0347-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-017-0347-8
This article is cited by
-
Composition and diversity of soil microbial communities change by introducing Phallus impudicus into a Gastrodia elata Bl.-based soil
BMC Microbiology (2024)
-
Whole genome sequencing and analysis of Armillaria gallica Jzi34 symbiotic with Gastrodia elata
BMC Genomics (2023)
-
Horizontal gene transfer explains unusual traits of Armillaria fungi
Nature Microbiology (2023)
-
Research Status and Application Prospects of the Medicinal Mushroom Armillaria mellea
Applied Biochemistry and Biotechnology (2023)
-
Nonribosomal peptide synthetase gene clusters and characteristics of predicted NRPS-dependent siderophore synthetases in Armillaria and other species in the Physalacriaceae
Current Genetics (2023)
