
Abstract
Tissue homeostasis and regeneration rely on resident stem cells (SCs), whose behaviour is regulated through niche-dependent crosstalk. The mechanisms underlying SC identity are still unfolding. Here, using spatiotemporal gene ablation in murine hair follicles, we uncover a critical role for the transcription factors (TFs) nuclear factor IB (NFIB) and IX (NFIX) in maintaining SC identity. Without NFI TFs, SCs lose their hair-regenerating capability, and produce skin bearing striking resemblance to irreversible human alopecia, which also displays reduced NFIs. Through single-cell transcriptomics, ATAC-Seq and ChIP-Seq profiling, we expose a key role for NFIB and NFIX in governing super-enhancer maintenance of the key hair follicle SC-specific TF genes. When NFIB and NFIX are genetically removed, the stemness epigenetic landscape is lost. Super-enhancers driving SC identity are decommissioned, while unwanted lineages are de-repressed ectopically. Together, our findings expose NFIB and NFIX as crucial rheostats of tissue homeostasis, functioning to safeguard the SC epigenome from a breach in lineage confinement that otherwise triggers irreversible tissue degeneration.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


Data availability
ChIP-Seq, ATAC-Seq, RNA-Seq and single-cell RNA-Seq data that support the findings of this study have been deposited in the Gene Expression Omnibus under accession codes GSE135142, GSE135143, GSE135144, GSE135145 and GSE135146 (super-series). Previously published sequencing data on bulge SC super-enhancers that were re-analysed here are available under accession code GSE61316. Source data for Figs. 1–3 and 6 and Extended Data Figs. 1, 2, 4, 5 and 8 are presented with the paper. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
Code availability
Codes for analysis of the figures related to single-cell RNA-Seq data are available at https://github.com/hanseulyang01/NFI-single-cell-analysis.
References
Avgustinova, A. & Benitah, S. A. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 17, 643–658 (2016).
Scadden, D. T. Nice neighborhood: emerging concepts of the stem cell niche. Cell 157, 41–50 (2014).
Gonzales, K. A. U. & Fuchs, E. Skin and its regenerative powers: an alliance between stem cells and their niche. Dev. Cell 43, 387–401 (2017).
Paksa, A. & Rajagopal, J. The epigenetic basis of cellular plasticity. Curr. Opin. Cell Biol. 49, 116–122 (2017).
Arwert, E. N., Hoste, E. & Watt, F. M. Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 12, 170–180 (2012).
Dvorak, H. F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659 (1986).
Hsu, Y.-C., Pasolli, H. A. & Fuchs, E. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell 144, 92–105 (2011).
Hsu, Y. C., Li, L. & Fuchs, E. Transit-amplifying cells orchestrate stem cell activity and tissue regeneration. Cell 157, 935–949 (2014).
Page, M. E., Lombard, P., Ng, F., Göttgens, B. & Jensen, K. B. The epidermis comprises autonomous compartments maintained by distinct stem cell populations. Cell Stem Cell 13, 471–482 (2013).
Ge, Y. et al. Stem cell lineage infidelity drives wound repair and cancer. Cell 169, 636–650 (2017).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Hnisz, D. et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 58, 362–370 (2015).
Adam, R. C. et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 521, 366–370 (2015).
Trompouki, E. et al. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell 147, 577–589 (2011).
Adam, R. C. et al. Temporal layering of signaling effectors drives chromatin remodeling during hair follicle stem cell lineage progression. Cell Stem Cell 22, 398–413 (2018).
Kadaja, M. et al. SOX9: a stem cell transcriptional regulator of secreted niche signaling factors. Genes Dev. 28, 328–341 (2014).
Folgueras, A. R. et al. Architectural niche organization by LHX2 is linked to hair follicle stem cell function. Cell Stem Cell 13, 314–327 (2013).
Lien, W. H. et al. In vivo transcriptional governance of hair follicle stem cells by canonical Wnt regulators. Nat. Cell Biol. 16, 179–190 (2014).
Horsley, V., Aliprantis, A. O., Polak, L., Glimcher, L. H. & Fuchs, E. NFATc1 balances quiescence and proliferation of skin stem cells. Cell 132, 299–310 (2008).
Genander, M. et al. BMP signaling and its pSMAD1/5 target genes differentially regulate hair follicle stem cell lineages. Cell Stem Cell 15, 619–633 (2014).
Lay, K., Kume, T. & Fuchs, E. FOXC1 maintains the hair follicle stem cell niche and governs stem cell quiescence to preserve long-term tissue-regenerating potential. Proc. Natl Acad. Sci. USA 113, E1506–E1515 (2016).
Wang, L., Siegenthaler, J. A., Dowell, R. D. & Yi, R. Foxc1 reinforces quiescence in self-renewing hair follicle stem cells. Science 351, 613–617 (2016).
Chang, C. Y. et al. NFIB is a governor of epithelial–melanocyte stem cell behaviour in a shared niche. Nature 495, 98–102 (2013).
Driller, K. et al. Nuclear factor IX deficiency causes brain malformation and severe skeletal defects. Mol. Cell Biol. 27, 3855–3867 (2007).
Matuzelski, E. et al. Transcriptional regulation of Nfix by NFIB drives astrocytic maturation within the developing spinal cord. Dev. Biol. 432, 286–297 (2017).
Steele-Perkins, G. et al. The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol. Cell. Biol. 25, 685–698 (2005).
Gründer, A. et al. Nuclear factor I-B (Nfib) deficient mice have severe lung hypoplasia. Mech. Dev. 112, 69–77 (2002).
Das Neves, L. et al. Disruption of the murine nuclear factor I-A gene (Nfia) results in perinatal lethality, hydrocephalus, and agenesis of the corpus callosum. Proc. Natl Acad. Sci. USA 96, 11946–11951 (1999).
Deneen, B. et al. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 52, 953–968 (2006).
Campbell, C. E. et al. The transcription factor Nfix is essential for normal brain development. BMC Dev. Biol. 13, 8–52 (2008).
Chen, K. S., Lim, J. W. C., Richards, L. J. & Bunt, J. The convergent roles of the nuclear factor I transcription factors in development and cancer. Cancer Lett. 410, 124–138 (2017).
Denny, S. K. et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell 166, 328–342 (2016).
Chávez, S. & Beato, M. Nucleosome-mediated synergism between transcription factors on the mouse mammary tumor virus promoter. Proc. Natl Acad. Sci. USA 94, 2885–2890 (1997).
Liu, R. et al. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell 106, 309–318 (2001).
Ferrari, S. et al. Chromatin domain boundaries delimited by a histone-binding protein in yeast. J. Biol. Chem. 279, 55520–55530 (2004).
Pjanic, M. et al. Nuclear factor I genomic binding associates with chromatin boundaries. BMC Genomics 14, 99 (2013).
Waki, H. et al. Global mapping of cell type-specific open chromatin by FAIRE-Seq reveals the regulatory role of the NFI family in adipocyte differentiation. PLoS Genet. 7, e1002311 (2011).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Meisterernst, M., Gander, I., Rogge, L. & Winnacker, E. L. A quantitative analysis of nuclear factor I/DNA interactions. Nucleic Acids Res. 16, 4419–4435 (1988).
Kruse, U. & Sippel, A. E. Transcription factor nuclear factor I proteins form stable homo- and heterodimers. FEBS Lett. 348, 46–50 (1994).
Soeda, T. et al. Sox9-expressing precursors are the cellular origin of the cruciate ligament of the knee joint and the limb tendons. Genesis 48, 635–644 (2010).
Pratt, C. H., King, L. E. Jr., Messenger, A. G., Christiano, A. M. & Sundberg, J. P. Alopecia areata. Nat. Rev. Dis. Primers 3, 17011 (2017).
Harries, M. J. & Paus, R. The pathogenesis of primary cicatricial alopecias. Am. J. Pathol. 177, 2152–2162 (2010).
Su, Y. et al. Cicatricial alopecia. IntechOpen https://doi.org/10.5772/intechopen.78971 (2018).
Harries, M., Hardman, J., Chaudhry, I., Poblet, E. & Paus, R. Profiling the human hair follicle immune system in lichen planopilaris and frontal fibrosing alopecia: can macrophage polarization differentiate these two conditions microscopically? Br. J. Dermatol. https://doi.org/10.1111/bjd.18854 (2019).
Volc-Platzer, B. et al. Accumulation of gamma delta T cells in chronic cutaneous lupus erythematosus. J. Invest. Dermatol. 100, 84S–91S (1993).
Lay, K. et al. Stem cells repurpose proliferation to contain a breach in their niche barrier. eLife 7, e41661 (2018).
Rosenblum, M. D. et al. Expression of CD200 on epithelial cells of the murine hair follicle: a role in tissue-specific immune tolerance? J. Invest. Dermatol. 123, 880–887 (2004).
Rigopoulos, D., Stamatios, G. & Ioannides, D. Primary scarring alopecias. Curr. Probl. Dermatol. 47, 76–86 (2015).
Anzai, A., Wang, E. H. C., Lee, E. Y., Aoki, V. & Christiano, A. M. Pathomechanisms of immune-mediated alopecia. Int. Immunol. 31, 439–447 (2019).
Jaks, V. et al. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet. 40, 1291–1299 (2008).
Picelli, S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013).
Brennecke, P. et al. Accounting for technical noise in single-cell RNA-Seq experiments. Nat. Methods 10, 1093–1095 (2013).
Snippert, H. J. et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 327, 1385–1389 (2010).
Yang, H., Adam, R. C., Ge, Y., Hua, Z. L. & Fuchs, E. Epithelial–mesenchymal micro-niches govern stem cell lineage choices. Cell 169, 483–496 (2017).
Karnik, P. et al. Hair follicle stem cell-specific PPARγ deletion causes scarring alopecia. J. Invest. Dermatol. 129, 1243–1257 (2009).
Zheng, Y. et al. Scd1 is expressed in sebaceous glands and is disrupted in the asebia mouse. Nat. Genet. 23, 268–270 (1999).
Yang, H. et al. ETS family transcriptional regulators drive chromatin dynamics and malignancy in squamous cell carcinomas. eLife 4, e10870 (2015).
Schmidl, C., Rendeiro, A. F., Sheffield, N. C. & Bock, C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods 12, 963–965 (2015).
Choi, H. M. et al. Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat. Biotechnol. 28, 1208–1212 (2010).
Beronja, S., Livshits, G., Williams, S. & Fuchs, E. Rapid functional dissection of genetic networks via tissue-specific transduction and RNAi in mouse embryos. Nat. Med. 16, 821–827 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 15, 550 (2014).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Robinson, J. T. et al. Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26 (2011).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Keyes, B. E. et al. Nfatc1 orchestrates aging in hair follicle stem cells. Proc. Natl Acad. Sci. USA 110, E4950–E4959 (2013).
Acknowledgements
We thank E. Wong, M. Nikolova, J. Racelis and P. Nasseir for technical assistance, and L. Polak, J. Levorse and L. Hidalgo for assistance with mouse handling and experiments. We thank The Rockefeller University FACS facility for cell sorting, the Rockefeller Genomics Resource Center and Weill Cornell Genomics Resource Center for high-throughput sequencing, and the Comparative Bioscience Center (AAALAC accredited) for the care of mice in accordance with National Institutes of Health guidelines. E.F. is an investigator of the Howard Hughes Medical Institute. R.C.A. was the recipient of an Anderson Cancer Center Graduate Fellowship. H.Y. was the recipient of a Kwanjeong Educational Foundation Graduate Fellowship. Y.G. was a postdoctoral fellow of the American Federation of Aging Research. N.R.I. is the recipient of a National Institutes of Health Predoctoral National Research Service Award F31 Fellowship from NIAMS. This work was supported by grants from the National Institutes of Health to E.F. (R01-AR31737).
Author information
Authors and Affiliations
Contributions
R.C.A., H.Y. and E.F. designed the experiments and wrote the manuscript. Y.G. performed the ATAC-Seq experiments. P.W., Y.Z. and D.Z. analysed the TF motifs of the ATAC-Seq data. N.R.I. performed the immunofluorescence quantifications and contributed to the single-cell RNA-Seq analysis. S.G.-C. performed the 16S bacterial FISH and spot analysis. Y.M. performed the flow cytometry analyses of immune cells. J.K., J.G.K., J.E.K., J.Y.K., S.S.P. and C.P.L. provided the human scalp samples. R.C.A. and H.Y. performed the rest of the experiments. R.M.G. provided the conditional Nfibfl/fl and Nfixfl/fl mice. E.F. supervised the project. All authors provided intellectual input and vetted and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
R.C.A. is currently employed at Regeneron Pharmaceuticals. E.F. is on the Scientific Advisory Board of L’Oreal and Arsenal Biosciences. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 NFI-TFs Maintain Bulge-SC Identity and Prevent Ectopic Differentiation.
a, Enrichment of NFIB within chromatin of super-enhancers, compared to typical enhancers. Comparisons were made with 377 randomly selected typical enhancers and their flanking sequence extended 5’ and 3’ to match the average length of super-enhancers (average of 3 analyses is shown). b, Table showing examples of bulge-SC super-enhancer regulated genes with NFIB ChIP-occupancy. c, mRNA expression levels (TPM) of NFI family members in bulge-SCs. Mean TPM from 2 independent replicates are shown. d, Validation of Nfib and Nfix gene knockout and antibody specificity. INTEGRIN β4 (β4) marks basal epithelial cells. Inner bulge cells are β4neg, adjacent to the hair shaft and are critical to anchor the hair. e, Images of WT and Nfix cKO mice at 2 months post-TAM. Nfix-ablation on its own has no impact on the hair coat. f, Immunofluorescence using K24 antibody to label bulge-SCs. Ablation of Nfib or Nfix alone does not affect bulge-SCs, whereas combined ablation results in a loss of K24+ bulge-SCs. g, Images of WT or NFI-dKO mice at 4 weeks post-TAM. h, Immunofluorescence comparing telogen HFs of WT, Nfib cKO, Nfix cKO and Nfib/Nfix-dKO mice. K5 marks basal epithelial cells. Note aberrant K10+ (differentiating) cells only in double Nfib/Nfix targeted (dKO) mice. i, In situ hybridization using scramble or mir-203 probes on mouse skin. Note expansion of signal for the epidermal differentiation microRNA, mir-203, into bulge of NFI-dKO skin. j, Tape strip assay to evaluate hair anchoring. Tape stripping applies a mild tug to the hairs, which will be released from the coat if anchorage is weak. All scale bars = 20 μm. Bu, bulge. Dashed lines, HF-dermal border. For d-j, at least three biological replicates were used; representative images are shown. See also Source Data.
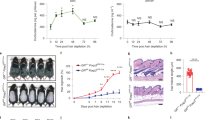
Extended Data Fig. 2 NFI-dKO Mice Exhibit Features of Primary Cicatricial Alopecia.
a, Immunofluorescence showing HF degeneration in NFI-dKO mice at 2 months post-TAM. YFP labels Sox9-CreER-targeted HFs. K6 labels inner bulge cells anchoring the hair. b, Immunofluorescence showing hyperkeratosis (K10) and follicular plugging of infundibulum in NFI-dKO HFs. K5 marks basal epithelial cells. c, Loss of PPARG, SCD1 and ADIPOPHILIN, lipid-related markers of mature sebocytes, in NFI-dKO follicles. Mean and standard deviation are shown. 30 HFs per genotype, pooled from n = 3 mice. P value is from unpaired, two-tailed t-test. d, Hematoxylin & Eosin image of NFI-dKO skin at 2 months post-TAM. Note follicular remnants and fibrous tracts (arrows). e, Immunofluorescence and quantifications of active CASPASE3+ (apoptotic) cells in NFI-dKO HFs. Mean and standard deviation are shown. For 2 weeks data, n = 80 HFs per genotype (total, pooled from 4 mice). For 2 months data, n = 59 HFs (WT) or n = 74 HFs (NFI-dKO), (total, pooled from 3 mice). P values are from unpaired, two-tailed t-test. f, (left) Flow cytometry analysis of immune cells at 2 months post-NFI deletion. Mean and standard deviation are shown. n = 4 mice/genotype. P values are from unpaired, two-tailed t-test. (right) Immunofluorescence analysis of FOXP3+ regulatory T-cells (Tregs) around the HF bulge niche. g, Fluorescence in situ hybridization (FISH) of pan-bacterial 16S rRNA (rainbow colors) in cleared skin whole-mounts, co-labeled for DAPI (gray) at 2 months post-TAM. Spot analysis of 16S-FISH signal was used to quantify bacterial load per μm3 of skin. Mean and standard deviation are shown. n = 10 (WT) and 16 (NFI-dKO) HFs from 2 biologically independent mice/genotype. Scale bars = 20 μm, unless otherwise specified. Bu, bulge. Inf, Infundibulum. Dashed lines, HF-dermal border. For a-d and f, at least three biological replicates were used; representative images are shown. See also Source Data.
Extended Data Fig. 3 Skin Immune Cell Profiling by Flow Cytometry.
a, Flow cytometry gating strategy for adaptive immune cell profiling. b, Flow cytometry gating strategy for innate immune cell profiling. Plots are shown for a representative WT mouse analyzed at 2 months post-NFI ablation. See Methods for details on immune cell identification.
Extended Data Fig. 4 Absence of Skin Immune Infiltration at 2 weeks post-NFI loss.
a, Fluorescence in situ hybridization (FISH) of pan-bacterial 16S rRNA (rainbow colors) in cleared skin whole-mounts, co-labeled for DAPI (gray) at 2 weeks post gene knockout. Spot analysis of 16S-FISH signal was used to quantify cutaneous bacterial load. Representative images of two biological replicates. b, Immunofluorescence showing skin immune cells (CD45+) are not changed at 2 weeks following Nfib/Nfix knockout (mean and standard deviation are shown). K5 marks basal epithelial cells. n = 3 mice. c, Flow cytometry analysis of immune cell composition at 2 weeks post-NFI deletion. Mean and standard deviation are shown. n = 4 mice/genotype. d, mRNA expression (from RNA-seq) of immune-related genes in bulge-SCs at 2 weeks post-TAM. All scale bars = 20 μm. Bu, bulge. Inf, Infundibulum. Dashed lines, HF-dermal border. See also Source Data.
Extended Data Fig. 5 Immunosuppression does not prevent bulge-SC loss in NFI-dKO mice.
All analyses in mice were done at 2 months post-NFI deletion. Dexamethasone (DEX) was administered continuously since gene deletion to evaluate the long-term effect of immunosuppression on bulge phenotypes. a, Immunofluorescence analysis of FOXP3+ regulatory T-cells (Tregs) around the HF bulge niche. K14 marks basal epithelial cells. b, Flow cytometry analysis of immune cell composition at 2 months post-NFI deletion. Mean and standard deviation are shown. n = 3 WT mice in PBS and DEX groups, n = 5 NFI-dKO mice in PBS group, n = 4 NFI-dKO mice in DEX group. P-values are from unpaired, two-tailed t-test. c, Immunofluorescence analysis of phosphorylated (active) NF-kB in the HF bulge. INTEGRIN β4 (β4) marks basal epithelial cells. d, Images of WT and NFI-dKO mice with or without DEX. Note DEX led to hair coat retention in NFI-dKO mice. e, Immunosuppression fails to rescue ectopic epidermal differentiation (K10) in the NFI-dKO bulge. f, Analysis of human scalp biopsies. Immunohistochemistry shows reduced expression of NFIB and NFIX in scarring alopecia patients compared to normal human scalp skin. g, Immunohistochemical analysis of human scalp biopsies using anti-K14 antibody, a marker of outer root sheath (ORS, progenitor) cells, where NFIB and NFIX are normally expressed. All scale bars = 20μm, unless otherwise indicated. Bu, bulge. Inf, Infundibulum. Dashed lines, HF-dermal border. For a, c- g, at least three biological replicates were used; representative images are shown. See also Source Data.
Extended Data Fig. 6 Bulk Transcriptome Analysis of NFI-dKO bulge-SCs.
a, b, Volcano plots showing differential gene expression of WT vs. NFI-dKO (a) or WT vs. Nfix cKO (b) bulge-SCs. Note that Nfix ablation on its own has little effect on bulge-SC transcriptomes. n=23491 genes were analyzed/genotype. c, Overlap of WT vs. NFI-dKO gene expression changes and NFIB ChIP-occupancy to identify transcriptional targets sensitive to NFIB levels. All transcriptome analyses were performed on 2 mice/genotype. Statistical analysis was performed using unpaired, two-tailed t-test and corrected using the Benjamini and Hochberg method.
Extended Data Fig. 7 Single Cell Transcriptome Analysis of Telogen Skin Epidermis.
a, FACS-purification of 2nd telogen skin progenitors from Sox9-CreER/Nfixfl/fl/R26-YFP (non-phenotypic) vs. NFI-dKO mice. Hair follicles were YFP+ and INTEGRIN α6+, while epidermal SCs (EpdSCs) were YFPneg, INTEGRIN α6+ and SCA1+. Representative plots for three biological replicates. b, Validation of FACS-purification strategy for single cell RNA-seq analysis. tSNE plot showing low Sox9 expression in the YFPneg cluster (EpdSC), whereas all YFP+ populations are Sox9+ (HF). n=2 mice per group. c, Correlation plots of single-cell RNA-seq libraries shows minimal batch to batch variation. n=2 mice per group. Correlation coefficients were calculated by Pearson’s method. d, tSNE plots showing expression of known epidermal lineage markers to determine the identity of individual clusters. n=2 mice per group. e, tSNE plot showing the unique cluster in NFI-dKO mice is Cd34neg. n=2 mice per group.
Extended Data Fig. 8 NFI-TFs are Required to Maintain Bulge-SC Identity.
a, Replicate analysis of ATAC-seq experiments show correlation coefficients of >0.94, indicating good reproducibility. Correlation coefficients were calculated by Pearson’s method. b, Reduction of chromatin accessibility at bulge-SC TF-bound loci upon loss of NFI-TFs. Note loci co-occupied by NFIB and bulge-SC TFs show a greater decrease of chromatin accessibility. In boxplots, the median (line), first and third quartiles (box), and whiskers (highest and lowest values) are shown. TF bindings sites are based on prior in vivo ChIP-seq on bulge-SCs13,16,17,18,20,72. ATAC-seq (this study) was used to determine differential accessibility at bulge-SC TF ChIP-bound loci. Statistics was analyzed using unpaired, two-tailed t-test. Number of peaks analyzed: SOX9: n=1813 (NFIB-bound), n=1175 (no NFIB binding); LHX2: n=1363 (NFIB-bound), n=1453 (no NFIB binding); NFATc1: n=1503 (NFIB-bound), n=4199 (no NFIB binding); pSMAD1: n=1547 (NFIB-bound), n=1778 (no NFIB binding); TCF3: n=7840 (NFIB-bound), n=7668 (no NFIB binding); TCF4: n=5346 (NFIB-bound), n=4371 (no NFIB binding); TLE: n=2901 (NFIB-bound), n=2840 (no NFIB-binding); bulge SE H3K27ac: n=970 (NFIB-bound), and n=2017 (no NFIB binding). c, Comparison of bulge-SC-TF ChIP-peaks reveals high co-occupancy with NFIB. d, Differential chromatin accessibility at NFIB ChIP-occupied super-enhancers in WT vs. Nfib/Nfix-dKO bulge-SCs (measured by ATAC-seq). e, Immunofluorescence analysis shows gradual reduction in bulge-SC marker LHX2 over time. Scale bars = 20 μm. Bu, bulge. Dashed lines, HF-dermal border. Representative images for three biological replicates. f, Replicate analysis of 2 independent ChIP-seq experiments show correlation coefficients (r) of >0.94, indicating good reproducibility. Number of peaks analyzed: H3K4me1: n=128538 (WT), n=119965 (NFI-dKO); H3K27ac: n=65493 (WT), and n=82522 (NFI-dKO). Correlation coefficients were calculated by Pearson’s method. g, Heatmap of H3K4me1, H3K27ac and H3K27me3 ChIP-seq read densities centered on NFIB-bound peaks, depicting how they change with NFI status in bulge-SC chromatin. Note that Nfib/Nfix ablation associates with reduced H3K4me1 and H2K27ac but not H3K27me3 at NFIB-bound loci. See also Source Data.
Extended Data Fig. 9 Model of NFIB and NFIX Function in the HF SC Niche.
Although our studies focused on using Sox9-CreER mice to ablate NFI proteins in the HF, we also show that LV-CreER ablation of NFI proteins in the epidermis does not affect its SCs or its differentiation program. Rather, NFIB and NFIX act on the bulge-SCs and without them, a primary scarring alopecia phenotype is generated. At the chromatin level, NFI proteins act in the bulge-SC niche to maintain chromatin accessibility of bulge-SC super-enhancers while repressing epidermal enhancers. When NFI proteins are absent, many bulge-SC super-enhancers are silenced while some epidermal enhancers become ectopically activated, leading to a lineage infidelity state.
Supplementary information
Supplementary Tables 1–8
Supplementary Table 1. Histopathological assessment of NFI dKO skin and comparison with human PCA. Supplementary Table 2. Human patient information and clinical diagnosis of primary scarring in patients with alopecia. Supplementary Table 3. List of differentially expressed genes (fold-change > 2; FDR < 0.01) in NFI dKO versus wild-type bulge SCs from bulk transcriptome analysis together with NFIB ChIP-Seq occupancy. n = 2 mice per group were analysed. P values were calculated from unpaired, two-tailed t-test and corrected using the Benjamini–Hochberg method. Supplementary Table 4. List of signature genes of cell clusters (fold-change > 2 and FDR < 0.1 versus other cells) in single-cell transcriptome analysis of skin progenitors. n = 2 mice per group were analysed. P values were calculated by unpaired, two-tailed t-test and corrected using the Benjamini–Hochberg method. Supplementary Table 5. List of ATAC peaks that are differentially accessible in wild-type and NFI dKO bulge SCs. Supplementary Table 6. List of super-enhancers in wild-type and NFI dKO bulge SCs. Supplementary Table 7. List of differentially expressed genes (fold-change > 2; FDR < 0.1) of the unique cell population in NFI dKO versus wild-type bulge SCs, from single-cell transcriptome analysis. n = 2 mice per group were analysed. P values were calculated by unpaired, two-tailed t-test and corrected using the Benjamini–Hochberg method. Supplementary Table 8. List of antibodies used in this study.
Source data
Source Data Fig. 1
Statistical source data
Source Data Fig. 2
Statistical source data
Source Data Fig. 3
Statistical source data
Source Data Fig. 6
Statistical source data
Source Data Extended Data Fig. 1
Statistical source data
Source Data Extended Data Fig. 2
Statistical source data
Source Data Extended Data Fig. 4
Statistical source data
Source Data Extended Data Fig. 5
Statistical source data
Source Data Extended Data Fig. 8
Statistical source data
Rights and permissions
About this article

Cite this article
Adam, R.C., Yang, H., Ge, Y. et al. NFI transcription factors provide chromatin access to maintain stem cell identity while preventing unintended lineage fate choices. Nat Cell Biol 22, 640–650 (2020). https://doi.org/10.1038/s41556-020-0513-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41556-020-0513-0
This article is cited by
-
Distinct regulatory networks control toxin gene expression in elapid and viperid snakes
BMC Genomics (2024)
-
DisP-seq reveals the genome-wide functional organization of DNA-associated disordered proteins
Nature Biotechnology (2024)
-
Deciphering the molecular mechanisms of stem cell dynamics in hair follicle regeneration
Experimental & Molecular Medicine (2024)
-
Downregulation of miR-182-5p by NFIB promotes NAD+ salvage synthesis in colorectal cancer by targeting NAMPT
Communications Biology (2023)
-
De novo mutations in the BMP signaling pathway in lambdoid craniosynostosis
Human Genetics (2023)
