
Abstract
Carnivorous plants exploit animals as a nutritional source and have inspired long-standing questions about the origin and evolution of carnivory-related traits. To investigate the molecular bases of carnivory, we sequenced the genome of the heterophyllous pitcher plant Cephalotus follicularis, in which we succeeded in regulating the developmental switch between carnivorous and non-carnivorous leaves. Transcriptome comparison of the two leaf types and gene repertoire analysis identified genetic changes associated with prey attraction, capture, digestion and nutrient absorption. Analysis of digestive fluid proteins from C. follicularis and three other carnivorous plants with independent carnivorous origins revealed repeated co-options of stress-responsive protein lineages coupled with convergent amino acid substitutions to acquire digestive physiology. These results imply constraints on the available routes to evolve plant carnivory.
Similar content being viewed by others

Carnivorous plants bear extensively modified leaves capable of attracting, trapping and digesting small animals, and absorbing the released nutrients1,2. Plant carnivory evolved independently in several lineages of flowering plants, providing a classic model for the study of convergent evolution3. Cephalotus follicularis (Cephalotus), a carnivorous plant native to southwest Australia that belongs to the monospecific family Cephalotaceae in the order Oxalidales, forms both carnivorous pitcher leaves and non-carnivorous flat leaves (Fig. 1). Co-existence of the two types of leaf in a single individual plant provides a unique opportunity to understand the genetic basis of plant carnivory through comparative analysis of these serially homologous organs. To this end, we sequenced the Cephalotus genome. A total of 305 Gb of Illumina reads were generated for contig assembly and scaffolding, and 17 Gb of PacBio reads for inter-contig gap filling (Supplementary Table 1). The resulting assembly consists of 16,307 scaffolds totalling 1.61 Gb with an N50 length of 287 kb (Supplementary Table 2), corresponding to 76% of the estimated genome size (Supplementary Fig. 1a). Long-terminal repeat retrotransposons account for 76% of the genome (Supplementary Tables 3 and 4). Syntenic block comparison with the robusta coffee genome, which maintained diploidy since the ancient split from the Cephalotus lineage4, reveals mostly one-to-one mappings (Fig. 1c and Supplementary Table 5), indicating that the Cephalotus genome has not experienced further whole genome duplications since the hexaploidy event at the origin of core eudicots5 (Supplementary Note 1). We annotated 36,503 protein-coding genes (Supplementary Fig. 1b–e), and 72 microRNA (miRNA) loci (Supplementary Table 6) and their potential targets (Supplementary Table 7) using RNA-sequencing (RNA-seq) data of representative tissues (Supplementary Tables 8–10). Orthologous gene groups (orthogroups) were defined using OrthoMCL6 for the complete gene sets of Cephalotus and eight eudicot species (Supplementary Tables 11 and 12). Analysis of shared singletons indicates that core eudicot genes are conserved in the Cephalotus genome (Supplementary Note 2 and Supplementary Table 13).

a, Pitcher and flat leaves. b, Flat and pitcher leaves predominantly produced at 15 °C and 25 °C, respectively, under continuous light conditions. Diamonds and error bars indicate means and standard deviations, respectively. Each filled circle represents an independent experiment with 45 plants. c, Synteny block matching of the Cephalotus genome against the coffee genome4 revealed a one-to-one matching in most genomic loci.
Maximum-likelihood gene gain and loss analysis detected lineage-specific expansion of 492 orthogroups in Cephalotus (Supplementary Table 14). Gene ontology (GO) enrichment analysis (Supplementary Tables 15–21) highlighted Cephalotus-expanded orthogroups containing purple acid phosphatases, known as a typical component of digestive fluids1,7 (Supplementary Table 17). RNase T2, also known as a constituent of digestive fluids1,8,9, is enriched among orthogroups composed only of genes from Cephalotus and another carnivorous plant Utricularia gibba (Supplementary Table 18). Also, the enriched GO term ‘cellular response to nitrogen levels’ included ten Cephalotus-specific singleton genes encoding dihydropyrimidinases, which have the potential function of acquired nitrogen recycling (Supplementary Table 19). Nitrogen is, in turn, known to be one of the primary limiting nutrients that carnivorous plants derive from prey1,10.
As we succeeded in regulating the developmental switch between pitcher and flat leaves by ambient temperature (Fig. 1b and Supplementary Fig. 1f,g), their transcriptomes were compared. The pitcher transcriptome was differentially enriched with cell cycle- and morphogenesis-related GO terms (Supplementary Table 22), which may reflect the morphological complexity of pitcher leaves. Although both developmental and thermoresponsive genes may change their expression in the temperature-dependent leaf switching, certain developmental regulators related to adaxial–abaxial polarity (for example, AS2, YAB5, and WOX1 orthologues) showed higher expression levels in shoot apices bearing pitchers than those terminating in flat leaves (Supplementary Fig. 2), implying the involvement of such factors in pitcher development and evolution. In contrast, the flat leaf transcriptome was enriched with photosynthesis-related GO terms (Supplementary Table 23). These results are compatible with the distinct functional specializations of carnivory-dominated pitcher leaves versus photosynthesis-dominated flat leaves.
Carnivorous plants attract potential prey by nectar, coloration and scent1,11,12. GO terms enriched in the pitcher transcriptome included ‘starch metabolic process’ and ‘sucrose metabolic process’ (Supplementary Table 22), which may be related to the production of attractive nectar. Indeed, we detected transcriptional upregulation of certain sucrose biosynthetic genes and members of sugar efflux carriers in pitcher leaves (Supplementary Fig. 3).
The epidermis of carnivorous pitfall traps often develops a slippery, waxy surface that promotes prey capture and prevents them from escaping1,13. A cytochrome P450 (CYP) orthogroup was expanded in the Cephalotus lineage (Supplementary Table 14). In a phylogenetic tree, these CYP genes belonged to a clade containing Arabidopsis genes involved in wax and cutin biosynthesis (CYP86 and CYP96A)14 (Supplementary Fig. 4). These genes, as well as wax ester synthase orthologues (WSD1)15, showed pitcher-predominant expression and are tandemly duplicated in the genome (Supplementary Fig. 4), suggesting possible co-regulated involvement of the clusters in slippery surface formation.
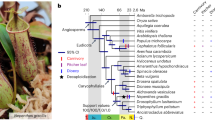
Carnivorous plants secrete digestive enzymes for degradation of trapped animals1,11,12. Previous studies on several digestive enzymes of Nepenthes spp., Drosera spp., Dionaea muscipula and Cephalotus indicate that pathogenesis-related proteins were co-opted for digestive function as well as for preventing microbial colonization of digestive fluid (refs 16–19 and refs in Supplementary Table 24). To further investigate the origin and evolution of digestive enzymes of Cephalotus and three other distantly related carnivorous plants (Drosera adelae, N. alata and Sarracenia purpurea), we sequenced fragments of digestive fluid proteins and identified 35 corresponding genes (Fig. 2a and Supplementary Tables 25–28). As Drosera and Nepenthes trace back to a common carnivorous origin in Caryophyllales3,20, the four species including Cephalotus therefore cover three independent origins of plant carnivory. Together with previously identified enzyme sequences including proteins from Dionaea (Supplementary Table 24), we inferred phylogenetic relationships among the digestive fluid proteins (Fig. 2b and Supplementary Fig. 5a–ah). Glycoside hydrolase family 19 (GH19) chitinase, β-1,3-glucanase, PR-1-like protein, thaumatin-like protein, purple acid phosphatase and RNase T2 genes showed orthologous relationships among carnivores despite their multiple origins. This result suggests that orthologous genes were repeatedly co-opted for digestive functions in independent carnivorous plant lineages.

a, Phylogenetic relationships of independently evolved carnivorous plants and, to their right, the digestive fluid proteins identified through proteomic analysis. Polytomy in the tree represents topological discrepancy between previously reported plastid and nuclear phylogenies (see Methods). Brackets connect protein variants likely to originate from the same gene. b, Phylogeny-based orthologue–paralogue classification. Branch colours denote species identities. Magenta on internal nodes indicates inferred duplication events. Gene numbers in collapsed clades are shown next to triangles. The collapsed clades do not contain genes encoding the digestive fluid proteins but may contain other Cephalotus genes as well as non-carnivorous plant genes. Complete trees are available in Supplementary Fig. 5. c,d, Transcriptome comparison of flat and pitcher organs in Cephalotus (c) and N. alata (d). Red numbers indicate positions of genes encoding digestive fluid proteins identified in this work (1–21, shown in a) and previous studies (22–25, Supplementary Table 24), several of which are outliers showing trap-specific expression.
To infer putative ancestral functions of these independently arisen digestive fluid proteins, we examined the expression patterns of their phylogenetically most closely related Arabidopsis genes (Supplementary Fig. 5a–ah). Compared with other genes in the same families, these Arabidopsis genes showed a significant tendency to be upregulated on various biotic and abiotic stresses (P < 0.02, randomization test) (Supplementary Fig. 5ai). This result suggests that co-option from stress-responsive proteins is a general evolutionary trend in the repeated evolution of carnivorous plant enzymes. Whether they are currently bifunctional—having both carnivorous and non-carnivorous roles—is unclear, but tissue-specific basal expression is probably optimized for carnivory in Cephalotus and N. alata, as the genes are preferentially expressed in their pitcher traps (Fig. 2c,d).
In Cephalotus, three aspartic proteases were identified in the digestive fluid proteome. We found three genomic clusters of aspartic protease genes containing both pitcher-preferential and constitutively expressed genes (Supplementary Fig. 5a–c). Together with the inferred tandem duplications of CYP, this result highlights the roles of gene duplication and subsequent functional divergence in carnivorous plant evolution.
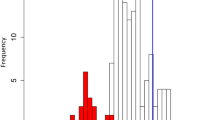
The repeated evolutionary utilization of similar genes may have been accompanied by convergent responses to carnivory-specific selective pressures at the amino acid substitution level. To test this, we developed a tree-based method for the detection of molecular convergence in multigene families, using phylogeny reconciliation between third codon position-derived gene trees and a consensus species tree (Supplementary Note 3, see Methods for the choice of a species tree). Using reconciled trees, the number of digestive enzyme-specific convergent substitutions was inferred on the basis of Bayesian ancestral sequence reconstructions (Fig. 3a,b and Supplementary Fig. 6). By comparing convergent substitution numbers and empirically calculated background-level expectations21, we found that GH19 chitinases (Fig. 3a,b), purple acid phosphatases (Supplementary Fig. 6i,j) and RNase T2s (Supplementary Fig. 6m,n) significantly accumulated convergent amino acid substitutions. For all three enzymes, two pitfall-type carnivorous pitcher plants, Cephalotus and N. alata, were associated as convergent branch pairs. Furthermore, for RNase T2, significant molecular convergence was also detected between Cephalotus and the common ancestor of the three Caryophyllales species, D. adelae, D. muscipula and N. alata, which produce sticky, snap and pitfall traps, respectively. Parsimonious inference of character evolution indicates that trapping strategy diversified after the establishment of carnivory in the Caryophyllales3,20. Therefore, molecular adaptation of RNase T2 probably occurred both during the evolution of carnivory and subsequently during the establishment of the specific capture strategy of pitfall traps. It is noteworthy that the Cephalotus RNase T2 and purple acid phosphatase genes are located adjacent to each other within a 40 kb interval of the Cephalotus genome (Supplementary Fig. 7). This placement could indicate an arrangement favoured by adaptive, positionally correlated co-expression22 of these modified carnivorous enzymes (Supplementary Note 4). In light of similar cases of convergent evolution shown for animal digestive enzymes23,24, we propose that major changes in nutritional strategy impose a selective pressure strong enough to override evolutionary contingency in both plants and animals.

a, GH19 chitinase phylogeny obtained from the phylogeny reconciliation. Identified digestive enzyme genes are indicated by trap illustrations. Magenta on internal nodes indicates inferred duplication events. The bar indicates 0.1 nucleotide substitutions per site. The complete tree is available in Supplementary Fig. 6q. b, Accumulation of convergent amino acid substitutions in GH19 chitinases. The positions of digestive enzyme branch pairs are indicated by red circles with corresponding numbers in a. Grey tones indicate branch pair density. The line shows a linear regression. c, Relationships between substitution processes and amino acid exposure in protein structures. As the convergent branch pairs in different families showed similar patterns (Supplementary Fig. 8e), data from GH19 chitinases, purple acid phosphatases and RNase T2s are pooled. The bottom panel shows posterior probabilities (PP) of convergent (pink) and divergent (light blue) substitution pairs. The top panel shows density distributions of convergent and divergent loci (PP ≥ 0.5, filled pink and light blue in the lower panel) as well as non-detected positions (PP < 0.5, filled grey with outline colour according to the substitution types). P value indicates a statistical difference of medians (vertical lines) (randomization test).
As protein structure imposes major constraints on amino acid substitutions25–27, we mapped amino acid residues identified as convergent onto corresponding 3D enzyme models. Convergent positions do not overlap with or cluster around catalytically essential amino acids (Supplementary Fig. 8). Instead, they tend to be located at exposed positions to an extent comparable to divergent substitutions (Fig. 3c), despite the prediction that more exposed positions result in lower convergence probability28. Exposed sites are structurally less constrained, and substitutions in such sites are likely to change their interactions with other molecules in solution, rather than changing protein conformation25–27. During the evolution of digestive enzymes, selective pressures may have come from the digestive fluid environment, which include the presence of insect-derived substrates, high endogenous proteolytic activity, low pH and microbial invasion or symbiosis1,11,12. As exposed residues constitute the protein–environment interface, the convergent amino acid substitutions may have been critical factors for the convergent establishment of carnivory across the angiosperms.
In the final phase of carnivorous plant physiology, digested molecules are absorbed into the plant body to promote growth and reproduction1,29. We found that various transporters were preferentially expressed in pitcher leaves (Supplementary Table 29). One pitcher-predominant transporter showed phylogenetic affinity to the AMMONIUM TRANSPORTER 1 (AMT1) subfamily (Supplementary Fig. 9), which contains the previously characterized carnivory-related D. muscipula gene DmAMT130. This result, together with the repeated co-option of digestive enzymes already described, indicates utilization of common genetic programs and evolutionary pathways in independently evolved carnivorous plant lineages.
The Cephalotus genome has allowed us to discover numerous genes associated with evolutionary transition to carnivory in plants. In particular, the high degree of convergent evolution in digestive enzymes indicates that there are few available evolutionary pathways for angiosperms to become carnivorous.
Methods
Plant materials and culture conditions
Axenically grown plants of C. follicularis were obtained from CZ Plants Nursery (Trebovice, Czech Republic) and were maintained in polycarbonate containers (60 × 60 × 100 mm) containing half-strength Murashige and Skoog solid medium31 supplemented with 3% sucrose, 1× Gamborg’s vitamins, 0.1% 2-(N-morpholino)ethanesulfonic acid, 0.05% Plant Preservative Mixture (Plant Cell Technology) and 0.3% Phytagel, at 25 °C in continuous light. For transcriptome sequencings, D. adelae was cultivated in a peat pot in an incubator at 25 °C in continuous light. N. alata was grown in soil in a greenhouse. S. purpurea was grown in peat-based soil and was maintained in a field. For digestive fluid sampling, C. follicularis, D. adelae, N. alata and S. purpurea were grown in a greenhouse.
Culture conditions for leaf fate regulation
Shoot apices with one or two expanded leaves were collected with fine forceps from plants grown at 25 °C and planted on medium. The plantlets were grown for 12 weeks under a light intensity of 20–40 μmol m−2 s−1. Numbers of youngest pitcher and flat leaves on main shoots were counted for each plantlet (Fig. 1b). Leaves with intermediate shapes were counted as either of the two categories based on morphological similarity.
DNA isolation
Total genomic DNA was isolated from young flat leaves and pitcher leaves of axenically grown plants. Collected leaves were homogenized in liquid nitrogen using a mortar and pestle. The homogenate was transferred into 2× CTAB extraction buffer (2% cetyltrimethylammonium bromide (CTAB), 1.4 M NaCl, 100 mM Tris-HCl (pH 8.0), 20 mM EDTA (pH 8.0)) preheated to 80 °C and was gently agitated at 60 °C for 1 h. An equal volume of chloroform:isoamyl alcohol (25:1) was added and agitated using a rotator at 20 r.p.m. for 10 min at room temperature. After centrifugation at 9,000 × g for 30 min at room temperature, supernatants were transferred to new tubes and supplemented with 1/10 volume of 10% CTAB and an equal volume of chloroform:isoamyl alcohol (25:1). The tubes were shaken with a rotator for 10 min. After centrifugation, supernatants were again transferred to new tubes and an equal volume of isopropanol was added. The tubes were centrifuged and supernatants were discarded. The crude DNA pellet was rinsed with 5 ml of 70% EtOH and air-dried for 10 min. The pellet was dissolved in 200 μl of TE (pH 8.0) containing 0.1 mg/ml RNase A, and gently agitated for 60 min at 37 °C. A 1/20 volume of 20 mg ml−1 Proteinase K was added, and tubes were incubated at 56 °C for 30 min. Subsequently, the DNA solution was further purified using Qiagen Genomic-tip, following the manufacturer’s instructions. DNA concentration was determined using fluorometry with Qubit 2.0 (Life Technologies).
Genome sequencing
Whole-genome shotgun short-read sequences were generated with an Illumina HiSeq 2000 to a depth of approximately 150-fold of the 2 Gb Cephalotus genome using paired-end and mate-pair protocols, according to the manufacturer’s instructions (Supplementary Table 1). For long read sequencing, genomic DNA samples were sheared to 6 kb or 10 kb using g-Tube (Covaris, Massachusetts). Libraries were prepared with DNA Template Prep Kit 2.0 (Pacific Biosciences, California) (3–10 kb) following the manufacturer’s instructions and sequencing was performed using PacBio RS with C2 chemistry, P2 polymerase and 45-min movies. Using 158 cells, a total of ca. 17 Gb were generated with a quality cut-off value of 0.75 (Supplementary Table 1).
Genome size estimation
The size of the Cephalotus genome was estimated by k-mer frequency analysis using JELLYFISH32 (Supplementary Fig. 1a).
Genome assembly
Illumina paired-end reads with all insert sizes, and mate-pair reads with insert sizes of 2 and 5 kb, were first assembled into 43,308 scaffolds using Allpaths-LG v4238133. Fragment filling was applied to paired-end libraries with insert sizes of 170 bp and 250 bp. Standard deviations of insert sizes were set to 10% of insert sizes. Gap filling and further scaffolding were performed by adding mate-pair reads with longer inserts using SSPACE34 and GapCloser35. PacBio reads were subjected to two rounds of error correction using Sprai v0.2.2.3 (http://zombie.cb.k.u-tokyo.ac.jp/sprai/) and used for four rounds of iterative gap filling with PBJelly v12.9.1436. The final assembly included 16,307 scaffolds with N50 of 287 kb (Supplementary Table 2).
Repeat identification
Repetitive elements of the Cephalotus genome were first identified and masked for gene prediction (Supplementary Tables 3 and 4). De novo prediction of transposable elements was performed using RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html) and LTR_FINDER (http://tlife.fudan.edu.cn/ltr_finder/). Known transposable elements were found using RepeatMasker and RepeatProteinMask (http://repeatmasker.org). Tandem repeat sequences were screened using Tandem Repeats Finder37.
RNA extraction
Plant materials were ground in liquid nitrogen using a mortar and pestle. Total RNA was extracted using the PureLink Plant RNA Reagent (Life Technologies) and subsequently purified using the RNeasy Mini Kit (QIAGEN). DNase treatment was performed during the column purification. Total RNA was qualified using a 2100 Bioanalyzer (Agilent).
Transcriptome sequencing
Extracted RNA was subjected to two rounds of mRNA enrichment using Dynabeads mRNA Purification Kit (Life Technologies) according to the manufacturer’s instructions. RNA-seq libraries were prepared using TruSeq RNA Sample Preparation kit v.2 (Illumina). Strand-specific mRNA libraries were constructed using the dUTP second-strand marking method38. These libraries were sequenced on an Illumina HiSeq 2000 with three biological replications (Supplementary Table 8).
Gene prediction
For gene predictions, we used homology-based, ab inito and transcript-based methods. Protein data sets of Arabidopsis thaliana, Linum usitatissimum, Manihot esculenta, Populus trichocarpa and Ricinus communis (Supplementary Table 11) were aligned to the Cephalotus genome using tblastn (cut-off: 1e−5) and then homology-based gene predictions were generated using GeneWise39. We also used Augustus (http://augustus.gobics.de/), GENSCAN (http://genes.mit.edu/GENSCAN.html), GlimmerHMM (https://ccb.jhu.edu/software/glimmerhmm/) and SNAP (http://korflab.ucdavis.edu/software.html) for ab initio predictions, with model parameters trained using 730 Cephalotus gene models that were well supported by homology evidence. RNA-seq data generated from 16 samples (Supplementary Table 8) were used for transcript-based predictions with the Bowtie–Tophat–Cufflinks pipeline40. These models were merged using GLEAN (http://glean-gene.sourceforge.net/). Finally, gene models that were not in the GLEAN non-redundant gene set but supported by both homology and RNA-seq evidences, or homology-based models (frame shift mutation not allowed and aligning rate >50%), or RNA-seq models encoding proteins ≧120 amino acids in length, were further added.
Gene annotation
Gene functions were assigned using BLAST searches (E-value cut-off of 10−5) against the following databases: KEGG (Release 58), nr (NCBI release 20130904), Swissprot and TrEMBL (Uniprot release 201203). Conserved protein domains were assessed by InterPro41 and InterProScan42 with applications including HMMPfam, HMMPanther, ProfileScan, HMMSmart, FPrintScan and BlastProDom.
Evaluation of genome assembly and gene prediction
Gene coverage of predicted gene sets was evaluated using CEGMA 2.443 (Supplementary Fig. 1b). Read mapping rates of 15 RNA-seq libraries from five tissues ranged from 74.4% to 83.6% (Supplementary Table 9), indicating consistency between the assembled genome and the sequenced transcriptome.
Small RNA extraction and sequencing
Plant tissues were ground in liquid nitrogen using a mortar and pestle. Total RNA was extracted using PureLink Plant RNA Reagent (Life Technologies) and subsequently purified using the miRNeasy kit (QIAGEN). DNase treatment was performed during the column purification. Briefly, for each sample, RNA of the desired size range (18–30 nucleotides) was size-fractionated and ligated with the 5' adapter and, subsequently, the 3' adapter. Ligated RNA was then subjected to PCR with reverse transcription (RT-PCR) to produce sequencing libraries. Small RNA-seq was performed on an Illumina HiSeq 2000 (Supplementary Table 10).
miRNA prediction and target prediction
Cephalotus miRNA loci were predicted in the genome by both transcriptome- and homology-based methods (Supplementary Table 6). Small RNA-seq reads were mapped onto genomic inverted repeats predicted by EMBOSS einverted44. miRNA loci were identified from the mapping results using ShortStack v1.2.345. For homology-based prediction, 7,385 mature miRNA sequences of Viridiplantae species were retrieved from miRbase release 2046. These miRNA sequences were mapped onto the Cephalotus genome using patscan47, allowing one mismatch. Putative loci mapped by less than five independent miRNAs were excluded. Secondary structures were identified from flanking regions of mapped loci (±350 bp) using RNAfold of Vienna RNA Package 2.048, and putative miRNA loci were predicted using miRcheck with default parameters49. When putative miRNAs were predicted on both strands of the same loci, the minor locus was collapsed. Putative targets of annotated miRNAs were identified using psRNATarget50 using default settings (Supplementary Table 7).
OrthoMCL gene classification
Orthologues were clustered by comparison of protein data sets among A. thaliana, C. follicularis, Theobroma cacao, Vitis vinifera, Prunus persica, Coffea canephora, Solanum lycopersicum, U. gibba and P. trichocarpa using BLASTP (cut-off: 10−5) and OrthoMCL6 (Supplementary Tables 11 and 12). Protein data sets of the nine genomes were BLAST searched against nr (NCBI release 20140407; BLASTP, E-value cut-off of 10−5). Functional terms (GO and enzyme codes) were then assigned to each query sequence using Blast2GO (https://www.blast2go.com/).
Maximum-likelihood inference of orthogroup gains and losses
We estimated the divergence times of the surveyed species using RAxML version 851, employing tree topologies published previously52–54. The reported placement of P. persica (Rosales) is discrepant between plastid-54 and nuclear-based analyses52,53. To account for that, we analysed phylogenetic relationships using the single-copy orthologue alignment (see below). Although the bootstrap supports were low, the maximum-likelihood tree supported the nuclear-based topology (Supplementary Fig. 1h), and therefore we placed P. persica as sister to the clade containing A. thaliana, T. cacao, P. trichocarpa and C. follicularis. The placement of V. vinifera is also different among previously published phylogenies52–54. To account for that, two alternative tree topologies with different placements of V. vinifera were assumed in this analysis. For that, we leveraged the amino acid sequence data of all single-copy orthologues, as defined by OrthoMCL (1,836 1:1 orthologues), after excluding all putative TE sequences identified in BLAST searches against different TE databases (TIGR Plant Repeat Databases55, TransposonPSI (http://transposonpsi.sourceforge.net) and NCBI's non-redundant (nr) protein database). We then aligned the sequences of each orthogroup with the program M-Coffee56 and used trimAl57 to automatically remove poorly aligned regions. The best-fit amino acid substitution model for each multiple sequence alignment was selected using ProtTest58 and specified in the RAxML analysis under a partitioned scheme. We finally used r8s59 to obtain the ultrametric trees required for the BadiRate60 analysis, by applying the nonparametric rate smoothing algorithm59 to the maximum-likelihood trees and fixing the age of the root to 113 Myr in both cases. This date, a compromise for the two trees we tested, was derived from the average of the 2 BEAST point estimates for the earliest split within the rosid clade (with Vitaceae as one sister lineage), as calculated in ref. 54 (their Fig. 1 and Table 2). The two trees tested are detailed below.
Tree 1:
((V_vinifera:92.251246,(((T_cacao:72.791098,A_thaliana:72.791098): 5.199433,(P_trichocarpa:72.150935,C_follicularis:72.150935):5.839595):5.054431,P_persica:83.044961):9.206285):20.748754,(U_gibba:101.840606,(C_canephora:89.804910,S_lycopersicum:89.804910):12.035696):11.159394).
Tree 2:
(((U_gibba:82.830331,(C_canephora:74.586612,S_lycopersicum:74.586612):8.243718):14.783045,(((T_cacao:74.611384,A_thaliana:74.611384):5.510359,(P_trichocarpa:73.737693,C_follicularis:73.737693):6.384050):5.848981,P_persica:85.970724):11.642652):15.386624,V_vinifera:113.000000).
To identify gene families specifically expanded in the Cephalotus genome, we followed the method implemented in refs 4 and 61, accepting a weighted Akaike information criterion (wAIC) ratio of 2.7 for the best-fit branch model to the second-best-fit model. We ran BadiRate60 twice, once for each of the two alternative topologies shown above. Only those families strongly supported as expanded (wAIC ratio >2.7) under both of the two alternative topologies were considered for further analyses (Supplementary Table 14).
GO enrichment analysis
Supplementary Table 12 shows the per species summary of orthogroups and singletons in nine plant species. Before BadiRate analyses, orthogroups containing sequences with significant similarity to transposable elements (resulting in E-values <10−15 in TBLASTX searches against sequences of the RepBase v19.12 database)62 were filtered out from all nine genomes. The functional categories (generic GO terms) differentially represented among 493, 495 and 492 Cephalotus-specific expanded genes families (grouping 2,560, 2,567 and 2,557 total genes, respectively), as identified in BadiRate analyses performed using tree 1, tree 2 and the intersection of both trees, are displayed in Supplementary Tables 15, 16 and 17, respectively. Similarly, differential representation of GO generic terms among 2,716 Cephalotus-specific singletons, 237 Cephalotus-specific two-gene families (474 total genes) and Cephalotus-specific 201 multigene families (1,714 total genes) are shown in Supplementary Tables 19, 20 and 21, respectively. Finally, differential representation of GO generic terms among five pairs of genes unique to Cephalotus and U. gibba is presented in Supplementary Table 18. We performed significance analyses of differential distribution of GO terms by comparing different subsets of genes with the entire complement of genes in the genome using Fisher’s exact test (seefor example, ref. 4). To control for multiple testing, the resulting P values were corrected according to ref. 63.
Selection of differentially expressed genes
Strand-specific RNA-seq reads were mapped to gene models on the genome assembly using Tophat264 with minimum and maximum intron lengths of 20 and 20,000 bp, respectively (Supplementary Table 9). Transcript abundances calculated by featureCounts65 were normalized using the iterative differentially expressed gene elimination strategy (iDEGES)66, which consists of sequential TMM-(edgeR-TMM)n normalization67,68. Using the normalized reads per million mapped reads (RPM) values, differentially expressed genes were identified by an exact test for a negative binomial distribution69 and subsequent multiple correction by adjusting the false discovery rate to q < 0.01 (ref. 63; Fig. 2c,d and Supplementary Figs 2–5 and 9). Normalized RPM values are used in Fig. 2c,d, whereas unnormalized RPM values are plotted in Supplementary Figs 2–5 and 9. The significantly differentially expressed genes were subjected to a subsequent GO-enrichment analysis (Supplementary Tables 22 and 23).
Protein sequencing of digestive fluids
Digestive fluids of C. follicularis, D. adelae, N. alata and S. purpurea were collected from soil-grown plants in a greenhouse. Fluids were freeze dried and stored at room temperature. Dried samples were dissolved in a protease inhibitor cocktail (cOmplete, Mini, EDTA-free, Roche), precipitated with 8% trichloroacetic acid (TCA) and then washed with 90% acetone. They were dissolved in SDS sample buffer (62.5 mM Tris-HCl, 2% SDS, 0.25% BPB, 10% glycerol, 5% 2-mercaptoethanol, pH 6.8), denatured at 95 °C for 3 min and then separated by 12% SDS-polyacrylamide gel electrophoresis. Negative staining was performed using the Gel-Negative Stain Kit (Nacalai Tesque) according to the manufacturer’s instructions. After destaining, proteins were transferred to polyvinylidene difluoride (PVDF) membranes. N-terminal sequences of each protein band were determined by the Edman degradation method using an ABI Procise 494-HT instrument (Applied Biosystems). To obtain internal protein sequences, protein bands were dissected from the gel, destained, dehydrated with 100% acetonitrile for 5 min, dried using an evaporator and then reduced by incubating in 10 mM DTT and 25 mM ammonium bicarbonate at 56 °C for 60 min. After washing with 25 mM ammonium bicarbonate, the proteins were alkylated in 55 mM iodoacetamide and 25 mM ammonium bicarbonate for 45 min at room temperature. After washing with 50% acetonitrile containing 25 mM ammonium bicarbonate, the samples were dried using an evaporator. The proteins were in-gel-digested with 10 ng μl−1 trypsin in 50 mM ammonium bicarbonate, 10 ng μl−1 lysyl endopeptidase in 25 mM Tris-HCl (pH 9.0) or 20 ng μl−1 V8 protease in 50 mM phosphate buffer (pH 7.8) at 37 °C overnight. The digested peptides were extracted twice by sonication in 50% acetonitrile containing 5% trifluoroacetic acid (TFA) for 10 min. The peptides were separated by high-performance liquid chromatography (HPLC) using the Pharmacia SMART System and a reverse-phase column (μRPC C2/C18 PC 3.2/3, GE Healthcare Life Sciences, or XBridge C8 5 μm 2.1×100 mm, Waters) under the following conditions: constant flow rate of 200 μl min−1; solvent A, 0.5% TFA, solvent B, acetonitrile containing 0.5% TFA; linear gradient from 10 to 40% (B over A in % (v/v)) over 30 min (1% min−1). Separated peptides were then used for protein sequencing by the Edman degradation method.
Transcriptome assembly and identification of transcripts encoding biochemically identified proteins
RNA-seq reads of D. adelae, N. alata, and S. purpurea (Supplementary Table 8) were assembled into transcripts using Trinity (version r2013-02-25)70 with a 200 bp minimum contig length cut-off. Partial amino acid sequences of digestive fluid proteins were subjected to TBLASTN searches71 against the transcriptome assemblies and the Cephalotus gene models to identify the corresponding transcripts (Supplementary Tables 25–28). Sequence variants within a Trinity’s component were considered as originating from the same gene.
Preparation of digestive fluid protein data sets
In addition to proteins identified in this study (Supplementary Tables 25–28), we obtained for phylogenetic analyses a number of previously published sequences of digestive fluid proteins8,9,72–78 (Supplementary Table 24). Although many protein and transcript sequences for possible digestive enzymes are available (for example, refs 17,79–84), we included only genes for which complete coding sequences were available and for which their presence in digestive fluid had been biochemically validated (Supplementary Table 24, last searched 20 January 2016).
Phylogenetic analyses of gene families
Phylogenetic relationships of digestive enzyme genes and other carnivory-related genes were analysed along with their homologues in the annotated genomes of ten angiosperm species (Supplementary Table 11). TBLASTX searches71 were performed against the above coding sequence (CDS) data sets with an E-value cut-off of 10. After sequence retrieval, multiple alignments were prepared using MAFFT 6.95685, and ambiguous codons were removed using trimAl57 implemented in Phylogears2-2.0.2013.03.15 (http://www.fifthdimension.jp/products/phylogears/) with the ‘gappyout’ option. Poorly aligned sequences were removed using MaxAlign86. Phylogenetic trees were reconstructed by the maximum-likelihood method using RAxML v8.0.2651 with the general time-reversible (GTR) model of nucleotide substitution and four discrete gamma categories of rate heterogeneity (‘GTRGAMMA’ option). Support for nodes was estimated by rapid bootstrapping with 100 replicates. Trees were rooted at the midpoint between the two most divergent genes. Gene duplication events shown in Figs 2b and 3a were inferred on the basis of species overlap between partitions87 using a Python package ‘ETE3’88. The trees were visualized using iTOL89.
Detection of orthologous relationships
Orthology of Cephalotus genes and digestive enzyme genes was inferred on the basis of tree topologies reconstructed by the maximum-likelihood method using the ten plant genomes described above (Supplementary Figs 2, 4 and 5). As we cannot exclude the possibility of parallel gene losses, a clade containing genes from at least five plant genomes was designated as a putative orthologous unit.
Expression profiling of Arabidopsis genes
Affymetrix ATH1 (25K) microarray data sets on stress-related experiments were retrieved from ArrayExpress90 if two or more replicates were available on wild-type Arabidopsis plants (Supplementary Table 30). Robust multi-array average normalized expression data91 were subjected to heatmap visualization using the R package ‘gplots’. Dendrograms were constructed using the furthest neighbour method with Euclidian distances. Significance of differential expression was analysed by a randomization test with 10,000 iterations in which resamplings were performed in each gene family and the sum of expression changes was compared with the original value.
Evaluation of detection methods for molecular convergence
To evaluate different tree reconstruction methods, simulated gene sequences were generated using the R package ‘Phylosim’92. We used publicly available simulated data sets for 16 fungi species93,94. These data sets contain 1,000 simulated tree topologies of gene families, each of which was generated under observed gene duplication and loss rates. Sequences of 300 codons were simulated on the tree topologies of the fungi data set. Codon usage was sampled from the actual frequencies in Saccharomyces cerevisiae95. The κ (transition/transversion rate) was set to 1. The ω (nonsynonymous / synonymous nucleotide substitution rate ratio (dN/dS)) of each codon position was randomly sampled from a gamma distribution (shape = 0.5, rate = 1). To mimic molecular convergence, two genes were randomly selected to be converged. In terminal branches of selected genes, codon usage of S. cerevisiae was replaced with a biased matrix in which frequencies of codons coding for two randomly selected amino acids were increased. Increased frequency was calculated by multiplying the original value by 100, and then total frequencies of all codons were scaled to 1.
Gene trees were inferred by the maximum-likelihood method51 using first, second, third and all codon positions as well as 300 nucleotide random sequences. To obtain a robust tree topology, the gene trees were reconciled with the species tree using Treefix 1.1.1094, which incorporates duplication-loss parsimony and a test statistic for likelihood equivalence. Reconciliation was accomplished using default settings for which 1,000 iterations of topology searches were performed and rearrangements were accepted when likelihood was not significantly reduced by the Shimodaira–Hasegawa test96 (P value threshold of 0.05). Branch lengths of reconciled trees were optimized using RAxML51. Finally, the numbers of convergent and divergent substitutions were estimated from the inferred tree topologies and the original simulated alignments using CodeMLancestral21 (Supplementary Fig. 10). Substitution pairs that result in the same descendant amino acid at the same alignment position in both branches were categorized as convergent changes, whereas the remaining substitution pairs were counted as divergent changes21,28.
Detection of molecular convergence in digestive fluid proteins
Genes encoding digestive fluid proteins identified in this study (Supplementary Tables 25–28) and previous research (Supplementary Table 24) were analysed. When corresponding gene sequences for a given species clustered together in the maximum-likelihood trees (Supplementary Fig. 5), they were considered to represent the same gene, whereafter we retained our own sequences to circumvent incorrect inference of gene duplication events in phylogeny reconciliation. A maximum-likelihood tree was reconstructed using third codon position sequences of the trimAl-processed alignments, and it was subsequently reconciled with a species tree prepared from a dated large-scale plastid phylogeny of flowering plants54 using Treefix94 with default parameters, except with the number of iterations increased to 1,000. Although the plastid-based topology54 is partly different from nuclear-based topology52,53 (Supplementary Fig. 1h), we employed it because of the necessity to include carnivorous lineages in which nuclear genome sequences are unavailable (for example, Drosera, Nepenthes and Sarracenia). Branch lengths of the reconciled trees were optimized against trimAl-processed CDS alignments using RAxML51. The trees were subsequently used for Bayesian ancestral state reconstruction using PhyloBayes97 over 12,000 generations (2,000 generations of burn-in) with an infinite mixture of GTR substitution models (CAT-GTR model) of amino acid substitution and five discrete gamma categories of rate heterogeneity to calculate posterior numbers of convergent and divergent substitution pairs. Background levels (null hypothesis) of convergent substitution pairs were estimated by a linear regression in which the posterior numbers of convergent changes were predicted by divergent changes21. Over-accumulation of convergent changes in a tree was examined by one-sided single-sample proportion tests98 with Yate’s continuity correction99 and subsequent Bonferroni adjustment for multiple comparisons100. Digestive enzyme branch pairs among independent carnivorous plant lineages were examined in the statistical test. Corrected P values are shown in Fig. 3b and Supplementary Fig. 6.
Homology modelling of protein structures
Protein structures of digestive enzymes were analysed using the SWISS-MODEL Workspace101. Template models were selected using the ‘Template Identification’ tool. SWISS-MODEL Template Library IDs of selected templates were 2dkv.1.A, 3zk4.1.A and 1dix.1.A for GH19 chitinases, purple acid phosphatases and RNase T2s, respectively. Predicted models were visualized using UCSF Chimera 1.10102. Relative exposure of amino acid surfaces was calculated by dividing solvent-accessible surface in protein structures by the theoretical maximum of corresponding amino acids in Gly-X-Gly tripeptide contexts103. The relative solvent-accessible surface area for a paired amino acid substitution was reported by averaging values in proteins constituting the two clades (Supplementary Fig. 8).
Data availability
The Cephalotus genome assembly and gene models are available from the DNA Data Bank of Japan (DDBJ) with the accession numbers BDDD01000001 to BDDD01016307. The genomic sequences, gene models and other source data are also available at CoGe (Genome ID = 29002) and Dryad (doi:10.5061/dryad.50tq3). The DDBJ accession numbers for DNA-seq (DRR053706–DRR053720), mRNA-seq (DRR053690–DRR051749; DRR029007–DRR29010) and small RNA-seq (DRR058704–DRR058708) are shown in Supplementary Tables 1, 8 and 10, respectively. DDBJ accessions and gene IDs for coding sequences of digestive fluid proteins are provided in Supplementary Tables 25–28.
Additional information
How to cite this article: Fukushima, K. et al. Genome of the pitcher plant Cephalotus reveals genetic changes associated with carnivory. Nat. Ecol. Evol. 1, 0059 (2017).
References
Juniper, B. E., Robins, R. J. & Joel, D. M. The Carnivorous Plants (Academic, 1989).
Darwin, C. Insectivorous Plants (D. Appleton and Company, 1875).
Albert, V. A., Williams, S. E. & Chase, M. W. Carnivorous plants: phylogeny and structural evolution. Science 257, 1491–1495 (1992).
Denoeud, F. et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345, 1181–1184 (2014).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Gallie, D. R. & Chang, S. C. Signal transduction in the carnivorous plant Sarracenia purpurea. Regulation of secretory hydrolase expression during development and in response to resources. Plant Physiol. 115, 1461–1471 (1997).
Okabe, T., Yoshimoto, I., Hitoshi, M., Ogawa, T. & Ohyama, T. An S-like ribonuclease gene is used to generate a trap-leaf enzyme in the carnivorous plant Drosera adelae . FEBS Lett. 579, 5729–5733 (2005).
Nishimura, E. et al. S-like ribonuclease gene expression in carnivorous plants. Planta 238, 955–967 (2013).
Ellison, A. M. Nutrient limitation and stoichiometry of carnivorous plants. Plant Biol. 8, 740–747 (2006).
Król, E. et al. Quite a few reasons for calling carnivores ‘the most wonderful plants in the world’. Ann. Bot. 109, 47–64 (2012).
Adlassnig, W., Peroutka, M. & Lendl, T. Traps of carnivorous pitcher plants as a habitat: composition of the fluid, biodiversity and mutualistic activities. Ann. Bot. 107, 181–194 (2011).
Whitney, H. M. & Federle, W. Biomechanics of plant–insect interactions. Curr. Opin. Plant Biol. 16, 105–111 (2013).
Bak, S. et al. Cytochromes P450. Arab. B. 9, e0144 (2011).
Li, F. et al. Identification of the wax ester synthase/acyl-coenzyme A: diacylglycerol acyltransferase WSD1 required for stem wax ester biosynthesis in Arabidopsis . Plant Physiol. 148, 97–107 (2008).
Buch, F. et al. Secreted pitfall-trap fluid of carnivorous Nepenthes plants is unsuitable for microbial growth. Ann. Bot. 111, 375–383 (2013).
Schulze, W. X. et al. The protein composition of the digestive fluid from the Venus flytrap sheds light on prey digestion mechanisms. Mol. Cell. Proteomics 11, 1306–1319 (2012).
Michalko, J. et al. Glucan-rich diet is digested and taken up by the carnivorous sundew (Drosera rotundifolia L.): implication for a novel role of plant β-1,3-glucanases. Planta 238, 715–725 (2013).
Bemm, F. et al. Venus flytrap carnivorous lifestyle builds on herbivore defense strategies. Genome Res. 26, 1–14 (2016).
Heubl, G., Bringmann, G. & Meimberg, H. Molecular phylogeny and character evolution of carnivorous plant families in Caryophyllales — revisited. Plant Biol. 8, 821–830 (2006).
Castoe, T. A. et al. Evidence for an ancient adaptive episode of convergent molecular evolution. Proc. Natl Acad. Sci. USA 106, 8986–8991 (2009).
Michalak, P. Coexpression, coregulation, and cofunctionality of neighboring genes in eukaryotic genomes. Genomics 91, 243–248 (2008).
Stewart, C. B., Schilling, J. W. & Wilson, A. C. Adaptive evolution in the stomach lysozymes of foregut fermenters. Nature 330, 401–404 (1987).
Zhang, J. Parallel adaptive origins of digestive RNases in Asian and African leaf monkeys. Nat. Genet. 38, 819–823 (2006).
Choi, S. S., Vallender, E. J. & Lahn, B. T. Systematically assessing the influence of 3-dimensional structural context on the molecular evolution of mammalian proteomes. Mol. Biol. Evol. 23, 2131–2133 (2006).
Bustamante, C. D., Townsend, J. P. & Hartl, D. L. Solvent accessibility and purifying selection within proteins of Escherichia coli and Salmonella enterica . Mol. Biol. Evol. 17, 301–308 (2000).
Goldman, N., Thorne, J. L. & Jones, D. T. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 149, 445–458 (1998).
Goldstein, R. A., Pollard, S. T., Shah, S. D. & Pollock, D. D. Nonadaptive amino acid convergence rates decrease over time. Mol. Biol. Evol. 32, 1373–1381 (2015).
Ellison, A. M. & Gotelli, N. J. Energetics and the evolution of carnivorous plants—Darwin’s ‘most wonderful plants in the world’. J. Exp. Bot. 60, 19–42 (2009).
Scherzer, S. et al. The Dionaea muscipula ammonium channel DmAMT1 provides NH4 + uptake associated with Venus flytrap’s prey digestion. Curr. Biol. 23, 1649–1657 (2013).
Murashige, T. & Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 15, 473–497 (1962).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA 108, 1513–1518 (2011).
Boetzer, M., Henkel, C. V, Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 18 (2012).
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7, e47768 (2012).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Levin, J. Z. et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 7, 709–715 (2010).
Birney, E. & Durbin, R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 10, 547–548 (2000).
Trapnell, C. et al. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221 (2014).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120 (2005).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European molecular biology open software suite. Trends Genet. 16, 276–277 (2000).
Axtell, M. J. ShortStack: comprehensive annotation and quantification of small RNA genes. RNA 19, 740–751 (2013).
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73 (2014).
Dsouza, M., Larsen, N. & Overbeek, R. Searching for patterns in genomic data. Trends Genet. 13, 497–498 (1997).
Lorenz, R. et al. ViennaRNA package 2.0. Algorithms Mol. Biol. 6, 26 (2011).
Jones-Rhoades, M. W. & Bartel, D. P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 14, 787–799 (2004).
Dai, X. & Zhao, P. X. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159 (2011).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Zeng, L. et al. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat. Commun. 5, 4956 (2014).
Wickett, N. J. et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl Acad. Sci. USA 111, E4859–E4868 (2014).
Bell, C. D., Soltis, D. E. & Soltis, P. S. The age and diversification of the angiosperms re-revisited. Am. J. Bot. 97, 1296–1303 (2010).
Ouyang, S. & Buell, C. R. The TIGR Plant Repeat Databases: a collective resource for the identification of repetitive sequences in plants. Nucleic Acids Res. 32, D360–D363 (2004).
Wallace, I. M., O’Sullivan, O., Higgins, D. G. & Notredame, C. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 34, 1692–1699 (2006).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Sanderson, M. J. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302 (2003).
Librado, P., Vieira, F. G. & Rozas, J. BadiRate: estimating family turnover rates by likelihood-based methods. Bioinformatics 28, 279–281 (2012).
Carretero-Paulet, L. et al. High gene family turnover rates and gene space adaptation in the compact genome of the carnivorous plant Utricularia gibba . Mol. Biol. Evol. 32, 1284–1295 (2015).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Sun, J., Nishiyama, T., Shimizu, K. & Kadota, K. TCC: an R package for comparing tag count data with robust normalization strategies. BMC Bioinformatics 14, 219 (2013).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, 2010–2011 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Robinson, M. D. & Smyth, G. K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9, 321–332 (2008).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Paszota, P. et al. Secreted major Venus flytrap chitinase enables digestion of Arthropod prey. Biochim. Biophys. Acta 2, 374–383 (2014).
Takahashi, K. et al. A cysteine endopeptidase (‘dionain’) is involved in the digestive fluid of Dionaea muscipula (Venus’s fly-trap). Biosci. Biotechnol. Biochem. 75, 346–348 (2011).
Athauda, S. B. et al. Enzymic and structural characterization of nepenthesin, a unique member of a novel subfamily of aspartic proteinases. Biochem. J. 381, 295–306 (2004).
Buch, F., Pauchet, Y., Rott, M. & Mithofer, A. Characterization and heterologous expression of a PR-1 protein from traps of the carnivorous plant Nepenthes mirabilis . Phytochemistry 100, 43–50 (2014).
Rottloff, S. et al. Functional characterization of a class III acid endochitinase from the traps of the carnivorous pitcher plant genus, Nepenthes . J. Exp. Bot. 62, 4639–4647 (2011).
Hatano, N. & Hamada, T. Proteome analysis of pitcher fluid of the carnivorous plant Nepenthes alata . J. Proteome Res. 7, 809–816 (2008).
Hatano, N. & Hamada, T. Proteomic analysis of secreted protein induced by a component of prey in pitcher fluid of the carnivorous plant Nepenthes alata . J. Proteomics 75, 4844–4852 (2012).
Renner, T. & Specht, C. D. Molecular and functional evolution of class I chitinases for plant carnivory in the Caryophyllales. Mol. Biol. Evol. 29, 2971–2985 (2012).
Eilenberg, H., Pnini-Cohen, S., Schuster, S., Movtchan, A. & Zilberstein, A. Isolation and characterization of chitinase genes from pitchers of the carnivorous plant Nepenthes khasiana . J. Exp. Bot. 57, 2775–2784 (2006).
Matusíková, I. et al. Tentacles of in vitro-grown round-leaf sundew (Drosera rotundifolia L.) show induction of chitinase activity upon mimicking the presence of prey. Planta 222, 1020–1027 (2005).
An, C.-I., Fukusaki, E. & Kobayashi, A. Aspartic proteinases are expressed in pitchers of the carnivorous plant Nepenthes alata Blanco. Planta 214, 661–667 (2002).
Srivastava, A., Rogers, W. L., Breton, C. M., Cai, L. & Malmberg, R. L. Transcriptome analysis of Sarracenia, an insectivorous plant. DNA Res. 18, 253–261 (2011).
Jensen, M. K. et al. Transcriptome and genome size analysis of the Venus flytrap. PLoS ONE 10, e0123887 (2015).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Gouveia-Oliveira, R., Sackett, P. W. & Pedersen, A. G. MaxAlign: maximizing usable data in an alignment. BMC Bioinformatics 8, 312 (2007).
Huerta-Cepas, J. et al. The human phylome. Genome Biol. 8, 934–941 (2007).
Huerta-Cepas, J., Serra, F. & Bork, P. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638 (2016).
Letunic, I. & Bork, P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478 (2011).
Kolesnikov, N. et al. ArrayExpress update—simplifying data submissions. Nucleic Acids Res. 43, D1113–D1116 (2015).
Irizarry, R. A. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003).
Sipos, B., Massingham, T., Jordan, G. E. & Goldman, N. PhyloSim - Monte Carlo simulation of sequence evolution in the R statistical computing environment. BMC Bioinformatics 12, 104 (2011).
Rasmussen, M. D. & Kellis, M. A Bayesian approach for fast and accurate gene tree reconstruction. Mol. Biol. Evol. 28, 273–290 (2011).
Wu, Y. C., Rasmussen, M. D., Bansal, M. S. & Kellis, M. TreeFix: statistically informed gene tree error correction using species trees. Syst. Biol. 62, 110–120 (2013).
Nakamura, Y., Gojobori, T. & Ikemura, T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 28, 292 (2000).
Shimodaira, H. & Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114–1116 (1999).
Lartillot, N., Lepage, T. & Blanquart, S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288 (2009).
Wilson, E. B. Probable inference, the law of succession, and statistical inference. J. Am. Stat. Assoc. 22, 209–212 (1927).
Yates, F. Contingency tables involving small numbers and the χ2 test. J. R. Stat. Soc. 1(suppl.) 217–235 (1934).
Neyman, J. & Pearson, E. S. On the use and interpretation of certain test criteria for purposes of statistical inference: part I. Biometrika 20A, 175–240 (1928).
Biasini, M. et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 (2014).
Pettersen, E. F. et al. UCSF Chimera —a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Tien, M. Z., Meyer, A. G., Sydykova, D. K., Spielman, S. J. & Wilke, C. O. Maximum allowed solvent accessibilites of residues in proteins. PLoS ONE 8, e80635 (2013).
Acknowledgements
We acknowledge the following sources for funding: MEXT/JSPS KAKENHI grant numbers 12J04926 (K.F.), 22128008 (T.N.), 221S0002 (M.K.), 16370102 (M.H.), 22128001 (M.H.) and 22128002 (M.H.); MECS CGL2013-45211 (J.R.), R01 GM097251 (D.D.P.), and the US NSF 0922742 and 1442190 (V.A.A.). The sequence data are archived at DDBJ and CoGe. This work was supported by MPRF-NIBB, I. Kajikawa and Y. Matsuzaki for cultivation, J. G. Conran and M. Waycott for locality observation and FGF-NIBB, Y. Makino, S. Ooi and K. Yamaguchi for experiments. Computations were partially performed on the NIG supercomputer and DIAF-NIBB.
Author information
Authors and Affiliations
Contributions
K.F., N.S. and M.H. maintained and collected samples. K.F. and M.H. established culture conditions to control the leaf dimorphism. K.F., X.F., T.N., S.L., and M.H. coordinated genome assembly and annotation. K.F., X.F., C.C., M.N., S.S. and M.X. prepared and sequenced Illumina libraries. M.N. coordinated PacBio sequencing. T.F.S. performed PacBio sequencing. T.I. and M.K. performed error correction of PacBio reads and inter-contig gap filling. K.F., X.F., H.C. and L.M. performed gene prediction and annotation. K.F. performed miRNA prediction and annotation. D.A.-P. performed singleton gene analysis. C.Z. and D.S. performed synteny analysis. P.L., A.S.-G and J.R. performed BadiRate analysis. L.C.-P. and V.A.A. performed GO-enrichment analysis. K.F., K.M.F., T.-H.C. and G.P. performed gene family analysis. T.F., Y. Hiwatashi, Y. Hoshi, H.M., N.S., T.U. and M.H. performed digestive fluid sampling and protein sequencing. K.F., S.T.P. and D.D.P. performed convergence analysis. K.F., V.A.A. and M.H. wrote the paper with input from all authors. M.H. initiated and directed the project. K.F., V.A.A., S.L. and M.H. are representatives of each group. K.F. and X.F. should be considered joint first authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary information
Supplementary Figures; Supplementary Tables; Supplementary Notes; Supplementary References (PDF 5174 kb)
Supplementary information
Supplementary Tables 1,2,6–10,12,14–23,29–30 (XLS 8269 kb)
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Fukushima, K., Fang, X., Alvarez-Ponce, D. et al. Genome of the pitcher plant Cephalotus reveals genetic changes associated with carnivory. Nat Ecol Evol 1, 0059 (2017). https://doi.org/10.1038/s41559-016-0059
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41559-016-0059
This article is cited by
-
A diffusible small-RNA-based Turing system dynamically coordinates organ polarity
Nature Plants (2024)
-
Micropropagation and genetic transformation of Byblis liniflora
In Vitro Cellular & Developmental Biology - Plant (2024)
-
Multi-integrated genomic data for Passiflora foetida provides insights into genome size evolution and floral development in Passiflora
Molecular Horticulture (2023)
-
Subgenome dominance shapes novel gene evolution in the decaploid pitcher plant Nepenthes gracilis
Nature Plants (2023)
-
Detecting macroevolutionary genotype–phenotype associations using error-corrected rates of protein convergence
Nature Ecology & Evolution (2023)
