
Abstract
Lateral-flow assays (LFAs) are rapid and inexpensive, yet they are nearly 1,000-fold less sensitive than laboratory-based tests. Here we show that plasmonically active antibody-conjugated fluorescent gold nanorods can make conventional LFAs ultrasensitive. With sample-to-answer times within 20 min, plasmonically enhanced LFAs read out via a standard benchtop fluorescence scanner attained about 30-fold improvements in dynamic range and in detection limits over 4-h-long gold-standard enzyme-linked immunosorbent assays, and achieved 95% clinical sensitivity and 100% specificity for antibodies in plasma and for antigens in nasopharyngeal swabs from individuals with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Comparable improvements in the assay’s performance can also be achieved via an inexpensive portable scanner, as we show for the detection of interleukin-6 in human serum samples and of the nucleocapsid protein of SARS-CoV-2 in nasopharyngeal samples. Plasmonically enhanced LFAs outperform standard laboratory tests in sensitivity, speed, dynamic range, ease of use and cost, and may provide advantages in point-of-care diagnostics.
Similar content being viewed by others


Main
Lateral-flow assays (LFAs) are among the simplest, fastest and cheapest point-of-care (POC) diagnostic methods, and offer broad potential for population-level screening for disease1,2. Although numerous LFAs for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) antibodies3,4,5 and antigens6,7 have been introduced, none has sensitivity and quantitation comparable to laboratory-based diagnostics such as real-time PCR with reverse transcription (RT–PCR) and enzyme-linked immunosorbent assay (ELISA)8,9,10. In general, conventional colorimetric LFAs are ~1,000-fold less sensitive than these standard laboratory tests11,12, and diagnosis using LFAs requires an additional confirmatory laboratory-based test to correctly establish negative results. Colorimetric LFAs are often inadequate for quantitative read-outs, owing to limited changes in colour with respect to the variation of the concentration of the target analyte13.
The coronavirus disease 2019 (COVID-19) pandemic highlighted the need for improved LFAs for precise and rapid clinical diagnoses, mass screenings and epidemiological studies14,15. RT–PCR16,17 and direct antigen tests18,19 have been the mainstay for the diagnosis of COVID-19, and serological assays are important for the determination of infection stage and vaccine efficacy and for epidemiological studies3,20,21. These diagnostic assays are available only in qualified microbiology laboratories and remain expert dependent, labour intensive and time intensive. These limitations have precluded the millions of tests per day that are needed during epidemiological surges22,23. Therefore, a critical need exists for diagnostic and screening tools that are not only as accurate as laboratory-based assays but also rapid, easy-to-use, inexpensive, readily available (for home-based and POC use) and scalable for rapid population-level screening.
Efforts to improve the bioanalytical performance of LFAs have included the use of fluorescent molecules or quantum dots as reporter elements24,25. Although fluorescent reporters improve quantification, their relatively weak signal intensity limits their sensitivity and POC diagnostic utility, and their low light absorption compared with conventional colloidal gold nanoparticles (AuNPs)26 precludes the direct visual detection that conventional LFAs allow. Moreover, they require the use of LFA readers with highly sensitive detectors or powerful excitation light sources. These considerations limit the utility of fluorescent LFAs in mass screening and resource-limited settings10.
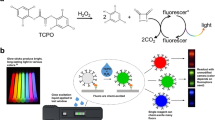
We envision a ‘bimodal’ LFA in which an initial screening can be performed with a visual test and subsequent quantitative testing can be performed when needed on the same LFA strip using a fluorescence reader. To achieve this, we used an ultrabright fluorescent nanoscale construct that we have recently introduced27, called plasmonic fluor, as a bimodal colorimetric and fluorescent reporter in LFAs (Fig. 1a). These nanoscale constructs harness plasmon-enhanced fluorescence28,29,30,31,32 to achieve nearly 7,000-fold brighter fluorescence signal compared with conventional molecular fluorophores. We conjugated plasmonic fluors with detection antibodies and used them to enable rapid and ultrasensitive colorimetric and fluorescent detection of analytes, using human interleukin-6 (IL-6) (limit of detection (LOD), 93 fg ml−1), SARS-CoV-2 S1 (subunit of the spike protein) antibodies (LOD, 185 pg ml−1) and SARS-CoV-2 antigen (nucleocapsid (N)) protein (LOD, 212 pg ml−1). We validated the clinical efficacy of the plasmonic-fluor-based LFAs (p-LFAs) by testing plasma, serum and nasopharyngeal (NP) swab samples for the detection of SARS-CoV-2 S1 antibodies, IL-6 and SARS-CoV-2 antigen, respectively, and achieved high clinical specificity and sensitivity. We also demonstrate the quantitative ability of p-LFA using a portable self-designed scanner compatible with plasmonic fluor to demonstrate its versatile POC application. Substantially, this technology can be readily deployed as an alternative to a laboratory-based test for the diagnosis of clinically relevant pathogenic infections.

a, Schematic illustration of plasmonic fluor, used as a bimodal nanolabel (colorimetric + fluorescent) in LFAs, comprising AuNR as plasmonic core, polymer layer as spacer, molecular fluorophores (800 CW) and biotin as recognition element. b,c, Transmission electron microscopy image of AuNPs (b) and plasmonic fluors (c). d, Mean grey values obtained from nitrocellulose membrane drop-casted with different concentrations of AuNPs. Inset: the 8-bit ImageJ-processed image of the nitrocellulose membrane. e,f, Fluorescence intensities obtained from nitrocellulose membrane drop-casted with different concentrations of plasmonic fluors (e) and molecular fluorophores (f). Insets: the corresponding fluorescence image of the nitrocellulose membrane. g, Mean grey values obtained from nitrocellulose membranes, with biotinylated BSA used as capture ligand at test sites, after exposure to different concentrations of streptavidin-conjugated AuNPs. Inset: schematic illustration of streptavidin-conjugated AuNPs. h, Fluorescence intensities obtained from nitrocellulose membranes, with biotinylated BSA as recognition elements at test sites, after exposure to different concentrations of streptavidin-conjugated plasmonic fluors. Inset: schematic illustration of streptavidin-conjugated plasmonic fluors. Purple arrows indicate the direction of flow of the nanoconjugates. Data are mean ± s.d.; n = 4 repeated tests. Schematics were created with BioRender.com.
Results and discussion
Plasmonic fluor increases the sensitivity of LFAs
Plasmonic fluors were first applied to overcome three fundamental limitations of the 30–40 nm AuNPs used as conventional colorimetric labels in LFAs. AuNPs have low capture rate (<5%), low signal-to-background ratio and thus relatively low sensitivity33,34. Even with the use of 100 nm AuNPs, shown recently to improve LFA sensitivity35, these problems persist. Because of these three limitations, colour changes in AuNP-based LFAs are limited to qualitative analysis or simply a binary output, indicating the presence or absence of the target analyte.
To assess whether plasmonic fluors (length 98 ± 8.7 nm, diameter 29.2 ± 3.1 nm) could overcome these limitations, we compared their performance with AuNPs (diameter 104 ± 13.4 nm) on a nitrocellulose membrane. The localized surface plasmon resonance wavelength of plasmonic fluors (and the gold nanorod (AuNR) core) was tuned to match the excitation and emission wavelengths of the molecular fluorophores27 by modifying their aspect ratios36,37, and the optimal dimensions of the nanostructures were chosen to maximize fluorescence enhancement, on the basis of our previous study38. We set out to determine the minimum number of AuNPs and plasmonic fluors required to produce a detectable visible or fluorescence signal. When serially diluted AuNPs (Fig. 1b and Supplementary Fig. 1) and plasmonic fluors (Fig. 1c and Supplementary Fig. 1) of known concentration were drop-casted onto nitrocellulose membrane, accumulations of ~106 AuNPs and plasmonic fluors were needed to produce a discernible visible signal (Fig. 1d and Supplementary Fig. 2). However, only ~102 plasmonic fluors were required to produce a detectable fluorescence signal (Fig. 1e and Supplementary Fig. 3). Furthermore, accumulations of ~0.6 × 106 molecular fluorophores (800 CW, the fluorescence unit of plasmonic fluors) were required to produce a detectable fluorescence signal (Fig. 1f), indicating ~6,000-fold lower concentration threshold for a detectable fluorescence signal with plasmonic fluors compared with molecular fluorophores.
Plasmonic fluors exhibited colorimetric signal nearly identical to that of AuNPs (Supplementary Fig. 4). The colorimetric signal enabled qualitative visual detection (by the naked eye), obviating the need for specialized read-out equipment at a relatively high concentration of the target analyte, while the fluorescence signal enabled ultrasensitive detection and quantification of low-abundance analytes. Thus, plasmonic fluors function as bimodal nanolabels (colorimetric + fluorescent) and offer ultrasensitive detection in a biological assay representative of LFAs.
Next, to compare the performance of plasmonic fluors and AuNPs in LFA format, we used the well-characterized biotin–streptavidin conjugate pairing, known to exhibit extremely high binding affinity39. Both AuNPs and plasmonic fluors were functionalized with streptavidin, and biotinylated bovine serum albumin (BSA) was used as a capture ligand. LFA strips were then subjected to different known concentrations of streptavidin-conjugated AuNPs and plasmonic fluors for 20 min (Supplementary Fig. 5). Nanolabels flow along the nitrocellulose membrane by capillary force and get captured by the capture ligand, leading to the accumulation of nanoparticles at the test spot. Accumulation of sufficient number of nanolabels converts the colour at the test site to red, indicating a positive result and the presence of the target analyte. The average greyscale intensity of the colorimetric signal at the test site with AuNPs and the fluorescence signal with plasmonic fluors monotonically increased with the concentration of the nanolabels (Fig. 1g,h). Notably, for both AuNPs and plasmonic fluors, ~107 nanoparticles are needed to produce a discernible visible signal; however, only ~103 plasmonic fluors are enough to produce a detectable fluorescence signal. The four orders of magnitude lower concentration threshold for a detectable signal with plasmonic fluors compared with AuNPs in the LFA format is consistent with the drop-casting approach discussed above. These results establish the fundamental basis that plasmonic fluors can serve as ultrabright nanolabels for ultrasensitive detection of target analytes in an LFA.
Bioanalytical parameters of p-LFA compared with LFA
We optimized the bioanalytical performance of LFA by tuning the concentration of the capture ligand and nanolabels. We used biotin–streptavidin as a model system. Both AuNPs and plasmonic fluors were biotin functionalized; streptavidin and biotinylated BSA were utilized as the target analyte and the capture ligand, respectively (Supplementary Fig. 6). It was observed that as the concentration of the capture ligand (that is, biotinylated BSA) increased, both mean greyscale intensity and fluorescence intensity of the test spot corresponding to AuNPs (Supplementary Fig. 7) and plasmonic fluors (Supplementary Fig. 8), respectively, increased. These results suggest that higher concentrations of the capture ligand result in better signal intensity. Furthermore, as the number of nanolabels increased, both mean greyscale intensity and fluorescence intensity of the test spot corresponding to AuNPs (Supplementary Fig. 9) and plasmonic fluors (Supplementary Fig. 10), respectively, increased, implying better signal intensity with higher number of nanolabels. However, in both cases, the background signal (signal from the LFA strip outside the capture spot) also increased with the number of nanolabels. Therefore, the optimum number of nanolabels for both AuNPs-based LFA and p-LFA was determined by subtracting the background signal from the test spot signal. As expected, the optimum number of plasmonic fluors (1.2 × 106) was four orders of magnitude lower than the optimum number of AuNPs (1.78 × 1010).
Next, we compared the bioanalytical parameters (LOD, limit of quantitation (LOQ) and dynamic range) of biotin–streptavidin AuNPs-based LFA and p-LFA. It is worth noting that colorimetric signal, obtained from the 8-bit ImageJ-processed images of LFA strips, from both AuNPs and plasmonic fluors exhibit similar LOD, suggesting no loss in visual detection capabilities in p-LFAs (Supplementary Fig. 11). The LOD (defined as mean + 3σ of the blank; σ is standard deviation) of colorimetric LFA was calculated to be 4.8 ng ml−1 (Supplementary Fig. 12, five-parameter logistic). In contrast, the fluorometric p-LFA enabled the detection down to 2.3 pg ml−1 (Supplementary Fig. 13, five-parameter logistic fit), representing ~2,000-fold improvement in the LOD. The LOQ (defined as mean + 10σ of the blank) of fluorometric p-LFA is ~2,500-fold better than the LOQ of colorimetric LFA. Furthermore, the fluorescent component of plasmonic fluor augmented the dynamic range of the assay by three orders of magnitude. Therefore, owing to the ultrabright fluorescence signal of the plasmonic fluors, the p-LFAs enable ultrasensitive detection of target analyte over a much broader range of analyte concentration.
p-LFA for quantitative detection of human IL-6
Cytokines are small (5–26 kDa) proteins involved in cell signalling and immuno-modulation and are critical indicators of health and disease40. Several diseases including cancer, sepsis, human immunodeficiency virus, chronic inflammation and auto-immune diseases are known to be associated with dysregulation of the immune system, leading to disruption of the subtle balance between pro-inflammatory and anti-inflammatory cytokines41,42. Pro-inflammatory cytokines include IL-1, IL-6, IL-12, tumour necrosis factor-α and interferon-γ, while the anti-inflammatory cytokines include transforming growth factor-β, IL-10 and IL-4. Rapid monitoring of the immune status by analysing serum cytokines and early diagnosis of these diseases is essential for prompt clinical intervention and for inhibiting disease progression. Although few LFAs for IL-6 detection have been introduced recently43,44, none provides sensitivity and quantitation comparable to gold-standard ELISA. Therefore, we used IL-6 as a model target analyte to investigate the applicability of our p-LFA.
Human IL-6 capture antibodies and sheep anti-immunoglobulin G (IgG) antibodies were immobilized on a nitrocellulose membrane to form test and control spots, respectively (Fig. 2a and Supplementary Fig. 14). The LOD of AuNP-based colorimetric LFA (Fig. 2b) and of molecular fluorophore-based LFA was calculated to be 166 pg ml−1 (Fig. 2c, five-parameter logistic fit) and 362 pg ml−1 (Supplementary Fig. 15), respectively. In contrast, the fluorometric p-LFA (Fig. 2d) enabled the detection down to 93 fg ml−1 (Fig. 2e, five-parameter logistic fit), which represents a 1,785-fold improvement in the LOD compared with conventional AuNP-based LFAs and at least an order of magnitude higher than the previously reported LFAs43,44,45,46. The LOQ of fluorometric p-LFA (298 fg ml−1) is 2,288-fold better than the LOQ of colorimetric LFA (682 pg ml−1). Furthermore, the plasmonic fluor improved the dynamic range of the LFA by nearly three orders of magnitude. The colorimetric signal from both AuNPs and p-LFA exhibited similar LODs, suggesting no loss in visual detection capabilities in p-LFAs (Fig. 2f,g and Supplementary Fig. 16). In addition, the fluorescence signal from the plasmonic fluors enabled ultrasensitive detection and quantitative analysis over a much broader range of analyte concentration (Fig. 2e,h).

a, Schematic illustration of IL-6 LFA strips comprising an IL-6 capture antibody test spot and a sheep IgG control spot. b,c, Schematic illustration of AuNP-based IL-6 LFA (b) and dose-dependent mean grey values (c), corresponding to different IL-6 concentrations, acquired from these AuNP-based LFAs. d,e Schematic illustration of IL-6 p-LFA (d) and dose-dependent fluorescence intensities of IL-6 p-LFA (e). f,g, Eight-bit, ImageJ-processed images of AuNP-based IL-6 LFAs (f) and IL-6 p-LFAs (g), depicting the visual read-out mode. h, Fluorescence images of the IL-6 p-LFA strips depicting the fluorescence read-out mode. i, RMC curves for ELISA, p-FLISA and p-LFA (see Supplementary Information for calculations). The dashed lines indicate RMC cut-offs at µ = 2 and µ = 5; intersections of dashed lines and RMC curves indicate the range of concentrations over which a specific quantitative performance of the assay is achieved. For IL-6 p-LFA, µ < 2 over a concentration range of 0.13–86.0 pg ml−1, suggesting that IL-6 p-LFA can distinguish signals corresponding to any two concentrations within that range that differ by at least 100% with at least 99% confidence. The relevant RMC parameters are listed in Supplementary Table 1. j, Stability of IL-6 p-LFA over 7 months, as evidenced by the error in concentration estimates of IL-6 concentration deduced using four different standard curves obtained over a span of 7 months. Schematics were created with BioRender.com.
We also compared the sensitivity and LOD of fluorometric p-LFA with gold-standard ELISA and plasmonic-fluor-linked immunosorbent assay (p-FLISA) implemented on a microtitre plate (Supplementary Fig. 17). The LOD of p-LFA is nearly 30-fold lower compared with conventional sandwich ELISA (2.9 pg ml−1) and only 5-fold inferior to that of p-FLISA (16.8 fg ml−1) (Supplementary Fig. 18). However, the sample-to-answer time for p-LFAs was 20 min, whereas ELISA and p-FLISA require 4 h.
To evaluate the ability of fluorometric p-LFA to accurately resolve changes in concentration of human IL-6, we quantified the resolution of molecular concentration (RMC), a recently introduced metric that indicates whether changes in analyte concentration can be discriminated with statistical significance47. This metric is complementary to LOD: whereas the low LOD represents the smallest analyte concentration that can be distinguished from the background, RMC represents the smallest fold change in concentration that can be discriminated with 99% certainty47. We compared the RMC of ELISA, p-FLISA and p-LFA for the resolution of twofold changes in concentration of human IL-6 (RMC parameter µ = 2, meaning a twofold change in concentration could be resolved). The RMC curves for p-LFA exhibited µ ≤ 2 over a concentration range of 0.13–86.1 pg ml−1, two orders of magnitude lower than that of ELISA (Fig. 2i) and nearly identical to that of p-FLISA. This suggests that IL-6 p-LFA can distinguish signals corresponding to two concentrations that differ by at least 100% within that range with at least 99% confidence. The RMC function and other bioanalytical parameters of p-FLISA and p-LFA, listed in Supplementary Table 1, indicate that the performance of the 20 min POC-compatible p-LFA is nearly identical to 4 h lab-based p-FLISA.
Next, to establish the stability of fluorometric p-LFA for quantitative detection without the use of standards, multiple IL-6 standard curves were acquired over a span of 7 months (Supplementary Fig. 19). All standard curves attained similar RMC (Supplementary Fig. 20) and bioanalytical parameters, suggesting excellent repeatability and reproducibility. Using these standard curves, IL-6 concentrations ranging from 1 pg ml−1 to 50 pg ml−1 were quantified with less than 20% deviation (Fig. 2j and Supplementary Fig. 21).
Overall, the POC assay showed performance comparable to that of the lab-based assay and showed the ability to accurately quantify the analyte concentration in a standard-free manner. This has not been reported previously with LFA technology and ascertains that p-LFAs overcome the long-standing limitations of LFAs—limited sensitivity, low accuracy and smaller analytical range compared with laboratory tests, and limited quantitation ability.
Ultrasensitive p-LFA for SARS-CoV-2 serology
To assess the potential for clinical translation of our p-LFA, we next optimized it for detection of SARS-CoV-2 antibodies. A pressing need persists for sensitive, rapid and POC serological assays for SARS-CoV-2, both for epidemiological studies and for studies on vaccine efficacy against SARS-CoV-23,20. Several LFAs3,4,48 and other assay methods49 use SARS-CoV-2 spike protein as the recognition element for detection of SARS-CoV-2 antibodies. Using p-LFA, our goal was to extend the sensitivity and LOD beyond the range possible with current assays and into the range of ELISA.
Recombinant SARS-CoV-2 S1 subunit of spike protein was immobilized at the test spot, and sheep IgG was used for control spot (Fig. 3a and Supplementary Fig. 22). We first determined the bioanalytical parameters of AuNP-based LFA (Fig. 3b) and p-LFA (Fig. 3d) for detection of SARS-CoV-2 S1 antibody. Using the colorimetric signal obtained from LFA strips, the LOD of AuNP-based LFA was determined to be ~1.05 µg ml−1 (Fig. 3c). In contrast, fluorometric p-LFA exhibited an LOD of 185 pg ml−1 (Fig. 3e, five-parameter logistic fit), which represents a nearly 5,675-fold improvement. Furthermore, as expected, the mean greyscale intensities obtained from both AuNP and p-LFA exhibited similar sensitivity, suggesting no compromise in the visual detection capabilities (Fig. 3f,g and Supplementary Fig. 23). However, the fluorescence signal from plasmonic fluors enabled ultrasensitive detection and quantitative analysis over a much broader (four orders of magnitude higher) range of analyte concentration (Fig. 3e,h). Fluorometric p-LFA showed 165-fold improvement in LOD compared with conventional sandwich ELISA (Fig. 3i) and comparable LOD to p-FLISA (Fig. 3j).

a, Schematic illustration of the SARS-CoV-2 S1 antibody LFA strips comprising recombinant SARS-CoV-2 S1 protein as capture element at the test spot and sheep IgG at the control spot. b,d, Schematic illustrations of AuNP-based SARS-CoV-2 S1 antibody LFA (b) and plasmonic-fluor SARS-CoV-2 S1 antibody LFA (d). c, Dose-dependent mean grey values, corresponding to different concentrations of SARS-CoV-2 S1 antibody, acquired from AuNP-based LFA. e, Dose-dependent SNR ratio of SARS-CoV-2 S1 antibody p-LFA performed in 20 min. f,g, Eight-bit ImageJ-processed images of AuNP-based SARS-CoV-2 S1 antibody LFA (f) and SARS-CoV-2 S1 antibody p-LFA (g), depicting the visual read-out mode. h, Fluorescence images of SARS-CoV-2 S1 antibody p-LFA strips, depicting the fluorescence read-out mode. i,j Dose-dependent optical densities and fluorescence intensities, corresponding to different SARS-CoV-2 S1 antibody concentrations, obtained by standard ELISA (i) and p-FLISA (j) implemented on a microtitre plate, performed in 4 h. Schematics were created with BioRender.com.
To assess the translational potential of fluorometric p-LFAs, we tested 79 plasma samples obtained from COVID-19-positive individuals and 48 archived de-identified serum or plasma samples which were collected pre-COVID-19 (March to October 2019)50 for the presence of SARS-CoV-2 S1 antibodies. All 127 plasma samples were diluted 500-fold and tested using fluorometric p-LFA. Out of 79 IgG-positive samples (tested positive by ELISA), 76 were tested positive (sample signal-to-noise ratio (SNR) ≥ blank SNR + 3σ of blank) with p-LFA, indicating 96.2% sensitivity. All pre-COVID-19 samples tested negative with LFA for SARS-CoV-2 S1 IgGs, indicating 100% specificity (Supplementary Table 2). Thus, the p-LFAs for detection of SARS-CoV-2 antibodies offers POC applicability with accuracy comparable to gold-standard ELISA and with potential applicability to vaccine efficacy and epidemiological studies.
p-LFA for SARS-CoV-2 antigen detection
Next, we evaluated the potential of p-LFAs to fill the critical need for a highly sensitive and specific POC SARS-CoV-2 antigen test. In serological testing of virus-specific immunoglobulins, the antibody responses to viral antigens are usually detected in the late stage of infection (7–14 days after virus exposure); therefore, serological antibody tests cannot achieve accurate screening of asymptomatic populations or early stages of infection51. Furthermore, RT–PCR, the current gold standard in diagnosing COVID-19, has proven highly successful in identifying individuals who have contracted the SARS-CoV-2 virus; however, they may fail to distinguish between infectious patients and non-infectious individuals and may yield false-positive results for months even after a patient has recovered from the disease52,53.
As antigens are expressed only when the virus is actively replicating, the antigen-based tests may have better correlation with infectiousness than RNA detection by RT–PCR. Current antigen detection tests for diagnosing COVID-19 are scalable and convenient but are limited by their low and wide-ranging accuracy54,55,56,57. LFAs for detection of SARS-CoV-2 antigens can be the most important tool in addressing the infection outbreaks owing to their ease of use, lower cost and better correlation with infectivity. Currently, several LFA-based antigen6,7,58 assays have been reported and are widely used, but none offers the optimal sensitivity59; thus, a negative result with such assays in a symptomatic patient requires a confirmatory RT–PCR test or frequent retesting. Therefore, there is an urgent need for a more sensitive POC antigen assay that would be just as reliable and accurate as the RT–PCR method.
p-LFA provided the accuracy and sensitivity needed for this in samples from patients who simultaneously had PCR tests performed. Our test focused on the detection of SARS-CoV-2 N protein. The test and the control spots on the LFA strips were prepared by immobilizing N protein capture antibodies and sheep IgG, respectively (Fig. 4a and Supplementary Fig. 24). Both colorimetric and fluorescence signals obtained from p-LFAs increased monotonically with an increase in the concentration of N protein standard (Fig. 4b,c). However, the LOD and LOQ of fluorometric p-LFA were calculated to be nearly 400-fold better than the colorimetric counterpart, ascertaining the importance of plasmonic fluors as ultrabright fluorescent nanolabels (Fig. 4d). Furthermore, fluorometric p-LFA showed 37-fold improvement in LOD compared with conventional sandwich ELISA and comparable LOD to p-FLISA (Fig. 4e and Supplementary Fig. 25).

a, Schematic illustration of the N protein p-LFA strips comprising N protein capture antibody as test spot and sheep IgG as control spot. b,c, Colorimetric (b) and fluorometric (c) read-out modes of p-LFA for N protein detection. d, Dose-dependent mean grey values, corresponding to different concentrations of N protein, acquired from colorimetric p-LFA (black) and dose-dependent SNR of N protein fluorometric p-LFA performed in 20 min (red). e, Dose-dependent optical densities and fluorescence intensities, corresponding to different N protein concentrations, obtained by standard ELISA (black) and p-FLISA (red) implemented on a microtitre plate, performed in 4 h. f, Comparison of fluorometric p-LFA (red) and commercial POC rapid antigen kit (BD Veritor) (black). g, N-protein SNR in PCR-positive NP swab samples (wild-type SARS-CoV-2) determined by colorimetric p-LFA (grey), fluorometric p-LFA (black) and BD Veritor. h, Comparison of colorimetric (grey) and fluorometric (black) p-LFA in terms of their ability to quantify N protein concentrations present in NP swab samples of 35 PCR-positive samples (19 wild-type SARS-CoV-2 and 16 Delta variant). ND, not detected. i, N protein SNR in NP swab samples tested negative for COVID-19 and positive for different seasonal coronaviruses and other respiratory viruses. Schematics were created with BioRender.com.
Next, to demonstrate the advantage of p-LFAs over an existing commercial US Food and Drug Administration (FDA) Emergency Use Authorization approved rapid, POC antigen testing method, we compared the analytical sensitivity of p-LFAs with the BD Veritor assay, which classified samples with concentrations below 50 ng ml−1 as ‘presumptive negative’ (Fig. 4f). This implies that the fluorometric p-LFA offers nearly 235-fold better analytical sensitivity compared with the commercial antigen test. p-LFA outperformed the FDA-approved BD Veritor antigen kit when analysing PCR-positive COVID-19 patient samples (wild-type SARS-CoV-2). BD Veritor antigen kit and colorimetric p-LFA correctly identified 8 out of 19 PCR-positive NP swab samples (analytical sensitivity, 42.1%), whereas fluorometric p-LFA correctly identified 18 out of 19 samples (analytical sensitivity, 94.7%) (Fig. 4g). Thirteen out of fourteen patient samples in the early stage of illness (<10 days since symptoms onset) were tested positive by fluorometric p-LFA (93% sensitivity), while only seven tested positive by BD Veritor (50% sensitivity) (Supplementary Table 3). Notably, the FDA-approved BD Veritor antigen kit can only be used in negative/positive format; however, fluorometric p-LFA enabled quantitative detection of the target analyte in patient samples (Supplementary Table 4). We also compared the quantitative performance of colorimetric and fluorometric p-LFA. While only 3 out of 19 samples were quantifiable (above LOQ) via colorimetric p-LFA, 18 out of 19 samples were quantifiable via fluorometric p-LFA (Fig. 4h).
To further substantiate the clinical translational potential of fluorometric p-LFAs for the detection of N protein, we tested 16 PCR-positive Delta B.1.617.2 variant (confirmed by gene sequencing) NP swab patient samples. Colorimetric p-LFA detected N protein in 7 out of 16 Delta variant positive samples, of which only 3 were quantifiable. However, fluorometric p-LFA detected N protein in all 16 samples, of which 15 were quantifiable (above LOQ) (Fig. 4h, Supplementary Fig. 26 and Supplementary Table 5). We also tested 17 PCR-positive Omicron BA.1 (confirmed by gene sequencing) samples and observed that fluorometric p-LFA returned positive results for 16 out of 17 Omicron (Supplementary Fig. 27 and Supplementary Table 6) variant samples. All Omicron-positive patient samples were collected within a short duration from the onset of symptoms (1–2 days), and all but one patient had very mild illness (Supplementary Table 7). These findings establish the efficacy of p-LFA for the early detection of N protein.
A total of 52 PCR-positive samples was tested. While only 15 out of 52 returned positive results with colorimetric p-LFA, indicating 28.8% clinical sensitivity, 50 out of 52 tested positive with fluorometric p-LFA (SNR > mean + 3σ), indicating 96.2% analytical sensitivity. The diagnostic sensitivity of p-LFA for samples with low viral load (cycle threshold (CT) values ≥25) was 91.7% (11 out of 12), and for samples with high viral load (CT values <25) was 97.5% (39 out of 40). This diagnostic sensitivity was substantially higher than those previously reported for rapid antigen/POC SARS-CoV-2 tests (~80% for samples with CT values <25 and 20–40% for samples with CT values ≥25)7,59,60,61.
Finally, to evaluate the specificity of p-LFA to SARS-CoV-2 N protein, we tested 19 PCR-negative NP swab samples. The negative NP swab samples comprised a mix of healthy samples and samples tested positive for seasonal coronaviruses and other respiratory viruses. All the 19 PCR-negative samples tested negative (SNR < mean of blank + 3σ) using p-LFA, suggesting 100% analytical specificity to COVID-19 N protein and no cross-reactivity with different seasonal coronaviruses and other viruses (Fig. 4i). These results substantiate that p-LFAs enable ultrasensitive, accurate, rapid, inexpensive and POC diagnosis of COVID-19 antigen and antibodies and can thus be a potential tool for rapid and quantitative diagnosis of symptomatic and asymptomatic infections.
POC p-LFA using an inexpensive, portable fluorescence scanner
Finally, to determine the applicability of this biodiagnostic technology in POC settings, we validated the performance of p-LFA using a portable, inexpensive fluorescence scanner. Note that the Stokes shift corresponding to plasmonic fluors is much smaller (~15 nm) than those corresponding to commonly used fluorescent nanoparticles such as quantum dots and europium nanoparticles (hundreds of nanometres)62,63. To the best of our knowledge, no inexpensive, portable fluorescence scanner compatible with plasmonic fluor is available commercially. Therefore, we developed an inexpensive portable fluorescence scanner for reading p-LFA using plasmonic fluors as nanolabels. The scanner prototype, with dimensions 25 × 25 × 19 cm (L × B × H), was built using routinely available, off-the-shelf optical components (see Methods for a detailed description; Fig. 5a and Supplementary Fig. 28). The total cost of the scanner is US$1,429, and the most expensive component is the laser, which costs US$919. It is worth noting that for all the measurements described in this work, the laser (excitation source) power was set to 1% of the maximum power to avoid fluorescence signal saturation. Thus, these components can be miniaturized and replaced with less expensive components for commercialization. Also, the portable scanner can run from a battery and thus can be immediately deployed in resource-limited settings.

a, Photograph of the portable fluorescence scanner. b, Schematic illustration of the LFA cassette used in the study and the workflow of p-LFA. S, T and C correspond to the sample pad, test line and control line, respectively. The blue arrow represents the direction of the fluorescence measurements made on the LFA cassette using the portable scanner. c, Representative positive (black) and negative (red) signals obtained using the portable scanner. d, Fluorescence intensities (black spheres) and area under the curve values (red spheres) obtained from LFA strips, drop-casted with different concentrations of plasmonic fluors, scanned using benchtop and portable scanners. e, Eight-bit ImageJ-processed image of the full-strip IL-6 colorimetric p-LFA depicting the visual read-out mode. f, Fluorescence image of the full-strip IL-6 fluorometric p-LFA depicting the fluorescence read-out mode. g, Dose-dependent signal of 15 min IL-6 fluorometric p-LFA measured by benchtop (black) and portable scanners (red). h, Linear regression plot of IL-6 concentration in serum samples determined by fluorometric p-LFA and measured using the benchtop and portable scanners. i, Linear regression plot of IL-6 concentration in serum samples determined by 4 h lab-based p-FLISA and a benchtop fluorescence scanner, compared with measurements made using 15 min fluorometric p-LFA and the portable scanner. j, Linear regression plot of N protein concentration in NP swab samples determined by fluorometric p-LFA and measured using the benchtop and portable scanners. Data are mean ± s.d.; n = 2 × 2 repeated tests. Schematic was created with BioRender.com.
We fabricated full-strip LFAs with separate sample and conjugate pads along with test membranes and absorbent pads. The assembled strip was embedded into a standard LFA cassette (Fig. 5b and Supplementary Fig. 29). Fluorescence measurements were performed by translating the cassette using travel actuator along the optical system of the portable scanner in the direction of the blue arrow in Fig. 5b. This produced a trace of pixel value (signal intensity), averaged from ten images, versus the travel length of the test membrane, taken in 100 µm increments (Fig. 5c). Consequently, a valid positive result has peaks at test and control lines, and a negative result has a peak only at the control line. A test without a peak at the control line is considered to be an invalid result (Supplementary Fig. 30).
First, to compare the performance of the portable scanner with the benchtop scanner, we determined the minimum number of nanolabels that could be detected by each scanner. When serially diluted plasmonic fluors (Fig. 5d and Supplementary Fig. 31) and 800 CW molecular fluorophores (Supplementary Figs. 32 and 33) of known concentration were drop-casted on the test membranes, accumulations of ~100 plasmonic fluors were needed to produce detectable fluorescence intensity (mean of blank + 3σ) when measured using the benchtop scanner, and ~200 plasmonic fluors were needed for the portable scanner (Fig. 5d). Accumulations of ~0.6 × 106 molecular fluorophores (not plasmonically enhanced) were needed to produce detectable fluorescence intensity when measured using either the benchtop or portable scanner. The data acquired by the portable scanner and the subsequent data processing methodology are discussed in detail in Supplementary Information (Supplementary Figs. 34 and 35). These observations indicate nearly identical performance of the benchtop and portable scanners in detecting the fluorescence signal from plasmonic fluors.
Next, to demonstrate the POC-compatible workflow of p-LFA and compare the performance of the portable and benchtop scanners, we used human IL-6 as a model analyte. Human IL-6 capture antibodies and sheep anti-IgG antibodies were printed on a nitrocellulose membrane to form test and control lines, respectively (Supplementary Fig. 36). The LOD of the colorimetric IL-6 p-LFA in full-strip format (Fig. 5e) was calculated to be ~526 pg ml−1 (Supplementary Fig. 37). In contrast, the fluorometric p-LFA (Fig. 5f and Supplementary Fig. 38) enabled the detection down to 813 fg ml−1 (Fig. 5g in black, five-parameter logistic fit), measured using the benchtop scanner. Notably, with the portable scanner, the IL-6 p-LFA exhibited similar LOD, 916 fg ml−1 (Fig. 5g in red, five-parameter logistic fit). The near-identical performance of benchtop and portable scanners was further confirmed by comparing the N protein dose–response curves (Extended Data Fig. 1 and Supplementary Figs. 39 and 40). Note that the LOD of the full-strip LFAs is higher compared with the half-strip format discussed above due to the shorter time (15 min versus 20 min) and smaller analyte volume (70 µl versus 100 µl) available for binding of the analytes to the capture antibody-conjugated nanolabels in the full-strip format.
Finally, to demonstrate the clinical translational potential and the possible POC application of p-LFA with the portable scanner, we tested 28 serum and 14 NP swab samples from COVID-19 PCR-positive individuals for detection of IL-6 (Extended Data Figs. 2 and 3) and N protein (Supplementary Figs. 41 and 42), respectively. These samples were tested by 15 min p-LFAs and measured using benchtop and portable scanners. Quantitative results from the benchtop and portable scanners exhibited excellent correlation with a Pearson’s r value of 0.97 for IL-6 (Fig. 5h) and 0.94 for N protein concentrations (Fig. 5j and Supplementary Table 8). Equally important, the IL-6 concentrations determined by 15 min p-LFA and measured by the portable scanner also exhibited excellent correlation with those determined by 4-h-long lab-based p-FLISA (Pearson’s r value of 0.91) (Fig. 5i and Supplementary Table 9).
This observation, along with the nearly identical bioanalytical parameters of the p-LFA standard curve generated using the benchtop and portable scanners for IL-6 and N protein, suggests that the sensitivity and quantitative detection ability of p-LFA is not compromised by the use of an inexpensive, portable fluorescence scanner. Results using this portable scanner were comparable to those obtained using the 4-h-long lab-based tests performed using the expensive, non-portable benchtop fluorescence scanner. These results highlight the simple workflow of p-LFA and its potential for biodiagnostics in POC settings.
In summary, we have shown that plasmonic fluors can serve as a bimodal (colorimetric + fluorescent) reporter element for overcoming the long-standing limitations of LFAs. Specifically, p-LFA overcomes the limited sensitivity, low accuracy, small dynamic range and limited quantitation ability of LFAs when compared with laboratory tests. Plasmonic fluors produced a discernible fluorescence signal at densities 10,000-fold lower than those needed in conventional colorimetric AuNPs. p-LFAs for various analytes (IL-6, SARS-CoV-2 S1 antibodies and SARS-CoV-2 antigens) exhibited ~1,000-fold improvement in bioanalytical parameters (LOD, LOQ and dynamic range) over conventional LFAs. p-LFAs offered standard-free quantitative detection with over 10-fold better sensitivity than that of gold-standard ELISA, with a much lower sample-to-answer time (20 min versus 4–6 h) and similar ability to resolve molecular concentration as lab-based tests. p-LFAs for the detection of COVID-19 antibodies and antigens present in plasma and NP swab samples achieved >95% sensitivity and 100% specificity, showing clinical applicability. The inexpensive and portable fluorescence scanner we developed and optimized for reading p-LFA was as effective as the benchtop scanner we used. When applied to human specimens of COVID-19-positive individuals, the concentrations of IL-6 and N protein measured for 15 min p-LFAs using the benchtop and portable scanners exhibited excellent correlation with each other and also with concentrations determined by lab-based 4 h p-FLISA. We believe that p-LFAs are highly attractive for realizing POC biodiagnostics that require accurate and quantitative detection of bioanalytes. The technology reported here can be readily adapted for the detection of other infectious pathogens and disease biomarkers and could complement or even replace laboratory-based tests for the diagnosis of pathogenic infections and other acute conditions.
Methods
Synthesis of plasmonic fluors
A plasmonic fluor consists of a plasmonically active core, a AuNR synthesized by seed-mediated method64, a polymer spacer layer, fluorophores and a universal biorecognition element (biotin). Plasmonic fluors were synthesized following a similar procedure described in our previous study27. The detailed stepwise procedure is discussed in Supplementary Information.
Synthesis of AuNPs
Citrate-stabilized AuNPs were synthesized using seed-mediated synthesis method and using citrate as reducing agent. Au seeds (~15 nm) were synthesized as described previously by ref. 65. Briefly, 20 ml of 0.25 mM of HAuCl4 (Sigma Aldrich, 520918) was brought to boil under vigorous stirring at 800 rpm. Immediately after the solution started boiling, 0.2 ml of 3% (w/v) sodium citrate (Sigma Aldrich, 1613859) aqueous solution was added and maintained under boiling condition until the solution colour changed to wine red, indicating the formation of Au seeds. Next, ~100 nm AuNPs were synthesized using hydroquinone (Sigma Aldrich, H9003) as reducing agent for reduction of ionic gold.
Materials characterization
Transmission electron microscopy images were obtained using a JEOL JEM-2100F field emission instrument. The extinction spectra of plasmonic nanostructures were obtained using a Shimadzu UV-1800 spectrophotometer. Fluorescence mappings were recorded using LI-COR Odyssey CLx imaging system. A digital camera (Sony cybershot DSC HX300) and imaging software ImageJ 1.53e were used to characterize mean grey intensities. SpectraMax iD3 (Molecular Devices) plate reader was used to measure the optical density in ELISA.
Functionalization of nanolabels
To functionalize nanolabels with streptavidin (Sigma Aldrich, SA101), 1 µl of 10 mg ml−1 of streptavidin (or BSA–biotin or detection antibody) was added to 1 ml OD1 of nanolabels and incubated for 1 h on a shaker at room temperature. To stabilize the particles, 1 µl of 10 mg ml−1 of BSA (Sigma Aldrich, A7030) was added to the solution and further incubated for 20 min. Unbound protein was removed by washing the solution 4 times with pH 10 nanopure water (1 µl NaOH in 10 ml water). Finally, nanolabels were redispersed in 1% BSA in 1× PBS solution for use in the LFAs. To functionalize nanolabels with antibodies (IL-6 and N protein detection antibody and anti-human IgG), a similar process was used.
LFA assembly and preparation procedures
Nitrocellulose test membrane and absorbent pads with adhesive backing material (GE Healthcare, FF120HP) were used for fabricating the LFA strips. The test membrane and absorbent pad was cut into 4-mm-wide strips using a paper trimmer. To prepare the LFA strip, biorecognition element (for example, capture antibody) solution was pipetted onto the test membrane and dried at room temperature for 30 min. Subsequently, the test membrane was blocked using 3% BSA in 1× PBS solution. Next, strips were washed with PBST (1× PBS and 0.5% Tween20 (Sigma Aldrich, P9416)), followed by drying at room temperature in a vacuum desiccator for 1 h. After drying, absorbent pads (GE Healthcare, CF5) were assembled onto the polystyrene adhesive backing next to the nitrocellulose test membrane. To ensure efficient transfer of the solution from the test membrane to the absorbent pad, we ensured an overlap of 1–2 mm between both strips. Experiments were performed by dipping LFAs into 96-well plates filled with 100 µl sample/standard solutions for 20 min. The visual signals of LFAs were obtained by a digital camera. The images were converted to 8-bit greyscale image using ImageJ. Mean grey values of the test spot were calculated by averaging the test spot greyscale intensities obtained from ImageJ. The fluorescence signals were obtained by averaging test dot fluorescence intensities obtained using LI-COR Odyssey CLx fluorescence scanner using the following scan parameters: laser power, ~L2; resolution, 21 µm; channel, 800 nm; height, 0 mm.
Optimization of LFA parameters
To determine the optimum concentration of biotinylated BSA on the test spot, different LFA strips with varying concentrations of biotinylated BSA (100 µg ml−1 to 5 mg ml−1) were prepared in duplicates. LFAs were then subjected to the same concentration of streptavidin (1,000 ng ml−1 for AuNP-LFA and 1 ng ml−1 for p-LFA) and biotinylated nanolabels. To determine the optimal concentration of the nanolabels, LFA strips with the same concentration of biotinylated BSA (5 mg ml−1) were prepared in duplicates. These LFA strips were then subjected to the same concentration of streptavidin (1,000 ng ml−1 for AuNPs and 1 ng ml−1 for plasmonic fluors) but different numbers of biotin-functionalized nanolabels (4.45 × 106 to 3.56 × 1010 for AuNPs and 1.2 × 104 to 6 × 106 for plasmonic fluors). The optimum number of nanolabels for colorimetric AuNPs-LFA and p-LFA, and fluorometric p-LFA was determined by subtracting the background signal from the test spot signal.
Biotin–streptavidin LFA
Test spots were formed by pipetting 0.5 µl of 5 mg ml−1 biotinylated BSA onto the nitrocellulose membrane. The LFA strips were assembled as described above. For AuNP-based and plasmonic-fluor-based biotin–streptavidin LFA, 1 µl biotinylated AuNPs and 1 µl biotinylated plasmonic fluors, respectively, were mixed with 99 µl of different concentrations of streptavidin standard solutions (0.1 pg ml−1 to 1,000 µg ml−1) in 96-well plates to allow the binding of streptavidin with the biotinylated nanolabels. LFA strips in duplicates were then exposed to the sample and standard solutions for 20 min.
Human IL-6 immunoassays
Human IL-6 DuoSet ELISA kit (R&D Systems, DY206) was utilized in the study. For AuNP-based IL-6 LFA, AuNPs were conjugated with IL-6 detection antibody for the test spot and with anti-sheep IgG (R&D Systems, BAF016) for the control spot. For p-LFA, plasmonic fluors were conjugated with IL-6 detection antibody for the test spot, and AuNPs were conjugated with anti-sheep IgG for the control spot, respectively. To prepare LFA strips for IL-6 immunoassay, 0.5 µl of 2 mg ml−1 IL-6 capture antibody and 0.5 µl of 2 mg ml−1 sheep IgG (R&D Systems, 5-001-A) was pipetted onto the nitrocellulose membrane at different spots to create test and control spot, respectively. Subsequently, similar steps mentioned above were followed for LFA preparation and assembly. For AuNP-based IL-6 LFA, 1 µl IL-6 detection antibody-conjugated AuNPs and 1 µl anti-sheep IgG-conjugated AuNPs for test and control spot, respectively, were mixed with 98 µl of different concentrations of human IL-6 standard solutions (64 fg ml−1 to 5 ng ml−1) in 96-well plates to allow the binding of the analyte with the detection antibody-conjugated nanolabels. LFA strips in duplicates were then exposed to the sample and standard solutions for 20 min. For IL-6 p-LFA, 1 µl IL-6 detection antibody-conjugated plasmonic fluors and 1 µl anti-sheep IgG-conjugated AuNPs were mixed with 98 µl human IL-6 standard solutions (1 fg ml−1 to 1 ng ml−1) in 96-well plates. Visual signals and the fluorescence signals were obtained according to the procedure described above.
Human IL-6 ELISA was carried out according to the procedure described in DuoSet ELISA kit manual and is discussed in detail in Supplementary Information. p-FLISA was performed by adopting a similar approach, except that the HRP-labelled streptavidin was replaced by streptavidin-functionalized plasmonic fluor. Instead of streptavidin–HRP, 100 µl streptavidin–plasmonic fluors (OD 1) was incubated for 30 min, and then the plate was washed 3 times with PBST. Both ELISA and p-FLISA were conducted in duplicates. The fluorescence signal was obtained by averaging the fluorescence intensities from the microtitre wells obtained using LI-COR Odyssey CLx with the following scan parameters: laser power, ~L2; resolution, 169 µm; channel, 800 nm; height, 4 mm.
LFA quantitation study
Four p-LFA IL-6 standard curves (1 fg ml−1 to 1 ng ml−1) were generated over a span of 6 months, and samples with varying IL-6 concentrations (0.5 pg ml−1 to 62.5 pg ml−1) were tested in duplicates in a standard-free manner. Their experimental concentrations were determined using each standard curve, and deviations from actual concentrations were calculated.
SARS-CoV-2 S1 antibody immunoassays
We pipetted 0.5 µl of 2 mg ml−1 recombinant SARS-CoV-2 S1 protein (R&D Systems, 10522-CV) and 0.5 µl of 2 mg ml−1 sheep IgG onto the nitrocellulose membrane as test and control spot, respectively. Subsequently, we followed the same steps described above to prepare the LFA strips. For detecting SARS-CoV-2 S1 antibodies, AuNP-LFA and p-LFA, AuNPs and plasmonic fluors were conjugated with biotinylated anti-human IgG (Rockland, 609-4617) for test spots, respectively. In both cases, AuNPs were conjugated with anti-sheep IgG for control spot. For AuNP-based SARS-CoV-2 S1 antibody LFA, 1 µl anti-human IgG-conjugated AuNPs and 1 µl anti-sheep IgG-conjugated AuNPs were mixed with different concentrations of standard solutions (16 pg ml−1 to 25 µg ml−1) in 96-well plates, before exposure to LFA strip for 20 min. For plasmonic-fluor-based SARS-CoV-2 S1 antibody LFA, 1 µl anti-human IgG-conjugated plasmonic fluors and 1 µl anti-sheep IgG-conjugated AuNPs were mixed with different concentrations of standard solutions (16 pg ml−1 to 1 µg ml−1) in 96-well plates, before exposure to LFA strip for 20 min. Plasma samples were diluted 500-fold in reagent diluent (1× PBS containing 3% BSA, 0.2 µm filtered) before use. All experiments were done in duplicates. Visual signals and the fluorescence signals were obtained by using the same procedure mentioned above.
SARS-CoV-2 S1 antibody ELISA was carried out according to the following procedure. Microtitre wells in duplicates were coated with 100 µl of 5 µg ml−1 (in 1× PBS) recombinant SARS-CoV-2 S1 protein via overnight incubation at room temperature. For blocking, 300 µl reagent diluent was added to the wells for a minimum of 1 h. Next, 100 µl serially diluted standard samples were incubated for 2 h, followed by incubation of 100 µl of 100 ng ml−1 biotinylated anti-human IgG for 2 h. Next, 100 µl of 500 ng ml−1 streptavidin-labelled HRP (Thermo Fisher Scientific, N100) was incubated for 20 min, followed by the addition of 100 µl substrate solution for 20 min. The reaction was stopped by addition of 50 µl of 2 N H2SO4 (R&D Systems, DY994) and immediately the optical density at 450 nm was measured using a microplate reader. p-FLISA was carried out by adopting a similar procedure, except that the HRP-labelled streptavidin was replaced by streptavidin functionalized-plasmonic fluor. Instead of HRP, 100 µl plasmonic fluors (OD 1) were incubated for 30 min, and then the plate was washed 3 times with PBST. The fluorescence signal was obtained by averaging the fluorescence intensities from the microtitre wells obtained using LI-COR Odyssey CLx.
SARS-CoV-2 antigen (N protein) immunoassays
We pipetted 0.5 µl of 2 mg ml−1 N protein capture antibodies (SinoBiologicals, 40143-MM08) and 0.5 µl of 2 mg ml−1 sheep IgG onto the nitrocellulose membrane as test and control spots, respectively. For N protein p-LFA, plasmonic fluors were conjugated with biotinylated N protein detection antibody (SinoBiologicals, 40143-R004) for the test spots. AuNPs conjugated with anti-sheep IgG were used for the control spot. Subsequently, similar steps mentioned above were followed to prepare and assemble the LFA strips. For plasmonic-fluor-based N protein LFA, 1 µl detection antibodies-conjugated plasmonic flours and 1 µl anti-sheep IgG-conjugated AuNPs were incubated with different concentrations of standard solution (12 pg ml−1 and 1 µg ml−1; SinoBiologicals, 40588-V08B) spiked in universal transport media (UTM) in 96-well plates before exposure to LFA strips for 20 min. p-LFAs were used for the detection of N protein present in patient NP swab samples. The NP swab samples were in UTM and were used without any dilution or processing. All experiments were performed in duplicates. The visual signals and the fluorescence signals were obtained using the same process described above.
N protein ELISA was carried out by first coating the microtitre wells in duplicates with 100 µl of 100 ng ml−1 N protein capture antibodies (in 1× PBS) via overnight incubation at room temperature. For blocking, 300 µl reagent diluent was added to the wells for a minimum of 1 h. Next, 100 µl serially diluted standard samples were incubated for 2 h, followed by incubation of 100 µl of 200 ng ml−1 biotinylated N protein detection antibody for 2 h. Next, 100 µl of 500 ng ml−1 streptavidin-labelled HRP (Thermo Fisher Scientific, N100) was incubated for 20 min, followed by the addition of 100 µl substrate solution for 20 min. The reaction was stopped by addition of 50 µl of 2 N H2SO4 (R&D Systems, DY994), and immediately the optical density at 450 nm was measured using a microplate reader. p-FLISA was carried out by adopting a similar procedure, except that the HRP-labelled streptavidin was replaced by streptavidin-functionalized plasmonic fluor. Instead of HRP, 100 µl plasmonic fluors (OD 1) were incubated for 30 min, and then the plate was washed 3 times with PBST. The fluorescence signal was obtained by averaging the fluorescence intensities from the microtitre wells obtained using LI-COR Odyssey CLx.
Commercial antigen test
BD Veritor kit, Veritor System, for rapid detection of SARS-CoV-2, was used to analyse the presence of N protein in the patient samples. BD Veritor System was used in conjunction with the BD Veritor Plus Analyzer. NP swabs were eluted in UTM and Aimes (ESwab) transport medium. Internal validation and the assay precision was conducted and deemed acceptable for testing on clinical samples by the Barnes Jewish Clinical Microbiology Laboratory.
Patient sample acquisition
The serum or plasma samples collected before COVID-19 breakout were collected under the study approved by the Human Research Protection Office at Washington University in St. Louis under HRPO 201102546. The clinical samples used in the study were acquired from the repository of saliva, serum, plasma and NP swab samples from individuals confirmed or suspected with COVID-19 disease, located at Washington University School of Medicine in St Louis, and from the Barnes Jewish Clinical Microbiology Laboratory; acquisition of samples was supported by: the Barnes-Jewish Hospital Foundation; the Siteman Cancer Center grant P30 CA091842 from the National Cancer Institute of the National Institutes of Health (NIH); and the Washington University Institute of Clinical and Translational Sciences grant UL1TR002345 from the National Center for Advancing Translational Sciences (NCATS) of the NIH. This repository was developed and is maintained by Jane O’Halloran, MD, PhD; Charles Goss, PhD; and Phillip Mudd, MD, PhD. Control NP swab samples from asymptomatic healthy volunteers were obtained with prior written consent. For evaluation of cross reactivity with seasonal coronaviruses, samples were obtained from adults at Barnes-Jewish Hospital who were tested positive with either of the four seasonal coronaviruses or respiratory diseases via clinically warranted NP samples tests. Washington University School of Medicine Human Research Protection Office approved the study. All clinical data pre-existed at the time of data collection. A prior waiver of consent was obtained for the clinical information and data on COVID-19 PCR results.
Preparation and assembly of full-strip LFAs
Full-strip p-LFA components include: NC membrane, FF80HP on polystyrene backing (catalogue number 10547020, from Whatman, Cytiva); sample pad, Fusion 5 (catalogue number 8151-9915, from Whatman, Cytiva); conjugate pad, Whatman Standard 14 (8133-2250, Cytiva); and absorption pad, CF5 (catalogue number 8115-2250, Cytiva). Sample and conjugate pads were subjected to the following pre-treatment process: sample pads were soaked in 5% BSA, 0.5% Tween 20 and 1× PBS and then dried in 37 °C oven for 2 h. Conjugate pads were soaked in 5% BSA, 10% sucrose, 0.5% Tween 20 and 1× PBS and then dried in 37 °C oven for 2 h. After pre-treatment, sample and conjugate pads were cut into strips of 15 mm × 25 mm and 13 mm × 25 mm dimensions, respectively. Absorption pads were used as received and were cut into 18 mm × 25 mm dimensions.
To prepare nanolabels for the test line, 1–3 µl of biotinylated SARS-CoV-2 N protein or human IL-6 detection antibody of 1 mg ml−1 concentration was added to 1 ml streptavidin functionalized plasmonic fluors of extinction 2. After 30 min incubation, 100 µl of 10% BSA in 1× PBS was added to this antibody-conjugated plasmonic-fluor solution. After another 30 min incubation, the conjugated nanolabel solution was centrifuged 3 times to remove unbound detection antibodies, and the subsequent solution was dispersed back to 2 mM sodium borate, pH 8.5 with 10% sucrose. For preparation of nanolabels for the control line, 1–5 µl biotinylated anti-goat IgG of 2 mg ml−1 concentration was added to 1 ml streptavidin plasmonic fluors of extinction 2. After 30 min incubation, 100 µl of 10% BSA in 1× PBS was added to this antibody–plasmonic fluors conjugate solution. After another 30 min incubation, the conjugated nanolabel solution was centrifuged 3 times and dispersed back to 2 mM sodium borate of pH 8.5 consisting of 10% sucrose.
Next, the nanolabels for test and control lines were mixed in 1:1 ratio. Thereafter, the resulting solution was sprayed on to the pre-treated conjugate pad. The nanolabel solution was air-jet sprayed with a dispense rate of 5 µl cm−1 using a reagent dispenser (XYZ Platform Dispenser HM3030, Kinbio). After being sprayed, conjugate pads were dried in 37 °C oven for 2 h. Next, the test membrane was prepared by printing the capture antibodies specific to the test and control lines. For the test line SARS-CoV-2 Ag and human IL-6 capture antibody of 1 mg ml−1 concentration and for control line goat IgG of 2 mg ml−1 concentration were simultaneously printed on FF80HP nitrocellulose test membrane at a dispense rate of 0.5 µl cm−1 and speed of 50 mm s−1 by a reagent dispenser (XYZ Platform Dispenser HM3030, Kinbio). Thereafter, the membranes were dried in 37 °C oven for 2 h.
Finally, the pre-treated sample pad, the conjugate pad after spraying of nanolabels and the membrane pad after printing of capture antibodies were assembled with a 2 mm overlap between each pad and cut to strips with a width of 3 mm using a strip cutter (Programmable Strip Cutter ZQ2002, Kinbio). For the schematic illustration of the design of p-LFA, refer to Supplementary Fig. 45.
Portable fluorescence scanner
An 80 mW 785 nm diode laser (Zlaser, Z80M18S3-F-785-pe) was used as an excitation source. The laser beam was attenuated with 1% neutral density filter and shaped into a 4-mm-wide line using the combination of the laser focus control and a 30 mm focal length cylinder plano-convex lens. Fluorescence was collected with a 30 mm focal length plano-convex lens (12.5 mm diameter) and passed through an 832/37 nm emission filter (Edmund Optics, 84-107). A 45 mm focal length achromatic doublet lens (Edmund Optics, 49-355) was used to form a ×1.5 magnified image of the lateral-flow strip on the sensor of the camera (ZWO ASI462MC). Fluorescence was measured at a 45° angle relative to excitation. Measurements from lateral-flow cassettes were carried out by translating the sample (using Actuonix L16-R 50 mm travel actuator) through the optical system at 1 mm s−1 while streaming the camera video. A video was made with 100 ms exposure (10 images per second). The average pixel value from each ten images was used for analysis and corresponded to one point in the trace produced by this instrument. A Raspberry Pi 4 single board computer was used for controlling all hardware components of the instrument.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and the Supplementary Information. All data generated in this study are available from figshare via the identifier https://doi.org/10.6084/m9.figshare.21556365.
Code availability
The code for the RMC µ and RMC parameter calculation and the instructions on how to use the code for Langmuir and five-parameter logistic fitting is available at https://github.com/seanwangsalad/PythonRMC. The code for processing the data from the portable scanner and the instructions on how to use the code is available at https://github.com/seanwangsalad/AreaUnderCurveForLFAReader.
References
Soh, J. H., Chan, H.-M. & Ying, J. Y. Strategies for developing sensitive and specific nanoparticle-based lateral flow assays as point-of-care diagnostic device. Nano Today 30, 100831 (2020).
Nguyen, V.-T., Song, S., Park, S. & Joo, C. Recent advances in high-sensitivity detection methods for paper-based lateral-flow assay. Biosens. Bioelectron. 152, 112015 (2020).
Li, Z. et al. Development and clinical application of a rapid IgM–IgG combined antibody test for SARS‐CoV‐2 infection diagnosis. J. Med. Virol. 92, 1518–1524 (2020).
Charlton, C. L. et al. Evaluation of six commercial mid-to high-volume antibody and six point-of-care lateral flow assays for detection of SARS-CoV-2 antibodies. J. Clin. Microbiol. 58, e01361-20 (2020).
Heggestad, J. T. et al. Multiplexed, quantitative serological profiling of COVID-19 from blood by a point-of-care test. Sci. Adv. 7, eabg4901 (2021).
Mertens, P. et al. Development and potential usefulness of the COVID-19 Ag Respi-Strip diagnostic assay in a pandemic context. Front. Med. 7, 225 (2020).
Porte, L. et al. Evaluation of a novel antigen-based rapid detection test for the diagnosis of SARS-CoV-2 in respiratory samples. Int. J. Infect. Dis. 99, 328–333 (2020).
Pickering, S. et al. Comparative assessment of multiple COVID-19 serological technologies supports continued evaluation of point-of-care lateral flow assays in hospital and community healthcare settings. PLoS Pathog. 16, e1008817 (2020).
Kontou, P. I., Braliou, G. G., Dimou, N. L., Nikolopoulos, G. & Bagos, P. G. Antibody tests in detecting SARS-CoV-2 infection: a meta-analysis. Diagnostics 10, 319 (2020).
Hu, J. et al. Colorimetric-fluorescent-magnetic nanosphere-based multimodal assay platform for Salmonella detection. Anal. Chem. 91, 1178–1184 (2018).
Posthuma-Trumpie, G. A., Korf, J. & van Amerongen, A. Lateral flow (immuno) assay: its strengths, weaknesses, opportunities and threats. A literature survey. Anal. Bioanal. Chem. 393, 569–582 (2009).
Hu, J. et al. Advances in paper-based point-of-care diagnostics. Biosens. Bioelectron. 54, 585–597 (2014).
Huang, L. et al. Hierarchical plasmonic-fluorescent labels for highly sensitive lateral flow immunoassay with flexible dual-modal switching. ACS Appl. Mater. Interfaces 12, 58149–58160 (2020).
García-Basteiro, A. L. et al. Monitoring the COVID-19 epidemic in the context of widespread local transmission. Lancet Respir. Med. 8, 440–442 (2020).
COVID-19 Weekly Epidemiological Update, 25 May 2021 (World Health Organization, 2021).
Pang, J. et al. Potential rapid diagnostics, vaccine and therapeutics for 2019 novel coronavirus (2019-nCoV): a systematic review. J. Clin. Med. 9, 623 (2020).
Yu, F. et al. Quantitative detection and viral load analysis of SARS-CoV-2 in infected patients. Clin. Infect. Dis. 71, 793–798 (2020).
Che, X.-y et al. Sensitive and specific monoclonal antibody-based capture enzyme immunoassay for detection of nucleocapsid antigen in sera from patients with severe acute respiratory syndrome. J. Clin. Microbiol. 42, 2629–2635 (2004).
Di, B. et al. Monoclonal antibody-based antigen capture enzyme-linked immunosorbent assay reveals high sensitivity of the nucleocapsid protein in acute-phase sera of severe acute respiratory syndrome patients. Clin. Vaccine Immunol. 12, 135–140 (2005).
Long, Q.-X. et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat. Med. 26, 845–848 (2020).
Peterhoff, D. et al. A highly specific and sensitive serological assay detects SARS-CoV-2 antibody levels in COVID-19 patients that correlate with neutralization. Infection 49, 75–82 (2021).
Tromberg, B. J. et al. Rapid scaling up of COVID-19 diagnostic testing in the United States—the NIH RADx initiative. N. Engl. J. Med. 383, 1071–1077 (2020).
Valera, E. et al. COVID-19 point-of-care diagnostics: present and future. ACS Nano 15, 7899–7906 (2021).
Hu, J. et al. Sensitive and quantitative detection of C-reaction protein based on immunofluorescent nanospheres coupled with lateral flow test strip. Anal. Chem. 88, 6577–6584 (2016).
Udugama, B. et al. Diagnosing COVID-19: the disease and tools for detection. ACS Nano 14, 3822–3835 (2020).
Jain, P. K., Lee, K. S., El-Sayed, I. H. & El-Sayed, M. A. Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: applications in biological imaging and biomedicine. J. Phys. Chem. B 110, 7238–7248 (2006).
Luan, J. et al. Ultrabright fluorescent nanoscale labels for the femtomolar detection of analytes with standard bioassays. Nat. Biomed. Eng. 4, 518–530 (2020).
Tam, F., Goodrich, G. P., Johnson, B. R. & Halas, N. J. Plasmonic enhancement of molecular fluorescence. Nano Lett. 7, 496–501 (2007).
Kinkhabwala, A. et al. Large single-molecule fluorescence enhancements produced by a bowtie nanoantenna. Nat. Photon. 3, 654–657 (2009).
Zhang, B., Kumar, R. B., Dai, H. & Feldman, B. J. A plasmonic chip for biomarker discovery and diagnosis of type 1 diabetes. Nat. Med. 20, 948–953 (2014).
Bardhan, R., Grady, N. K., Cole, J. R., Joshi, A. & Halas, N. J. Fluorescence enhancement by Au nanostructures: nanoshells and nanorods. ACS Nano 3, 744–752 (2009).
Rycenga, M. et al. Controlling the synthesis and assembly of silver nanostructures for plasmonic applications. Chem. Rev. 111, 3669–3712 (2011).
Moghadam, B. Y., Connelly, K. T. & Posner, J. D. Two orders of magnitude improvement in detection limit of lateral flow assays using isotachophoresis. Anal. Chem. 87, 1009–1017 (2015).
Mosley, G. L., Nguyen, P., Wu, B. M. & Kamei, D. T. Development of quantitative radioactive methodologies on paper to determine important lateral-flow immunoassay parameters. Lab Chip 16, 2871–2881 (2016).
Zhan, L. et al. The role of nanoparticle design in determining analytical performance of lateral flow immunoassays. Nano Lett. 17, 7207–7212 (2017).
Chen, H., Shao, L., Li, Q. & Wang, J. Gold nanorods and their plasmonic properties. Chem. Soc. Rev. 42, 2679–2724 (2013).
Dreaden, E. C., Alkilany, A. M., Huang, X., Murphy, C. J. & El-Sayed, M. A. The golden age: gold nanoparticles for biomedicine. Chem. Soc. Rev. 41, 2740–2779 (2012).
Liang, C. et al. Gold nanorod size-dependent fluorescence enhancement for ultrasensitive fluoroimmunoassays. ACS Appl. Mater. Interfaces 13, 11414–11423 (2021).
Weber, P. C., Ohlendorf, D. H., Wendoloski, J. & Salemme, F. Structural origins of high-affinity biotin binding to streptavidin. Science 243, 85–88 (1989).
Schett, G., Elewaut, D., McInnes, I. B., Dayer, J.-M. & Neurath, M. F. How cytokine networks fuel inflammation: toward a cytokine-based disease taxonomy. Nat. Med. 19, 822–824 (2013).
Coussens, L. M. & Werb, Z. Inflammation and cancer. Nature 420, 860–867 (2002).
McInnes, I. B. & Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 7, 429–442 (2007).
de Souza Sene, I. et al. A point of care lateral flow assay for rapid and colorimetric detection of interleukin 6 and perspectives in bedside diagnostics. J. Clin. Med. Res. 2, 1–16 (2020).
Huang, D. et al. Rapid and sensitive detection of interleukin-6 in serum via time-resolved lateral flow immunoassay. Anal. Biochem. 588, 113468 (2020).
Swanson, C. & D’Andrea, A. Lateral flow assay with near-infrared dye for multiplex detection. Clin. Chem. 59, 641–648 (2013).
Tang, J., Wu, L., Lin, J., Zhang, E. & Luo, Y. Development of quantum dot‐based fluorescence lateral flow immunoassay strip for rapid and quantitative detection of serum interleukin‐6. J. Clin. Lab. Anal. 35, e23752 (2021).
Wilson, B. D., Eisenstein, M. & Soh, H. T. Comparing assays via the resolution of molecular concentration. Nat. Biomed. Eng. 6, 227–231 (2022).
Huang, C., Wen, T., Shi, F.-J., Zeng, X.-Y. & Jiao, Y.-J. Rapid detection of IgM antibodies against the SARS-CoV-2 virus via colloidal gold nanoparticle-based lateral-flow assay. ACS Omega 5, 12550–12556 (2020).
Amanat, F. et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 26, 1033–1036 (2020).
Fischer, P. U. et al. Evaluation of commercial rapid lateral flow tests, alone or in combination, for SARS-CoV-2 antibody testing. Am. J. Trop. Med. Hyg. 105, 378–386 (2021).
Grant, B. D. et al. SARS-CoV-2 coronavirus nucleocapsid antigen-detecting half-strip lateral flow assay toward the development of point of care tests using commercially available reagents. Anal. Chem. 92, 11305–11309 (2020).
Mina, M. J., Parker, R. & Larremore, D. B. Rethinking COVID-19 test sensitivity—a strategy for containment. N. Engl. J. Med. 383, e120 (2020).
He, X. et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 26, 672–675 (2020).
Peeling, R. W., Olliaro, P. L., Boeras, D. I. & Fongwen, N. Scaling up COVID-19 rapid antigen tests: promises and challenges. Lancet Infect. Dis. 21, E290–E295 (2021).
La Scola, B. et al. Viral RNA load as determined by cell culture as a management tool for discharge of SARS-CoV-2 patients from infectious disease wards. Eur. J. Clin. Microbiol. Infect. Dis. 39, 1059–1061 (2020).
McKay, S. L. et al. Performance evaluation of serial SARS-CoV-2 rapid antigen testing during a nursing home outbreak. Ann. Intern. Med. 174, 945–951 (2021).
Wölfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020).
Krüger, L. J. et al. Accuracy and ease-of-use of seven point-of-care SARS-CoV-2 antigen-detecting tests: a multi-centre clinical evaluation. EBioMedicine 75, 103774 (2022).
Mattiuzzi, C., Henry, B. M. & Lippi, G. Making sense of rapid antigen testing in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) diagnostics. Diagnosis 8, 27–31 (2021).
Dinnes, J. et al. Rapid, point‐of‐care antigen and molecular‐based tests for diagnosis of SARS‐CoV‐2 infection. Cochrane Database Syst. Rev. 3, CD013705 (2021).
Liotti, F. M. et al. Performance of a novel diagnostic assay for rapid SARS-CoV-2 antigen detection in nasopharynx samples. Clin. Microbiol. Infect. 27, 487–488 (2020).
Syamchand, S. & Sony, G. Europium enabled luminescent nanoparticles for biomedical applications. J. Lumin. 165, 190–215 (2015).
Kundu, J., Ghosh, Y., Dennis, A. M., Htoon, H. & Hollingsworth, J. A. Giant nanocrystal quantum dots: stable down-conversion phosphors that exploit a large Stokes shift and efficient shell-to-core energy relaxation. Nano Lett. 12, 3031–3037 (2012).
Gupta, R. et al. Refreshable nanobiosensor based on organosilica encapsulation of biorecognition elements. ACS Appl. Mater. Interfaces 12, 5420–5428 (2020).
Frens, G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nat. Phys. Sci. 241, 20–22 (1973).
Acknowledgements
We acknowledge support from the National Science Foundation (CBET-2027145, CBET-2029105 and CMMI 1548571) and the National Cancer Institute-Innovative Molecular Analysis Technologies (R21CA236652 and R21CA236652-S1). Research reported in this publication was supported by the Washington University Institute of Clinical and Translational Sciences grant UL1TR002345 from the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH). We also thank Nano Research Facility (NRF) and Institute of Materials Science and Engineering (IMSE) at Washington University for providing access to electron microscopy facilities. Part of schematic illustrations depicted in Figs. 1a, 2a, 3a, 4a and 5b were created in BioRender.com. We thank H. Baldi for his help with BioRender images. We thank M. S. Diamond, A. Ellebedy, D. H. Fremont and S. P. J. Whelan of the Washington University School of Medicine and J. Luan of the Washington University McKelvey School of Engineering for helpful discussions. We thank G. J. Weil and P. U. Fischer, Washington University School of Medicine, for kindly providing pre-COVID-19 samples. The samples used in this study were obtained from the Washington University School of Medicine’s COVID-19 biorepository, which is supported by the Barnes-Jewish Hospital Foundation; the Siteman Cancer Center grant P30 CA091842 from the National Cancer Institute of the NIH; and the Washington University Institute of Clinical and Translational Sciences grant UL1TR002345 from NCATS of the NIH. This repository was developed and is maintained by J. O’Halloran, C. Goss and P. Mudd. The content is solely the responsibility of the authors and does not necessarily represent the view of the NIH.
Author information
Authors and Affiliations
Contributions
S.S. and R.G. conceived the project. R.G., S.S., I.G., S.G., G.A.S., P.S., B.A.P., J.J.M. and G.M.G. designed the experiments. R.G. synthesized the nanolabels and performed the experiments. R.G. and P.G. configured the LFA strips. S.W. and R.G. did the mathematical calculations for RMC and wrote the required code for RMC parameter calculations. A.M. developed the handheld/portable scanner. Q.J. configured the full-strip LFAs and required nanolabels. R.G. and J.J.M. performed SARS-CoV-2 antibody lateral-flow immunoassays. R.G. and Z.W. performed SARS-CoV-2 antibody immunoassays. R.G. and A.S. performed SARS-CoV-2 antigen immunoassays and analysed nasal swab samples. R.G. and S.W. performed the experiments with the handheld scanner and the subsequent data processing. R.G., S.S., I.G., S.G., G.A.S., B.A.P., J.J.M. and G.M.G. analysed the results. R.G., S.S. and G.M.G. wrote the paper. All authors reviewed and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
J.J.M. and S.S. are inventors on provisional patent related to plasmonic-fluor technology, and the technology has been licensed by the Office of Technology Management at Washington University in St. Louis to Auragent Bioscience LLC, which is developing plasmonic-fluor products. J.J.M. and S.S. are co-founders and shareholders of Auragent Bioscience LLC. J.J.M. and S.S. along with Washington University may have financial gain through Auragent Bioscience LLC through this licensing agreement. A.M., Q.J. and A.S. currently work with Auragent Bioscience LLC. These potential conflicts of interest have been disclosed and are being managed by Washington University in St. Louis. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks David Walt and Daming Wang for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Benchtop vs portable scanner N protein p-LFA.
(a) Dose-dependent fluorescence images of the nitrocellulose membrane corresponding to different concentrations of N protein solutions acquired from plasmonic-fluor-based N protein LFA and measured by (b) benchtop scanner and (c) portable scanner.
Extended Data Fig. 2 Schematic illustration of the full strip IL-6 p-LFA.
Schematic illustration of the full strip IL-6 p-LFA employed for the quantitative detection of IL-6 in the serum of COVID-19 positive (PCR confirmed) individuals.
Extended Data Fig. 3 Representative examples of portable scanner data.
Representative examples of data processing conducted on data acquired by portable scanner from IL-6 p-LFA strips exposed to the (a, b) serum sample of the PCR-positive individuals and (c, d) blank for quantitative analysis of IL-6 concentration.
Supplementary information
Supplementary Information
Supplementary methods, figures, tables and references.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gupta, R., Gupta, P., Wang, S. et al. Ultrasensitive lateral-flow assays via plasmonically active antibody-conjugated fluorescent nanoparticles. Nat. Biomed. Eng 7, 1556–1570 (2023). https://doi.org/10.1038/s41551-022-01001-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-022-01001-1
This article is cited by
-
The development of a novel zeolite-based assay for efficient and deep plasma proteomic profiling
Journal of Nanobiotechnology (2024)
-
Afterglow Nanoprobes for In-vitro Background-free Biomarker Analysis
Chemical Research in Chinese Universities (2024)
-
Biosensors based on potent miniprotein binder for sensitive testing of SARS-CoV‑2 variants of concern
Microchimica Acta (2024)
-
Amplified fluorogenic immunoassay for early diagnosis and monitoring of Alzheimer’s disease from tear fluid
Nature Communications (2023)
